

Abstract
Positive allosteric modulators (PAM) binding to the transmembrane (TM) domain of metabotropic glutamate receptor 5 (mGluR5) are promising therapeutic agents for psychiatric disorders and traumatic brain injury (TBI). Novel PAMs based on a trans-2-phenylcyclopropane amide scaffold have been designed and synthesized. Facilitating ligand design and allowing estimation of binding affinities to the mGluR5 TM domain was the novel computational strategy, site identification by ligand competitive saturation (SILCS). The potential protective activity of the new compounds was evaluated using nitric oxide (NO) production in BV2 microglial cell cultures treated with lipopolysachharide (LPS), and the toxicity of the new compounds tested using a cell viability assay. One of the new compounds, 3a, indicated promising activity with potency of 30 μM, which is 4.5-fold more potent than its lead compound 3,3′-difluorobenzaldazine (DFB), and showed no detectable toxicity with concentrations as high as 1000 μM. Thus this compound represents a new lead for possible development as treatment for TBI and related neurodegenerative disorders.
Keywords: Traumatic brain injury, Metabotropic glutamate receptor, Neuroprotection, Computer-aided drug design, Positive allosteric modulator
Graphical Abstract
Traumatic brain injury (TBI) is a highly prevalent neurodegenerative disorder with no proven neuroprotective therapies.1 TBI induces chronic neuroinflammation associated with microglial activation,2–4 which contributes to delayed neuronal cell death and functional disabilities.5–10 Recent experimental evidence has shown that such secondary injury in the central nervous system (CNS) may last for months to even years, associated with progressive neurodegeneration.11–13 Metabotropic glutamate receptor 5 (mGluR5) is commonly found in neurons and astrocytes, and is highly expressed in microglial cells.14 Recent work has shown that activation of mGluR5 can effectively inhibit microglial activation as late as one month after experimental trauma.10 Activation of mGluR5 can also block the neurotoxicity of activated microglia in vitro and in vivo.10,15,16 Therefore mGluR5 has emerged as a promising neuroprotective drug target for TBI.
The structure of mGluR5 includes an N-terminal ligand-binding domain (LBD) and a seven-helical transmembrane (TM) domain. Although numerous orthosteric mGluR5 agonists are known,17–20 none have been used in the clinic largely due to the challenge in identifying selective21 and CNS permeable agonists of the receptor. Recent advances in the development of positive allosteric modulators (PAMs), by targeting the seven-helical trans-membrane (TM) of mGluR5, have provided new opportunities for discovery of therapeutic agents for TBI. Because the TM domains mGluRs are less conserved and the ligands to the hydrophobic TM domain do not require the charged amino acid character as for mGluR5 agonists, PAMs have greater potential to achieve specificity for mGluR5 and have a higher potential for CNS penetration compared to LBD binders. Numerous mGluR5 PAMs have been reported.12,13,22–27 Of these mGluR5 PAM VU0360172 has shown promising in vivo efficacy for TBI,16 and in vivo efficacy in rodent models for anxiety and psychosis.28
In previous studies we found that mGluR5 PAM 3,3′-difluorobenzaldazine (DFB, Figure 1) showed potential protective activity (IC50 = for 136 μM NO production).29 However, DFB, along with other tested PAMs, have limitations such as modest efficacy, significant cellular toxicity, and poor aqueous solubility. In addition, the azo group of DFB is light sensitive. Here we describe the design, synthesis and evaluation of mGluR5 PAMs (1-3) based on a trans-2-phenylcyclopropyl amide scaffold. The chemical structures of compounds 1-3 were chosen to mimic the planar (1E,2E)-1,2-dibenzylidenehydrazine core of DFB while maintaining favorable interactions with the receptor based on computer-aided drug design (CADD, see below). We hypothesize that improved PAMs can be achieved by replacing the azo linker of DFB with a photo stable trans-cyclopropyl amide group that is commonly used by natural and synthetic drugs (Figure 1). The trans-cyclopropyl moiety is selected to break the planar configuration of the compounds. The efforts also included testing the effects of the orientation of the amide linker for the neuroprotective potency of new compounds.
Figure 1.

Chemical structures of DFB and compounds 1-3. Only one of the two enantiomers of the racemic mixture is shown for compounds 1-3.
To facilitate ligand design we undertook CADD analysis of the PAM binding region of the mGluR5 TM domain. CADD analysis involved the site identification by ligand competitive saturation (SILCS) approach30 on a homology model of the TM domain of mGluR5 derived from the mGluR1 crystal structure (PDB: 4OR2).31 SILCS calculations and ligand modeling used the CHARMM36 and CGenFF force field along with the programs Modeller, CHARMM, and Gromacs. SILCS is a method that maps the functional group affinity patterns of a protein. The method accounts for both protein flexibility and desolvation contributions by running molecular dynamics (MD) of the protein in an aqueous solution of the small solute molecules representative of different chemical functional groups.32 To sample the partially occluded ligand binding pocket of the mGluR5, we applied an extension of the SILCS method that involves an iterative Grand Canonical Monte Carlo/MD (GCMC/MD) methodology.33,34 From the simulations, discretized probability distributions of the fragment atoms that are normalized by their bulk values are obtained and then converted to free energies based on a Boltzmann distribution, yielding Grid Free Energy (GFE) FragMaps. The maps thus represent the 3D free energy distribution of functional group binding at the ligand binding pocket and may be used both qualitatively and quantitatively to direct ligand design. In the current work, eight representative solutes with different chemical functionalities: benzene, propane, acetaldehyde, methanol, formamide, imidazole, acetate and methylammonium were chosen to probe the ligand binding pocket of mGluR5. Benzene and propane serve as probes for nonpolar functionalities. Methanol, formamide, imidazole and acetaldehyde are neutral molecules that participate in hydrogen bonding. The positively charged methylammonium and negatively charged acetate molecules serve as probes for charged donor and acceptors, respectively. The voxel occupancies of the eleven atom types were merged in the following manner to create five generic FragMap types: (1) generic nonpolar, APOLAR (benzene and propane carbons); (2) generic neutral hydrogen bond donor, HBDON (methanol, formamide and imidazole polar hydrogens); (3) generic neutral hydrogen bond acceptor, HBACC (methanol, formamide, and acetaldehyde oxygen and imidazole unprotonated nitrogen) (4) positive donor, POS (methylammonium polar hydrogens); and (5) negative acceptor, NEG (acetate oxygens). The FragMaps used in the present work were those prepared for our previous study, which includes details of the computational methods.33,35
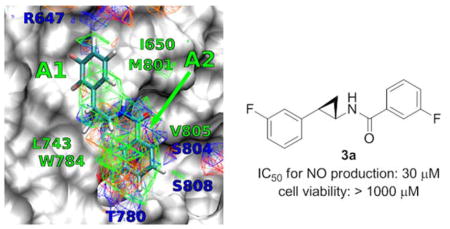
Favorable FragMap affinities were found near residues R647, L743, T780 and W784, previously identified through mutational studies to be important for ligand binding and activity.36 Presented in Figure 2A is DFB docked into the PAM binding site using Autodock-Vina37 directed by the SILCS FragMaps. The phenyl moiety overlaps with the APOLAR FragMaps in the proximity of L743, W784 and V805 (marked A2 in Figure 2B) indicating this region of the model to be important for binding.
Figure 2.

FragMaps overlaid on the PAM binding site of mGluR5 with ligands A) DFB, B) Compound 1a. Receptor atoms occluding the view of the binding pocket were removed to facilitate visualization. The color for nonpolar (APOLAR), neutral donor (HBDON), neutral acceptor (HBACC), negative acceptor (NEG) and positive donor (POS) FragMaps are green, blue, red, orange and cyan, respectively. APOLAR, HBACC and HBDON FragMaps are set to a cutoff of −0.5 kcal/mol, while NEG and POS are set to −1.2 kcal/mol. Distinct FragMap affinities that overlap with the functional groups of the ligands are indicated by arrows colored the same as the FragMaps. C) Satisfactory correlation was observed between the LGFE and the ΔGbind when ligands 1e, 1f and 2f were not considered in the R2 and PI calculations.
This information motivated the design of the novel scaffolds (compounds 1-3) based on a trans-2-phenylcyclopropyl amide scaffold. Docking of 1 into the SILCS FragMaps was then performed with the resulting orientation shown in Figure 2B. In addition to the overlap of the phenyl ring with the APOLAR FragMaps is the overlap of the cyclopropyl moiety and of the amide carbonyl oxygen with a HBACC FragMap, interactions that may improve binding. A collection of 15 derivatives of 1, 2 and 3 were then designed and synthesized based on the overlap with FragMaps in the region of the hydrophobic cavity and the donor and acceptor FragMaps in the proximity of T780, S804 and S808 (Table 1). Quantitative predictions of the binding of DFB, compound 1, and its derivatives in the pocket were then performed using Ligand Grid Free Energy (LGFE) scoring 32. LGFE is based on the overlap of atoms in the ligand functional moieties with their respective GFE FragMap types and was calculated as a Boltzmann averages over 25 runs with 200,000 steps of MC sampling of each of the ligands in the field of FragMaps. Individual MC sampling was performed for all the possible enantiomers of the ortho- and meta-substituted compounds. Presented in Table 1 are the resulting LGFE scores. Notably, all the designed compounds were predicted to have improved affinity over DFB, indicating that the design strategy would yield improved analogs. The following section describes the synthesis of all the compounds in Table 1 and subsequent biological evaluation.
Table 1.
Comparison of structure features, calculated properties, potency and cell viability of compounds
| Cmpd | LGFE (kcal/mol) | IC50c (μM) | Viabilityc,d (μM) | Selective Index (viability/IC50) |
|---|---|---|---|---|
| DFB | −24.7 | 136 | 500 | 3.68 |
| 1a | −33.08 | 210 | 1000 | 4.76 |
| 1b | −31.22 | 160 | 1000 | 24.6 |
| 1c | −32.75 | 180 | 1000 | 5.68 |
| 1d | −34.24 | 46 | 1000 | 21.8 |
| 1e | −29.17 | 46 | 300 | 6.49 |
| 1f | −30.09 | 21 | 300 | 14.6 |
| 2b | −33.73 | 58 | 1000 | 17.4 |
| 2c | −33.43 | 71 | 1000 | 14.1 |
| 2d | −32.04 | 230 | 1000 | 4.37 |
| 2f | −29.76 | 67 | 400 | 5.96 |
| 3a | −33.08 | 30 | 1000 | 33.0 |
| 3b | −33.79 | 200 | 1000 | 5.03 |
| 3c | −33.28 | 130 | 400 | 3.01 |
| 3d | −29.83 | 200 | 200 | 1.02 |
| 3e | −32.03 | 170 | 800 | 4.60 |
The listed result was the average of three independent experiments.
The highest concentration of the tested compound at which no obvious cytotoxicity was observed.
The new compounds (1-3) were synthesized as shown in Schemes 1–2.38 Rh(II)-catalyzed cyclopropanation of 1-substituted-3-vinylbenzenes (4a-b) produced cyclopropyl compounds 5a-b as cis/trans mixtures in good yields (Scheme 1).39 Ethyl esters 5a-b were treated with NaOCH3 in refluxing ethanol to induce epimerization, generating the thermodynamically more stable trans isomers, which were hydrolyzed using aqueous LiOH to yield compounds 6a-b in good yields.39 Finally, the carboxylic acid groups of compounds 6a-b were coupled to various aromatic amines mediated by either ethyl(dimethylaminopropyl) carbodiimide (EDC)/4-(dimethylamino) pyridine (DMAP) or O-(N-succinimidyl)-1,1,3,3-tetramethyl uranium tetrafluoroborate (TSTU)/diisopropylethyl amine (DIPEA) to give compounds 1-3 in modest to good yields.
Scheme 1.

Synthesis of compounds 1-2.a
aReagents and conditions: (a) (i) EtO2CCHN2, Rh2(OAc)4, toluene, 85 °C, 12 h, (ii) NaOCH3 in EtOH, reflux, 18 h, 45–60% for two steps; (b) LiOH, MeOH/H2O, 70 °C, 12 h, 71–75%; (c) aromatic amine, EDC, DMAP, CH2Cl2, r.t., 16 h, 23–77%.
Scheme 2.

Synthesis of compounds 3a-e.a
a Reagents and conditions: (a) diphenyl phosphorazidate, triethylamine, t-BuOH, 85 °C, 48 h, 75–82%; (d) TFA/CH2Cl2, r.t., 1 h; (c) aromatic amine, EDC, DMAP, CH2Cl2, r.t., 16 h, 65–88%; (d) aromatic amine, TSTU, DIPEA, DMF, r.t., 16 h, 43–46%
The synthesis of compounds 3 began with compound 6a (Scheme 2).40 The carboxylic acid group of compound 6a was converted to Boc-protected amine (7a) using Curtius rearrangement in good yields.40 Next, the Boc-protecting group of 7a was removed in trifluoroacetic acid (TFA) to provide (±)-trans-8 as a TFA salt in high yields. Compound 8 was coupled to five different carboxylic acids using either EDC and DMAP or TSTU as the coupling reagents to give compounds 3a-e in good yields.
The anti-inflammatory activities of compounds 1-3 were measured for their ability to inhibit NO production (Table 1). The toxicity of the synthesized compounds to BV2 microglial cells has also been evaluated. Comparing to DFB, N-(2-fluorophenyl)-2-(3-fluorophenyl) cyclopropane-1-carboxamide (1a) and its 4-fluorophenyl (1b) and 2-cholorphenyl (1c) analogs showed decreased anti-inflammatory potency, however, these new cyclopropyl-containing compounds indicated no obvious cell toxicity at concentrations as high as 1000 μM. N-(4-Bromo-2-methoxyphenyl)-2-(3-fluorophenyl)cyclopropane-1-carboxamide (1d) was 3-fold more potent than DFB with excellent BV2 microglial cell viability. N-(Benzo[d]thiazol-2-yl)-2-(3-fluorophenyl)cyclopropane-1-carboxamide (1e) and N-(1H-benzo[d]imidazol-2-yl)-2-(3-fluorophenyl)cyclopropane-1-carboxamide (1f) indicated superior potency to DFB, however, these compounds became toxic to BV2 cells at concentrations higher than 300 μM. 2-(3-Chlorophenyl)-N-(4-fluorophenyl) cyclopropane-1-carboxamide (2b) and N-(2-chlorophenyl)-2-(3-chlorophenyl)cyclopropane-1-carboxamide (2c) indicated an 2.3- and 1.9-fold increase in potency, respectively. These compounds showed no toxicity to BV2 cells at concentrations as high as 1000 μM. N-(4-Bromo-2-methoxyphenyl)-2-(3-chlorophenyl)cyclopropane-1-carboxamide (2d) showed decreased potency. Similar to compound 1f, compound 2f showed improved potency, however, it indicated significant toxicity to cells at concentrations >300 μM. With a reversed-amide linker, 3-fluoro-N-(2-(3-fluorophenyl)cyclopropyl)benzamide (3a) indicated a 4.5-fold increase in potency comparing to DFB, with no detectable toxicity at 1000 μM. Replacement of the 3-fluoro with a chlorine substituent led to compound 3b with a 6.7-fold decrease in potency. Both 3-nitro (3c) and 4-nitro (3d) analogs of compound 3a indicated significant BV2 cell toxicity. To achieve compounds with improved aqueous solubility, N-(2-(3-fluorophenyl)cyclopropyl) picolinamide (3e) was synthesized and tested. However, this compound showed similar protective potency as DFB.
Given the availability of the biological data, further analysis of the utility of the SILCS LGFE data was undertaken. Conversion of the IC50 values to binding free energies (ΔGbind=kBTlog(IC50), kB is the Boltzmann constant, T is the temperature) allows for correlation analysis and calculation of the predictive index (PI)41 with respect to the LGFE scores.
Ligand 1d had the most favorable LGFE score and docked into a conformation such that the fluorobenzene moiety occupied the hydrophobic cavity formed between residues L710 of TM4 and V739, V740, P742 of TM5 (marked A1 in Fig. 2B). The 4-bromo-2-methoxyphenyl moiety occupied a second hydrophobic cavity defined by residues L743 of TM5, W784 of TM6 and V805 of TM7 (marked A2 in Fig. 2B). In comparison, LGFE of 2d with a chlorobenzene moiety that docked in a conformation similar to 1d, was less favorable, due to poor GFE scoring of the meta-substituted chlorine. This is because the fluorine of 1d had better overlap with the acceptor FragMaps in the proximity of R647, compared to chlorine’s overlap with apolar FragMaps at that position. Differences in LGFE scores between these two ligands correlated well the corresponding experimentally measured ΔGbind differences.
Ligand 3a also docked in a conformation similar to 1d, such that the second fluorobenzene moiety of 3a occupied A2 marked in Fig. 2B. The favorable LGFE score of 3a correlated well with its high binding affinity. LGFE scoring was also useful in identifying the favorable ring position of a particular substituent. For instance, -NO2 at the meta-substituted position in 3c scored better than in the para-substituted position of 3d due to better overlaps with the acceptor affinities in the vicinity of T780 in the model. On the other hand, ligands 1b and 2b preferred a binding mode where the para-substituted fluorine occupied the first hydrophobic cavity marked A1 and the meta-substituted chlorobenzene occupied the second site marked by A2 in Fig. 2B. Consequently, in this case, the meta-substituted chlorine had better overlap with the apolar FragMaps compared to the overlap of fluorine with acceptor FragMaps at that site. These differences in LGFE scores across the meta- and para- substituted positions between 3a & 3c and 1b & 2b also correlated well with the experimentally measured ΔGbind differences.
Three of the analogs, 1e, 1f and 2f, containing benzo[d]thiazol and benzimidazole moieties were poorly predicted by the LGFE scores. All these compounds docked such that the larger benzo[d]thiazol and benzimidazole moieties occupied the A2 site. Although the heterocycle carbons had good overlap with the apolar FragMaps of A2, polar nitrogens had poorer overlap with their respective FragMaps, leading to a decrease in the LGFE scores. These structures were hence not included in the LGFE vs. ΔGbind correlation calculation, yielding a predictive index (PI)41 of 0.56 and R2 ~ 0.26. Improved structures of the mGluR5 TM region, versus the presently used homology models, are anticipated to further improve the predictability of the SILCS based modeling.
Along with ranking ligands that are known to bind to a pocket, identifying a favorable ring position of a particular substituent, SILCS FragMaps could also be used to guide ligand optimization studies. LGFE, ΔGbind, toxicity and potency calculations point to the therapeutic potential of ligand 3a in TBI and other neurodegenerative diseases therapy. Binding affinity of this ligand could be further increased through a meta-substituted aliphatic extension to the second fluorobenzene in A2 so as to overlap with the favorable apolar FragMaps in that site. Additionally, a hydroxyl group could be added to the para-substituted position to overlap well with the donor and acceptor FragMaps in the proximity of T780. Future efforts will address these and other possible modifications of the presented compounds.
Conclusion
In summary, we have described the design and synthesis of novel cyclopropyl-containing compounds as potential neuroprotective agents by targeting mGluR5. The synthesized compounds were shown to inhibit LPS stimulated NO production, likely through actions at mGluR. One of the compounds, 3a, indicated an IC50 value of 30 μM, with excellent cell viability. Further inhibition activity of 3a on primary rat cortical neurons and microglia is being investigated and, guided by the SILCS analysis of the PAM binding site, further refinement of the chemical series exemplified by compound 3a will be undertaken.
Supplementary Material
Acknowledgments
This work was supported by the UMB Pilot & Exploratory Interdisciplinary Research (IDR) and University of Maryland Computer-Aided Drug Design Center and NIH Grant R43GM109635.
References
- 1.Guskiewicz KM, Marshall SW, Bailes J, McCrea M, Cantu RC, Randolph C, Jordan BD. Neurosurgery. 2005;57:719. doi: 10.1093/neurosurgery/57.4.719. [DOI] [PubMed] [Google Scholar]
- 2.Spranger M, Fontana A. Neuroscientist. 1996;2:293. [Google Scholar]
- 3.Byrnes KR, Garay J, Di Giovanni S, De Biase A, Knoblach SM, Hoffman EP, Movsesyan V, Faden AI. Glia. 2006;53:420. doi: 10.1002/glia.20295. [DOI] [PubMed] [Google Scholar]
- 4.De Biase A, Knoblach SM, Di Giovanni S, Fan CG, Molon A, Hoffman EP, Faden AI. Physiol Genomics. 2005;22:368. doi: 10.1152/physiolgenomics.00081.2005. [DOI] [PubMed] [Google Scholar]
- 5.Demediuk P, Daly MP, Faden AI. J Neurochem. 1989;52:1529. doi: 10.1111/j.1471-4159.1989.tb09204.x. [DOI] [PubMed] [Google Scholar]
- 6.Faden A, Demediuk P, Panter S, Vink R. Science. 1989;244:798. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 7.Panter SS, Faden AI. Neurosci Lett. 1992;136:165. doi: 10.1016/0304-3940(92)90040-e. [DOI] [PubMed] [Google Scholar]
- 8.Popovich PG, Guan Z, McGaughy V, Fisher L, Hickey WF, Basso DM. J Neuropath Exp Neur. 2002;61:623. doi: 10.1093/jnen/61.7.623. [DOI] [PubMed] [Google Scholar]
- 9.Keane RW, Davis AR, Dietrich WD. J Neurotrauma. 2006;23:335. doi: 10.1089/neu.2006.23.335. [DOI] [PubMed] [Google Scholar]
- 10.Byrnes KR, Loane DJ, Stoica BA, Zhang J, Faden AI. J Neuroinflammation. 2012;9:43. doi: 10.1186/1742-2094-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall ED, Springer JE. NeuroRx. 2004;1:80. doi: 10.1602/neurorx.1.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bramlett HM, Dietrich WD. Acta Neuropathol. 2002;103:607. doi: 10.1007/s00401-001-0510-8. [DOI] [PubMed] [Google Scholar]
- 13.Bramlett HM, Dietrich WD. Prog Brain Res. 2007;161:125. doi: 10.1016/S0079-6123(06)61009-1. [DOI] [PubMed] [Google Scholar]
- 14.Byrnes KR, Garay J, Di Giovanni S, De Biase A, Knoblach SM, Hoffman EP, Movsesyan V, Faden AI. Glia. 2006;53:420. doi: 10.1002/glia.20295. [DOI] [PubMed] [Google Scholar]
- 15.Loane DJ, Stoica BA, Pajoohesh-Ganji A, Byrnes KR, Faden AI. J Biol Chem. 2009;284:15629. doi: 10.1074/jbc.M806139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loane DJ, Stoica BA, Tchantchou F, Kumar A, Barrett JP, Akintola T, Xue F, Conn PJ, Faden AI. Neurotherapeutics. 2014;11:857. doi: 10.1007/s13311-014-0298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sekiyama N, Hayashi Y, Nakanishi S, Jane DE, Tse HW, Birse EF, Watkins JC. Br J Pharmacol. 1996;117:1493. doi: 10.1111/j.1476-5381.1996.tb15312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brabet I, Mary S, Bockaert J, Pin JP. Neuropharmacology. 1995;34:895. doi: 10.1016/0028-3908(95)00079-l. [DOI] [PubMed] [Google Scholar]
- 19.Doherty AJ, Palmer MJ, Henley JM, Collingridge GL, Jane DE. Neuropharmacology. 1997;36:265. doi: 10.1016/s0028-3908(97)00001-4. [DOI] [PubMed] [Google Scholar]
- 20.Wisniewski K, Car H. CNS Drug Rev. 2002;8:101. doi: 10.1111/j.1527-3458.2002.tb00218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakkaraju SK, Xue F, Faden A, MacKerell AD. J Chem Info Model. 2013;53:1337. doi: 10.1021/ci400160x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Brien JA, Lemaire W, Chen TB, Chang RSL, Jacobson MA, Ha SN, Lindsley CW, Schaffhauser HJ, Sur C, Pettibone DJ, Conn PJ, Williams DL. Mol Pharmacol. 2003;64:731. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- 23.O’Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser HJ, Rowe B, Sur C, Duggan ME, Pettibone DJ, Conn PJ, Williams DL. J Pharmacol Exp Ther. 2004;309:568. doi: 10.1124/jpet.103.061747. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Z, Wisnoski DD, O’Brien JA, Lemaire W, Williams DL, Jr, Jacobson MA, Wittman M, Ha SN, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Bioorg Med Chem Lett. 2007;17:1386. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 25.Engers DW, Rodriguez AL, Williams R, Hammond AS, Venable D, Oluwatola O, Sulikowski GA, Conn PJ, Lindsley CW. Chem Med Chem. 2009;4:505. doi: 10.1002/cmdc.200800357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma S, Kedrowski J, Rook JM, Smith RL, Jones CK, Rodriguez AL, Conn PJ, Lindsley CW. J Medicinal Chem. 2009;52:4103. doi: 10.1021/jm900654c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ritzén A, Sindet R, Hentzer M, Svendsen N, Brodbeck RM, Bundgaard C. Bioorg Med Chem Lett. 2009;19:3275. doi: 10.1016/j.bmcl.2009.04.095. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon UN, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Mol Pharmacol. 2010;78:1105. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xue F, Stoica BA, Hanscom M, Kabadi SV, Faden AI. CNS & Neurol Disord drug targets. 2014;13:558. doi: 10.2174/18715273113126660199. [DOI] [PubMed] [Google Scholar]
- 30.Guvench O, MacKerell AD. PLOS Comput Biol. 2009;5:e1000435. doi: 10.1371/journal.pcbi.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, Niswender CM, Katritch V, Meiler J, Cherezov V. Science. 2014;344:58. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raman EP, Yu W, Lakkaraju SK, MacKerell AD., Jr J Chem Info Model. 2013;53:3384. doi: 10.1021/ci4005628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lakkaraju SK, Yu W, Raman EP, Hershfeld A, Fang L, Deshpande DA, MacKerell AD. J Chem Info Model. 2015;55:700. doi: 10.1021/ci500729k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lakkaraju SK, Raman EP, Yu W, MacKerell AD. J Chem Theory Comput. 2014;10:2281. doi: 10.1021/ct500201y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He X, Lakkaraju SK, Hanscom M, Zhao Z, Wu J, Stoica B, MacKerell AD, Jr, Faden AI, Xue F. Bioorg Med Chem. 2015 doi: 10.1016/j.bmc.2015.02.054. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, Fischer C, Porter RH. Mol Pharmacol. 2003;64:823. doi: 10.1124/mol.64.4.823. [DOI] [PubMed] [Google Scholar]
- 37.Trott O, Olson AJ. J Comput Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McPhillie MJ, Trowbridge R, Mariner KR, O’Neill AJ, Johnson AP, Chopra I, Fishwick CWG. ACS Med Chem Lett. 2011;2:729. doi: 10.1021/ml200087m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pryde DC, Cook AS, Burring DJ, Jones LH, Foll S, Platts MY, Sanderson V, Corless M, Stobie A, Middleton DS, Foster L, Barker L, Van der Graaf P, Stacey P, Kohl C, Coggon S, Beaumont K. Bioorg Med Chem. 2007;15:142. doi: 10.1016/j.bmc.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 40.Li HY, Xue FT, Kraus JM, Ji HT, Labby KJ, Mataka J, Delker SL, Martasek P, Roman LJ, Poulos TL, Silverman RB. Bioorg Med Chem. 2013;21:1333. doi: 10.1016/j.bmc.2012.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pearlman DA, Charifson PS. J Med Chem. 2001;44:3417. doi: 10.1021/jm0100279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.