

Abstract
Retinitis pigmentosa (RP) is a highly heterogeneous group of disorders characterized by degeneration of the retinal photoreceptor cells and progressive loss of vision. While hundreds of mutations in more than 100 genes have been reported to cause RP, discovering the causative mutations in many patients remains a significant challenge. Exome sequencing in an individual affected with non-syndromic RP revealed two plausibly disease-causing variants in TRNT1, a gene encoding a nucleotidyltransferase critical for tRNA processing. A total of 727 additional unrelated individuals with molecularly uncharacterized RP were completely screened for TRNT1 coding sequence variants, and a second family was identified with two members who exhibited a phenotype that was remarkably similar to the index patient. Inactivating mutations in TRNT1 have been previously shown to cause a severe congenital syndrome of sideroblastic anemia, B-cell immunodeficiency, recurrent fevers and developmental delay (SIFD). Complete blood counts of all three of our patients revealed red blood cell microcytosis and anisocytosis with only mild anemia. Characterization of TRNT1 in patient-derived cell lines revealed reduced but detectable TRNT1 protein, consistent with partial function. Suppression of trnt1 expression in zebrafish recapitulated several features of the human SIFD syndrome, including anemia and sensory organ defects. When levels of trnt1 were titrated, visual dysfunction was found in the absence of other phenotypes. The visual defects in the trnt1-knockdown zebrafish were ameliorated by the addition of exogenous human TRNT1 RNA. Our findings indicate that hypomorphic TRNT1 mutations can cause a recessive disease that is almost entirely limited to the retina.
Introduction
Retinitis pigmentosa (RP) is a highly heterogeneous group of disorders characterized by progressive photoreceptor degeneration, bone spicule-like pigmentation, poor night vision and visual field defects. RP can occur alone or in combination with one or more extraocular findings such as deafness, obesity, polydactyly, renal disease, diabetes, developmental delay, ataxia and progressive central nervous system degeneration. To date, mutations in more than 190 genes have been associated with an RP-like phenotype (1), and the incomplete sensitivity of even the most sophisticated of our current genetic investigations suggests that more RP-causing genes and mutations remain to be identified. A practical effect of this extraordinary degree of genetic heterogeneity is that an average RP gene is the cause of <1% of cases of a disease that in total affects 1 in 4000 individuals in the population. Thus, one must clinically identify and molecularly screen hundreds of patients with RP to be in a position to observe even two or three examples of disease-causing mutations in most RP genes.
The rod photoreceptor cells of the retina are among the most metabolically active cells in the body (2). Each day, these photoreceptor cells shed and regenerate ∼10% of their outer segments, the specialized extension of the primary cilia that are sensitive enough to detect even single photons. The very large amount of protein synthesis required to replace 10% of the rod outer segment per day, coupled with the post-mitotic nature of these cells, may make them more susceptible to abnormalities of protein synthesis than other cell types. Indeed, many genes such as RP9, PRPF3, PRPF8, PRPF31 and SNRNP200 are ubiquitously expressed and involved in very basic functions of protein synthesis, and yet when mutated, cause selective photoreceptor death without affecting other cells in the body (3).
TRNT1 is a nucleotidyltransferase involved in tRNA processing. This enzyme is responsible for adding the CCA trinucleotide to the 3′ end of tRNAs and is required for both mitochondrial and cytoplasmic translations (4,5). TRNT1 is an essential gene in yeast (CCA1), and siRNA knockdown of TRNT1 in wild-type (WT) human fibroblasts causes cytotoxicity and apoptosis (6). Recently, mutations in TRNT1 were found to cause a syndrome characterized by congenital sideroblastic anemia, B-cell immunodeficiency, recurrent fevers and developmental delay (referred to as SIFD) (6,7). However, some SIFD patients also display sensorineural hearing loss, ataxia and RP (7). SIFD is a severe multi-organ syndrome with life-threatening complications, and many SIFD patients die in the first decade of life.
In this study, we report two non-syndromic RP pedigrees with segregating mutations in TRNT1. In both families, affected individuals harbored asymmetric genotypes consisting of one truncating mutation and one hypomorphic mutation that allowed some residual protein function. The combination of clinical and molecular evaluations of a large cohort of RP patients, functional studies of patient-derived fibroblasts and disease modeling in zebrafish (where expression levels can be titrated to recapitulate features of the human disease) is a reusable strategy for identifying the many other rare causes of heritable human blindness that remain to be discovered.
Results
Whole-exome sequencing reveals compound heterozygous mutations in TRNT1
Exome sequencing of an otherwise healthy 19-year-old male college student (P1) recently diagnosed with non-syndromic RP revealed only two one-allele findings (CRB1, Val398Ile; TTC21B, Arg476His) among genes known to cause photoreceptor disease. Potential two-allele findings were discovered in each of the 39 genes not previously associated with non-syndromic photoreceptor disease. In TRNT1, a deletion of one base of a mononucleotide repeat in the final exon was discovered (c.1246A[6]), which would be expected to result in a protein with 10 incorrect amino acids that is truncated by 7 amino acids. A three-base-pair exonic deletion (c.126_128delAGA) was also identified, and this variant is present in the ExAC database at a frequency of 0.008% (8). The Sanger sequencing of the proband's parents revealed these variants to lie on separate alleles. The three-base-pair deletion would result in the loss of the Glu43 residue. This residue is located on the surface of the TRNT1 structure within the so-called head domain, in a helix-turn-helix secondary structure motif. The structure can accommodate deletion of Glu43 by shortening the first helix by one residue along with slight changes to the turn that leads into the second helix (Fig. 1). Although this analysis does not address thermodynamic or kinetic consequences of Glu43 deletion on TRNT1, the structural evidence is consistent with the view this variant would have a relatively mild effect on TRNT1 function and thus explain a phenotype that was much milder than the SIFD syndrome previously associated with mutations in this gene (6,7).
Figure 1.

Experimental structure of TRNT1 (gray) with overlay (green) showing a computational estimate of Glu43 deletion. Inset: area around Glu43 deletion with side chain of Glu43 shown in the experimental structure. The minor change in the modeled structure is consistent with a small functional impact of the allele.
TRNT1 is a rare cause of non-syndromic RP
In an attempt to strengthen this genotype–phenotype correlation with additional subjects, we conducted stepwise screening of additional RP patients. In all, 1729 unrelated probands with RP but without plausible mutations in known RP genes were screened for TRNT1 mutations. These 1729 probands were the negative result of screening over 3400 RP probands for mutations in known RP genes and would therefore be expected to be enriched almost 2-fold for mutations in currently undiscovered genes. The first screen of these 1729 patients was designed to identify individuals with mutations that were similar to those in the index case. A single-strand conformational polymorphism (SSCP) assay was designed for TRNT1 Exons 2 and 8 and shown to detect the mutations present in the index subject. This assay was used to rapidly screen the 1729 RP probands, and one plausible disease-causing allele was identified in each of two different families. The first of these was a heterozygous missense change, Lys417Thr. Sequencing of the remaining exons of TRNT1 in this individual failed to reveal another disease-causing allele, and subsequent exome sequencing revealed a previously described homozygous disease-causing allele in a different RP gene (BBS1). The second family consisted of two affected brothers (P2 and P3) who each carried a heterozygous frameshift mutation that was later found in a SIFD patient (c.1246A[8]). This variant is present at a frequency of 0.07% in the European population in the ExAC database (8) and is predicted to result in a protein with 6 incorrect amino acids that is truncated by 9 amino acids. Sequencing of the remaining TRNT1 exons in these two brothers identified a putative splice variant (c.609-26T > C) on the allele opposite their frameshift mutation. This variant was neither present in the 1000 genomes database (9), ExAC (8), nor in 257 normal individuals ascertained in Iowa. The entire cohort of 1729 RP probands was then screened for variations in Exon 6 using SSCP, but no additional subjects were found to harbor a variant in this exon. A 727-patient subset of the 1729-member RP cohort was then screened for mutations in the complete coding sequence of TRNT1 using a combination of the Fluidigm Access Array platform, next-generation sequencing and sequence analysis (for Exon 5 which failed to amplify well on the Fludigm platform). No additional plausible disease-causing genotypes were observed during this phase of the screening. Finally, during the course of this study, whole-exome sequencing was performed in an additional 195 unrelated probands with RP who were ascertained after the creation of the original 1729-patient cohort. No additional disease-causing TRNT1 genotypes were identified in this group.
Clinical findings in TRNT1-associated RP patients
P1 first presented at age 19 years after a routine eye examination revealed pigmentary changes in the retina. He first noticed nyctalopia at age 16 years, when he began driving. His visual acuity with a low myopic correction was 20/20 OU. His anterior segment examination was unremarkable OU. His optic nerve heads were pink, with a cup-to-disc ratio of 0.1 OU. There was a small epiretinal membrane in the macula OD, and mild arteriolar attenuation OU. There were scant bone spicule-like pigmentary changes in the periphery of the retina OU (Fig. 2A and B). The Goldmann perimetry revealed a partial ring scotoma to the V4e isopter OU (Supplementary Material, Fig. S1A and B). Optical coherence tomography (OCT) of the macula (Fig. 2C) revealed diffuse outer retinal atrophy with foveal preservation and without intraretinal fluid OU. His past medical history was significant for juvenile rheumatoid arthritis affecting the hip and neck that began in the first year of life. He experienced bouts of fever >40°C that occurred two to three times per year. He was treated with methotrexate between ages 4 and 7 years and has not had a recurrence of the fever or arthritis since. In view of the anemia associated with SIFD syndrome, we obtained a complete blood count (CBC), which revealed a low normal hemoglobin (13 g/dl; normal = 13–18 g/dl) and hematocrit (41%; normal = 38.5–52%), but abnormally small red blood cells (mean corpuscular volume 65.9 fl; normal = 80–98 fl) and significant anisocytosis (erythrocyte distribution width 20.2%; normal = 11–14%; Fig. 3). The white blood cells were normal in number (8400/µl; normal = 3000–11 600/µl) and differential composition. The platelet count (226 000/µl; normal = 130 000–400 000/µl) and reticulocyte count (1.43%; normal = 0.8–2.1%) were also normal. Iron studies revealed a normal ferritin (113 ng/ml; normal 24–336 ng/ml), low serum iron (23 µg/dl; normal = 35–150 µg/dl), normal transferrin (207 mg/dl; normal = 180–329 mg/dl), normal iron binding capacity (289 µg/dl; normal = 250–450 µg/dl) and low iron saturation (8%; normal = 15–55%). The peripheral blood smear revealed microcytosis, hypochromia, anisocytosis and poikiocytosis with numerous eliptocytes (Fig. 3).
Figure 2.

Color fundus photographs (A, B, D, E, G, H) and OCT (C, F, I) from individuals P1 (A–C), P2 (D–F) and P3 (G–I) affected with TRNT1-associated autosomal-recessive RP. The color photographs reveal vascular narrowing in all three individuals, subtle bone spicule-like pigmentation in P1 and cystoid macular edema in P2 and P3. OCT reveals thinning of the photoreceptor layers in all three individuals and marked cystoid macular edema in P2 and P3.
Figure 3.
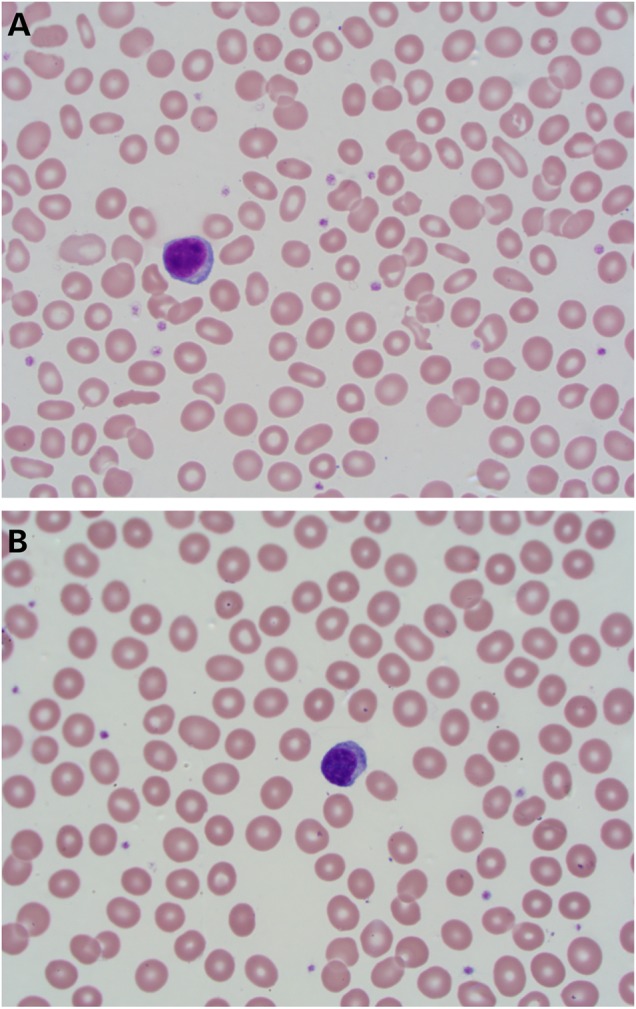
Photomicrograph of a Wright's stained peripheral blood smear from Patient 1 (A) demonstrating microcytosis, hypochromia, anisocytosis and numerous eliptocytes. A normal blood smear (B) is provided for comparison (100×).
P2 presented to The University of Iowa at age 21 years. He reported that his night vision had always been poor and that he first began bumping into objects as a child. He denied photophobia or poor color vision. His visual acuity with a high myopic correction was 20/40 OD and 20/32 OS. His anterior segment examination was unremarkable OU. The optic nerve heads demonstrated mild pallor and a cup-to-disc ratio of 0.25 OU. There was significant macular edema OU. The retinal arterioles were mildly attenuated (Fig. 2D and E). There was diffuse atrophy of the retinal pigment epithelium in the periphery OU. The Goldmann kinetic perimetry revealed mild generalized depression OU (Supplementary Material, Fig. S1C and D). OCT of the maculae demonstrated significant intraretinal cystoid changes and diffuse outer retinal atrophy with extrafoveal preservation OU (Fig. 2F). A trial of topical dorzolamide did not improve the patient's vision or intraretinal fluid. P2 had no history of arthritis or unusual febrile illnesses. His CBC revealed a near normal hemoglobin (13.4 g/dl; normal = 13.8–17.5 g/dl) and a low normal hematocrit (42.2%; normal = 42–52%), with microcytosis (66.7 fl; normal = 81–97 fl) and anisocytosis (erythrocyte distribution width 16.8%; normal = 11.6–14.8%); almost identical to P1. Like P1, the white blood cells were normal in number (8000/µl; normal = 4600–10 500/µl), and differential composition and the platelet count were also normal (259 000/µl; normal = 140 000–420 000/µl). Iron studies revealed a normal ferritin (94.8 ng/ml; normal 22–332 ng/ml), normal serum iron (113 µg/dl; normal = 65–175 µg/dl), normal unsaturated iron binding capacity (223 mg/dl; normal = 130–375 µg/dl), normal iron binding capacity (336 µg/dl; normal = 260–400 µg/dl) and normal iron saturation (34%; normal = 13–45%).
Patient 3 (P3), the younger brother of P2, presented at age 18 years. He first noted nyctalopia at approximately age 13 years. An electroretinogram performed locally revealed absent scotopic responses and severely diminished photopic responses OU. His visual acuity with contact lenses was 20/40 OD and 20/32 OS. His anterior segment examination was unremarkable OU. The optic nerve heads demonstrated mild pallor and a cup-to-disc ratio of 0.2 OU. There was significant macular edema OU. The retinal arterioles were mildly attenuated, and the retinal pigment epithelium showed diffuse atrophy OU (Fig. 2G–I). His visual fields showed a mild generalized depression OU (Supplementary Material, Fig. S1E and F), and OCT of his maculae was very similar to Patient 2 (Fig. 2I). He underwent a trial of topical dorzolamide without significant improvement. P3 had no history of arthritis or unusual febrile illnesses. His CBC revealed a near normal hemoglobin (12.8 g/dl; normal = 13.2–17.7 g/dl) and hematocrit (39%; normal = 40–52%), with microcytosis (68 fl; normal = 82–99 fl) and an elevated erythrocyte distribution width (19.6%; normal = 9.0–14.5%); almost identical to P1 and P2. The white blood cells were normal in number (5000/µl; normal = 3700–10 500/µl) and differential composition, and the platelets were also normal in number (214 000/µl; normal = 150 000–400 000/µl). Iron studies revealed a slightly elevated serum iron (162 µg/dl; normal = 87–150 ug/dl) and soluble transferrin receptor (1.8 ng/ml; normal = <1.5 ng/ml), normal transferrin (268 mg/dl; normal = 200–360 mg/dl) and normal iron saturation (42%; normal = 27–44%).
None of these three patients had any history or signs of developmental delay, deafness, ataxia, seizures or cardiac disease.
Abnormal splicing and reduced protein levels in patient fibroblasts
In order to further assess the potential pathogenicity of the putative splice variant identified in Family 2, RNA was extracted from the keratinocytes of P2, as well as from unaffected control individuals and unaffected donor retina. Sequences corresponding to Exons 4–6 were amplified by reverse transcriptase-polymerase chain reaction (RT-PCR) (Supplementary Material, Fig. S2). An additional 14 bp polymerase chain reaction (PCR) product was observed that was absent from control cells, indicating abnormal splicing and supporting the pathogenicity of the variant. Bidirectional sequencing of this PCR product revealed the addition of 14 nucleotides to the spliced RNA, which would be expected to result in the addition of four incorrect amino acids followed by premature termination in the protein translated from this splice variant.
Western blots of protein extracted from the fibroblasts of P1, P2 and P3 showed significantly decreased levels of TRNT1 protein in patients relative to controls (Fig. 4). Control samples show a single, major band at 50 kDa, while cells from the three affected patients show a TRNT1-reactive band at 50 kDa and a second band at a slightly lower molecular weight (47 kDa), the combination of which is significantly diminished compared with the WT TRNT1 in the control fibroblasts (Fig. 4A and B) but less so than in the published cases of SIFD (4). These lower-molecular-weight bands are qualitatively consistent with the premature termination of translation caused by the frameshift mutations observed in the final exons of both families. To further demonstrate that the control samples contained a single TRNT band only (i.e. that the single band detected was not due to the presence of two bands that were blended together due to an over-abundance of protein loading), a western blot experiment was performed in which decreasing amounts of total protein was loaded (i.e. 15, 10 and 5 µg). As shown in the Supplementary Material, Fig. S2D, regardless of the amount of protein loaded onto each western blot, only a single TRNT1 product could be detected in either of the normal or RP control samples.
Figure 4.

Expression of WT and mutant TRNT1 proteins in control and patient fibroblasts. (A) Western blot analysis depicting TRNT1 expression in dermal fibroblasts obtained from a normal control, an RP control (i.e. patient with non-TRNT1-associated RP) and three compound heterozygous patients (gel was loaded with 20 µg of total protein). GAPDH served as loading control. (B) Quantification by densitometry of lysates between groups for TRNT1 (n = 4) normalized to GAPDH. Data presented as mean ± SEM. Statistical significance between control and patient fibroblasts determined by one-way ANOVA–Tukey, post hoc test, (*) P-value < 0.05, n = 4. An overall diminution of TRNT1 protein and a second 47 kDa band present only in cells from patients with TRNT1 mutations was detected (see the Supplementary Material, Fig. S2D).
Zebrafish trnt1 expression during development
To evaluate the role of tRNA nucleotidyl transferase in vertebrate development and visual function, we characterized trnt1 in zebrafish (Fig. 5A). Zebrafish trnt1 is highly conserved with human TRNT1 (Supplementary Material, Fig. S3) and is expressed in the adult retina (Supplementary Material, Fig. S4). To evaluate spatial expression, antisense probes (and sense control) were used for whole mount in situ hybridization. Zebrafish trnt1 is expressed ubiquitously during early embryogenesis (Fig. 5B and Supplementary Material, Fig. S5). By 2 days post-fertilization (dpf), expression is enriched in the brain, cardiovascular region, pectoral fin buds and in proliferative cells of the retina (Fig. 5C and Supplementary Material, Figs S6 and S7). One day later (3 dpf), trnt1 transcript expression is maintained in the brain, retina (Supplementary Material, Fig. S4D) and cardiac regions. By 5 dpf, expression is observed in the head and trunk neuromasts, which are zebrafish sensory structures analogous to the auditory system of mammals (Fig. 5E and Supplementary Material, Fig. S7) (10). Thus, zebrafish trnt1 expression is found in all of the organs affected in human SIFD and RP.
Figure 5.

trnt1 expression and knockdown in zebrafish embryos. (A) Zebrafish trnt1 gene structure noting the location of MOs used for knockdown. (B) Zebrafish trnt1 transcript is broadly expressed throughout the embryo up to 1 dpf. (C) By 2 dpf, trnt1 becomes enriched in the brain, pectoral fin, blood and heart (inset). (D) Section through the central retina of 3 dpf larva showing trnt1 expression. (E) trnt1 expression in blood forming regions and the neuromast. (F) RT-PCR of RNA isolated from control and morphant embryos showing altered splicing in MO-injected embryos through 5 dpf. Uninjected WT embryos are used as control. Phenotypic range of morphological defects observed in trnt1 morphants at 5 dpf. (G) WT-like, (H) reduced eye size and blood flow and (I) eye and cardiovascular defects. Blue bar represents the penetrance of morphological defects with MO dose: 6.5 ng generating 80% defective, 4.5 ng generating 60% defective and 1.5 ng inducing defects in 15% of injected embryos.
Trnt1 knockdown generates visual phenotypes as well as phenotypes resembling SIFD
To test the hypothesis that the degree of TRNT1 activity influences phenotypic severity, we utilized morpholino oligonucleotides (MO) to reduce endogenous Trnt1 activity in the zebrafish. Three different MOs were generated that target translation (MO1) and splicing (MO2 and MO3) (Fig. 5A). The efficacy of the splice block MOs was assessed by RT-PCR. Both MO2 and MO3 altered splicing and caused premature termination of the Trnt1 protein at amino acids 22 and 87, respectively. To determine the duration of knockdown, 2.5 ng of MO2 was injected into zebrafish, RNA was extracted at different stages and followed by RT-PCR. Altered splicing of trnt1 was detected out to 5 dpf (Fig. 5F). Morphological analysis of the MO-injected embryos (morphants) revealed cardiovascular defects and reduced eye size (Fig. 5H and I). To investigate the phenotypic spectrum associated with increasing amounts of functional Trnt1 protein, we titrated the MO dose to yield >75% WT morphology (Fig. 5G).
SIFD patients have anemia and variable hearing abnormalities (7) in addition to retinal degeneration. We evaluated the erythroid composition in morphant embryos by o-dianisidine staining (11). Morphants exhibited reduced staining of hemoglobin-containing red blood cells (Supplementary Material, Fig. S7). The zebrafish sensory system that is analogous to the human auditory system consists of neuromasts, which detect vibrations in the environment, including the sense of touch (12). Embryos with reduced Trnt1 activity show significantly reduced touch responses (Supplementary Material, Fig. S8).
Reduced CCA incorporation in zebrafish morphants
To verify Trnt1 enzymatic activity in vivo and to investigate whether hypomorphic knockdown alters tRNA processing, RNA sequencing was performed on zebrafish embryos. Size-fractionated RNA pools (73–94 nt) collected from morphant and control embryos were sequenced. Reads that aligned to annotated zebrafish tRNAs were inspected for inclusion of a CCA in the last three bases. Morphants had a significantly lower CCA incorporation efficiency than controls (P < 1 × 10−6, χ2 test) (Fig. 6A), indicating that MO knockdown of trnt1 affects tRNA maturation.
Figure 6.

trnt1 knockdown affects tRNA maturation and visual behaviors. (A) RNA-Seq analysis of control and morphant embryos evaluating nucleotide triplet CCA addition of all annotated tRNAs. (B) Vision Startle Reflex Response. (C) Violin plot showing the distribution of the number of responses for control, knockdown and hTRNT1 RNA-injected larvae. ***P-value < 1e-6 MO versus WT; *P-value = 0.001032 MO versus MO+RNA; **P-value = 1.492e-6 MO+RNA versus WT by the Mann–Whitney test.
Human TRNT1 suppresses the knockdown defects
When exposed to rapid changes in light intensity, zebrafish display a natural escape response. In the vision startle assay, an embryo in a brightly lit petri dish is exposed to five short interruptions of the light and is monitored for movement. Visually responsive embryos change swimming directions in response to the changes in light (13) (Fig. 6B). WT embryos respond on average 3 times with a distribution weighted between 3 and 5 (Fig. 6C). In order to evaluate vision, trnt1 morphants with normal gross morphology that retained touch response were used. trnt1 morphants show a significantly lower response rate than controls with a distribution weighted around 1 (P < 1 × 10−6 by the Mann–Whitney test) (Fig. 6C). Like our human patients, these fish exhibit reduced visual function in the absence of detectable syndromic features.
Human TRNT1 was expressed in embryos to determine the extent to which the knockdown defect could be rescued. We first established that myc-tagged human TRNT1 RNA injected into 2–4-cell-stage embryos is translated and expressed through 5 dpf (Supplementary Material, Fig. S9). Expression of hTRNT1 alone did not disrupt normal visual function (Fig. 6C). A sequential injection of trnt1MO followed by hTRNT1 RNA resulted in a significantly higher response compared with morphant siblings (P = 0.001 by the Mann–Whitney test). However, the group of morphants receiving the RNA still showed a significantly reduced response compared with uninjected embryos (P = 1.49 × 10−6 by the Mann–Whitney test), indicating that the visual defect is only partially rescued by human TRNT1. It should be noted that the touch response defect was rescued to WT levels (Supplementary Material, Fig. S8). Taken together, these data suggest that the visual response is more sensitive to Trnt1 levels than the other MO-induced defects, which is compatible with the findings in human patients.
Discussion
The three patients described in this report nicely illustrate both the opportunities and the challenges of the new era of precision medicine. On the opportunity side, whole-exome sequencing correctly identified the disease-causing genotype in a single individual, P1, which in turn led to the identification of two additional affected individuals (P2 and P3) whose disease-causing genotypes would have been missed by exome sequencing alone. Review of the medical literature revealed a severe developmental and hematologic syndrome caused by mutations in the same gene, which led to the discovery of an unsuspected hematologic manifestation in these patients. On the challenge side, TRNT1-associated autosomal-recessive RP with erythrocytic microcytosis was detected in only 2 of the 3400 families with RP we have studied suggesting that it occurs in <1 in 6 million people in the general population or ∼55 people total in the United States. Only ∼25% of these 55 people would be expected to have sufficiently advanced disease to be detected clinically while still retaining a sufficient number of viable photoreceptors to be reasonable candidates for gene replacement therapy. Although the coding sequence of TRNT1 is small enough to fit into the well-proven AAV gene augmentation delivery system, the costs of developing, testing and delivering such a treatment would need to be dramatically reduced from current levels before physicians will be able to offer such treatments to patients affected with diseases that are this uncommon. Patient-derived induced pluripotent stem cells will likely play an important role in this cost reduction by serving as an inexpensive but biologically relevant platform for investigating genotype-specific disease mechanisms and testing gene therapies (14–21). Such cells may also provide a means of restoring vision to older individuals who have already lost so many photoreceptor cells that gene therapy alone would be insufficient (17,22,23). CRISPR correction of the disease-causing mutations (22–24) prior to transplantation would be no more difficult or expensive for an extremely rare condition like TRNT1-associated RP than it would for a condition that is 1000 times more common.
Many of the RP genes that remain to be discovered will likely cause a similarly small fraction of the disease, and this will pose a challenge not only for treatment but also for diagnosis. Although whole-exome and whole-genome sequencing strategies are extraordinarily sensitive, they are also quite nonspecific, yielding hundreds to tens of thousands of plausible disease-causing variations in each individual depending on the phenotypic and genotypic breadth of the pretest hypothesis. For example, a pretest hypothesis of autosomal-recessive Leber congenital amaurosis, Bardet Biedl syndrome or Joubert syndrome would constrain the primary analysis to <20 genes and would require the identification of two plausible disease-causing variations, one demonstrably inherited from each parent. In our experience, <1 in 1000 individuals would harbor such a genotype by chance. In contrast, a pretest hypothesis of RP with unknown inheritance pattern would require the consideration of almost 200 genes (1) and the possibility that heterozygous variants could be disease-causing. With a pretest hypothesis of this breadth, almost every individual will be found to harbor at least one plausible disease-causing genotype at the whole-exome level and over 1000 at the whole-genome level. The solution to this lack of specificity in the sequencing dimension is the incredible specificity of some clinical observations and of focused functional studies in animal models and patient-derived tissues. The recently developed methods for deriving almost any adult tissue from a patient's own cells have made the latter strategy for genotype validation available to all scientists, even those who study historically inaccessible tissues like the retina and brain (15,20,25). In the present study, whole-exome sequencing of P1 with an autosomal-recessive non-syndromic RP analysis model was the hypothesis-generating initial experiment. This identified variations in TRNT1 as the most likely disease-causing genotype, even though at that time there were no published reports associating that gene with a human photoreceptor degeneration. Validation of this observation proceeded along three lines: (1) an attempt to identify other individuals with a similar genotype and phenotype (in our patients and in the published literature), (2) investigation of the effect of incremental loss of TRNT1 function using morpholino-based gene knockdown and WT RNA rescue in zebrafish and (3) investigation of the effects of the observed genotypes on TRNT1 mRNA and protein in patient-derived cells. The first of these approaches quickly identified a second family with two affected individuals with a very similar ophthalmic phenotype. The identification by others of mutations in TRNT1 as the cause of SIFD (6) and the recognition of retinal degeneration as a component of that severe syndrome (7) strengthened our belief in TRNT1 as the disease-causing gene in these families and also prompted us to examine their blood for abnormalities of erythrocyte maturation and iron metabolism. The identification of a striking microcytosis and anisocytosis in all three otherwise healthy individuals is extraordinarily unlikely to occur by chance and is an excellent example of the power of post hoc literature review and focused clinical examination to strengthen a next-generation sequencing result. The zebrafish is an excellent model organism for testing at least two kinds of hypotheses: (1) that abnormalities of a given gene may cause disease in a given set of tissues and (2) that a range of residual function may result in a range of phenotypes similar to that observed in human patients. In this case, we observed that relatively mild knockdown of trnt1 caused a vision-selective phenotype, while more complete knockdown caused abnormalities of eye development, heart development and erythroid maturation. Examination of patient-derived cells showed that all three patients could translate significant amounts of normal-sized TRNT1 protein and that the mutation discovered on the most normal TRNT1 allele of Family 2 does indeed alter splicing in a subset of transcripts. It is important to note that this intronic mutation in Family 2 would not have been annotated as potentially disease-causing in a typical exome sequencing analysis and was only detected because of a focused study of this gene in a cohort of over 1700 unrelated individuals with RP. To put this in another way, TRNT1-associated RP is probably the rarest disease that can be detected and confirmed when one's research cohort consists of 3400 unrelated families with the disease. A smaller cohort or a rarer disease may not have yielded a confirmatory family.
TRNT1 encodes an evolutionarily ancient enzymatic activity that is essential for all protein synthesis. No eukaryotes are known that completely lack this function. All patients with the recently described SIFD syndrome have some residual TRNT1 function mediated by at least one hypomorphic mutation. The genotypes of the three patients in this report likely result in even greater TRNT1 function. The loss of a single, poorly conserved (Table 1; Supplementary Material, Fig. S3) codon in P1 is predicted to have a mild structural impact on the protein (Fig. 1). It is also reasonable to imagine that the loss of some fraction of otherwise normal transcripts in P2 and P3 would be phenotypically milder than missense mutations occurring in phylogenetically invariant residues within the active site of the protein that are seen in SIFD patients (Supplementary Material, Fig. S3).
Table 1.
Reported pathogenic variants in TRNT1
| HG19 | NT | PRO | Exon | References |
|---|---|---|---|---|
| chr3:3170850:AGA>- | c.126_128delAGA | p.E43 inframe | Exon 2 | This study |
| chr3:3179013_3179014:TA>Tins22A | c.218_219ins22 | p.V73fs | Exon 3 | (6) |
| chr3:3182234:A>G | c.383A>G | p.D128G | Exon 4 | (4) |
| chr3:3182294:C>T | c.443C>T | p.A148V | Exon 4 | (4) |
| chr3:3182312:C>T | c.461C>T | p.T154I | Exon 4 | (6) |
| chr3:3182323:A>G | c.472A>G | p.M158V | Exon 4 | (6) |
| chr3:3186283:T>C | c.497T>C | p.L166S | Exon 5 | (6) |
| chr3:3186304:A>T | c.518A>T | p.Y173F | Exon 5 | (4) |
| chr3:3186355:G>T | c.569G>T | p.R190I | Exon 5 | (6) |
| chr3:3186395:G>T | c.608+1G>T | IVS5 | (6) | |
| chr3:3188088:T>C | c.609-26T>C | IVS5 | This study | |
| chr3:3188173:T>C | c.668T>C | p.I223T | Exon 6 | (6) |
| chr3:3189308:T>C | c.977T>C | p.I326T | Exon 7 | (6) |
| chr3:3189385_3189397:GATGTAAGTATAT>- | c.1054_1056+10delGATGTAAGTATAT | p.D352 inframe | Exon 7 | (6) |
| chr3:3189583:C>G | c.1057-7C>G | IVS7 | (6) | |
| chr3:3189674_3189675:TG>TATGTG | c.1142insATGT | p.W381fs | Exon 8 | (6) |
| chr3:3189785_3189786:AG>AAG | c.1246A[8] | p.S418fs | Exon 8 | (6), This study |
| chr3:3189779:A>G | c.1246A>G | p.K416E | Exon 8 | (6) |
| chr3:3189786:A>- | c.1246A[6] | p.S418fs | Exon 8 | This study |
The phenomenon of increasing amounts of residual protein function giving rise to increasingly mild phenotypes is not uncommon in biology, nor indeed in inherited retinal disease. The gene that is perhaps closest to the behavior of TRNT1 in humans is CEP290 in which very severe genotypes cause the lethal disorder Meckel Gruber syndrome, intermediate genotypes cause developmental brain and/or kidney abnormalities known as the Joubert syndrome and the Senior Loken syndrome, while the mildest genotypes affect only the photoreceptors and is known as Leber congenital amaurosis. There are also a number of genes involved in RNA splicing that if completely lost would likely be lethal but in a hypomorphic state cause only RP (3). The reason for this may be that rod photoreceptors have perhaps the greatest demand for continuous protein synthesis—the entire outer segment of the cell turns over every 10 days—of any post-mitotic human cell. This demand for protein synthesis in the retina is reflected in the enriched expression of trnt1 in the zebrafish retina (Fig. 5). The synthesis of globin by erythroid precursor cells is quite sensitive to translational regulation (26), and it is therefore interesting that the forms of RP that are associated with defects in RNA splicing do not also have erythrocyte abnormalities similar to those we observed in our three patients. The hematological findings of microcytosis and hypochromia seen in our patients are suggestive of either impaired iron utilization (similar to findings seen in patients with SIFD (7)) or thalassemia. Laboratory studies in our patients were not suggestive of iron deficiency, and no thalassemia-causing mutations were observed in the whole-exome sequence data from P1. We speculate that defective tRNA processing caused by TRNT1 mutations may lead to imbalanced synthesis of alpha or beta globin chains (a thalassemia-like condition) in hematopoietic cells, with a secondary abnormality in iron utilization (27). Patients with SIFD appear to be at risk for end organ damage due to iron overload (7). Serum iron studies in our patients did not suggest a markedly elevated iron status, but such patients could be easily monitored for iron overload and treated with iron chelation therapy if necessary.
In summary, we have identified a previously unrecognized TRNT1 phenotype, autosomal-recessive RP with erythrocytic microcytosis, using a combination of next-generation sequencing, focused screening of a large validation cohort, recursive clinical investigation, gene knockdown in zebrafish and molecular analysis of patient-derived cells. In so doing, we identified an unsuspected and treatable component of the disease in the three patients we studied. This multidisciplinary and iterative approach will be increasingly common as the field seeks to identify all remaining causes of inherited retinal disease for the purpose of delivering genotype-specific therapy to all affected individuals.
Materials and Methods
Human subjects
All subjects provided written informed consent for this study, which was approved by the Institutional Review Board at The University of Iowa and adhered to the tenets set forth in the Declaration of Helsinki.
Sequencing and high-throughput screening
Whole-exome sequencing was performed on DNA using the Agilent v5 kit and an Illumina HiSeq 2000/2500 instrument. The resulting reads were aligned to the genome using the burrows-wheeler aligner (BWA), variants were called using the genome analysis toolkit (GATK) and Conifer and annotated using a custom pipeline (28–30). Variants were prioritized for follow-up using methods we have described previously (14,31). Briefly, variants were first filtered to retain those predicted to alter coding sequence or impact a splice site. Variants that were seen in at least 1% of the 1000 genomes project samples or 0.6% of the exome variant server cohort were assumed to be too common to cause disease and were removed. After variants in known retinal disease-causing genes were excluded, genes harboring two alleles passing the previously mention filters were considered. For each of these genes, the Sanger sequencing in the proband and parents was conducted in an attempt to confirm the existence and phase of the variant. The Fluidigm AccessArray was used to prepare barcoded libraries from 727 RP patients to screen for variants in TRNT1 using the primers provided in the Supplementary Material, Table S1. The barcoded libraries were pooled and sequenced on an Illumina HiSeq 2000. Primer sequences were removed from reads using Btrim64, and the reads were subsequently aligned to the genome using BWA, and variants were called using GATK (28,30,32). Single-strand conformational polymorphism analysis (SSCP) was used to screen the cohort of 1729 RP individuals for variations in Exons 2, 6 and 8 as described previously (33). Greater than 92% of the 5187 SSCP reactions produced interpretable data.
Sequence comparison of TRNT1 homologs
Homologs of the human TRNT1 gene (NM_182916.2) were identified using PSI-BLAST (34) and aligned using ClustalW2 (35). Conserved domains were annotated using the National Center for Biotechnology Information's Conserved Domain Database (36).
Patient fibroblast isolation and culture
Patient dermal fibroblasts were isolated from 3 mm dermal punch biopsies via explant culture as described previously (37). Briefly dermal biopsies were minced, spread onto 6-well synthamax-coated tissue culture plates and allowed to air dry for 10–15 min before adding culture media. Tissue was subsequently cultured in complete biopsy medium [MEMα (Life Technologies, Grand Island, NY) containing 10% fetal bovine serum (FBS) (Life Technologies) and 2% Primocin (Life Technologies)] to allow fibroblasts to emerge. Upon reaching confluence (∼2–3 weeks post-plating), cells were passaged onto fresh 6-well synthamax-coated tissue culture plates at a density of 200 000 cells per well using 0.25% trypsin/ethylenediaminetetraacetic acid (Life Technologies).
RNA isolation and RT-PCR
Total RNA was extracted using the RNeasy Mini-kit (Qiagen, Valencia, CA) following the manufacturer’s instructions. Briefly, cells were lysed, homogenized and diluted in 70% ethanol to adjust binding conditions. Samples were spun through RNeasy spin columns, washed and RNA was eluted with RNase-free water. One microgram of total RNA was reverse transcribed into cDNA using the random hexamer (Invitrogen, Carlsbad, CA) priming method. All PCRs were performed in a 25 μl reaction containing 1× PCR buffer, 1.5 mm MgCl2, 0.2 mm dNTPs, 100 ng of DNA, 1.0 U of Platnium Taq (Invitrogen) and 20 pmol of each gene-specific primer (Integrated DNA Technologies, Coralville, IA). All cycling profiles incorporated an initial denaturation temperature of 50°C for 30 min followed by 35 amplification cycles (94°C for 15 s, 30 s at 56°C and 1 min at 68°C) and a final extension at 68°C for 5 min. PCR products were separated by gel electrophoresis using 4% agarose e-gels (Invitrogen).
Abnormal length products were cut from the gel and purified using the QIAquick Gel Extraction Kit (Qiagen). Purified PCR products were then TA cloned into the pCR®2.1 TOPO® Vector using the TOPO® TA Cloning® Kit (Invitrogen, Grand Island, NY) as per the manufacturer's instructions. Cloning reactions were then transformed into One Shot® TOP10 chemically competent cells (Invitrogen) and cultured onto plates containing lysogeny broth (LB) agar, ampicillin, isopropyl β-D-1-thiogalactopyranoside and X-gal. The pCR®2.1 TOPO® Vector contains beta-galactosidase that enables blue (if vector only) and white (if product is cloned into the vector) colony discrimination. White colonies were selected for PCR screening using the primers M13-20 forward (5′ TGTAAAACGACGGCCAGT 3′) and M13 reverse (5′ CAGGAAACAGCTATGACC 3′). Using the M13-20 forward and M13 reverse primers for the PCR adds 263 bp to the amplified product, making the band of interest for TRNT1 Exons 1–3 596 bp. Positive clones were cultured overnight in LB broth containing ampicillin at 37°C with shaking. The following day, bacterial DNA was purified using the QIAprep® Spin Miniprep Kit (Qiagen) and used for the Sanger sequencing analysis and diagnostic enzyme digests. Both cloned and uncloned PCR fragments were sequenced.
Western blot analysis
For fibroblast lysate preparation, fibroblasts from each cell strain (control human foreskin, P1, P2 and P3) were individually grown on six-well culture dishes in biopsy medium (MEMalpha media supplemented with 10% FBS and 1% Primocin). Samples from each well were lysed using 200 μl of lysis buffer [phosphate-buffered saline (PBS) +1% Triton-X with protease inhibitor tablet (cOmplete) and phosphatase inhibitor tablet (Roche Diagnostics Corporation, Indianapolis, IN)]. The lysate supernatant was collected after centrifugation at 11 000 rpm for 15 min at 4°C. Protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Life Technologies). Protein (20 μg) for all the cell strains and protein (5, 10 and 20 μg) for the amount-dependent analysis of control fibroblasts were individually resolved using Novex® 4–20% Tris-Glycine Mini Protein Gel electrophoresis (Life Technologies) and transferred to Immuno-Blot PVDF membranes (BioRad, Hercules, CA). Blocking and antibody incubations were done in SuperBlock (Life Technologies), and washes were in PBS + 0.1% Tween 20. Membranes were incubated overnight at 4°C with rabbit polyclonal anti-TRNT1 (NBP1-86589, Novus Biologicals, LLC, Littleton, CO) or mouse monoclonal anti-GAPDH (NB300-221, Novus Biologicals). The blots were probed for 1 h at room temperature with either horseradish peroxidase (HRP)-conjugated secondary antibody (SC-2004, goat anti-rabbit IgG HRP, Santa Cruz Biotechnology, Inc., Dallas, Texas) for TRNT1 or with Alexa Fluor® 488 (A11001, goat anti-mouse, Life Technologies) for GAPDH. Detection of HRP-labeled secondary antibody was done with ECL SuperSignal West Femto Maximum Sensitivity Substrate (Pierce Biotechnology). The bands were identified by autoradiography using the Carestream Kodak Biomax XAR film (Sigma-Aldrich, St. Louis, MO) and developed in the SRX-101A Konika Minolta Developer (Z & Z Medical, Cedar Falls, IA). For detecting the secondary fluorescent bands, the multi-wavelength imaging system BioRad VersaDoc (BioRad) and Quantity One Imaging software (BioRad) were utilized. Immunoreactivity was quantified using the ImageJ spot densitometry software.
Structural analysis of TRNT1
Beginning from a human TRNT1 structure determined by X-ray crystallography (4X4W) (38), the Glu43 residue was deleted followed by local optimization using the polarizable AMOEBA force field (39) in the program Force Field X (40).
Animal care
Collection of eggs from naturally spawning zebrafish adults and staging of the embryos were conducted using standard protocols (41,42).
Morpholinos, expression constructs and primers
Morpholinos designed to target the trnt1 start codon (5′ AGCTGCATCGTCAATAAAGCCCTGC), the Exon 2 splice acceptor (5′ GTGGACCTTAAAACAAGAGAGTCGT) and the Exon 3 splice donor (5′ AACCTATGCTTCACTCACCCTGGC) were purchased from Gene Tools. One- to two-cell-stage embryos were air-pressure injected with MO concentrations ranging from 1.5 to 6.5 ng. The 1.5 ng dose was used for the hypomorphic analyses.
For rescue experiments, 1.5 ng of trnt1 MO was injected directly into the cell at the 1 cell stage immediately followed by sequential injection of hTRNT1-myc RNA (200 pg) at the 1–2 cell stage. This was repeated in reverse order, RNA first then MO. Embryos were evaluated for morphological defects; touch sensitivity and visual function with the investigator masked to the experimental condition.
Myc-tagged human TRNT1 plasmid was linearized, and in vitro transcription was performed using the mMessage mMachine in vitro transcription kit (Ambion, Life Technologies, Carlsbad, CA).
The efficiencies of the SpMO (MO2 and MO3) were tested by carrying out RT-PCRs on total RNA (MMLV Advantage RT Kit, Clonetech) extracted from zebrafish embryos at the stages of interest (TRIzol, Life Technologies). PCR primers used 5′ GCAGGGCTTTATTGACGATG 3′ and 5′ CGTTTTCTCTTATTGCCTCCA 3′ for zebrafish trnt1 with Choice Taq DNA polymerase (Denville Scientific, Inc.) in 35 cycles with the annealing temperature of 59°C. Products were electrophoresed on a 2% agarose gels, purified from the gel band and cloned into pCR™II Vector (Life Technologies) for sequencing.
In situ hybridization
Whole mount in situ hybridization was performed according to previously described protocols (43). Embryos were fixed in 4% paraformaldehyde in 1× PBS. Digoxigenin-UTP-labeled trnt1 RNA riboprobes were made from linearized construct using the MAXIscript in vitro transcription kit (Ambion, Life Technologies, Carlsbad, CA). Retina sections (10 μm) were collected from postfixed embryos by cryosectioning as previously described (44).
Western blot, zebrafish
RNA-injected embryos were lysed, and processed protein samples were electrophoresed on a pre-cast 4–12% NuPAGE Novex Bis-Tris gel (Invitrogen). Protein was transferred to a PVDF membrane (Amersham). The membrane was blocked and then probed with either mouse anti-myc antibody (9E10; Santa Cruz Biotechnology, Santa Cruz, CA at 1:10 000) or rabbit polyclonal anti-β-actin (1:2000, Sigma, St Louis, MO) antibody.
Vision startle response assay
The vision-evoked startle response assay is a standard behavioral test that we and others have described previously (13,45–47). Briefly, light-adapted 5 dpf fish are subjected to a 1 s block of the bright-light stimulus to simulate a shadow cast by a predator. Abrupt changes in motion are recorded as a positive response. After five trials spaced 30 s apart, a blunt needle is used to mechanically stimulate a response from the fish. Fish that fail to move after mechanical stimulation are removed from the analysis to reduce the possibility of motor and neural defects confounding the evaluation of vision. Data were plotted as the average number out of five trials that an animal was scored as positive. Comparisons between cohorts were made using the Mann–Whitney test (sample size: uninjected 95, MO only 92, RNA only 48, MO plus RNA 59).
Whole embryo staining for globin expression
o-Dianisidine (Sigma) staining was used to study the expression of globin. Dechorionated embryos (trnt1 morphant and wt) were stained for 15 min in the dark in o-dianisidine (0.6 mg/ml), 0.01 M sodium acetate (pH 4.5), 0.65% H2O2 and 40% (vol/vol) ethanol. After staining, embryos were postfixed in 4% paraformaldehyde.
tRNA-seq and analysis
RNA samples from WT and trnt1-knockdown animals were size selected to capture tRNAs (73–94 nt), prepared for sequencing using the TruSeq Small RNA library preparation kit (Illumina, San Diego, CA) and sequenced on the Illumina MiSeq platform at the Genomics Division of the Iowa Institute for Human Genetics. Adapter and barcode sequences were removed from reads using cutadapt (ver. 1.8.dev0) (48), retaining reads between 65 and 95 nucleotides in length. To build a database of tRNA sequences, we added an artificial CCA to the 3′ end of the predicted sequences annotated in the Genomic tRNA Database (http://gtrnadb.ucsc.edu/Dreri/) (49). Because of the high rate of duplication of tRNA sequences, we collapsed highly similar sequences using uicluster (ver. 3.0.5) (50). Trimmed RNA-Seq reads were mapped to the set of predicted tRNAs using blat (ver. 35) (51).
Blat output files were filtered so that (1) there were one or fewer mismatches between a read query and the predicted tRNA target, (2) there were no gaps, (3) at least 95% of the read query length matched the predicted tRNA, (4) the matching portion of the read query extended to the end of the read, (5) the aligned read fell within the last four nucleotides of the predicted tRNA target, (6) the read aligned to the plus strand of the predicted tRNA, (7) the read covered 95% or more of the predicted tRNA and (8) the longest match for a read aligned to a single predicted tRNA.
After this processing, tRNAs that correctly underwent the posttranscriptional modification to add the CCA would align to the end of the sequence, where tRNAs escaping the modification would produce a read ending 3 bp before the end of the target sequence. Using Pearson's χ2 test with Yates’ continuity correction (implemented in R), we compared the number of reads with and without a complete CCA in both groups (Fig. 6A). The raw data from this experiment are available in the Sequence Read Archive (SRP056603).
Supplementary Material
Funding
This study was supported in part by National Institutes of Health grants DP2OD007483 and EY024605 and the Stephen A. Wynn Foundation.
Supplementary Material
Acknowledgements
The authors wish to acknowledge Armin Avdic, Carol Holman and Shemin Zeng for assistance in data gathering and analysis. The authors also wish to thank the patients and their families for their participation in this research study and their partnership in identifying the causes and cures of retinal degenerations.
Conflict of Interest statement. None declared.
References
- 1.Daiger S.P., Sullivan L.S., Bowne S.J. (2013) Genes and mutations causing retinitis pigmentosa. Clin. Genet., 84, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wangsa-Wirawan N.D., Linsenmeier R.A. (2003) Retinal oxygen: fundamental and clinical aspects. Arch. Ophthalmol., 121, 547–557. [DOI] [PubMed] [Google Scholar]
- 3.Poulos M.G., Batra R., Charizanis K., Swanson M.S. (2011) Developments in RNA splicing and disease. Cold Spring Harb. Perspect. Biol., 3, a000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sasarman F., Thiffault I., Weraarpachai W., Salomon S., Maftei C., Gauthier J., Ellazam B., Webb N., Antonicka H., Janer A. et al. (2015) The 3′ addition of CCA to mitochondrial tRNASer(AGY) is specifically impaired in patients with mutations in the tRNA nucleotidyl transferase TRNT1. Hum. Mol. Genet., 24, 2841–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Betat H., Rammelt C., Mörl M. (2010) tRNA nucleotidyltransferases, ancient catalysts with an unusual mechanism of polymerization. Cell. Mol. Life Sci., 67, 1447–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakraborty P.K., Schmitz-Abe K., Kennedy E.K., Mamady H., Naas T., Durie D., Campagna D.R., Lau A., Sendamarai A.K., Wiseman D.H. et al. (2014) Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood, 124, 2867–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiseman D.H., May A., Jolles S., Connor P., Powell C., Heeney M.M., Giardina P.J., Klaassen R.J., Chakraborty P., Geraghty M.T. et al. (2013) A novel syndrome of congenital sideroblastic anemia, B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD). Blood, 122, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Exome Aggregation Consortium (ExAC). Cambridge, MA: http://exac.broadinstitute.org. [Google Scholar]
- 9.1000 Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lush M.E., Piotrowski T. (2014) Sensory hair cell regeneration in the zebrafish lateral line. Dev. Dyn., 243, 1187–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paffett-Lugassy N.N., Zon L.I. (2005) Analysis of hematopoietic development in the zebrafish. Methods Mol. Med., 105, 171–198. [DOI] [PubMed] [Google Scholar]
- 12.Whitfield T.T. (2002) Zebrafish as a model for hearing and deafness. J. Neurobiol., 53, 157–171. [DOI] [PubMed] [Google Scholar]
- 13.Easter S.S., Nicola G.N. (1996) The development of vision in the zebrafish (Danio rerio). Dev. Biol., 180, 646–663. [DOI] [PubMed] [Google Scholar]
- 14.Tucker B.A., Scheetz T.E., Mullins R.F., DeLuca A.P., Hoffmann J.M., Johnston R.M., Jacobson S.G., Sheffield V.C., Stone E.M. (2011) Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc. Natl Acad. Sci. U S A, 108, E569–E576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tucker B.A., Mullins R.F., Streb L.M., Anfinson K., Eyestone M.E., Kaalberg E., Riker M.J., Drack A.V., Braun T.A., Stone E.M. (2013) Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. eLife, 2, e00824–e00824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drivas T.G., Wojno A.P., Tucker B.A., Stone E.M., Bennett J. (2015) Basal exon skipping and genetic pleiotropy, A predictive model of disease pathogenesis. Sci. Transl. Med., 7, 291ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiley L.A., Burnight E.R., Songstad A.E., Drack A.V., Mullins R.F., Stone E.M., Tucker B.A. (2015) Patient-specific induced pluripotent stem cells (iPSCs) for the study and treatment of retinal degenerative diseases. Prog. Retin. Eye Res., 44, 15–35. [DOI] [PubMed] [Google Scholar]
- 18.Burnight E.R., Wiley L.A., Drack A.V., Braun T.A., Anfinson K.R., Kaalberg E.E., Halder J.A., Affatigato L.M., Mullins R.F., Stone E.M. et al. (2014) CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther., 21, 662–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamao H., Mandai M., Okamoto S., Sakai N., Suga A., Sugita S., Kiryu J., Takahashi M. (2014) Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Reports., 2, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamba D.A., Karl M.O., Ware C.B., Reh T.A. (2006) Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc. Natl Acad. Sci. U S A, 103, 12769–12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamba D.A., McUsic A., Hirata R.K., Wang P.-R., Russell D., Reh T.A. (2010) Generation, purification and transplantation of photoreceptors derived from human induced pluripotent stem cells. PLoS ONE, 5, e8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burnight E.R., Wiley L.A., Mullins R.F., Stone E.M., Tucker B.A. (2015) Gene therapy using stem cells. Cold Spring Harb. Perspect. Med., 5, a017434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tucker B.A., Mullins R.F., Stone E.M. (2014) Stem cells for investigation and treatment of inherited retinal disease. Hum. Mol. Genet., 23, R9–R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sander J.D., Joung J.K. (2014) CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol., 32, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131, 861–872. [DOI] [PubMed] [Google Scholar]
- 26.Han A.P., Yu C., Lu L., Fujiwara Y., Browne C., Chin G., Fleming M., Leboulch P., Orkin S.H., Chen J.J. (2001) Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J., 20, 6909–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taher A.T., Viprakasit V., Musallam K.M., Cappellini M.D. (2013) Treating iron overload in patients with non-transfusion-dependent thalassemia. Am. J. Hematol., 88, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krumm N., Sudmant P.H., Ko A., O'Roak B.J., Malig M., Coe B.P.; Nhlbi Exome Sequencing Project, Quinlan A.R., Nickerson D.A., Eichler E.E. (2012) Copy number variation detection and genotyping from exome sequence data. Genome Res., 22, 1525–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner A.H., Taylor K.R., DeLuca A.P., Casavant T.L., Mullins R.F., Stone E.M., Scheetz T.E., Braun T.A. (2013) Prioritization of retinal disease genes: an integrative approach. Hum. Mutat., 34, 853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kong Y. (2011) Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics, 98, 152–153. [DOI] [PubMed] [Google Scholar]
- 33.Webster A.R., Héon E., Lotery A.J., Vandenburgh K., Casavant T.L., Oh K.T., Beck G., Fishman G.A., Lam B.L., Levin A. et al. (2001) An analysis of allelic variation in the ABCA4 gene. Invest. Ophthalmol. Vis. Sci., 42, 1179–1189. [PubMed] [Google Scholar]
- 34.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R et al. , (2007) Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- 36.Marchler-Bauer A., Derbyshire M.K., Gonzales N.R., Lu S., Chitsaz F., Geer L.Y., Geer R.C., He J., Gwadz M., Hurwitz D.I. et al. (2015) CDD: NCBI's conserved domain database. Nucleic Acids Res., 43, D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tucker B.A., Anfinson K.R., Mullins R.F., Stone E.M., Young M.J. (2013) Use of a synthetic xeno-free culture substrate for induced pluripotent stem cell induction and retinal differentiation. Stem Cells Transl Med., 2, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuhn C.-D., Wilusz J.E., Zheng Y., Beal P.A., Joshua-Tor L. (2015) On-enzyme refolding permits small RNA and tRNA surveillance by the CCA-adding enzyme. Cell, 160, 644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ponder J.W., Wu C., Ren P., Pande V.S., Chodera J.D., Schnieders M.J., Haque I., Mobley D.L., Lambrecht D.S., DiStasio R.A. Jr et al. (2010) Current status of the AMOEBA polarizable force field. J. Phys. Chem. B, 114, 2549–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fenn T.D., Schnieders M.J. (2011) Polarizable atomic multipole X-ray refinement, weighting schemes for macromolecular diffraction. Acta Cryst., D67, 957–965 doi: 10.1107/S0907444911039060. [DOI] [PubMed] [Google Scholar]
- 41.Westerfield M. (2000) The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio), 4th edn. University of Oregon Press, Eugene. [Google Scholar]
- 42.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn., 203, 253–310. [DOI] [PubMed] [Google Scholar]
- 43.Westfall T.A., Hjertos B., Slusarski D.C. (2003) Requirement for intracellular calcium modulation in zebrafish dorsal-ventral patterning. Dev. Biol., 259, 380–391. [DOI] [PubMed] [Google Scholar]
- 44.Mei X., Wu S., Bassuk A.G., Slusarski D.C. (2013) Mechanisms of prickle1a function in zebrafish epilepsy and retinal neurogenesis. Dis. Model Mech., 6, 679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishimura D.Y., Baye L.M., Perveen R., Searby C.C., Avila-Fernandez A., Pereiro I., Ayuso C., Valverde D., Bishop P.N., Manson F.D.C. et al. (2010) Discovery and functional analysis of a retinitis pigmentosa gene, C2ORF71. Am. J. Hum. Genet., 86, 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pretorius P.R., Baye L.M., Nishimura D.Y., Searby C.C., Bugge K., Yang B., Mullins R.F., Stone E.M., Sheffield V.C., Slusarski D.C. (2010) Identification and functional analysis of the vision-specific BBS3 (ARL6) long isoform. PLoS Genet., 6, e1000884–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baye L.M., Patrinostro X., Swaminathan S., Beck J.S., Zhang Y., Stone E.M., Sheffield V.C., Slusarski D.C. (2011) The N-terminal region of centrosomal protein 290 (CEP290) restores vision in a zebrafish model of human blindness. Hum. Mol. Genet., 20, 1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin M. (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal, 17, 10–12. [Google Scholar]
- 49.Lowe T.M., Eddy S.R. (1997) tRNAscan-SE, a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res., 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chun C.K., Scheetz T.E., Bonaldo M., de F., Brown B., Clemens A., Crookes-Goodson W.J., Crouch K., DeMartini T., Eyestone M., Goodson M.S. et al. (2006) An annotated cDNA library of juvenile Euprymna scolopes with and without colonization by the symbiont Vibrio fischeri. BMC Genomics, 7, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kent W. (2002) BLAT-the BLAST-like alignment tool. Genome Res., 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.