

Abstract
Increasing evidence supports an important role for the brain’s reward circuitry in controlling mood under normal conditions and contributing importantly to the pathophysiology and symptomatology of a range of mood disorders, such as depression. Here we focus on the nucleus accumbens (NAc), a critical component of the brain’s reward circuitry, in depression and other stress-related disorders. The prominence of anhedonia, reduced motivation, and decreased energy level in most individuals with depression supports the involvement of the NAc in these conditions. We concentrate on several transcription factors (CREB, ΔFosB, SRF, NFκB, and β-catenin), which are altered in the NAc in rodent depression models—and in some cases in the NAc of depressed humans, and which produce robust depression- or antidepressant-like effects when manipulated in the NAc in animal models. These studies of the NAc have established novel approaches toward modeling key symptoms of depression in animals and could enable the development of antidepressant medications with fundamentally new mechanisms of action.
1. INTRODUCTION
Depression and related mood disorders are among the world’s greatest public health problems. While there are many effective treatments of depression, roughly half of affected individuals are inadequately treated by available medications and psychotherapeutic approaches (Trivedi & Daly, 2008). In addition, virtually all existing antidepressant medications, which act initially on the brain’s serotonergic or noradrenergic systems, are based on serendipitous discoveries made more than a half-century ago (Berton & Nestler, 2006). Despite tremendous effort, the field has not yet succeeded in developing fundamentally new antidepressants with distinct mechanisms of action. One reason for this lack of progress is that we still lack a comprehensive understanding of the neural circuitry that malfunctions in depression as well as of the molecular pathology that drives abnormal circuit function. A stark reminder of this fact is that, if we had an opportunity to biopsy the brains of patients with depression, it is not at all clear which brain regions should be biopsied. The likelihood that depression comprises numerous, distinct disease states, and the lack of any clear distinction between depression and other stress-related disorders such as posttraumatic stress disorder and anxiety disorders, also have contributed to our lack of progress. In fact, this heterogeneity of clinical syndromes raises the possibility that different subtypes of depression and related conditions may be mediated by molecular pathology localized to different brain areas, which might be responsive to very different types of treatments.
Earlier work on depression focused on the hippocampus, frontal regions of cerebral cortex, and amygdala, among other regions. This impressive body of work is reviewed elsewhere (Duman & Monteggia, 2006; Miller & Hen, 2014; Nestler et al., 2002; Turner, Watson, & Akil, 2012). In more recent years, we and other groups have been interested in a role for the brain’s reward regions in also contributing to depression and antidepressant treatment. Studies from the drug addiction field have identified the nucleus accumbens (NAc), part of the ventral striatum, and its dopaminergic inputs from the ventral tegmental area (VTA) of the mid-brain, as the one of the most important anatomical substrates for drug reward as well as for natural rewards, such as food, sex, and social interactions (Koob & Le Moal, 2001; Wise, 1998). A striking observation related to brain reward is the extent to which abnormalities in reward and motivation are seen in depression and related disorders. For example, most depressed patients prominently exhibit a reduced ability to experience pleasure (anhedonia) and loss of motivation, as well as abnormalities in several neurovegetative functions such as appetite, sleep, energy level, and circadian rhythms (American Psychiatric Association, 2013).
Of course, these various brain areas cannot be thought of as distinct, since they function in highly overlapping and interacting circuits (Fig. 1). For example, the VTA and NAc receive strong glutamatergic inputs from several frontal cortical regions, hippocampus, and amygdala (see Everitt & Wolf, 2002; Hyman, Malenka, & Nestler, 2006; Kalivas, 2004). All of these regions, in turn, receive innervation from VTA dopamine neurons, where dopaminergic transmission has been shown to profoundly affect the functioning of these regions in electrophysiological and behavioral paradigms (e.g., see Arnsten, Wang, & Paspalas, 2012; Pezze & Feldon, 2004; Wittmann et al., 2005).
Figure 1. The neural circuitry of mood.
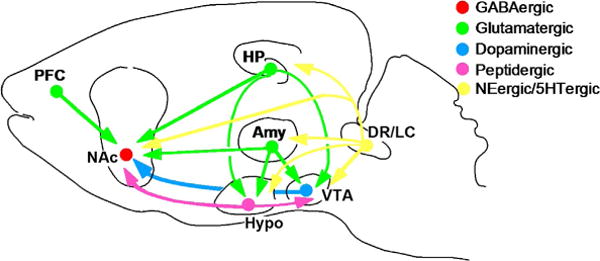
The figure shows a highly simplified summary of a series of neural circuits in the brain that are believed to contribute to the regulation of mood. While most research in the depression field until recently has focused on hippo-campus (HP) and cerebral cortex (e.g., prefrontal cortex or PFC), there is the increasing realization that several subcortical structures implicated in reward, fear, and motivation are also critically involved. These include the nucleus accumbens (NAc), amygdala (Amy), and hypothalamus (Hypo). The figure shows only a subset of the many known interconnections among these various brain regions. The figure also shows the innervation of several of these brain regions by monoaminergic neurons. The ventral teg-mental area (VTA) provides dopaminergic input to the NAc; inputs to most of the other brain areas are not shown in the figure. Norepinephrine (NE, from the locus coeruleus or LC) and serotonin (5HT from the dorsal raphe and other raphe nuclei) innervate all of the regions shown in the figure. In addition, strong connections between the hypothalamus and VTA–NAc pathway have been established in recent years. From Nestler and Carlezon (2006) with permission.
The objective of this review is to summarize the growing evidence for a role of the NAc in the regulation of mood and motivation under normal conditions, and in mediating many of the prominent behavioral abnormalities seen in depression and other mood disorders, with a particular focus on transcriptional mechanisms that operate in this brain region and have been implicated in depression and its treatment. We highlight how such studies of the NAc may provide novel targets for the development of new antidepressant treatments.
2. THE NAc REWARD CIRCUIT IN MOOD REGULATION
The involvement of the NAc in mood regulation and depression is supported by an increasing literature. The notion that midbrain dopamine systems contribute to depression-like behaviors was first proposed on the basis of studies with dopamine receptor antagonists (Wise, 1982). The ensuing several decades saw only sporadic publications reporting an association between the two (reviewed by Nestler & Carlezon, 2006). These early studies showed that stress, in the context of animal models of depression, potently activates VTA dopamine neurons and stimulates dopaminergic transmission to its limbic targets including the NAc. There were also reports that antidepressant treatments alter dopaminergic activity in the VTA or its targets, and that experimental manipulation of dopaminergic transmission in the VTA–NAc pathway can regulate depression-like behavior in acute stress assays in rodents.
The pace of progress has increased dramatically over the past decade. First, as will be seen below, the manipulation of a host of genes within the NAc was demonstrated to exert dramatic effects in more sophisticated animal models of depression. Second, in parallel to these preclinical studies, brain imaging investigations of humans documented abnormal functioning of the VTA–NAc in depression (Pizzagalli, 2014; Silbersweig, 2013; Tremblay et al., 2005). As just one example, depressed patients show reduced activation of the NAc as measured by functional magnetic resonance imaging in response to rewarding stimuli, direct evidence of a “reward deficit” in this syndrome. Third, advances in neural circuitry studies, enabled by track tracing tools and optogenetic approaches, have begun to define subsets of VTA and NAc neurons that play different roles in reward and stress responses. Distinct subsets of VTA dopamine neurons, for example, display very different responses to stressful stimuli: while some neurons are activated, others are suppressed. And there is increasing evidence that these subsets may be “wired” differently in the brain, with distinct afferent inputs and efferent projections (Chaudhury et al., 2013; Lammel, Lim, & Malenka, 2014; Tye et al., 2013). Similarly, the two major subtypes of medium spiny neurons (MSNs) in the NAc, those predominantly express D1 dopamine receptors versus those that express D2 receptors, which together comprise ~95% of all neurons in this region, differently control stress responsiveness (Francis et al., 2014; Lammel et al., 2014).
3. TRANSCRIPTIONAL AND EPIGENETIC MECHANISMS IN THE NAc IN MOOD REGULATION
There is now robust evidence for a role of several specific molecular pathways in the NAc, most of which were first implicated in regulating drug and natural reward in this circuit, in animal models of depression and antidepressant action. Prominent among such pathways are proteins important in the control of gene transcription and the regulation of chromatin. Representative examples are discussed here.
3.1 CREB-Mediated Transcription in the NAc in Mood Regulation
The transcription factor CREB (cAMP response element-binding protein) is stimulated in the NAc by exposure to several types of drugs of abuse or stress. This effect seems to be shared by both D1- and D2-type MSNs, and numerous studies have established that CREB activity in the NAc has a profound effect on an animal’s responsiveness to emotional stimuli (Blendy, 2006; Carlezon, Duman, & Nestler, 2005; Conti & Blendy, 2004). CREB function in the NAc is normally regulated by glutamatergic and dopaminergic inputs (Dudman et al., 2003), suggesting that—by determining the set point of NAc neurons (Dong et al., 2006)—it represents an emotional gate for behavioral responsivity. This view is now supported by a large body of data. Elevations of CREB activity within the NAc, achieved via viral-mediated gene transfer or in inducible transgenic mice, reduce the rewarding effects of cocaine, morphine, and sucrose (Barrot et al., 2002; Carlezon et al., 1998; McClung & Nestler, 2003; Pliakas et al., 2001), which indicates that a sustained elevation of CREB activity in the NAc produces anhedonia-like signs. In fact, this CREB phenotype appears to reflect a generalized numbing of behavioral responses to emotional stimuli, since animals with increased CREB function in the NAc also show reduced responses to a wide range of aversive conditions (Barrot et al., 2002). Elevations of CREB activity produce pro-depression-like symptoms in several acute and sub-chronic stress models (Newton et al., 2002; Pliakas et al., 2001).
Conversely, reductions in CREB activity in the rat NAc, through viral-or transgenic-mediated expression of a dominant negative CREB mutant (mCREB) or through a local knockout of CREB from the NAc of floxed CREB mice, increases the rewarding effects of cocaine, morphine, and sucrose (Barrot et al., 2002; Carlezon et al., 1998; DiNieri et al., 2009) and produces antidepressant-like effects in several stress assays (Conti, Cryan, Dalvi, Lucki, & Blendy, 2002; Newton et al., 2002; Pliakas et al., 2001), including chronic social defeat stress (Covington et al., 2011)—an ethologically validated model of depression (Berton et al., 2006; Krishnan et al., 2007).
CREB function in the NAc also has a profound effect on anxiety-like behavior in rodents. Disruption of CREB function within the NAc, achieved by viral overexpression of mCREB, produces anxiety-like effects, whereas increased CREB function causes opposite changes (Barrot et al., 2002, 2005; Wallace et al., 2009). On the other hand, global knockdown of CREB reduces anxiety-like behavior (Valverde et al., 2004), perhaps owing to CREB actions outside the NAc. The notion that elevated CREB function in NAc causes certain depression-like symptoms, while reduced CREB function in this region causes anxiety-like behavior, may seem paradoxical, but can be understood within the hypothesized role CREB plays in this reward circuit under normal conditions. Our hypothesis is that CREB in the NAc is a key regulator of the reactivity of brain reward circuits and thereby regulates individual sensitivity to emotional stimuli (Carlezon et al., 2005). Short-term increases in CREB activity in NAc, induced by normal rewarding or aversive stimuli, dampen responses to subsequent stimuli and facilitate the ability to actively deal with the situation at hand (e.g., consumption of reward and escape from danger). Under more pathological conditions, however, larger and more sustained increases in CREB activity, induced by drugs of abuse or excessive stress, would lead to an excessive dampening of emotional reactivity and to the behavioral phenotype outlined above. Conversely, sustained reductions in CREB activity, which are seen under conditions of prolonged social isolation (Barrot et al., 2005; Wallace et al., 2009), heighten emotional reactivity, and in the extreme induce a state of anxiety. This work highlights the notion that extreme increases or decreases in CREB function in NAc may be detrimental and contribute to the symptomology of different mood disorders (Carlezon et al., 2005).
This role for CREB in the NAc in depression models is in stark contrast to CREB’s activity in the hippocampus and other regions in many of the same behavioral models. In hippocampus, for example, CREB is an important mediator of antidepressant effects (Duman & Duman, 2005). These findings underscore the importance of identifying the region-specific target genes through which CREB exerts these various effects, as such targets could be mined in antidepressant drug discovery efforts.
3.2 ΔFosB and SRF in the NAc Pathway in Mood Regulation
A large literature has established that ΔFosB, a Fos family transcription factor, is induced in the NAc uniquely in response to chronic exposure to virtually any drug of abuse (Nestler, 2008). Moreover, once induced, the accumulated ΔFosB persists in the NAc for several weeks during a period of drug withdrawal due to the unusual stability of the protein. Such drug induction of ΔFosB in NAc is selective for D1-type MSNs with the one exception of opiate drugs of abuse which induce the protein equally in D1- and D2-type neurons (Lobo et al., 2013). A wealth of evidence now supports the notion that induction of ΔFosB in D1 MSNs promotes natural reward and enhances behavioral responses, including self-administration, of drugs of abuse, while inhibition of ΔFosB function in this cell type exerts the opposite effects (Nestler, 2008).
A role for ΔFosB in stress responses was first implicated in 2004, when chronic exposure to restraint stress was found to induce ΔFosB in the NAc (Perrotti et al., 2004). However, it was not until 2010 when information became available about the functional significance of this phenomenon. Chronic social defeat stress was found to induce ΔFosB predominantly in mice resistant to the deleterious effects of the stress, so-called resilient mice (Vialou, Robison, et al., 2010). Such induction is specific to D1-type MSNs (Lobo et al., 2013), consistent with our knowledge that ΔFosB in this cell type promotes reward and motivation. By contrast, a lower level of induction of ΔFosB in the NAc of susceptible mice (those that succumb to the deleterious effects of chronic stress) is seen in D2-type MSNs only (Lobo et al., 2013). While the functional consequences of ΔFosB induction in D2 MSNs remain poorly understood, these data suggest it might reduce hedonic responses.
Moreover, selective overexpression of ΔFosB in D1 MSNs of NAc was shown to promote resilience in the social defeat paradigm and exert antidepressant-like responses in mice susceptible to the stress (Donahue, Muschamp, Russo, Nestler, & Carlezon, 2014; Muschamp, Nemeth, Robison, Nestler, & Carlezon, 2012; Vialou, Robison, et al., 2010). In contrast, antagonism of ΔFosB activity, achieved via overexpression of a dominant negative mutant, made mice more susceptible. Interestingly, another type of chronic stress, prolonged social isolation in adulthood, decreases ΔFosB levels in the NAc and overexpression of ΔFosB in this region of isolated mice reverses the depression-like behavioral abnormalities exhibited by the animals (Vialou, Robison, et al., 2010). Depressed humans, like isolated mice, display lower levels of ΔFosB in the NAc, lending important clinical validation to the importance of ΔFosB in regulating depression-related behavioral abnormalities.
These behavioral effects of ΔFosB in the NAc, like those of CREB, show regional specificity. In the medial prefrontal cortex (mPFC), ΔFosB is induced selectively in susceptible mice after chronic social defeat stress, and in this region, ΔFosB overexpression promotes susceptibility while blocking ΔFosB function exerts the opposite effect (Vialou et al., 2014). Interestingly, we found that ΔFosB’s prosusceptible effect in the mPFC is mediated in part by controlling the output of this region to the NAc. These region-specific effects of ΔFosB once again point to the importance of identifying target genes of this transcription factor that predominate in one region or another.
3.3 Other Transcription Factors in the NAc in Mood Regulation
Several other transcription factors in the NAc have been implicated in depression. Studies of ΔFosB have led to the discovery that another transcription factor, serum response factor (SRF), also exerts proresilience effects through actions in the NAc. ΔFosB induction in the NAc of resilient mice, after a period of chronic social defeat stress, is mediated via SRF, which binds to the FosB promoter and increases transcription of the gene (Vialou, Maze, et al., 2010). Interestingly, CREB, which plays a role in mediating ΔFosB induction in response to chronic cocaine, has no role in stress induction of ΔFosB (Vialou et al., 2012). Further research is needed to understand why CREB—which is induced in D1 NAc MSNs in response to chronic stress—does not control FosB transcription in the context of stress, while it does so in the context of cocaine exposure. Further research is also needed to identify additional targets of SRF, which like ΔFosB might contribute to behavioral resilience. The importance of this effort is underscored by the observation that SRF levels are reduced in the NAc of depressed humans (Vialou, Maze, et al., 2010).
Another transcription factor implicated in depression is β-catenin, which we demonstrated recently displays reduced activity in the NAc of susceptible mice after chronic social defeat stress, with resilient mice exhibiting a robust induction of β-catenin activity (Fig. 2) (Dias et al., 2014; Wilkinson et al., 2011). This regulation predominates in D2 MSNs. Importantly, depressed humans, like stressed mice, show reduced levels of β-catenin activity in the NAc. Viral-mediated expression of a dominant negative β-catenin mutant, or local knockdown of β-catenin from the NAc of floxed β-catenin mice, induces heightened susceptibility to defeat stress, whereas overexpression of β-catenin—in D2 MSNs selectively—exerts a proresilient effect (Dias et al., 2014). As will be discussed below, genome-wide analysis of β-catenin targets in the NAc has revealed numerous target genes through which the transcription factor mediates this interesting behavioral phenotype.
Figure 2. β-Catenin mediates stress resilience through Dicer1/miRNA regulation.
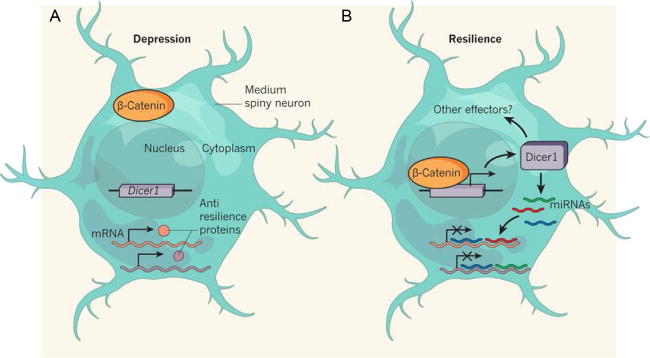
(A) D2-type MSNs are less activated in chronically stressed mice. As a consequence, β-catenin protein remains in the cytoplasm in these cells, unable to enter the nucleus, and the Dicer1 gene is thus inactive. Antiresilience (or prosusceptible) proteins may therefore be produced from mRNAs that would otherwise have been inhibited by miRNAs generated by the DICER1. (B) In resilient mice, β-catenin enters the nucleus of activated D2 MSNs, thereby turning on Dicer1 transcription. Elevated levels of DICER1 increase production of miRNAs and possibly other effectors of resilience. This might, in turn, inhibit the production of antiresilience proteins, because of binding and inhibition of mRNA by miRNAs. From Schratt (2014) with permission.
NFκB (nuclear factor κB) is a transcription factor that is best studied in the immune system, although it has been shown to regulate neural function in more recent years. Interest in NFκB function in the NAc was sparked by the finding that it is a target gene for ΔFosB and is induced in this brain region by chronic cocaine administration (Ang et al., 2001). Years later, it was found that viral-mediated expression of NFκB in the NAc promotes behavioral responses to cocaine, whereas expression of a dominant negative mutant exerts the opposite effect (Russo et al., 2009). It was surprising then to find that NFκB induction in the NAc occurs selectively in susceptible mice after a course of chronic social defeat stress and that NFκB expression in this region promotes susceptibility while inhibition of NFκB activity promotes resilience (Christoffel et al., 2011). On the other hand, NFκB activity in the NAc appears to exert opposite functional effects in the NAc of female mice (LaPlant et al., 2009), something not examined by Christoffel et al. (2011). The cell type in NAc where NFκB exerts these various addiction- and depression-related phenotypes remains unknown and is an active topic of current research.
3.4 Epigenetic Mechanisms in the NAc in Mood Regulation
Advances in chromatin biology have made it possible for the first time to investigate the epigenetic changes that occur in concert with transcriptional regulation in the context of depression models (Peña, Bagot, Labonté, & Nestler, 2014; Sun, Kennedy, & Nestler, 2013; Vialou, Feng, Robison, & Nestler, 2013). Thus, a large literature has demonstrated the important role of histone acetylation and methylation and DNA methylation in controlling gene expression, and each of these chromatin regulatory mechanisms has been implicated in depression and its treatment (Fig. 3). Most studied to date are HDACs (histone deacetylases), which generally inhibit gene expression. Sustained inhibition of HDACs selectively in the NAc exerts a potent antidepressant-like effect in several acute and chronic stress assays, which is stronger and more consistent than that elicited by standard antidepressants such as fluoxetine (Covington et al., 2009). Genome-wide analysis of gene expression changes induced by such HDAC inhibition identified a set of genes, only a portion of which were similarly regulated by chronic fluoxetine administration, which reveal paths for the generation of novel antidepressants. The HDAC subtype mediating this effect is not known, since knockout of HDAC5, albeit from all brain regions, makes mice more susceptible to social defeat (Renthal et al., 2007). Less is known about how chronic stress influences HDACs in the NAc. Expression of HDAC2 and HDAC5 is reduced in the NAc of mice subjected to chronic social defeat stress, with similar effects seen in depressed humans (Covington et al., 2009; Renthal et al., 2007). In contrast, total HDAC activity is reportedly increased in the NAc of mice subjected to early life stress, effects reversed by antidepressant administration, although levels of expression of various HDAC isoforms was not examined in this study (Réus et al., 2013). Future work is needed to resolve these discrepant findings.
Figure 3. Examples of chromatin modifications regulated in the NAc by stress or antidepressant treatment.
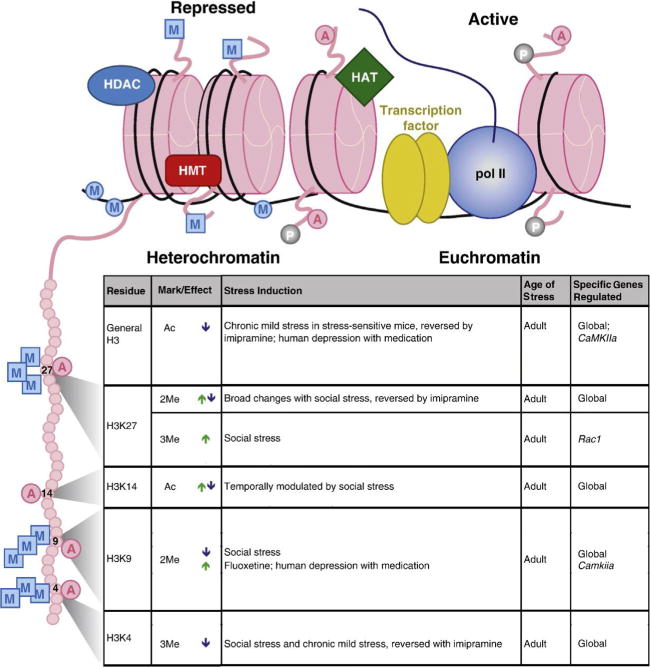
Illustration (top) indicates histone octamers (pink (light gray in the print version)) in heterochromatin (left) and euchromatin (right), along with associated proteins and histone tail/DNA modifications. Table (bottom) lists histone tail modifications of specific residues—depicted on the expanded histone tail illustration (left)—that are regulated by various stress paradigms or antidepressant treatments within the NAc. Arrows indicate an increase (green (light gray in the print version)) or decrease (blue (dark gray in the print version)) in specific modifications. Abbreviations: A, acetylation; P, phosphorylation; M (in a square), histone methylation; M (in a circle), DNA methylation; HAT, histone acetyltransferase; HDAC, histone deacetylase; HMT, histone methyltransferase; pol II, RNA polymerase II. See Peña et al. (2014) for references. Modified from Peña et al. (2014) with permission.
Histone methylation in the NAc is also implicated in depression. Chronic social defeat stress decreases expression of G9a and GLP (G9a-like protein), two histone methyltransferases that catalyze the dimethylation of Lys9 of histone H3 (H3K9me2) (Covington et al., 2011), a mark associated with gene repression. This effect contributes to depression-related behav-ioral abnormalities, since local knockdown of G9a from the NAc of floxed G9a mice increases susceptibility to social defeat stress, while viral-mediated G9a overexpression in this region promotes resilience. An earlier genome-wide study of changes in H3K9me2 in NAc of susceptible and resilient mice identified numerous genes that might mediate these behavioral effects (Wilkinson et al., 2009). Several additional histone methyltransferases are altered in the NAc upon exposure to chronic stress; however, their influence on stress vulnerability has not yet been investigated (see Peña et al., 2014).
Several forms of chronic stress, including chronic social defeat stress and early life stress, have been shown to increase the expression of DNMTs (DNA methyltransferases) in the NAc (Anier et al., 2014; LaPlant et al., 2010). Importantly, overexpression of one of the regulated DNMTs, DNMT3a, in the NAc makes mice more susceptible to social defeat stress, while intra-NAc administration of a DNMT inhibitor (RG108) exerts antidepressant-like effects (LaPlant et al., 2010).
A recent study has demonstrated that another type of epigenetic mechanism—namely, microRNAs (miRNAs), also act in the NAc to control depression-related behavior. miRNAs are small (20 nt) noncoding RNAs which exert powerful control over the translation of mRNAs into proteins. Our interest in miRNAs was sparked by the finding, alluded to above, that genome-wide characterization of β-catenin target genes in the NAc identified Dicer1—the protein product of which catalyzes a crucial step in the bio-genesis of miRNAs—as a prominent target that is induced in this brain region of resilient mice (Dias et al., 2014). Indeed, viral-mediated DICER1 expression in NAc makes mice more resilient. This led to a genome-wide study of all miRNAs that are regulated in the NAc of susceptible versus resilient mice and whose regulation is mediated by β-catenin. The identified miRNAs now become interesting targets for future antidepressant drug discovery research (Dias et al., 2014).
While the unusually potent pro- or antidepressant effects of these various epigenetic mechanisms, acting in the NAc, have stoked interest in mining these discoveries in the development of new treatments, a major caveat is that all of these mechanisms are ubiquitous. Consequently, it is likely that direct manipulations of HDACs, histone methyltransferases, or DNMTs would produce side effects that render them unsuitable as antidepressants. This once again emphasizes the importance of identifying the target genes that mediated these actions. Still, it is possible that the particularly potent behavioral actions of these mechanisms are precisely because they alter numerous genes at once. This points to the potential unique utility of miRNAs, the manipulation of which would regulate multiple mRNAs but a relatively small number, and thus might be amenable to drug discovery efforts.
3.5 Identification and Characterization of Target Genes for Specific Transcription Factors in the NAc
The discussion above highlights the importance of going beyond transcriptional and epigenetic mechanisms per se in the quest to develop improved antidepressant treatments based on our increasing appreciation of the role of the NAc in depression. In general, two approaches have been used in the field: a candidate gene approach whereby investigators have focused on a small number of genes based on a priori hypotheses of their role in depression models and unbiased genome-wide efforts to search for novel target genes.
Candidate gene approaches have been fruitful. Based on the knowledge that the opioid peptide dynorphin—which activates κ-opioid receptors—is a CREB target gene in other systems, we and others have established that dynorphin is one CREB target that is induced in the NAc by chronic stress and contributes to depression-related behavioral abnormalities (Fig. 4) (Bruchas, Land, & Chavkin, 2010; Van’t Veer & Carlezon, 2013). This has led to interest in κ-opioid antagonists for the treatment of depression. In contrast, there is evidence that ΔFosB suppresses dynorphin expression in the NAc (Zachariou et al., 2006), which might contribute to its proresilience effects. BDNF (brain-derived neurotrophic factor) and its signaling pathways are another example. BDNF, like dynorphin, is a CREB target and local synthesis of BDNF likely contributes to stress susceptibility (Berton et al., 2006; Krishnan et al., 2007). Moreover, genes encoding proteins that mediate the actions of BDNF show reduced binding of H3K9me2, leading to induction of those genes, enhanced BDNF signaling, and prosusceptible effects (Covington et al., 2011). Finally, the GluA2 AMPA glutamate receptor subunit is induced in the NAc of resilient mice only, an action mediated by ΔFosB, and direct manipulation of this subunit within the NAc confirms that its upregulation promotes behavioral resilience (Vialou, Robison, et al., 2010).
Figure 4. CREB and dynorphin in the NAc in depression.
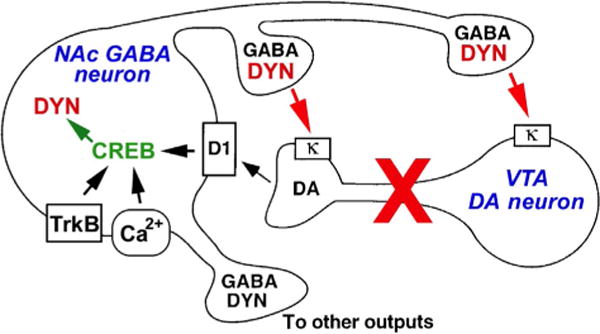
The figure shows a simplified hypothetical scheme by which CREB induction of dynorphin (DYN) in the NAc contributes to certain symptoms of depression. CREB is activated by D1 dopamine receptors (through activation of the cAMP pathway) or by Ca2+- or TrkB-regulated signal transduction pathways, which leads to increased expression of DYN. DYN feeds back on κ-opioid receptors located on the terminals and cell bodies/dendrites of VTA dopamine (DA) neurons. Stimulation of these κ receptors inhibits the VTA neurons, which may contribute to anhedonia and related symptoms of depression. Antagonists of κ receptors may thus block the consequences of CREB-induced increases in DYN activity, and exert antidepressant activity in some individuals. From Nestler and Carlezon (2006) with permission.
The limitation of candidate gene approaches is that we still know relatively little about depression and hence are limited in our choice of specific genes to investigate. The discovery of DICER1 and miRNAs as downstream mediators of the proresilience effects of β-catenin is a good illustration of the power of unbiased, open-ended approaches. ChIP-seq (chromatin immunoprecipitation followed by deep sequencing) is the method used to identify targets of a transcription factor or chromatin modification genome wide and was used to characterize β-catenin targets (Dias et al., 2014). An earlier variant of ChIP-seq, termed ChIP-chip (ChIP followed by analysis on promoter chips) was used to study CREB and H3K9me2 targets in the NAc in the chronic social defeat stress and social isolation paradigms (Wilkinson et al., 2009). However, ChIP-chip is far less quantitative than ChIP-seq and covers only promoter regions of genes, in contrast to ChIP-seq which provides a truly genome-wide measure. A major need of future research, therefore, is to carry out ChIP-seq for the increasing number of transcription factors and chromatin regulatory mechanisms that are being shown to control depression-related behavior via actions in the NAc. As such data are generated, advanced bioinformatic tools will be used to identify the molecular pathways that are most highly regulated in a state of susceptibility versus resilience, or in depressed human NAc, findings which can then be mined for fundamentally novel approaches to antidepressant drug discovery efforts (Maze et al., 2014).
4. FUTURE DIRECTIONS
A major need of future research is to better define how the numerous and diverse molecular pathways discussed above influence the several cell types in the NAc and how that altered function controls mood and depression-related behavioral abnormalities. Early steps of this work have been achieved for CREB (Dong et al., 2006; Huang et al., 2008) and ΔFosB (Grueter, Robison, Neve, Nestler, & Malenka, 2013): CREB controls the intrinsic excitability and NMDA responses of both D1 and D2 MSNs, while ΔFosB exerts selective effects on AMPA glutamate responses in D1 MSNs only. Likewise, optogenetic tools have been used to demonstrate opposite effects of D1 versus D2 MSN activation in controlling stress responses (Francis et al., 2014). A goal of current research is to define the various outputs of D1 and D2 NAc MSNs in mediating these effects.
A surprising finding to date is the complex relationship between the brain’s reward circuitry and mood disorders. One might have assumed that “more reward” equals “less depression”: for example, that a protein acting in a certain brain structure that increase cocaine’s rewarding effects would exert an antidepressant-like effect based on the assumption that the protein also boosts natural reward (Russo & Nestler, 2013). To put it another way: one might expect that the influence of a protein in depression models is predictable based on its effects in addiction models and vice versa. However, this is clearly not the case: there is no predictable relationship between the effects of a given protein in the NAc in depression models versus drug addiction models (Table 1); this is despite the considerable comorbidity between depression and addiction syndromes. One complicating factor is that addiction likely involves adaptations that impair brain reward and others that promote reward-related memories (Russo & Nestler, 2013). The heterogeneity of cell types within the NAc is another likely source of different actions observed for a protein in stress versus drug models. However, cell type is not the only explanation. For instance, CREB appears to have very different target genes in the NAc in depression versus addiction models, even within the same cell types (Covington et al., 2011; McClung & Nestler, 2003; Wallace et al., 2009; Wilkinson et al., 2009). This suggests that the stimulus itself (i.e., stress or drugs) can engage different intracellular pathways, thereby regulating chromatin structure and gene expression in their own unique ways. Future studies using cell-selective molecular profiling and viral gene transfer approaches will be crucial for shedding light on these complex stimulus-specific effects on reward-related behavior and depression.
Table 1.
Effect of Molecular Mediators in the NAc on Depression- Versus Addiction-Like Behavior
| Protein | Effect in Depression Models | Effect in Addiction Models |
|---|---|---|
| CREB | ↑ Susceptibility | ↓ Drug reward |
| ΔFosB | ↓ Susceptibility | ↑ Drug reward |
| SRF | ↓ Susceptibility | 0 Drug reward |
| NFκB | ↑ Susceptibility | ↑ Drug reward |
| β-Catenin | ↓ Susceptibility | N/A |
| HDAC inhibition | ↓ Susceptibility | ↑ Drug reward |
| G9a inhibition | ↑ Susceptibility | ↑ Drug reward |
| DNMT inhibition | ↓ Susceptibility | ↑ Drug reward |
| BDNF-TrkB | ↑ Susceptibility | ↑ Cocaine reward ↓Morphine reward |
| GluA2 | ↓ Susceptibility | ↑ Drug reward |
| Dynorphin | ↑ Susceptibility | ↓ Drug reward |
The table illustrates examples from our laboratory which indicate that there is not a clear relationship between an effect of a protein in the NAc on stress susceptibility and on drug reward. One complicating factor may be differential effects of a protein in D1-type versus D2-type NAc MSNs, which have only recently begun to be parsed. Another possible explanation is complex combinatorial effects unique to a given condition. For example, we know that a given transcription factor (e.g., CREB) has very different target genes in NAc under stress or cocaine conditions, even after different types of stress (Wilkinson et al., 2009), presumably due to a host of other factors induced uniquely in each situation.
Adapted from Russo and Nestler (2013).
Acknowledgments
This work was supported by grants from the National Institute of Mental Health (P50MH096890 and R01MH051399).
References
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th. Washington, DC: American Psychiatric Press; 2013. [Google Scholar]
- Ang E, Chen JS, Zagouras P, Magna H, Holland J, Schaeffer E, et al. Induction of NFκB in nucleus accumbens by chronic cocaine administration. Journal of Neurochemistry. 2001;79:221–224. doi: 10.1046/j.1471-4159.2001.00563.x. [DOI] [PubMed] [Google Scholar]
- Anier K, Malinovskaja K, Pruus K, Aonurm-Helm A, Zharkovsky A, Kalda A. Maternal separation is associated with DNA methylation and behavioural changes in adult rats. European Neuropsychopharmacology. 2014;24:459–468. doi: 10.1016/j.euroneuro.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Wang MJ, Paspalas CD. Neuromodulation of thought: Flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron. 2012;76:223–239. doi: 10.1016/j.neuron.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrot M, Olivier JDA, Perrotti LI, Impey S, Storm DR, Neve RL, et al. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrot M, Wallace-Black D, Bolanos CA, Graham D, Perrotti LI, Neve RL, et al. Regulation of anxiety and initiation of sexual anxiety by CREB in the nucleus accumbens. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8357–8362. doi: 10.1073/pnas.0500587102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, McClung CA, DiLeone RJ, Krishnan V, Russo S, Graham D, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Berton O, Nestler EJ. New approaches to antidepressant drug discovery: Beyond monoamines. Nature Reviews. Neuroscience. 2006;7:137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- Blendy JA. The role of CREB in depression and antidepressant treatment. Biological Psychiatry. 2006;59:1144–1150. doi: 10.1016/j.biopsych.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Research. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends in Neurosciences. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, et al. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493:532–536. doi: 10.1038/nature11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF, et al. IκB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. Journal of Neuroscience. 2011;31:314–321. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti AC, Blendy JA. Regulation of antidepressant activity by cAMP response element binding proteins. Molecular Neurobiology. 2004;30:143–155. doi: 10.1385/MN:30:2:143. [DOI] [PubMed] [Google Scholar]
- Conti AC, Cryan JF, Dalvi A, Lucki L, Blendy JA. cAMP response element-binding protein is essential for the upregulation of brain-derived neurotrophic factor transcription, but not the behavioral or endocrine responses to antidepressant drugs. Journal of Neuroscience. 2002;22:3262–3268. doi: 10.1523/JNEUROSCI.22-08-03262.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, III, Maze I, LaPlant QC, Vialou VF, Yoshinori ON, Berton O, et al. Antidepressant actions of HDAC inhibitors. Journal of Neuroscience. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, III, Maze I, Sun HS, Wu EY, Dietz D, Lobo MK, et al. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron. 2011;71:656–670. doi: 10.1016/j.neuron.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias C, Feng J, Sun HS, Shao NY, Mazei-Robison MS, Damez-Werno D, et al. β-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature. 2014;516:51–55. doi: 10.1038/nature13976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNieri JA, Nemeth C, Parsegian A, Carle T, Gurevich VV, Gurevich E, et al. Altered sensitivity to rewarding and aversive drugs in mice with inducible disruption of cAMP response element-binding protein function within the nucleus accumbens. Journal of Neuroscience. 2009;29:1855–1859. doi: 10.1523/JNEUROSCI.5104-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue RJ, Muschamp JW, Russo SJ, Nestler EJ, Carlezon WA., Jr Effects of striatal ΔFosB overexpression and ketamine on social defeat stress-induced anhedonia in mice. Biological Psychiatry. 2014;76:550–558. doi: 10.1016/j.biopsych.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, et al. CREB modulates excitability of nucleus accumbens neurons. Nature Neuroscience. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- Dudman JT, Eaton ME, Rajadhyaksha A, Macías W, Taher M, Barczak A, et al. Dopamine D1 receptors mediate CREB phosphorylation via phosphory-lation of the NMDA receptor at Ser897-NR1. Journal of Neurochemistry. 2003;87:922–934. doi: 10.1046/j.1471-4159.2003.02067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman CH, Duman RS. Neurobiology and treatment of anxiety: Signal transduction and neural plasticity. Handbook of Experimental Pharmacology. 2005:305–334. doi: 10.1007/3-540-28082-0_11. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biological Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Wolf ME. Psychomotor stimulant addiction: A neural systems perspective. Journal of Neuroscience. 2002;22:3312–3320. doi: 10.1523/JNEUROSCI.22-09-03312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis TC, Chandra R, Friend DM, Finkel E, Dayrit G, Miranda J, et al. Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biological Psychiatry. 2014;77:212–222. doi: 10.1016/j.biopsych.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Robison AJ, Neve RL, Nestler EJ, Malenka RC. ΔFosB differentially modulates nucleus accumbens direct and indirect pathway function. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1923–1928. doi: 10.1073/pnas.1221742110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Brown TE, Han MH, Saal DB, Neve RL, et al. CREB modulates the functional output of nucleus accumbens neurons: A critical role of N-methyl-D-aspartate glutamate receptor (NMDAR) receptors. Journal of Biological Chemistry. 2008;283:2751–2760. doi: 10.1074/jbc.M706578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: The role of reward-related learning and memory. Annual Review of Neuroscience. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. Glutamate systems in cocaine addiction. Current Opinion in Pharmacology. 2004;4:23–29. doi: 10.1016/j.coph.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Malenka RC. Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology. 2014;76(Pt. B):351–359. doi: 10.1016/j.neuropharm.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlant Q, Chakravarty S, Vialou V, Mukherjee S, Koo JW, Kalahasti G, et al. Role of NFκB in ovarian hormone mediated stress hypersensitivity in female mice. Biological Psychiatry. 2009;65:874–880. doi: 10.1016/j.biopsych.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington HE, Dumitriu D, Feng J, Warren B, et al. DNMT3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nature Neuroscience. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Zaman S, Damez-Werno DM, Koo JW, Bagot RC, DiNieri JA, et al. ΔFosB induction in striatal medium spiny neuron subtypes in response to chronic pharmacological, emotional, and optogenetic stimuli. Journal of Neuroscience. 2013;33:18381–18395. doi: 10.1523/JNEUROSCI.1875-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Shen L, Zhang B, Garcia BA, Shao NY, Mitchell A, et al. Analytical tools and current challenges in the modern era of neuroepigenomics. Nature Neuroscience. 2014;17:1476–1490. doi: 10.1038/nn.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and ΔFosB. Nature Neuroscience. 2003;11:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- Miller BR, Hen R. The current state of the neurogenic theory of depression and anxiety. Current Opinion in Neurobiology. 2014;30C:51–58. doi: 10.1016/j.conb.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschamp JW, Nemeth CL, Robison AJ, Nestler EJ, Carlezon WA., Jr ΔFosB enhances the rewarding effects of cocaine while reducing the pro-depressive effects of the kappa-opioid receptor agonist U50488. Biological Psychiatry. 2012;71:44–50. doi: 10.1016/j.biopsych.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Transcriptional mechanisms of addiction: Role of deltaFosB. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 2008;363:3245–3255. doi: 10.1098/rstb.2008.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biological Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Newton SS, Thome J, Wallace T, Shirayama Y, Dow A, Schlesinger L, et al. Inhibition of CREB or dynorphin in the nucleus accumbens produces an antidepressant-like effect. Journal of Neuroscience. 2002;22:10883–10890. doi: 10.1523/JNEUROSCI.22-24-10883.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña CJ, Bagot RC, Labonté B, Nestler EJ. Epigenetic signaling in psychiatric disorders. Journal of Molecular Biology. 2014;426:3389–3412. doi: 10.1016/j.jmb.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotti LI, Hadeishi Y, Ulery P, Barrot M, Monteggia L, Duman RS, et al. Induction of deltaFosB in reward-related brain structures after chronic stress. Journal of Neuroscience. 2004;24:10594–10602. doi: 10.1523/JNEUROSCI.2542-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezze MA, Feldon J. Mesolimbic dopaminergic pathways in fear conditioning. Progress in Neurobiology. 2004;74:301–320. doi: 10.1016/j.pneurobio.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Pizzagalli DA. Depression, stress, and anhedonia: Toward a synthesis and integrated model. Annual Review of Clinical Psychology. 2014;10:393–423. doi: 10.1146/annurev-clinpsy-050212-185606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA., Jr Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated CREB expression in the nucleus accumbens. Journal of Neuroscience. 2001;21:7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, Xiao GH, Kumar A, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Réus GZ, Abelaira HM, dos Santos MA, Carlessi AS, Tomaz DB, Neotti MV, et al. Ketamine and imipramine in the nucleus accumbens regulate histone deacetylation induced by maternal deprivation and are critical for associated behaviors. Behavioural Brain Research. 2013;256:451–456. doi: 10.1016/j.bbr.2013.08.041. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nature Reviews. Neuroscience. 2013;14:609–625. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, et al. Nuclear factor κB signaling regulates neuronal morphology and cocaine reward. Journal of Neuroscience. 2009;29:3529–3537. doi: 10.1523/JNEUROSCI.6173-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt G. A molecular knife to dice depression. Nature. 2014;516:45–46. doi: 10.1038/nature13942. [DOI] [PubMed] [Google Scholar]
- Silbersweig D. Default mode subnetworks, connectivity, depression and its treatment: Toward brain-based biomarker development. Biological Psychiatry. 2013;74:5–6. doi: 10.1016/j.biopsych.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Sun H, Kennedy PJ, Nestler EJ. Epigenetics of the depressed brain: Role of histone acetylation and methylation. Neuropsychopharmacology. 2013;38:124–137. doi: 10.1038/npp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay LK, Naranjo CA, Graham SJ, Herrmann N, Mayberg HS, Hevenor S, et al. Functional neuroanatomical substrates of altered reward processing in major depressive disorder revealed by a dopaminergic probe. Archives of General Psychiatry. 2005;62:1228–1236. doi: 10.1001/archpsyc.62.11.1228. [DOI] [PubMed] [Google Scholar]
- Trivedi MH, Daly EJ. Treatment strategies to improve and sustain remission in major depressive disorder. Dialogues in Clinical Neuroscience. 2008;10:377–384. doi: 10.31887/DCNS.2008.10.4/mhtrivedi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CA, Watson SJ, Akil H. The fibroblast growth factor family: Neuromodulation of affective behavior. Neuron. 2012;76:160–174. doi: 10.1016/j.neuron.2012.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Mirzabekov JJ, Warden MR, Ferenczi E, Tsai HC, Finkelstein J, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013;493:537–541. doi: 10.1038/nature11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde O, Mantamadiotis T, Torrecilla M, Ugedo L, Pineda J, Bleckmann S, et al. Modulation of anxiety-like behavior and morphine dependence in CREB-deficient mice. Neuropsychopharmacology. 2004;29:1122–1133. doi: 10.1038/sj.npp.1300416. [DOI] [PubMed] [Google Scholar]
- Van’t Veer A, Carlezon WA., Jr Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology. 2013;229:435–452. doi: 10.1007/s00213-013-3195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Bagot RC, Cahill ME, Ferguson D, Robison AJ, Dietz DM, et al. Prefrontal cortical circuit for depression- and anxiety-related behaviors mediated by cholecystokinin: Role of ΔFosB. Journal of Neuroscience. 2014;34:3878–3887. doi: 10.1523/JNEUROSCI.1787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Feng J, Robison AJ, Ku SM, Ferguson D, Scobie KN, et al. Serum response factor and cAMP response element binding protein are both required for cocaine induction of ΔFosB. Journal of Neuroscience. 2012;32:7577–7584. doi: 10.1523/JNEUROSCI.1381-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Feng J, Robison AJ, Nestler EJ. Epigenetic mechanisms of depression and antidepressant action. Annual Review of Pharmacology and Toxicology. 2013;53:59–87. doi: 10.1146/annurev-pharmtox-010611-134540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Maze I, Renthal W, LaPlant QC, Watts EL, Mouzon E, et al. Serum response factor promotes resilience to chronic social stress through the induction of DeltaFosB. Journal of Neuroscience. 2010;30:14585–14592. doi: 10.1523/JNEUROSCI.2496-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Robison AJ, LaPlant QC, Covington HE, III, Dietz DM, Ohnishi YN, et al. DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nature Neuroscience. 2010;13:745–752. doi: 10.1038/nn.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DL, Han MH, Graham DL, Green TA, Vialou V, Iniguez SD, et al. CREB regulation of nucleus accumbens excitability mediates social isolation-induced behavioral deficits. Nature Neuroscience. 2009;12:200–209. doi: 10.1038/nn.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MB, Dias C, Magida J, Mazei-Robison M, Lobo MK, Kennedy P, et al. A novel role of the WNT-dishevelled-GSK3β signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. Journal of Neuroscience. 2011;31:9084–9092. doi: 10.1523/JNEUROSCI.0039-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MB, Xiao GH, Kumar A, LaPlant Q, Renthal W, Sikder D, et al. Imipramine treatment and resiliency exhibit similar chromatin regulation in a key brain reward region. Journal of Neuroscience. 2009;29:7820–7832. doi: 10.1523/JNEUROSCI.0932-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. Neuroleptics and operant behavior: The anhedonia hypothesis. Behavioral and Brain Sciences. 1982;5:39–87. [Google Scholar]
- Wise RA. Drug-activation of brain reward pathways. Drug and Alcohol Dependence. 1998;51:13–22. doi: 10.1016/s0376-8716(98)00063-5. [DOI] [PubMed] [Google Scholar]
- Wittmann BC, Schott BH, Guderian S, Frey JU, Heinze HJ, Duzel E. Reward-related fMRI activation of dopaminergic midbrain is associated with enhanced hippocampus-dependent long-term memory formation. Neuron. 2005;45:459–467. doi: 10.1016/j.neuron.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Zachariou V, Bolanos CA, Selley DE, Theobald D, Cassidy MP, Kelz MB, et al. ΔFosB: An essential role for ΔFosB in the nucleus accumbens in morphine action. Nature Neuroscience. 2006;9:205–211. doi: 10.1038/nn1636. [DOI] [PubMed] [Google Scholar]