

Abstract
Diverse repertoires of antigen-receptor genes that result from combinatorial splicing of coding segments by V(D)J recombination are hallmarks of vertebrate immunity. The (RAG1-RAG2)2 recombinase (RAG) recognizes recombination signal sequences (RSSs) containing a heptamer, a spacer of 12 or 23 base pairs, and a nonamer (12-RSS or 23-RSS) and introduces precise breaks at RSS-coding segment junctions. RAG forms synaptic complexes only with one 12-RSS and one 23-RSS, a dogma known as the 12/23 rule that governs the recombination fidelity. We report cryo-electron microscopy structures of synaptic RAG complexes at up to 3.4 Å resolution, which reveal a closed conformation with base flipping and base-specific recognition of RSSs. Distortion at RSS-coding segment junctions and base flipping in coding segments uncover the two-metal-ion catalytic mechanism. Induced asymmetry involving tilting of the nonamer-binding domain dimer of RAG1 upon binding of HMGB1-bent 12-RSS or 23-RSS underlies the molecular mechanism for the 12/23 rule.
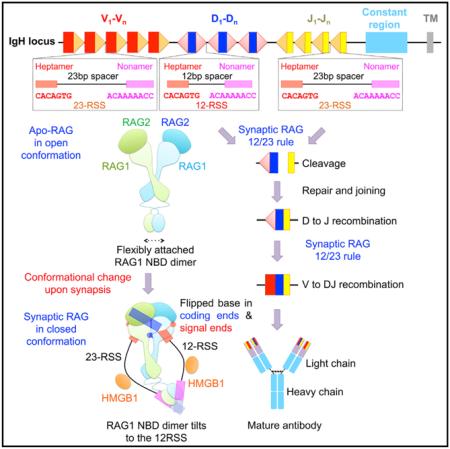
Graphical Abstract
INTRODUCTION
For optimal host defense, jawed vertebrates have evolved an elegant combinatorial mechanism to generate large repertoires of antibody and antigen-receptor genes. The V(D)J recombination process cleaves and splices variable (V), diversity (D), and joining (J) non-contiguous immunoglobulin (Ig) segments in the genome (Fanning et al., 1996; Tonegawa, 1983). Ig heavy chains and T cell receptor (TCR) b chains are formed by sequential steps of D-J and V-DJ recombination, while Ig light chains and TCR a chains are generated by direct VJ recombination. The critical cleavage step in V(D)J recombination is executed by the lymphocyte-specific enzyme containing the multi-domain proteins recombination-activating gene 1 and 2 (RAG1 and RAG2) (Oettinger et al., 1990; Schatz et al., 1989) (Figure 1A). RAG recognizes specific recombination signal sequences (RSSs) flanking the 3′ end of the V, D, and J segments, which are composed of a conserved heptamer, a spacer of either 12 or 23 base pairs (bp), and a conserved nonamer (Akira et al., 1987; Ramsden et al., 1994) (Figures 1B and 1C). These RSSs are designated as 12-RSS or 23-RSS after the length of the spacer. Splicing can only occur between one gene coding segment flanked by a 12-RSS and another segment flanked by a 23-RSS, establishing the 12/23 rule (Schatz and Swanson, 2011). Because V, D, and J segments are flanked by different RSSs such as in the IgH locus (Figure 1C), the 12/23 rule helps to ensure recombination between V, D, and J, but not within homotypic gene segments.
Figure 1. Cryo-EM Structure Determination.

(A) Domain organization of zebrafish RAG1 and RAG2 (zRAG1 and zRAG2). NTD, N-terminal domain; RING ZnA, RING domain and zinc finger A domain; NBD, nonamer binding domain; DDBD, dimerization and DNA binding domain; PreR, pre-RNase domain; RNH, RNase H-like domain; ID/ZnB, insertion domain/zinc binding domain B; CTD, C-terminal domain; CTT, C-terminal tail; WD40, WD40 repeat domain; and PHD, plant homeodomain. Approximate domain boundaries are shown by residue numbers. Potential active site residues are indicated as red dots in RNH.
(B) Recombination signal sequences used in this study. Heptamer sequences (red) are numbered from 1 to 7 for the forward strand and from 1* to 7* for the reverse strand. Coding segment ends are numbered from −16 to −1 for the forward strand and from −16* to −1* for the reverse strand. The nicking site is between A-1 (coding end) and C1 (signal end) with a 3′-OH on the A-1.
(C) Overview of V(D)J recombination. In the red box, RSSs flanking V, D, and J segments in the IgH locus are illustrated. The dimeric (RAG1-RAG2)2 complex is shown as stacked cyan and lemon green trapezoids. Briefly, RAG binds a single RSS in the presence of HMGB1 to form a signal complex (SC), either 12-SC or 23-SC, which can undergo nicking at the coding end-signal end junction in the presence of Mg2+. Synapsis of one 12-RSS and one 23-RSS in the same RAG dimer forms the paired complex (PC), followed by generation of the cleaved signal complex with hairpin coding end and cleaved signal end. Hairpin release produces the signal end complex (SEC). Further processing by enzymes in the non-homologous end-joining DNA repair pathway results in ligation of the coding ends and circularization of the signal ends.
(D) Representative 2D class averages of cryo-EM particles of the SEC.
(E and F) Flow charts of cryo-EM structure determination for the SEC (E) and the PC (F). The two numbers in the blue box are the resolutions of the entire molecule and the NBD region only, respectively.
See also Figure S1.
The RAG complex catalyzes two consecutive reactions, nicking (strand cleavage) and hairpin formation (strand transfer), without dissociation. First, it binds either a 12-RSS substrate or a 23-RSS substrate and introduces a nick precisely at the junction between the coding segment and the RSS. Interactions with both the conserved heptamer and nonamer are required for optimal RAG activity because considerable sequence variation in endogenous RSSs substantially affects RAG binding affinity and recombination frequency (Schatz and Swanson, 2011). When a 12-RSS and a 23-RSS are bound to the same RAG, a synaptic, paired complex (PC) is formed (Figure 1C). Second, upon PC formation, the free 3′-hydroxyl released from the nicking step attacks the opposing strand to create a hairpin coding segment and a blunt signal end, generating the cleaved signal complex (CSC) (Figure 1C). Dissociation of gene segment hairpins results in a signal end complex (SEC) (Figure 1C). Proteins in the classical nonhomologous end joining (NHEJ) DNA repair pathway are recruited to the RAG complex to process and join the coding segments (Lieber, 2010). In vitro, high-mobility group (HMG) proteins such as HMGB1 have been shown to stimulate RAG’s activity in DNA binding, nicking, and hairpin formation, presumably by inducing RSS bending (Schatz and Swanson, 2011).
Many RAG mutations have been identified in humans that are associated with a spectrum of genetic disorders ranging from severe combined immunodeficiency (SCID) to milder variants, such as Omenn syndrome (OS), RAG deficiency with gd T cell expansion, granuloma formation, or maternofetal engraftment (Lee et al., 2014; Schatz and Swanson, 2011). Aberrant V(D)J recombination is an important mechanism responsible for chromosomal translocations in cancer and autoimmunity (Brandt and Roth, 2009). Despite extensive structural pursuits, the only known RAG structure in complex with DNA is that of the isolated nonamer-binding domain (NBD) dimer with a nonamer sequence (Yin et al., 2009). Here, we report cryo-electron microscopy (cryo-EM) structures of the core RAG complex in the absence of DNA and in the presence of RSS intermediates and products. These structures, representing the apo-form, the nicked paired complex, and the cleaved signal end complex, capture snapshots in RAG-mediated catalysis with additional implications for mechanistically related transposases and integrases.
RESULTS
Cryo-EM Structure Determination
Previous biochemical studies on the RAG complex almost exclusively utilized the mouse recombinant proteins (Schatz and Swanson, 2011). To tackle the long-standing structural questions on RAG, we screened RAG1 and RAG2 from different vertebrate species using both insect and mammalian cell expression systems. We selected insect cell-expressed zebrafish RAG (zRAG) for further studies due to its higher expression level and favorable behavior in solution. We reconstituted the apo-RAG1-RAG2 complex (Apo-RAG) containing RAG1 (271–1031) and RAG2 (full-length) (Figure 1A), its complex with 12-RSS and 23-RSS signal ends (SEC) in the presence of Mg2+, and its complex with paired, nicked 12-RSS and 23-RSS intermediates (PC) in the presence of Ca2+, which was reported to inhibit RSS cleavage by RAG (Grundy et al., 2009) (Figure 1B). Consistently, our PC sample showed only nicked RSSs on a urea PAGE (Figure S1A). Human HMGB1, which is nearly identical with zebrafish HMGB1 (Figure S1B), was included in both SEC and PC reconstitutions.
We collected cryo-EM images for the SEC and PC samples, and two-dimensional (2D) class averages showed a distribution of different views of the complex (Figures 1D and S1C). Cryo-EM structure determination using multiple rounds of three-dimensional (3D) classification and refinement resulted in a number of density maps, including 2-fold symmetrized Apo-RAG, PC, and SEC at 9.0, 3.7, and 3.4 Å resolutions, respectively, and non-symmetrized PC at an overall resolution of 4.6 Å with an NBD/nonamer region resolution of 7.2 Å (Figures 1E, 1F, S1D, and S1E). The Apo-RAG maps refined with and without applying symmetry appeared similar, supporting the validity of the 2-fold symmetry (Figure S1F). While the Apo-RAG adopts a similarly open configuration as the mouse RAG1-RAG2 (mRAG1-RAG2) crystal structure (Kim et al., 2015), the SEC and PC complexes with DNA appear in a closed and more compact conformation (see below in Figure 3).
Figure 3. Open Conformation of Apo-RAG and Closed Conformation of Synaptic Complexes.

(A) Side and top views of RAG in complex with the complete 12-RSS and 23-RSS intermediates in the PC.
(B) Superimposition of RAG1-RAG2 monomers between the PC model (cyan and blue) and the crystal structure (PDB 4wwx, yellow).
(C) Superimposition of NBD dimers between the PC (cyan and lemon green) and the crystal structure (magenta and red, PDB: 4wwx).
(D) Top view of superimposed PC dimer in the closed conformation and Apo-RAG dimer in the open conformation. One RAG1-RAG2 monomer is aligned, and the difference in the location of the symmetric monomer is evident. The magnitude of displacement required to switch between the two conformations is indicated.
(E) Rigid body fit of the mRAG dimer crystal structure (yellow) in the 9.0 Å Apo-RAG map (light blue).
(F) Top and bottom views of the new interactions in the closed conformation between RAG1 of one monomer and RAG2 of the symmetric monomer and between two RAG1 subunits. Potential interactions are displayed as yellow dashed lines.
(G) The RAG1 β4-β5 loop and the RAG2 β27-β28 loop involved in interactions in the closed conformation are disordered in mRAG in the open conformation. A potential charged interaction is highlighted, including residue R860 that is equivalent to R841 mutated in human RAG patients.
See also Figure S3.
Atomic models were built into the 3.4 Å symmetrized SEC, 3.7 Å symmetrized PC, and 4.6 Å non-symmetrized PC maps using the crystal structures of the mRAG1-RAG2 complex (Kim et al., 2015) (PDB 4wwx) and the isolated nonamer-bound NBD dimer as references (Yin et al., 2009) (PDB 3gna), and refined (Table S1). The 9.0 Å resolution Apo-RAG map was first fitted with the refined RAG1-RAG2 monomer from SEC and later refitted directly with the mRAG1-RAG2 dimer from the crystal structure (Kim et al., 2015). Because the structures of SEC and PC reconstructions are largely identical, the cryo-EM particles were also grouped together to increase the data size and generated a structure at 3.3 Å resolution (Figures S1E, S1G, and S1H). These maps were used for cross-references in model building. The map/model Fourier shell correlation (FSC) curves and local resolution estimations are consistent with the gold-standard resolutions from FSC curves between half maps of split data (Figures S1D, S1E, and S2A–S2D).
Overview of PC and SEC Maps and Models
The cryo-EM maps revealed dimeric (RAG1-RAG2)2 structures with DNA chains running through the entire lengths of the complexes (Figures 2A–2C, S2E, and S2F). The RAG1 and RAG2 densities lacked the N-terminal RING and the C-terminal plant homeodomain (PHD) in the constructs, respectively (Figure 1A), likely due to the flexibility of these domains relative to the core regions. For symmetrized PC and SEC, in which the NBD is largely invisible, the final models contain residues 480–1,029 of zRAG1 (the active site region in Figure 1A), residues 1–351 of zRAG2 (the core region in Figure 1A), and 124 and 116 nucleotides, respectively (Figures 2A, 2C, S2E, and S2F). The excellent superimposition of cryo-EM densities with the refined model is shown for representative regions of the SEC map (Figure 2D). For non-symmetrized PC, by tracing from the nonamer DNA in the superimposed, previous crystal structure of the nonamer-bound NBD dimer (Yin et al., 2009), we could unambiguously fit the DNA sequences in which all the nucleotides were counted for. The final model contains residues 408–1,029 of zRAG1 (the core region in Figure 1A), residues 1–351 of zRAG2, and the 222 nucleotides in the entire 12-RSS and 23-RSS intermediates (Figure 2B and Movie S1).
Figure 2. Overview of Synaptic RAG Complexes.

(A) Orthogonal views of the symmetrized 3.4 Å resolution SEC map. One RAG1 is omitted in order to present the bound DNAs (red and orange) inside the protein clearly. For the side view, one RAG2 is also omitted to display the density for the flipped C1 base at the RSS.
(B and C) Atomic models superimposed with the non-symmetrized 4.6 Å resolution PC map (B) and the symmetrized 3.7 Å resolution PC map (C), respectively.
(D) Representative regions of the SEC map superimposed with the final model.
(E) Side-by-side comparison of the DNA models in symmetrized PC (orange) and SEC (cyan). Both schematic and ribbon models are shown. Locations of flipped bases are labeled.
(F) Orthogonal views of superimposed symmetrized SEC (cyan), symmetrized PC (orange), and non-symmetrized PC (slate) structures.
See also Figure S2.
Although no coding end DNA was included in the preparation, the SEC map contains density above the active site that mimics the coding end DNA as seen in the PC (Figures 2A and S2E). We reasoned that the density comes from RSSs non-specifically bound to the coding end binding site. This is supported by the variation on the lengths and protruding angles of the DNA duplex bound at this site among the 3D classes of the SEC complex due to the different lengths of RSSs (34 bp and 45 bp) available for this interaction (Figure 1E). By contrast, the coding end DNA (16 bp) of the PC complex appears to protrude to a homogeneous length (Figure 1F). In the 3.4 Å symmetrized SEC map, 14 base pairs appear bound symmetrically and therefore visible at this coding end mimic. Similarly, in the crystal structure of the eukaryotic transposase Mos1, precleaved transposon signal ends mimicked non-specific interactions by the flanking DNA (Richardson et al., 2009).
Although nicked DNA and blunt-ended DNA are present in the PC and the SEC, respectively (Figure 2E), a superposition showed that the overall structures of PC and SEC are highly similar (Figure 2F). The only gross difference is the visible NBD dimer and the longer RSS in the non-symmetrized PC (Figures 2B and 2F). The RAG protein dimer has a previously described Y shape (Kim et al., 2015). In non-symmetrized RAG/DNA complexes, this shape now resembles a butterfly with DNA chains at the top as antennas and DNA chains at the bottom that emphasize the tail (Figure 3A). This butterfly shape is also visible in some 2D averages of SEC and PC particles (Figures 1D and S1C). Because the 2-fold axis of the NBD dimer does not coincide with that of the RAG active site dimer and due to lack of symmetry at the bound 12-RSS and 23-RSS, symmetrized PC and SEC both lacked the butterfly tail (Figures 2A and 2C). The active site regions of RAG1-RAG2 monomers in all RAG structures are highly conserved with pairwise RMSDs of ~0.6 Å . When compared with a RAG1-RAG2 monomer from the mRAG1-RAG2 crystal structure (Kim et al., 2015), the NBD of RAG1 in the PC exhibits a dramatically different orientation (Figure 3B). The NBD dimers alone align well with each other (Figure 3C), suggesting a rigid body movement of the NBD dimer upon DNA binding. When the NBD region was excluded, individual RAG1-RAG2 monomers in PC and SEC aligned separately with the crystal structure at pairwise RMSDs of ~1.5 Å (Figure 3B). Local differences, in particular near the active site, are apparent (see below), as also shown by changes in secondary structures (Figures S3A and S3B).
Closure of the RAG Dimer upon DNA Binding
The RAG1-RAG2 monomers have a highly conserved mode of dimerization in the DNA-bound complex structures as the dimers superimpose to pairwise root-mean-square deviations (RMSDs) of ~0.8 Å (Figure 2F). However, this conserved relative orientation between the RAG1-RAG2 monomers is dramatically different from that in the Apo-RAG (Figure 3D). Up to ~27 Å movements in Cα positions are observed, which draw the two halves of the dimer closer together (Figure 3D). Consequently, the Apo-RAG is much more open, as shown by the gap between the two subunits (Figure S3C), which is closed upon DNA binding (Figure 3D). Therefore, we refer to Apo-RAG as the open conformation and DNA-bound RAG complexes as the closed conformation (Figure 3D). We found that the crystal structure of mRAG1-RAG2 (Kim et al., 2015) fits well with the symmetrized Apo-RAG cryo-EM density (Figure 3E). These observations suggest that, although 12-RSS and 23-RSS were both present in washed crystals, the mRAG1-RAG2 complex was crystallized in an apo-form, explaining the lack of DNA density. The low resolution of the Apo-RAG cryo-EM structure also suggests a highly dynamic property in the absence of DNA interaction.
Extensive RSS-induced interactions are observed between RAG1 from one RAG monomer and RAG2 from the symmetric RAG monomer and between the two RAG1 subunits (Figure 3F), which are completely absent in Apo-RAG. The RAG1-RAG2 interaction is mostly mediated by polar contacts between the α15 helix in RAG1 and the α1 helix and the β27-β28 loop in RAG2 (Figures 3F, S3A, and S3B). The RAG1-RAG1 interaction is mediated by the β4-β5 loop, α15, and the α15-α16 loop (Figures 3F and S3A). The β4-β5 loop is in the RNH domain, which is involved in RSS recognition and harbors the catalytic residues (see below). The interfacial residues in the closed conformation are largely conserved across species (Figures S3A and S3B). RAG disease mutations R841Q and R841W, equivalent to mutations on R860 in zRAG1, are associated with combined immune deficiency with granuloma and/or autoimmunity (Lee et al., 2014) (Figure S3A and Table S2). R860 resides on α15 at the closed dimerization interface and likely participates in charged interactions with E627 in the β4-β5 loop of RAG1 and E334 in the β27-β28 loop of RAG2 (Figures 3F and 3G). The β4-β5 loop of RAG1 and the β27-β28 loop in RAG2 are mostly disordered in Apo-RAG (Figure 3G). Because binding of each RSS requires both RAG1 monomers, the RSS interaction induces the dimer closure, may be sufficient to stabilize the closed formation, and is critically important for catalysis (Figure 4).
Figure 4. RSS Recognition at the Heptamer and the Spacer.

(A) Electrostatic surface of the RAG dimer from the symmetrized SEC model with bound RSSs (orange) and coding end mimics (magenta) in ribbons. The cyan oval shows the RAG surface for one RSS.
(B) Side and bottom views of the RAG dimer displayed as molecular surfaces with bound DNAs displayed in ribbons. For the reference RSS, nucleotides with base recognition by RAG are highlighted in blue, whereas others are shown in orange. Domains involved in RSS interactions are colored as in Figure 1A, except that one DDBD at the bottom view is shown in blue to distinguish it from the neighboring DDBD. Two RAG2 molecules are shown in gray. A prime symbol is added to domain names for the symmetric subunit that interacts with the reference RSS.
(C) Top and side views of RAG1 regions (magenta) that are in contact with the heptamer DNA. RSSs are colored as in (B). RAG2 subunits are omitted.
(D) A bound RSS colored as in (B) superimposed with an ideal B-form DNA (light blue), showing the widening of the minor groove. The α23-α24 loop inserted into the minor groove of the RSS is highlighted in magenta.
(E) Schematic depiction of the detailed interactions between RAG and the RSS. RAG1 residues from one and the symmetric subunits are displayed as cyan and lemon green ovals, respectively. These ovals are bounded in blue lines, no lines, and red lines, respectively, for side-chain, main-chain, and side- and main-chain interactions. Slate, green, magenta, and gray arrows indicate interactions at the major groove, minor groove, flipped base, and the backbone. Heptamer bases that have multiple contacts with RAG are highlighted in red, and those with a single contact are in pink.
(F) Detailed interactions between RAG and the heptamer DNA. Direct base interactions are highlighted in magenta for the protein and in green for the bases. Potential interactions are displayed as yellow dashed lines.
(G) Superimposed apo-like RAG crystal structure (magenta and red, PDB: 4wwx) and SEC (lemon green and cyan) at the heptamer-binding site. One RAG1 subunit (red and cyan) is aligned, showing the different orientations of the symmetric RAG1 (magenta and lemon green), especially at the β4-β5 loop that was disordered in the crystal structure, and the α23-α24 loop.
(H) Detailed interactions between RAG and the flipped C1 of the heptamer DNA.
See also Figure S4.
Cooperative RSS Recognition by RAG1 with Base Flipping
The RAG/DNA complex structures provide the first glimpse of RSS recognition at the heptamer. Because nearly identical RSS interactions are seen in the SEC and the PC (Figure S4A), we used the symmetrized SEC at 3.4 Å resolution for the structural description. The bound 12-RSS and 23-RSS are visible at the first 15 positions, suggesting that these positions of the RSSs are symmetrically arranged in the RAG dimer (Figures 2E and S2E). The first 12 positions of the RSS, including the heptamer and the first 5 positions of the spacer, form the region that directly contacts RAG. Consistently, the first 5 positions in the spacer of both 12-RSS and 23-RSS represent the most consecutively conserved spacer segments across genomes (Ramsden et al., 1994). Complementary electrostatics is displayed at the RSS-binding site of RAG1 (Figure 4A) with extensive sugar phosphate backbone contacts (Figures S4B and S4C).
An RSS is recognized by both subunits of RAG1 in the RAG dimer, with the beginning part mainly recognized by the insertion domain (ID) and RNase H-like domain (RNH) of one subunit and the more distal part recognized by the RNH, dimerization and DNA binding domain (DDBD), and C-terminal domain (CTD) of the symmetric subunit (Figures 4B and 4C). The interactions are mostly at the minor groove of the RSS, with significant widening in this region (Figure 4D). Specific base contacts are restricted to the heptamer only. Multiple interactions are observed at the first three positions (Figure 4E), explaining the perfect sequence conservation of these nucleotides across genomes (Ramsden et al., 1994). Helices α16 and α23 (Figure S3A) of one RAG1 subunit interact with beginning positions of the heptamer at the major and minor grooves, respectively (Figures 4C, 4E, and 4F). The α23-α24 loop of the symmetric subunit recognizes bases at more distal positions of the heptamer at the minor groove (Figures 4C, 4E, 4G, and S4D).
Grafting a bound RSS to one RAG1 subunit in the apo-like mRAG crystal structure (Kim et al., 2015) shows that the symmetric RAG1 needs to pivot in order to interact with the RSS (Figure 4G), a movement that is also evident in the comparison between Apo-RAG and DNA-bound RAG conformations (Figure 3D). Upon the RSS binding-induced structural shift, the β23-β24 loop of the symmetric RAG1 moves in position to contact multiple bases at the minor groove (Figures 4E, 4G, and S4D). The β4-β5 loop of the RNH domain, which was disordered in the Apo-RAG crystal structure, becomes ordered and interacts with the RSS (Figure 4G) and contributes to the dimer closure (Figures 3F and 3G). Thus, binding of one RSS induces conformational changes in both RAG1 monomers, and this cooperativity might facilitate formation of the 12-RSS and 23-RSS paired complex. Notably, the catalytic residue E984 in the active site situates on α23. Therefore, the binding of RSS may also induce catalytically competent conformations in RAG1 (see below in Figure 6).
Figure 6. Two-Metal-Ion Catalysis and Conformational Remodeling.

(A) Difference density between the PC cryo-EM map and the model without metal ions showing the proposed Ca2+ sites. Ca2+ sites A and B are shown as slate spheres and active site residues highlighted in magenta.
(B) Active site of the PC model around the Ca2+ sites (slate spheres, A and B). Ca2+ A and B are jointly coordinated by the scissile phosphate of the flipped nucleotide T-1* and D620. A is additionally coordinated by O3′ of G1* (the leaving group), E984, and the carbonyl oxygen of G621, and B is coordinated by 3′-OH of A-1 (the nucleophile), E684, and D730. Potential coordination of the metal ions is displayed using yellow dashed lines.
(C) Proposed pentacovalent phosphate intermediate in two-metal-ion catalysis of hairpin formation by RAG.
(D) Active sites of the SEC models. Mg2+ ions in SEC are presented as blue spheres. Briefly, A is coordinated by five ligands, 3′-OH of G1*, 3′-OH of the non-specifically bound DNA, D620, E984, and the carbonyl oxygen of G621. B is coordinated by four ligands, 3′-OH of the non-specifically bound DNA, E684, D730, and the carbonyl oxygen of G621.
(E) Superimposed PC and SEC models at the RNH domain, showing the shifted metal ions, the aligned −1 nucleotides, and the different positions of the −1* nucleotides in PC and SEC.
(F) Superimposed active sites of the PC (cyan) and the apo-like mRAG crystal structure (PDB: 4wwx, yellow). The D(E)DE motif side chains are shown. The E984 residue changes by ~3.2 Å in Cα position from its location on a disordered loop in the apo-conformation to its location on α23 in the PC. Active site residues in the PC and the crystal structure are highlighted in magenta and blue, respectively. Red arrows indicate regions of local conformational changes.
See also Figure S6.
A surprising observation is that the base of nucleotide C1 of the heptamer, which is either nicked or cleaved at this position in PC and SEC, flips out from the duplex (Figures 4E and 4H). It is recognized extensively by the region of β18-β19 in the ID of RAG1, including K912, P913, and R916 on the β18 helix; S917 and T918 on the β18-β19 loop; and D923 on β19 (Figures 4E and 4H). Multiple potential interactions are present at both the cytosine base and the ribose, putting a specific anchor at this position (Figure 4H). Inspection of the structure suggests that the base flipping is also necessary to avoid a clash with the closed conformation of RAG, which would have occurred if nicked C1 stayed in the duplex form (Figure 5A).
Figure 5. Interactions at the Coding Segment Ends.

(A) Superposition of an ideal B-form DNA (light blue) with the nicked RSS DNA (orange) from the symmetrized PC model. The distorted base T-1* is highlighted in blue. Parts of α14, α16, and the α15-α16 loop (yellow) of RAG1 clash with the B-form DNA.
(B) Cryo-EM density maps superimposed with the final DNA models (show in sticks) in the symmetrized PC (orange) and SEC (cyan).
(C) Side view of RAG/DNA interactions in the PC. Only one RAG1-RAG2 monomer is shown and displayed as a molecular surface. Nicked RSS intermediate (orange) is shown as ribbons. Domains in RAG1 involved in the interactions are colored as in Figure 1A.
(D) Electrostatic surface of one RAG1-RAG2 monomer in the PC model. The cyan ovals denote the positively charged patches in RAG1 and RAG2, respectively.
(E) Schematic depiction of the detailed interactions between RAG and the coding end in the PC. RAG1 and RAG2 residues are displayed as lemon green and green ovals, respectively. These ovals are bounded in blue lines, no lines, and red lines, respectively, for side-chain, main-chain, and side- and main-chain interactions. Green, magenta, and gray arrows indicate interactions at the minor groove, flipped base, and the backbone.
(F) Detailed interactions at the flipped T-1* base in the nicked DNA intermediate. T-1* is highlighted in marine blue. Residues that have direct contacts with T-1* and nearby nucleotides are shown in cyan. Potential interactions are displayed as yellow dashed lines.
(G) Top view of the flipped T-1* bases (magenta surfaces) in a deep pocket of the RAG dimer (surfaces in cyan, blue, lemon green, and green). The coding ends (orange and red) are shown as ribbons.
See also Figure S5.
Distortion and Base Flipping in Coding End DNA Recognition
From an overview of the PC model containing nicked DNA, the RSS and the coding segment almost appear continuous. When a standard B-form DNA duplex is superimposed with the RSS, it extends into the coding end binding site (Figure 5A), suggesting that an intact RSS substrate with both coding DNA and RSS may bind in a similar fashion. However, as discussed above, the observed clash of the modeled duplex with RAG suggests that a RAG complex with an intact RSS substrate may be in a partially, instead of a fully closed, conformation. In addition to sharing structural features at the RSS-binding site, the nicked PC and the cleaved SEC structures possess similarities at the coding ends, with clearly defined electron densities (Figure 5B). Superposition of the symmetrized PC and SEC structures shows that the PC coding end DNA is almost identical with the SEC coding end mimic, displaying only local deviations in the duplex positioning (Figure 2F).
Unlike RSS recognition, in which both RAG subunits of the dimer participate, each RAG1-RAG2 monomer interacts with one coding end DNA chain exclusively. The beginning of the coding end close to the active site interacts mostly with a highly positively charged patch on ID and RNH of RAG1, while the more distal part of the coding segments interacts with a highly positively charged patch on RAG2 (Figures 5C and 5D). Therefore, RAG2 is also important in DNA binding, a role that was not ascribed previously. The coding end DNA is recognized mostly by electrostatic interactions with few base pair interactions (Figures 5E, S5A, and S5D), which is consistent with lack of sequence conservation in the DNA.
In the nicked RSS intermediates in the PC, there is significant distortion in the DNA conformation at the junction between the RSS and the coding end, when the continuous strand of the DNA abruptly breaks off from the duplex trajectory (Figures 5A and 5B). The single-stranded junction essentially unwinds completely, and the terminal base T-1* of the coding end is flipped to the opposite side of the DNA chain (Figures 2E and 5B). This distortion places the scissile phosphate of T-1*, which is to be attacked by the free 3′-hydroxyl of A-1 for hairpin formation, to the already formed active site that generated the nicking at the opposing strand. A number of previous biochemical studies suggested that T-1* flips out of the coding end duplex during hairpin formation (Bischerour et al., 2009; Schatz and Swanson, 2011). Models for the mechanism of flipping led to proposals involving specific aromatic stacking, for example, by residues of mRAG1 equivalent to W915, Y957, W978, and F993 of zRAG1 (Schatz and Swanson, 2011).
Surprisingly, in the PC structure, the flipped base T-1* not only has no stacking interactions but also does not appear to form any specific polar interactions. Only van der Waals contacts to the M869 side chain and the aliphatic portion of the R870 side chain were observed (Figure 5F). The previously identified aromatic residues are located away from the T-1* nucleotide (Figures S5E–S5G). Only Y957 potentially interacts with the bound DNA at the phosphate group of A-1 (Figure 5F). The base of A-1 is stacked with the guanidinium group of R870 (Figure 5F). Instead of using base stacking, the position of the T-1* nucleotide is held by interactions at the ribose and the phosphate group, in particular, to R870 and H817 (Figure 5F). Because T-1* is a variable nucleotide at the coding end, it is reasonable that the conformation of the nucleotide is stabilized by interactions with the sugar phosphate backbone. The pocket for the flipped T-1* base locates near the 2-fold axis and is deep, highly solvent accessible, and able to accommodate larger bases such as purines (Figure 5G). Previously, an alternative model not involving aromatic stacking correctly predicted the accommodation of the flipped base in a non-specific pocket (Bischerour et al., 2009), as we observed here.
Metal Ion Binding and Catalytic Mechanisms
RAG1 contains a metal ion binding catalytic D(E)DE motif (D620, E684, D730 and E984) within the split RNH domain (Figure 1A) and belongs to the DDE family recombinases (Montanño and Rice, 2011). We used Mg2+ (the catalytic ion) and Ca2+ (a replacement ion), respectively, in the reconstitution of the SEC and the PC. Cryo-EM difference density suggested the presence of at least two metal ions in these structures (sites A and B) (Figures 6A and S6A). The positions of A and B in the PC structure (Figures 6A–6C) and in the SEC structure (Figures 6D and 6E) are somewhat shifted, likely because the coding end mimic duplex is not an authentic intermediate in the reaction. When compared with the nicked DNA, the coding end mimic occupies a similar −1 nucleotide position but a completely different −1* nucleotide position (Figures 6E and 1B).
In the PC structure containing the authentic nicked RSS reaction intermediate, the metal ions A and B are jointly coordinated by the scissile phosphate of the flipped nucleotide T-1* and the D(E)DE motif. A and B are additionally coordinated by O3′ of G1* and 3′-hydroxyl of A-1, which are, respectively, the leaving group and the attacking nucleophile in hairpin formation (Figure 6B). Therefore, while ion A stabilizes the leaving group and B activates the nucleophile, A and B together stabilize the pentacovalent intermediate (Figure 6C), poised for hairpin formation. The coordination environment of A and B in the PC is analogous to that in a substrate-bound RNase H structure but with switched roles of A and B (Nowotny et al., 2005) (Figure S6B). We propose that A and B oscillate their roles between the two RAG-catalyzed consecutive phosphoryl transfer reactions, the nicking step and the hairpin formation step, as similarly shown in other systems (Nowotny et al., 2005).
The catalytic D(E)DE motif appears to undergo significant conformational changes upon DNA binding. In the apo-like crystal structure of mRAG1-RAG2 (Kim et al., 2015), the D(E)DE motif residues are not properly positioned for metal ion coordination as seen in the PC structure (Figure 6F). Especially, the E962 (equivalent to zRAG1 E984) catalytic residue situates in a loop immediately preceding the α23 helix. In SEC or PC, α23 is much longer and E984 situates on α23, with 3.2 Å distance away in the Ca position from the equivalent E962 (Figure 6F). Of note, mRAG1 and zRAG1 have exactly the same sequence in the region around the α23 helix (Figure S3A). Two cooperative interactions, one from α23 of one RAG1 and the other from the α23-α24 loop of the symmetric RAG1 (Figure 4C), may be responsible for the coupled RSS binding-induced active site formation.
NBD Dimer Conformation and HMGB1-Induced RSS Bending
We used non-symmetrized PC structure to analyze the location of the NBD dimer and the highly bent conformations of bound 12-RSS and 23-RSS intermediates (Figure 7A). Similar observations were seen in the non-symmetrized reconstructions from merged datasets (Figure S1H). The coding segment, heptamer, and beginning of the spacer of an RSS are essentially 2-fold symmetric. Starting from the remainder of the spacer, the 12-RSS and 23-RSS each assume a different chain trajectory. While the 12-RSS spacer traverses about one turn as a DNA duplex, the 23-RSS spacer traverses about two turns, creating asymmetric conformations at the nonamer (Figure 7A). Both 12-RSS and 23-RSS duplexes are exceedingly bent, by ~60° for the 12-RSS and ~120° for the 23-RSS (Figures 7A and S7A). The bends occur most severely near the end of the spacer for the 12-RSS and near the middle of the spacer for the 23-RSS.
Figure 7. NBD Flexibility and RSS Bending by HMGB1 Underlie the 12/23 Rule.

(A) Surface representation of the non-symmetrized synaptic PC model in two orientations, with indicated bending at the 12-RSS and 23-RSS spacers. Color coding for regions of the RSSs is shown.
(B) Superimposition of three non-symmetrized NBD-containing reconstructions at 4.6 Å , 4.2 Å , and 4.4 Å resolutions (Figures 1F and S1G) in gray, yellow, and salmon, respectively. The variable positions of RSS are indicated by the arrows in corresponding colors.
(C) The crystal structure of the NBD/nonamer complex (PDB: 3gna, light blue for NBD and blue for DNA) is superimposed onto the synaptic PC model.
(D) The NBD dimer in the synaptic RAG complex tilts to the direction of 12-RSS. Superposition of the RAG active site dimer of the PC with that of the apo-like RAG crystal structure (PDB: 4wwx, magenta and red) shows that the NBD dimer needs to swing by ~76° to accommodate the bound RSS.
(E) Quantitative SDS-PAGE of the PC sample used for cryo-EM studies, showing the ratio of one HMGB1 per RAG monomer.
(F) Superimposed non-symmetrized PC model and PC map filtered at 5.5 Å resolution. Extra densities along 12-RSS and 23-RSS are highlighted by magenta and purple ovals, for which HMG box1 and box2 were fit, respectively, in (G) and (H).
(G) The difference density between the PC map and the PC model showed that the extra pieces of density in the magenta ovals in (F) could be fitted with residues 10-48 (a pair of helices) from the first HMG box of a tandem HMG box NMR structure (PDB: 2gzk) (Stott et al., 2006). Left, for 12-RSS; right, for 23-RSS.
(H) Modeled PC/HMGB1 complex. One HMG box is bound at the 12-RSS, and both HMG boxes of an HMGB1 molecule are bound at the 23-RSS. The fitting of the second HMG box was done by superimposing the DNA in the NMR structure and the DNA in the region of the second piece of extra density. HMG box-bound DNA segments in the NMR structure (PDB 2gzk) (Stott et al., 2006) are shown in blue.
(I) TBP causes similar bending in DNA as HMGB1. The TBP-DNA complex (PDB: 1tgh) was superimposed with 12-RSS and 23-RSS. Protein and DNA in the TBP-DNA complex are shown in magenta and blue, respectively.
(J) Structure-derived insights on RAG-mediated cleavage pathway in V(D)J recombination. RAG1 (light green and light cyan), RAG2 (green and cyan), and HMGB1 (orange) are represented as cartoons. Coding ends, heptamers, nonamers, and 3′ extension DNAs are shown by semi-transparent blue, red, magenta, and light slate rectangles, respectively. 12 and 23 bp spacers are shown as black lines. Briefly, without binding to an RSS, Apo-RAG is in an open conformation with the flexibly attached NBD dimer. When bound to either 12-RSS or 23-RSS intact DNA in the presence of HMGB1, a partially closed conformation of RAG may be induced. The NBD dimer is tilted through the interaction with either the 12-RSS or 23-RSS nonamer. Then a nick is produced at the junction between the coding end and the signal end, and the first base at the cleaved end of the heptamer is likely flipped out (red filled circle). Subsequent synapsis with a different RSS substrate leads to the formation of the PC, which assumes a fully closed conformation with a flipped base (blue filled circle) on the coding end of both nicked RSS intermediates. The coding ends are linked into hairpin DNA to form the CSC. NHEJ factors are recruited to dissociate the hairpin coding ends, leaving the SEC with only the bound signal ends.
See also Figure S7.
In non-symmetrized Apo-RAG map, no density is visible for the NBD region, suggesting that the NBD dimer is flexibly linked to the RAG active site dimer (Figure S1F). In the apo-like RAG crystal structure, the conformation of the NBD dimer may have been defined by crystal packing (Kim et al., 2015). The flexibility of the NBD dimer is evident even when it is bound to the nonamer DNA, as shown by conformational fluctuations in three superimposed reconstructions (Figures 7B, 1F, and S1H), which may allow ± 1 variation of the spacer lengths in both 12-RSS and 23-RSS in a minor population of recombination sites (Fanning et al., 1996). The flexibility may have also compromised the resolution of the NBD region to ~7.2 Å (Figures 1F and S1E). The dimeric NBD/nonamer complex in the PC aligns well with the previous crystal structure of the isolated NBD/nonamer complex (Yin et al., 2009) (Figure 7C), suggesting that the NBD dimer mostly oscillates as a rigid body. When the PC is superimposed with the apo-like RAG dimer using one of the RAG1-RAG2 monomers, the associated NBD dimer in the apo-like conformation needs to swing about 76° to reach the RSS-bound orientation (Figure 7D). The rotation moves the NBD closer to the 12-RSS but away from the 23-RSS.
Using known molar ratios of Apo-RAG and HMGB1 as standards, we showed by SDS-PAGE that our reconstituted PC sample contained one HMGB1 per RAG monomer (Figure 7E). This composition is different from that of a previously reconstituted SEC under a different condition, which showed one HMGB1 per RAG dimer (Grundy et al., 2009; Kim et al., 2015). HMGB1 contains two HMG boxes, each composed of a pair of helices and a long, slightly bent third helix, which interacts with DNA at the minor groove to generate bending (Stott et al., 2006) (PDB 2gzk). To determine if the PC cryo-EM density contained HMGB1, we displayed the map with the complete model superimposed. While one piece of unassigned density was revealed at the 12-RSS spacer, two were seen at the 23-RSS spacer (Figure 7F), suggesting that the former is bound with one HMG box and the latter with both HMG boxes in HMGB1.
The difference map between the cryo-EM reconstruction and the model clearly revealed densities with a two-cylinder shape at the 12-RSS and the 23-RSS that could be fitted with the pair of helices in an HMG box (Figure 7G). The fitting allowed positioning of one HMG box each to the 12-RSS and the 23-RSS (Figures 7G, 7H, and S7B). Although no clear density was apparent for the third helix of the HMG box bound to either 12-RSS or 23-RSS, the generated mode of DNA interaction is similar to that in the NMR structure of the HMG/DNA complex (Stott et al., 2006) (Figures S7C and S7D), supporting the HMG/RSS model. The second piece of density at the 23-RSS was not sufficiently clear to enable direct HMG fitting. However, the distance between the two pieces of density is similar to the distance between the two HMG boxes in the NMR structure, suggesting that the second piece of density represents the second HMG box of the same bound HMGB1 molecule (Figures 7H and S7B). Because the spacer DNA of 12-RSS is short, it is likely that only one HMG box of HMGB1 is bound.
DISCUSSION
Induced Asymmetry as the Structural Basis for the 12/23 Rule of Recombination
Recognition of two different signal sequences is unique for RAG, as most other recombinases interact with and cleave a pair of identical signal sequences. Our cryo-EM structures suggest an induced asymmetry mechanism that requires flexibility of NBD dimer orientation and plasticity of RSS conformation in the execution of the 12/23 rule. An RSS must bind RAG using both the heptamer and the nonamer for optimal affinity and recombination efficiency. The interactions at the heptamer are symmetric and relatively fixed. When a 12-RSS is bound at both the heptamer and the nonamer in an HMGB1-bent conformation, the NBD dimer tilts toward the shorter 12-RSS, leaving the same NBD dimer with the ability to synapse with only a 23-RSS, also in an HMGB1-bent conformation. Conversely, when a 23-RSS is bound, the NBD dimer tilts away from the longer 23-RSS, leaving the same NBD dimer with the ability to combine with only a 12-RSS. Modeling a pair of 12-RSSs or a pair of 23-RSSs onto RAG showed that the supposed interaction sites on NBD and the RSS are ~40 Å away (Figures S7E and S7F). Utilizing the spatial difference between the 12 bp and 23 bp spacers, it appears that nature has evolved one effective solution to RSS pairing to enable recombination fidelity. The synapsis of 12-RSS and 23-RSS also promotes effective chemical catalysis through coupled conformational changes at the active sites, further enhancing the 12/23 rule.
To contemplate whether other DNA sequences may also be paired by RAG, we used NBD dimer conformations in the apo-like crystal structure (Kim et al., 2015) and the PC as two possible extremes of NBD tilt angles. We then modeled hypothetical B-form RSS duplexes onto the RAG-bound RSSs by superimposing the heptamer regions (Figure S7G). This exercise suggests the possibility that, if the spacer lengths are varied, there may be additional ways of symmetrical and asymmetrical RSS synapsis, causing pathological DNA double-strand breaks and chromosomal translocations in cancer and autoimmunity, especially when facilitated by spatial proximity (Zhang et al., 2012). In cells, it is thus far unclear what causes bending of the spacer in an RSS to promote recombination. HMG proteins are abundant in the nucleus (Shirakata et al., 1991). Other nuclear proteins may also fulfill the function of RSS-bending, such as the TATA binding protein (TBP) that is known to dramatically bend DNA (Nikolov et al., 1996) (PDB: 1tgh) (Figure 7I).
RAG-Mediated Catalytic Pathway
We propose the following molecular mechanism in RAG-catalyzed cleavage phase of V(D)J recombination based on structures of Apo-RAG and synaptic complexes, as well as our conjecture on steps involving singly RSS-bound forms that still lack structural information (Figure 7J). First, our Apo-RAG structure shows a dynamic open conformation in the absence of RSS binding, with its NBD dimer flexibly attached to the RAG active site dimer. Second, because both RAG monomers are required for binding of each RSS, we posit that, upon interaction with an intact RSS substrate, immediate closure of the RAG dimer may ensue to bring the RAG monomers closer together, forming the signal complex (SC). Because the closed conformation of synaptic complexes would have been in clash with an intact RSS substrate, we suspect that an SC may be in a partially closed conformation. Nicking at the RSS-coding end junction can proceed in the SC; however, we do not yet know the associated conformational changes and whether base flipping of C1 and T-1* in synaptic complexes occurs in singly RSS-bound forms. Third, due to dimerization-mediated cooperativity, binding and nicking at one active site of the RAG dimer likely enhances the binding affinity of the symmetric active site to another RSS substrate for formation of the synaptic PC, leading to further closure of the RAG dimer. Fourth, the nicked RSS in PC is severely distorted at the RSS-coding end junction with local unwinding and base flipping of both C1 and T-1*. This conformation, together with the bound metal ions, promotes hairpin formation at the coding end. Simultaneous dissociation of the hairpin coding ends in the presence of proper DNA repair proteins, ensures joining fidelity, and leads to formation of the SEC.
Insights into Human RAG Disease Mutations
Over 100 disease mutations on human RAG1 and RAG2 have been identified (Lee et al., 2014; Schatz and Swanson, 2011). Mapping these mutations onto the crystal structure of mRAG has previously identified those that destabilize the tertiary structure or interfere with the conserved quaternary interaction within the RAG1-RAG2 complex monomer (Kim et al., 2015). The RAG1 mutations R841W/Q may disrupt RSS-induced RAG dimer closure because the equivalent zebrafish residue R860 appears central in stabilizing the closed conformation (Figures 3F and 3G and Table S2). We found that quite a few RAG1 mutations are on residues directly involved in heptamer recognition in the synaptic complexes (Table S2), explaining their deleterious effects on the recombinase. Mutations on residues that participate in non-specific spacer DNA interaction and coding end DNA interaction are also present (Table S2). Interestingly, analysis on the characteristics of rearrangements by hypomorphic RAG mutants revealed occurrence of preferential use of certain gene segments, which generates qualitative differences in the patient T and B cell repertoire when compared with normal controls (Lee et al., 2014). We hypothesize that mutations on residues that directly interact with RSSs may alter the specificity of V(D)J recombination, making some RSSs better substrates than others and leading to the wide spectrum of the disease.
Implications on Other DDE Family Enzymes
Despite having different structural architectures, many transposases and integrases also belong to the DDE family recombinases and catalyze multi-step strand cleavage and strand transfer reactions (Montanño and Rice, 2011). In general, these enzymes excise an insertion element by binding and breaking the DNA at the flanking signal sequences. The cleaved signal sequences with the insertion element located in between are then bound and become ligated to a target DNA. Only limited structural information is available on these enzymes, likely due to their fairly large size and complexity. Our RAG structures with DNA intermediates and products therefore provide insights into their mechanism of catalysis, especially for the excision step that bears a direct parallel to RAG-mediated cleavage. Specifically, cooperative conformational changes upon DNA synapsis, DNA distortion at the junction with the signal sequence, and based flipping at the nicked signal sequence may all be general features in transposases and integrases as well.
EXPERIMENTAL PROCEDURES
Cloning, Protein Expression, and Purification
The zRAG1 (271–1031) and full-length zRAG2 constructs containing N-terminal His-MBP tags were expressed using the Bac-to-Bac baculovirus-insect cell system. RAG1 was purified sequentially by amylose affinity, heparin, and size-exclusion chromatography, and RAG2 was purified by amylose affinity, anion exchange, and size-exclusion chromatography. The zRAG1-RAG2 complex was reconstituted by mixing zRAG1, zRAG2, and the H3K4Me3 peptide in a 1:1.2:1.2 molar ratio, followed by removal of the His-MBP tags and size-exclusion chromatography. C-terminally His-tagged HMGB1 was expressed in E. coli and purified using Ni-NTA affinity and size-exclusion chromatography. The DNAs were synthesized as oligos, annealed appropriately to generate nicked or cleaved RSSs, and purified by gel filtration. Synaptic RAG complexes were reconstituted by incubating the zRAG1-RAG2 complex, 12-RSS, 23-RSS, and HMGB1 in a 2:1:1:3 molar ratio in the presence of Ca2+ or Mg2+, followed by gel filtration.
Cryo-EM Data Acquisition, Image Processing, Model Building, and Refinement
Cryo-EM images were collected using an FEI TF30 Polara electron microscope and a Gatan K2 Summit direct electron detector in super-resolution counting mode. Simplified Application Managing Utilities for EM Labs (SAMUEL) scripts were used for image preprocessing, particle picking, 2D classification, and 3D initial model building. 3D classification and refinement were carried out in Relion (Scheres, 2012). All refinements follow the gold-standard procedure, in which two half datasets were refined independently. RELION “post-processing” was used to estimate resolution based on the Fourier shell correlation (FSC) = 0.143 criterion. The mRAG1-RAG2 complex (Kim et al., 2015) and the NBD-DNA complex structures (Yin et al., 2009) were used as starting points for model building. The atomic models were refined first in real space and then in reciprocal space using phase restrains, electron scattering factors, and artificial unit cells.
Supplementary Material
Highlights.
Cryo-EM structures of synaptic RAG complexes reveal a closed dimer conformation
RAG cooperatively recognizes the RSSs with base flipping in the nicked signal end
Distortion and base flipping in coding end DNA facilitate hairpin formation
Induced asymmetry and HMGB1-induced RSS bending underlie the 12/23 rule
In Brief.
Cryo-EM structures of synaptic RAG complexes reveal a recombination signal sequence (RSS)-induced closed conformation that enables catalytic activation and explain the molecular basis for the 12/23 rule, a dogma in which 12-RSS and 23-RSS flanking the V, D, and J segments are synapsed.
ACKNOWLEDGMENTS
We thank Dr. Wei Mi for help with sample freezing for cryo-EM studies, Drs. Liron David and Maria Ericsson for help with negative-stain EM experiments, Drs. Yang Li and Wei Ding for suggestions on model building and refinement, and the support of the Cancer Research Institute Irvington Postdoctoral Fellowship (to H.R.).
Footnotes
ACCESSION NUMBERS
The cryo-EM maps of symmetrized SEC, symmetrized PC, and non-symmetrized PC at 3.4 Å , 3.7 Å , and 4.6 Å resolutions, respectively, have been deposited in the EMDataBank under accession codes of EMD-6487, EMD-6488, and EMD-6489. The corresponding refined structural models have been deposited in the Protein Data Bank with PDB: 3JBX, 3JBY, and 3JBW, respectively. The cryo-EM maps of symmetrized Apo-RAG at 9.0 Å resolution and of symmetrized synaptic RAG at 3.3 Å resolution reconstructed from mixed PC and SEC particles have been deposited in the EMDataBank under accession codes of EMD-6490 and EMD-6491, respectively.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, two tables, and one movie and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2015.10.055.
AUTHOR CONTRIBUTIONS
H.R. and H.W. conceived the project. H.W. and H.R. designed the biochemical experiments. H.R. prepared Apo-RAG and all RAG complexes and performed biochemical and initial negative-stain EM experiments. M.G.C. carried out negative-stain EM analysis and characterization of cryo-EM conditions. M.L. and M.G.C. performed cryo-EM data collection. M.L. carried out cryo-EM data processing. H.R. and H.W. performed model building and refinement. T.-M.F. helped with model building and refinement, and A.B.T. calculated FSC curves and local resolution distributions. H.W., H.R., and M.L. performed data analysis, result discussion, and interpretation. H.W., H.R., and M.L. wrote the manuscript.
REFERENCES
- Akira S, Okazaki K, Sakano H. Two pairs of recombination signals are sufficient to cause immunoglobulin V-(D)-J joining. Science. 1987;238:1134–1138. doi: 10.1126/science.3120312. [DOI] [PubMed] [Google Scholar]
- Bischerour J, Lu C, Roth DB, Chalmers R. Base flipping in V(D) J recombination: insights into the mechanism of hairpin formation, the 12/23 rule, and the coordination of double-strand breaks. Mol. Cell. Biol. 2009;29:5889–5899. doi: 10.1128/MCB.00187-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt VL, Roth DB. Recent insights into the formation of RAG-induced chromosomal translocations. Adv. Exp. Med. Biol. 2009;650:32–45. doi: 10.1007/978-1-4419-0296-2_3. [DOI] [PubMed] [Google Scholar]
- Fanning L, Connor A, Baetz K, Ramsden D, Wu GE. Mouse RSS spacer sequences affect the rate of V(D)J recombination. Immunogenetics. 1996;44:146–150. doi: 10.1007/BF02660064. [DOI] [PubMed] [Google Scholar]
- Grundy GJ, Ramón-Maiques S, Dimitriadis EK, Kotova S, Biertümpfel C, Heymann JB, Steven AC, Gellert M, Yang W. Initial stages of V(D)J recombination: the organization of RAG1/2 and RSS DNA in the post-cleavage complex. Mol. Cell. 2009;35:217–227. doi: 10.1016/j.molcel.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Lapkouski M, Yang W, Gellert M. Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature. 2015;518:507–511. doi: 10.1038/nature14174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, Al-Herz W, Haddad E, LeDeist F, Bleesing JH, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J. Allergy Clin. Immunol. 2014;133:1099–1108. doi: 10.1016/j.jaci.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaño SP, Rice PA. Moving DNA around: DNA transposition and retroviral integration. Curr. Opin. Struct. Biol. 2011;21:370–378. doi: 10.1016/j.sbi.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov DB, Chen H, Halay ED, Hoffman A, Roeder RG, Burley SK. Crystal structure of a human TATA box-binding protein/TATA element complex. Proc. Natl. Acad. Sci. USA. 1996;93:4862–4867. doi: 10.1073/pnas.93.10.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 1990;248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- Ramsden DA, Baetz K, Wu GE. Conservation of sequence in recombination signal sequence spacers. Nucleic Acids Res. 1994;22:1785–1796. doi: 10.1093/nar/22.10.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JM, Colloms SD, Finnegan DJ, Walkinshaw MD. Molecular architecture of the Mos1 paired-end complex: the structural basis of DNA transposition in a eukaryote. Cell. 2009;138:1096–1108. doi: 10.1016/j.cell.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu. Rev. Genet. 2011;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakata M, Hüppi K, Usuda S, Okazaki K, Yoshida K, Sakano H. HMG1-related DNA-binding protein isolated with V-(D)-J recombination signal probes. Mol. Cell. Biol. 1991;11:4528–4536. doi: 10.1128/mcb.11.9.4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott K, Tang GS, Lee KB, Thomas JO. Structure of a complex of tandem HMG boxes and DNA. J. Mol. Biol. 2006;360:90–104. doi: 10.1016/j.jmb.2006.04.059. [DOI] [PubMed] [Google Scholar]
- Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- Yin FF, Bailey S, Innis CA, Ciubotaru M, Kamtekar S, Steitz TA, Schatz DG. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nat. Struct. Mol. Biol. 2009;16:499–508. doi: 10.1038/nsmb.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.