

Abstract
Hundreds of genomic loci have been associated with a significant number of immune-mediated diseases, and a large proportion of these associated loci are shared among traits. Both the molecular mechanisms by which these loci confer disease susceptibility and the extent to which shared loci are implicated in a common pathogenesis are unknown. We therefore sought to dissect the functional components at loci shared between two autoimmune diseases: coeliac disease (CeD) and rheumatoid arthritis (RA). We used a cohort of 12 381 CeD cases and 7827 controls, and another cohort of 13 819 RA cases and 12 897 controls, all genotyped with the Immunochip platform. In the joint analysis, we replicated 19 previously identified loci shared by CeD and RA and discovered five new non-HLA loci shared by CeD and RA. Our fine-mapping results indicate that in nine of 24 shared loci the associated variants are distinct in the two diseases. Using cell-type-specific histone markers, we observed that loci which pointed to the same variants in both diseases were enriched for marks of promoters active in CD14+ and CD34+ immune cells (P < 0.001), while loci pointing to distinct variants in one of the two diseases showed enrichment for marks of more specialized cell types, like CD4+ regulatory T cells in CeD (P < 0.0001) compared with Th17 and CD15+ in RA (P = 0.0029).
Introduction
Over the last decade, genome-wide analyses and fine-mapping efforts have shown that different autoimmune diseases share associations with many common genetic loci (1–9), a finding which suggests a common molecular background between these diseases. The most striking example of this is the HLA locus, which is associated with different autoimmune diseases. However, although the association signal to HLA is common to the majority of the autoimmune diseases, most autoimmune diseases are associated with a specific HLA-allele or haplotype. For example, both coeliac disease (CeD) and rheumatoid arthritis (RA) show strong association with the HLA-DR/DQ locus, with the strongest effect in CeD caused by the HLA-DQA1*05:01-HLA-DQB1*02:01 haplotype, while the RA association is to the HLA-DRB1*04 alleles (10,11). Furthermore, both CeD and RA also share other features. They are polygenic, complex and heterogeneous diseases (12). Also, both diseases show infiltration of T cells in specific primary target organs. Therefore, we sought to study the shared genetic components between RA and CeD to further our understanding of the common biological and molecular pathways involved in disease development.
Previous studies have already identified 26 shared genetic loci associated with CeD and RA (8,13–15) (Supplementary Material, Table S1). However, the co-morbidity of CeD and RA in a single patient is only marginally increased (16), which suggests that shared associated loci might affect different downstream pathways. This suggestion is further substantiated by the observation that the alleles of two of the established shared loci (TAGAP and LBH) show opposite allelic effects, whereby the risk allele for one disease is actually protective for the other disease.
In this work, we aimed to identify additional shared loci between CeD and RA, to perform comprehensive fine-mapping of the established and new shared loci by the analysis of densely genotyped regions present in the Immunochip platform and integrating different sources of information (genetic association, expression quantitative trait loci (eQTL) and cell-type-specific enrichment). For this study, we made use of two large case–control cohorts, each of which was genotyped on the Immunochip (17). Subsequently, we analysed the downstream effect of CeD- and RA-associated variants using eQTLs and by assessing their overlap with cell-type-specific histone modifications.
Results
The cross-disease meta-analysis was performed by combining CeD and RA cohorts (Fig. 1). After quality control and removing duplicates, the CeD dataset comprised 12 381 cases and 7827 controls and the RA dataset comprised 13 819 cases and 12 927 controls. A genotyping rate of >98% was used in both datasets. Each dataset consisted of multiple populations, mainly of Caucasian origin (Table 1). Quality control was performed per trait and per population in order to take into account possible intra- and interpopulation structures. Inspection of the principal components (PCs) showed uniform clusters of cases and controls across the populations (Supplementary Material, Fig. S1). After correcting for the first three PCs, we did not observe significant genomic inflation in the data (Supplementary Material, Fig. S2).
Figure 1.
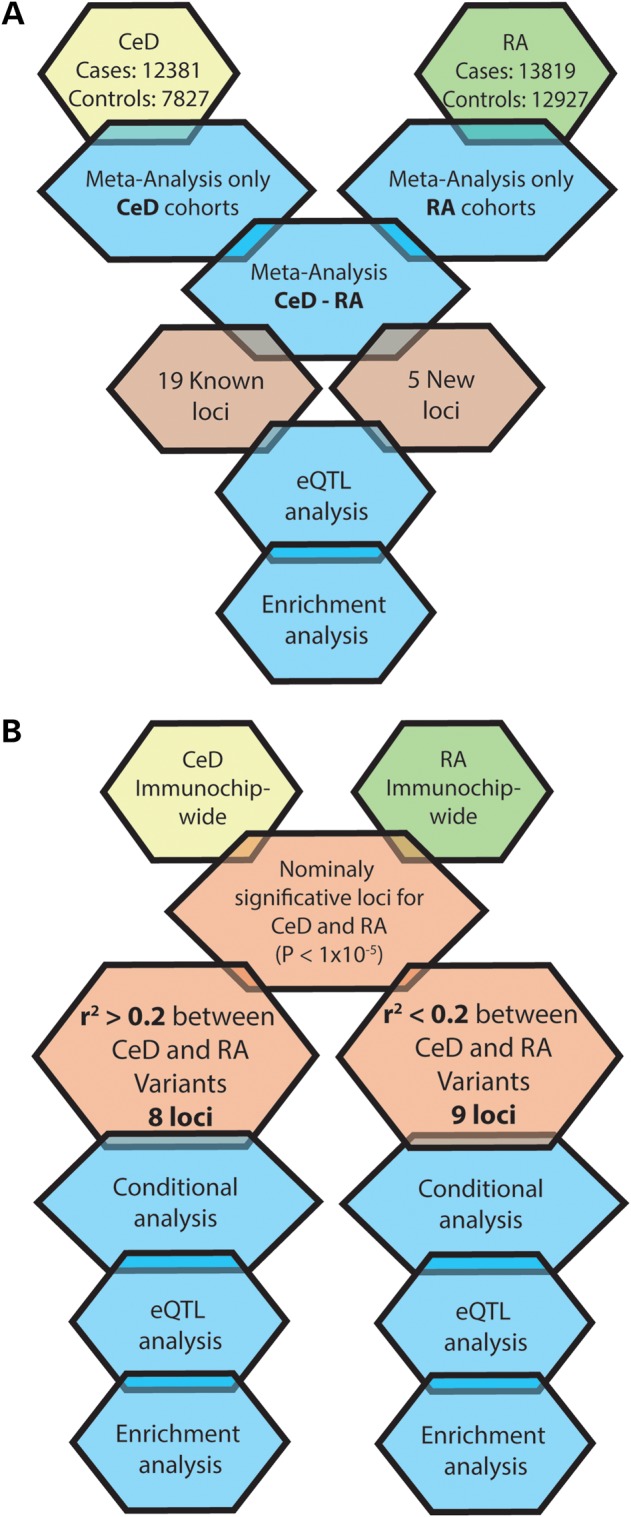
Analysis process. (A) Meta-analysis of CeD and RA datasets and subsequent functional in silico analysis. (B) Based on the results of the meta-analysis, regions with nominal P-values for both CeD and RA were determined. The LD between the CeD and RA SNPs associated in each region was tested with a subsequent functional in silico analysis in each group of SNPs.
Table 1.
Populations included as part of the meta-analysis and corrected inflation factors
| Collections | Cases | Controls | Females (%) | λ1000 | |
|---|---|---|---|---|---|
| CeD | India | 371 | 509 | 31.4 | 1 |
| Netherlands | 1150 | 716 | 40.8 | 1 | |
| Poland | 521 | 541 | 52.4 | 1 | |
| Spain | 1131 | 662 | 40.2 | 1 | |
| Italy | 1486 | 1270 | 26.1 | 1 | |
| UK | 7722 | 4129 | 64.5 | 1.04 | |
| Total | 12381 | 7827 | 37.1 | ||
| RA | Spain | 804 | 398 | 71.3 | 1.11 |
| Netherlands | 645 | 1004 | 49.7 | 1 | |
| Swedish EIRA | 2762 | 1939 | 71.4 | 1 | |
| Swedish Umea | 852 | 963 | 69 | 1 | |
| UK | 3857 | 4291 | 62.8 | 1.03 | |
| US | 4899 | 4332 | 43.3 | 1.02 | |
| Total | 13819 | 12927 | 57.6 | ||
| Total Meta-analysis | 26200 | 20754 | 65 |
Meta-analysis identifies five novel associations shared between CeD and RA
The results of our meta-analysis replicated (P-valueMeta < 5 × 10−8) 19 loci previously reported as shared between CeD and RA (Table 2) (8,13–15). Consistent with the previous reports, 17 of these loci showed an effect to the same allele, while two showed an opposite effect (13,15). We also identified associations with five new loci, all with the same directional effects (Table 2). All of the new loci have been previously associated with either CeD or RA (8,14).
Table 2.
Loci associated with a genome-wide significant threshold after the meta-analysis of CeD-RA in both the direct and the opposite effect of association. The SNP rs6679677 (PTPN22) presents a high significance in RA, which is boosting the P meta association, however it meets our criteria od shared loci
| CHR | SNP | Position (hg19) | Chromosome localization | Meta P-value | Meta OR | CeD P-value | CeD OR | RA P-value | RA OR | Q P-value | I2 | Previous Associated Trait |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Direct effect - Known associations | ||||||||||||
| 1 | rs4073285 | 2539796 | 1p36.32 | 2.39E-13 | 0.90 | 1.17E-05 | 0.91 | 3.70E-09 | 0.89 | 0.63 | 0 | Class 3 |
| 1 | rs6696533 | 198733567 | 1q32.1 | 2.70E-08 | 1.08 | 0.000203 | 1.08 | 3.49E-05 | 1.08 | 0.27 | 16.39 | Class 3 |
| 2 | rs10203477 | 61104985 | 2p16.1 | 3.88E-08 | 0.93 | 1.24E-06 | 0.90 | 2.11E-03 | 0.95 | 0.23 | 20.7 | Class 3 |
| 2 | rs11123810 | 100759285 | 2q11.2 | 7.40E-12 | 0.91 | 3.78E-06 | 0.91 | 4.28E-07 | 0.91 | 0.22 | 21.72 | Class 3 |
| 2 | rs6749371 | 191902184 | 2q32.3 | 6.92E-11 | 0.84 | 2.32E-07 | 0.81 | 3.42E-05 | 0.87 | 0.27 | 17.25 | Class 3 |
| 2 | rs1980422 | 204610396 | 2q33.2 | 1.95E-17 | 1.14 | 3.52E-11 | 1.17 | 3.99E-08 | 1.12 | 0.12 | 31.7 | Class 3 |
| 4 | rs62323881 | 123038295 | 4q27 | 6.05E-09 | 1.16 | 2.12E-05 | 1.18 | 6.11E-05 | 1.15 | 0.89 | 0 | Class 3 |
| 6 | rs72928038 | 90976768 | 6q15 | 3.91E-11 | 1.13 | 3.34E-06 | 1.14 | 2.37E-06 | 1.12 | 0.15 | 29.08 | Class 3 |
| 6 | rs17264332 | 138005515 | 6q23.3 | 2.51E-27 | 1.19 | 2.35E-23 | 1.28 | 1.13E-08 | 1.13 | 0.30 | 17.13 | Class 3 |
| 7 | rs74830391 | 37427289 | 7p14.1 | 2.70E-09 | 1.14 | 0.0001362 | 1.13 | 4.98E-06 | 1.14 | 0.36 | 8.44 | Class 3 |
| 8 | rs6651252 | 129567181 | 8q24.21 | 7.88E-09 | 0.89 | 0.0001265 | 0.89 | 1.60E-05 | 0.89 | 0.39 | 5.99 | Class 3 |
| 10 | rs947474 | 6390450 | 10p15.1 | 8.33E-11 | 0.89 | 7.61E-07 | 0.88 | 1.67E-05 | 0.90 | 0.92 | 0 | Class 3 |
| 11 | rs11217040 | 118680648 | 11q23.3 | 1.52E-11 | 0.89 | 2.01E-09 | 0.86 | 0.000199 | 0.92 | 0.62 | 0 | Class 3 |
| 12 | rs11066188 | 112610714 | 12q24.13 | 8.99E-10 | 1.09 | 2.36E-08 | 1.12 | 0.001112 | 1.06 | 0.39 | 5.86 | Class 3 |
| 18 | rs80209296 | 12860801 | 18p11.21 | 1.36E-12 | 1.14 | 3.71E-07 | 1.15 | 6.45E-07 | 1.13 | 0.13 | 30.23 | Class 3 |
| 21 | rs1893592 | 43855067 | 21q22.3 | 7.86E-11 | 0.91 | 4.41E-08 | 0.88 | 0.000107 | 0.93 | 0.11 | 33.36 | Class 3 |
| 22 | rs4821124 | 21979289 | 22q11.21 | 2.31E-11 | 1.12 | 5.28E-08 | 1.15 | 3.52E-05 | 1.10 | 0.83 | 0 | Class 3 |
| Opposite effect - Known associations | ||||||||||||
| 2 | rs7579944 | 30445026 | 2p23.1 | 1.81E-08 | 1.08 | 7.63E-05 | 1.09 | 5.85E-05 | 1.08 | 0.60 | 0 | Class 3 |
| 6 | rs212400 | 159473574 | 6q25.3 | 6.15E-11 | 1.10 | 6.87E-10 | 1.14 | 0.000977 | 1.06 | 0.48 | 0 | Class 3 |
| Direct effect - Novel associations | ||||||||||||
| 1 | rs6679677 | 114303808 | 1p13.2 | 1.22E-53 | 1.41 | 0.0004456 | 1.14 | 2.63E-62 | 1.60 | 0.00 | 79.8 | Class 1 |
| 1 | rs17849502 | 183532580 | 1q25.3 | 1.30E-14 | 1.26 | 1.66E-13 | 1.40 | 0.000145 | 1.16 | 0.36 | 8.35 | Class 2 |
| 2 | rs16867384 | 182111206 | 2q31.3 | 5.89E-09 | 1.10 | 9.49E-08 | 1.14 | 0.002204 | 1.07 | 0.19 | 24.54 | Class 2 |
| 3 | rs67676925 | 46274259 | 3p21.31 | 2.31E-11 | 1.18 | 6.72E-11 | 1.28 | 0.001568 | 1.11 | 0.20 | 23.47 | Class 2 |
| 21 | rs9979383 | 36715761 | 21q22.12 | 1.20E-08 | 0.92 | 0.004455 | 0.94 | 3.69E-07 | 0.91 | 0.31 | 13.46 | Class 1 |
Fine-mapping of association signals at shared loci defines shared and independent effects
Given that the Immunochip was designed as a fine-mapping platform for loci previously associated with autoimmune diseases, we aimed to ‘zoom-in’ on each of the 189 Immunochip loci (we did not included the MHC-HLA region) to refine the association patterns. In total, we selected the 17 loci (of which 14 overlap with loci identified via meta-analysis) that showed suggestive association with CeD and RA individually (P-valueTrait < 5 × 10−4), regardless of whether the association was observed to the same or to different SNP (Fig. 1B). We first determined if the top SNP in each of the 17 loci was the same for CeD and RA and found that in all the loci it was different (Table 3). Next we investigated, for each locus, if the CeD- and RA-associated SNPs were correlated by calculating the linkage disequilibrium (LD) between them. We identified that in three loci (harbouring MMEL1, ELMO1 and PLKCQ), the top CeD and RA signals were in strong LD with each other (r2 > 0.8). For another five loci (harbouring AFF3, BACH2, PVT1, PTPN2 and UBE2L3), the LD was intermediate (0.2 < r2 < 0.8), while in the remaining nine loci the CeD and RA SNPs were in low LD with each other (r2 < 0.2) (harbouring PUS10, STAT4, CD28-CTL4, CCR1-CCR3, IL12A, IL2-IL21, OLIG3-TNFAIP3, CD6 and MMP9-CD40).
Table 3.
Shared loci between CeD and RA
| CeD |
CeD conditioned on RA | RA | RA conditioned on CeD | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CHR | Locus | SNP | Position | Meta P-value | Distance | r2 | D’ | SNP | Position | Meta P-value | ||
| 1 | MMEL1 | rs10797437 | 2539006 | 6.74E-08 | 0.0001 | 790 | 0.90 | 0.98 | rs4073285 | 2539796 | 3.70E-09 | 0.0003 |
| 2 | AFF3 | rs11681227 | 100754997 | 3.31E-06 | 0.0044 | 82305 | 0.41 | 0.89 | rs10209110 | 100672692 | 7.36E-09 | 0.0013 |
| 6 | BACH2 | rs2174281 | 90987872 | 2.45E-06 | 0.0048 | 11104 | 0.26 | 1 | rs72928038 | 90976768 | 2.37E-06 | 2.15E-06 |
| 7 | ELMO1 | rs60600003 | 37382465 | 2.37E-06 | 0.0052 | 51735 | 0.85 | 0.95 | rs28482033 | 37434200 | 3.17E-06 | 0.0042 |
| 8 | MIR1208/PVT1 | rs10089519 | 129532118 | 9.50E-05 | 0.0177 | 35063 | 0.22 | 0.85 | rs6651252 | 129567181 | 1.60E-05 | 0.0035 |
| 10 | PRKCQ | rs2387397 | 6390192 | 1.46E-07 | 0.0518 | 258 | 0.81 | 0.99 | rs947474 | 6390450 | 1.67E-05 | 0.0543 |
| 22 | UBE2L3 | rs4821116 | 21973319 | 5.04E-08 | 0.0006 | 5777 | 0.75 | 0.98 | rs11089637 | 21979096 | 3.45E-05 | 0.2083 |
| 18 | PTPN2 | rs2847260 | 12775591 | 7.84E-08 | 0.0015 | 104615 | 0.43 | 0.67 | rs7241016 | 12880206 | 4.21E-07 | 0.0012 |
| 2 | REL/PUS10 | rs13003464 | 61186829 | 4.93E-10 | 6.02E-10 | 279774 | 0.01 | 0.52 | rs78404002 | 61466603 | 8.72E-06 | 6.14E-06 |
| 2 | STAT4 | rs7574608 | 191909864 | 4.09E-09 | 1.56E-08 | 23390 | 0.02 | 0.99 | rs13426947 | 191933254 | 4.65E-07 | 3.14E-06 |
| 2 | CD28/CTLA4 | rs2162610 | 204718729 | 1.49E-12 | 3.08E-08 | 20190 | 0.16 | 0.95 | rs3087243 | 204738919 | 9.48E-09 | 7.14E-07 |
| 3 | CCR1/CCR3 | rs6441963 | 46354839 | 1.51E-13 | 4.27E-13 | 373624 | 0.00 | 0.03 | rs17078454 | 45981215 | 4.17E-05 | 3.59E-05 |
| 3 | IL12A | rs17753641 | 159647674 | 8.23E-23 | 3.74E-24 | 62327 | 0.09 | 0.96 | rs582054 | 159710001 | 6.21E-05 | 4.18E-04 |
| 4 | IL2/IL21 | rs13132308 | 123551114 | 8.16E-29 | 9.86E-27 | 512819 | 0.02 | 0.96 | rs62323881 | 123038295 | 6.11E-05 | 1.45E-04 |
| 6 | OLIG3/TNFAIP3 | rs17264332 | 138005515 | 2.35E-23 | 1.58E-23 | 238224 | 0.02 | 0.46 | rs58721818 | 138243739 | 3.68E-09 | 3.77E-07 |
| 11 | CD6 | rs11230544 | 60746892 | 2.61E-05 | 2.97E-05 | 29592 | 0.01 | 0.39 | rs2074226 | 60776484 | 3.83E-06 | 2.63E-06 |
| 20 | MMP9/CD40 | rs6017715 | 44599839 | 6.07E-07 | 1.10E-06 | 149412 | 0.02 | 1 | rs4239702 | 44749251 | 7.35E-08 | 1.28E-07 |
Upper panel shows the regions with non-indepenpent (r2>0.2) association to both diseases. Lower panel shows the regions with independent (r2<0.2) associations in each of the loci analysed. Meta P-value columns in both CeD and RA are the significance values for the meta-analysis of the populations conforming each of the datasets. Distance refers to the amount of base-pairs between the CeD and RA SNPs. D’ and r2 are the LD mesuares between the CeD and RA top SNPs in the region.
Next, for each of the loci, we performed an association analysis in the CeD dataset, conditioning for the top RA SNP in the locus and vice versa, to test if the signals are independent. For the loci with strong or moderate LD, the associations dropped remarkably after the conditional analysis, with the exception of the RA association in the BACH2 locus, which remained after conditioning on the CeD top SNP (Table 4). The association of the SNPs in the nine loci with low LD (r2 < 0.2) remained significant after conditional analysis. From this analysis, we concluded that in 9 out of 17 loci with association to both CeD and RA, the CeD- and RA-associated SNPs were independent of each other and tag different haplotypes.
Table 4.
Downstream functional effect of shared loci between CeD and RA
| CeD |
RA |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CHR | locus | r2 | SNP | Risk allele (freq) | eQTL | Beta coefficient | eQTL P-value | SNP | Risk allele (freq) | eQTL | Beta coefficient | eQTL P-value |
| 1 | MMEL1 | 0.90 | rs10797437 | C(0.3283) | MMEL1 | −0.46 | 2.16E-35 | rs4073285 | T(0.3191) | MMEL1 | −0.44 | 2.19E-32 |
| 2 | AFF3 | 0.41 | rs11681227 | G(0.3499) | AFF3 | −0.24 | 6.15E-10 | rs10209110 | T(0.4847) | AFF3 | −0.4 | 5.61E-26 |
| 6 | BACH2 | 0.26 | rs2174281 | C(0.4754) | BACH2 | −0.14 | 0.0004023 | rs72928038 | A(0.1711) | BACH2 | −0.24 | 2.24E-10 |
| 7 | ELMO1 | 0.85 | rs60600003 | G(0.1077) | ELMO1 | −0.29 | 4.90E-14 | rs28482033 | C(0.1162) | ELMO1 | −0.29 | 3.99E-14 |
| 8 | MIR1208/PVT1 | 0.22 | rs10089519 | − | no eQTL | − | − | rs6651252 | − | no eQTL | − | − |
| 10 | PRKCQ | 0.81 | rs2387397 | G(0.2175) | DKFZP667F0711 | −0.21 | 7.42E-08 | rs947474 | G(0.1808) | DKFZP667F0711 | −0.21 | 2.67E-08 |
| 22 | UBE2L3 | 0.75 | rs4821116 | T(0.2029) | UBE2L3 | 0.46 | 1.40E-34 | rs11089637 | C(0.1751) | UBE2L3 | 0.4 | 5.63E-26 |
| 18 | PTPN2 | 0.43 | rs2847260 | − | no eQTL | − | − | rs7241016 | − | no eQTL | − | − |
| 2 | REL/PUS10 | 0.01 | rs13003464 | G(0.4076) | AHSA2 | −0.41 | 5.25E-28 | rs78404002 | T(0.04844) | KIAA1841,C2orf74 | −0.16 | 2.39E-05 |
| 2 | STAT4 | 0.02 | rs7574608 | − | no eQTL | − | − | rs13426947 | − | no eQTL | − | − |
| 2 | CD28/CTLA4 | 0.16 | rs2162610 | C(0.2008) | AC125238.1, AC125238.4 | 0.19 | 4.54E-07 | rs3087243 | A(0.4256) | AC016903.2 | 0.11 | 0.0036585 |
| 3 | CCR1/CCR3 | 0.00 | rs6441963 | T (0.3785) | CCR3 | 0.22 | 2.30E-08 | rs17078454 | A (0.2129) | FYCO1 | 0.22 | 7.96E-09 |
| 3 | IL12A | 0.09 | rs17753641 | − | no eQTL | − | − | rs582054 | − | no eQTL | − | − |
| 4 | IL2/IL21 | 0.02 | rs13132308 | − | no eQTL | − | − | rs62323881 | − | no eQTL | − | − |
| 6 | OLIG3/TNFAIP3 | 0.02 | rs17264332 | − | no eQTL | − | − | rs58721818 | − | no eQTL | − | − |
| 11 | CD6 | 0.01 | rs11230544 | − | no eQTL | − | − | rs2074226 | G (0.3111) | CD6 | 0.16 | 4.18E-05 |
| 20 | MMP9/CD40 | 0.02 | rs6017715 | G(0.05023) | PLTP | −0.13 | 0.0006454 | rs4239702 | T(0.2682) | CD40, PLTP | −0.33; 0.21 | 2.1E-18; 6.4E-8 |
Upper panel shows the regions with non-indepenpent association (r2>0.2) and lower panel shows the regions with independent associations (r2<0.2). R2 refers to the LD between the CeD and RA top SNPs in each of the regions in the table. Risk allele shows the allele associated in each of the diseases with the frequency calculated. The beta coefficient corresponds to the change in the expression calculated for the variant in each locus regarding to the gene with eQTL effect.
Associations in shared loci exhibit functional effects that can be different depending on the primary disease associated
We next investigated whether the SNPs found in the previous analysis also elicited different downstream effects by performing eQTL analysis, by analysing transcription factor binding sites and cell-type-specific histone marks. To explore all the shared regions with genome-wide P-values, we analysed 24 shared loci, of which 17 showed a significant eQTL effect (Table 5). In five out of the nine loci with low LD between CeD and RA SNPs (i.e. those harbouring independent SNPs), the SNPs for both diseases showed nominal significant eQTL effects that affected different transcripts or opposite effects between disease SNPs (i.e. CD40, CD6, REL, CTLA4 and CCR1/CCR3, see Table 4). For example, in the MMP9-CD40 locus (chr 20q13.12), the rs6017715-G allele (which confers risk for CeD) is correlated with decreased expression of the PLTP gene (P-valueeQTL = 6.5 × 10−4), whereas rs4239702 (which confers risk to RA, is not in LD with rs6017715 and is located in the same locus) strongly increases the expression of CD40 (P-valueeQTL = 2 × 10−18) and PLTP (P-valueeQTL = 6.4 × 10−8).
Table 5.
Downstream functional effects of common SNPs (and proxies) associated in CeD and RA
| LIFELINES eQTL effects | Blood eQTL effect (Westra et al. 2013) | 1000 genomes eQTLs (Geuvadis) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CHR | SNP | Position (hg19) | Chromosome localization | Variant localization | Gene by proximity (Annovar) | Gene | Gene | Gen | |
| Direct effect | |||||||||
| Known loci | 1 | rs4073285 | 2539796 | 1p36.32 | intronic | MMEL1 | MMEL1/FAM213B | TNFRSF14/PLCH2 | TTC34/MMEL1/FAM213B/RP3-395M20.8/TNFRSF14 |
| 1 | rs6696533 | 198733567 | 1q32.1 | intergenic | PTPRC/MIR181A1HG | no eQTL | no eQTL | RP11-16L9.3 | |
| 2 | rs10203477 | 61104985 | 2p16.1 | ncRNA_intronic | LINC01185/REL/PUS10 | AHSA2 | no eQTL | PUS10 | |
| 2 | rs11123810 | 100759285 | 2q11.2 | upstream | AFF3 | AFF3 | no eQTL | no eQTL | |
| 2 | rs6749371 | 191902184 | 2q32.3 | intronic | STAT4 | DNAJB1P1 | no eQTL | no eQTL | |
| 2 | rs1980422 | 204610396 | 2q33.2 | intergenic | CD28/CTLA4 | AC125238.1 (miRNA)/AC125238.4 (miRNA) | CD28 | AC125238.4 (miRNA)/CD28/AC010138.3 | |
| 4 | rs62323881 | 123038295 | 4q27 | intergenic | TRPC3/KIAA1109/IL2/IL21 | no eQTL | no eQTL | no eQTL | |
| 6 | rs72928038 | 90976768 | 6q15 | intronic | BACH2 | BACH2/RP3-512E2.2 | BACH2 | BACH2 | |
| 6 | rs17264332 | 138005515 | 6q23.3 | intergenic | OLIG3/RP11-356I2.4/TNFAIP3 | no eQTL | no eQTL | no eQTL | |
| 7 | rs74830391 | 37427289 | 7p14.1 | intronic | ELMO1 | ELMO1/ELMO1-AS1 | no eQTL | no eQTL | |
| 8 | rs6651252 | 129567181 | 8q24.21 | intergenic | MIR1208/LINC00977/PVT1 | no eQTL | no eQTL | no eQTL | |
| 10 | rs947474 | 6390450 | 10p15.1 | intergenic | RP11-563J2.2/PRKCQ | DKFZP667F0711 | No probe | no eQTL | |
| 11 | rs11217040 | 118680648 | 11q23.3 | intergenic | DDX6/CXCR5/UBE4A | DDX6/AP002954.4/BCL9L/MIR4492 | TRAPPC4/CXCR5 | ARCN1 | |
| 12 | rs11066188 | 112610714 | 12q24.13 | intronic | HECTD4/SH2B3 | ALDH2/RP11-162P23.2/SH2B3/TMEM116 | TMEM116 | ADAM1B/7SK | |
| 18 | rs80209296 | 12860801 | 18p11.21 | intronic | PTPN2 | no eQTL | no eQTL | no eQTL | |
| 21 | rs1893592 | 43855067 | 21q22.3 | intronic | UBASH3A | UBASH3A | UBASH3A | UBASH3A | |
| 22 | rs4821124 | 21979289 | 22q11.21 | downstream | UBE2L3 | UBE2L3/CCDC116 | No probe | UBE2L3/CCDC116 | |
| Novel loci | 1 | rs6679677 | 114303808 | 1p13.2 | downstream | RSBN1 | DCLRE1B/AP4B1/PHTF1 | PTPN22 | PHTF1/AP4B1-AS1/DCLRE1B/MAGI3/AP4B1 |
| 1 | rs17849502 | 183532580 | 1q25.3 | exonic | NCF2 | no eQTL | no eQTL | SMG7 | |
| 2 | rs16867384 | 182111206 | 2q31.3 | intergenic | UBE2E3/MIR4437 | UBE2E3 | UBE2E3 | UBE2E3 | |
| 3 | rs67676925 | 46274259 | 3p21.31 | intergenic | CCR1/CCR3 | CCR3/FLT1P1/RP11-24F11.2/CXCR6 | CCR2/CCR3/CXCR6/FYCO1 | FLT1P1/CCR3/CCRL2/RP11-24F11.2R | |
| 21 | rs9979383 | 36715761 | 21q22.12 | intergenic | RUNX1/LOC100506403 | no eQTL | no eQTL | no eQTL | |
| Opposite effect | |||||||||
| Known | 2 | rs7579944 | 30445026 | 2p23.1 | intergenic | YPEL5/LBH | no eQTL | A1CF (Trans) | no eQTL |
| 6 | rs212400 | 159473574 | 6q25.3 | intergenic | TAGAP/FNDC1 | no eQTL | RSPH3/TAGAP | TAGAP | |
The eQTL effects were calculated using the LifeLines Deep dataset (22), Westra et al. Blood eQTL (23) and Geuvadis (24) datasets.
Shared regions with independent signals point to involvement of multiple CD4+ T cell subsets
We assessed if variants from loci with shared and with independent signals overlap with H3K4me3 marks in the same or distinct cell types by investigating 130 different cell types. In those loci where the association was shared among traits, the enrichment was suggested as associated to CD14+ primary cells (P = 0.0088), CD34+ cultured cells (P = 0.0016) and embryonic stem cells (P = 0.0034), while the SNPs independently associated with CeD and RA pointed towards more specialized subsets of T cells. The strongest enrichment in CeD pointed to stimulated CD4+CD25-IL17- T cells (P < 0.0001), CD4+CD25-CD45RO+ primary memory cells (P = 0.0014), primary CD4+CD25- Th cells (P = 0.0058) and regulatory T cells (P = 0.0083). In RA, the analysis indicated enrichment in stimulated CD4+CD25-IL17+ T cells (P = 0.0001), in CD15+ (P = 0.0029) and regulatory T cells (P = 0.0005). These results suggest that loci capturing variants with independent effects in the two diseases may play a role in shared and distinct subsets of specialized CD4+ cells.
Discussion
CeD and RA share biological features that lead to an inappropriate immune response and ultimate to tissue destruction (21). In this meta-analysis, we confirmed 19 loci outside the HLA locus that have been previously associated with both diseases and discovered five new associations shared between CeD and RA. This brings the number of shared CeD-RA loci to 31, if we include seven loci reported in both diseases by the original Immunochip studies, but not captured in our analysis (8,13–15,22) (Table 2 and Supplementary Material, Table S1).
Within the shared loci, we observed a set of nine loci that first appeared to be shared between CeD and RA but which, after careful inspection, actually appear to have independent signals for each disease, as the associated SNPs reside on different haplotypes. The eQTL information from blood tissue confirmed our observations because for five out of nine loci we were able to show that the SNPs associated with CeD and RA have a different downstream effect on gene expression. One interesting example is the 20q13.12 (MMP9-CD40) locus. In this locus, the allele associated with CeD was related to decreased expression of the gene PLTP, whereas the allele associated with RA showed an increased expression of PLTP and a decreased effect on a second gene, CD40 (Table 4). The PLTP protein (phospholipid transfer protein) has an important impact on high-density lipoprotein (HDL) metabolism by facilitating the fusion of medium-sized HDL particles leading to enlargement of HDL and therefore proper functionality (23,24). HDLs are also involved in innate and adaptive immune responses. In the acute phase responses, a reduced antioxidant activity of HDLs and alterations in its composition in blood have been observed (25), whereas in the adaptive immune response, HDL regulates T- and B-cell activation. The latter occurs by changing the lipid raft composition in the membrane of cells, leading to a lower expression of HLA class II molecules on macrophages and dendritic cells, thereby lowering the expression and activity of T- and B-cell receptors (26–28). The amount of HDL in blood can differ depending on the autoimmune disease, with elevated levels observed in multiple sclerosis and reduced levels observed in systemic lupus erythematosus, Sjogren's diseases, ankylosing spondylitis, psoriatic arthritis, inflammatory bowel disease and RA (21,29–32). It is interesting to note that a previous analysis also pointed to shared genetics between regions involved in blood lipid levels and autoimmune diseases (33). The results from our analysis are in agreement with these functional and genetic findings. The reduced expression of PLTP that we observe for CeD may lead to reduced fusion of small HDL molecules and to a subsequent decrease in regulation of T- and B-cell activity. The increased expression of PLTP we saw in RA could be related to the reduced number of small HDL molecules observed in the disease, to its high levels of C-reactive protein and to RA patient risk for other comorbidities such as cardiovascular disease (34).
Another example of differential eQTL effects was observed in the locus on 11q12.2. In this locus, the variant rs2074226*G is associated with RA and correlates with increased expression of CD6. In the same region, rs11230544 is associated with CeD and does not affect CD6 expression. Interestingly, the CD6 gene is strongly expressed in synovial fluid and anti-CD6 medication is used in RA treatment (35,36). Despite our findings support the eQTL effect of some of the variants associated in this analysis, it should be taken into account that the eQTL information is derived from blood tissue. eQTL analyses in relevant tissues and cell types would be helpful in identifying disease-specific changes.
The analysis of tissue-specific signatures in the independent CeD and RA associations suggested differential cell-type enrichment for shared and disease-specific SNPs. Interestingly, the enrichment pointed to by the group of SNPs representing the shared loci indicates a group of cell types involved in the early stages of the immune response such as innate immunity and haematopoiesis. In contrast, the enrichment observed in CeD- and RA-specific SNPs indicates the role of more specialized pathways. For the CeD SNPs, we observed a suggestive enrichment of a cell line already recognized in the pathogenesis of the disease: CD4+ memory primary cells (37). In the RA group of SNPs, the enrichment was in Th17 primary cells, which are recognized as being involved in RA (38). In general, these results suggest the idea that common factors among autoimmune traits might be involved in early stages of the pathogenesis of autoimmunity, while specific genetic factors will lead to disease-specialized pathways. However, stronger signal of enrichment are necessary to determine the real functional implication of specific cell types.
Our analysis of these two autoimmune diseases has shown an interesting feature: genetic loci that seem to be commonly associated between two phenotypically similar traits could actually be contributing into the functional heterogeneity of the diseases, which might be related with the specific pathways that lead to the damage of specific organs. This observation is well known for associations in the HLA, where the same locus but different haplotypes are associated with various autoimmune diseases (10,11). Our results indicated shared genetic loci with variants associated within the same haplotype with histone enrichment pointing to cells involved in early stages of the immune response, but we also demonstrate that there are common loci with variants located on different haplotypes, suggesting the role of different pathways depending on the analysed disease. Depending on the point of view, CeD and RA could be phenotypically similar diseases (because they are autoimmune traits) or could be in different parts of the autoimmune spectrum with a different phenotypic outcome. Based on our findings, it is tempting to speculate that most of the shared loci pointing to common pathways might be related to the initial steps of any autoimmune process, reflecting the start of a snowball effect that will ultimately roll into the range of specific pathways that, depending on the genetic background and/or environmental triggers of each individual, will lead to the development of phenotypically specific traits.
We acknowledge that some of these nine shared loci lack significant genome-wide associations, however these loci have been associated in each disease separately, adding more confidence to our results (8,14). That the associations were not found in the meta-analysis is likely because of the fact that they reside on different haplotypes. These results also show that many autoimmune loci may contain several independently associated alleles for different autoimmune diseases, bringing complexity to the story through heterogeneity independent of other factors such as population- or cohort-specific association. Overall, our results indicate that dense fine-mapping in associated loci can point to disease-specific mechanisms and lead to a better understanding of disease-specific pathways.
Materials and Methods
Ethical statement
This study was approved by the respective institutional and university ethical committees. Informed written consent was received from all participants.
Study populations
Our CeD dataset included 12 387 cases and 12 429 controls. The study population were originally from India (n = 880), Italy (n = 2756), the Netherlands (n = 2323), Poland (n = 1062), Spain (n = 1793) and the UK (n = 16 002). For all cohorts, patients were diagnosed according to standard clinical, serological and histopathological criteria following the ESPGHAN criteria (39). These cohorts were previously described in Trynka et al. (8).
Our RA dataset included 13 849 cases and 18 068 controls. The study population were from Spain (n = 1206), the Netherlands (n = 2652), Sweden (n = 6517), USA (n = 9242) and UK (n = 12 300). All patients fulfilled the American College of Rheumatology 1987 classification criteria and have been described in previous studies (14).
Quality control
Both datasets were genotyped with the Illumina Immunochip Bead Array. In addition to the quality control steps described by Trynka et al. (8) and Eyre et al. (14), we identified all duplicates across cohorts, as well as first and second degree relatives, and we randomly removed one individual of each pair from the cohort. This analysis was performed with the software KING v1.4 (40). Based on the diverse composition of the datasets, and in order to determine possible population outliers, we performed a principal component analysis per dataset and per population, separately, using a set of 32 369 SNPs obtained after removing the HLA region (chr 6 25–32 Mb) and LD pruning using r2 ≤ 0.2 as a threshold calculated in a window of 50 SNPs and shifting the window by 5 SNPs in each step. This analysis was run using Plink v1.9 (41,42).
We also made sure that both datasets were in the same strand, taking into account allele frequencies and LD patterns using Genotype Harmonizer (43). In total, 156 376 SNPs in the CeD dataset and 111 780 SNPs in the RA dataset passed our quality control and had a genotypic rate >0.98; 109 572 SNPs overlapped in both diseases and were included in the final meta-analysis.
Statistical analysis
For each of the populations in each of the disease datasets, we calculated association using logistic regression including gender and the first three population-specific PCs. Using the coefficients and standard errors from the regressions obtained in each of the populations, we performed an inverse-variance-weighted meta-analysis for each of the disease datasets (within traits meta-analysis), and then jointly across diseases (across traits meta-analysis) to identify shared immune loci.
The association in the CeD-RA meta-analysis was calculated in two ways taking into account the direction of the association per trait: we first calculated the directional allelic effect (assuming that the same allele confers risk to both diseases) and then the opposite allelic effect (assuming that the same allele can confer risk to one disease while being protective for another). In practice, for the opposite allelic effect, we calculated the inverse of the coefficients from the logistic regression analysis for all the populations present in the RA dataset. To classify an association as significant, either in the same or opposite allelic effect analysis, we required a genome-wide significant P-value (P-valueMeta ≤ 5 × 10−8) in the CeD-RA meta-analysis, and a P-valueTrait ≤ 5 × 10−3 in each of the traits analysed (the latter condition prevents an association being driven by only one phenotype).
Assessing the disease-specific effects at shared loci
The above approach will fail to detect the effect of those loci where variants associated with individual diseases map to distinct haplotypes within the same region. Therefore, we undertook an additional step to identify such disease-specific signatures in shared regions. Because the content of the Immunochip was designed following a fine-mapping strategy targeting 189 loci previously reported to associate with autoimmune diseases (17), we investigated the association signals in these loci individually. To be considered in the analysis, we required an SNP association of P < 5 × 10−4 for both diseases. For loci with different SNPs associated with CeD and RA, we determined if the signals were independent from each other by calculating the LD between them in the controls of each dataset and by performing a conditional analysis of the RA-associated SNP in the CeD dataset and vice versa including the first three PCs corresponding to each dataset as covariates in the analysis. We designated a given locus as one with disease-independent effects if SNPs associated between the diseases showed low LD (r2 < 0.2) and the disease-associated variant for each disease remained significant when conditioning on the variant associated with the other disease. Similarly, we designated a locus as one with shared effect if the LD was higher than r2 > 0.2 and we observed loss of significant association in the conditional analysis.
Functional interpretation of associated variants
Identifying plausible functional genes
We assessed the functional consequences for the shared loci which had the same variant associated with both diseases and for those loci where the associated variants for each disease were independent. The list of SNPs were used to determine the expression signals (eQTL) in blood RNAseq data from 629 individuals from the LifeLines-DEEP population cohort (18), blood eQTL data from Westra et al. (19) and the Geuvadis dataset (20). Briefly, cis-eQTL analysis was performed on gene–SNP combinations for which the distance from the beginning of the gene to the genomic location of the SNP was ≤250 kb, while eQTLs at a distance >5 Mb were defined as trans-eQTLs. Associations were tested by the non-parametric Spearman's rank correlation test and a FDR significance threshold below 0.05; the significance was defined based on the number of SNPs.
Enrichment with tissue-specific histone modifications
To assess if CeD-RA loci that tag the same (shared effects) and distinct signals (independent effects) can imply the function in specific cell types, we performed an enrichment analysis looking for cell-type-specific overlap with histone 3 lysine 4 trimethylation marks (H3K4me3). We used H3K4me3 as it indicates active gene promoters and has previously been shown to be informative for this type of analysis (44). We used 118 primary cell types and tissues assayed by the Roadmap Epigenomics Project (45) and estimated the significance of cell-type-specific enrichment using 10 000 permutations. Peaks were called using MACS v2 as described (46). For loci that share effects between the diseases, we used eight top SNPs from the meta-analysis, while at nine loci-tagging distinct variants (r2 < 0.2) we used the most significant SNP per disease.
Supplementary Material
Authors’ contributions
J.G.-A., A.Z., G.T. and C.W. conceived the project, performed analyses, interpreted the data and wrote the paper. M.M.Z., I.R.-P., D.V.Z. and L.F. performed analyses and contributed to the writing. D.D. and S.R. performed the sample collection.
Funding
This work was supported by a grant from the Coeliac Disease Consortium, an Innovative Cluster approved by the Netherlands Genomics Initiative, to C.W.; a European Research Council advanced grant (FP/2007-2013/ERC grant 2012-322698) to C.W.; a grant from the Dutch Reumafonds (11-1-101) to A.Z. and a Rosalind Franklin Fellowship from the University of Groningen to A.Z. Part of the experimental work was made possible by BBMRI-NL complementation projects (CP2011-20). G.T. is supported by the Wellcome Trust Sanger Institute (WT098051).
Supplementary Material
Acknowledgements
We thank Jackie Senior and Kate McIntyre for carefully reading the manuscript.
Conflict of Interest statement. None declared.
References
- 1.Okada Y., Han B., Tsoi L.C., Stuart P.E., Ellinghaus E., Tejasvi T., Chandran V., Pellett F., Pollock R., Bowcock A.M. et al. (2014) Fine mapping major histocompatibility complex associations in psoriasis and its clinical subtypes. Am. J. Hum. Genet., 95, 162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooper J.D., Simmonds M.J., Walker N.M., Burren O., Brand O.J., Guo H., Wallace C., Stevens H., Coleman G., Wellcome Trust Case Control Consortium. et al. (2012) Seven newly identified loci for autoimmune thyroid disease. Hum. Mol. Genet., 21, 5202–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinks A., Cobb J., Marion M.C., Prahalad S., Sudman M., Bowes J., Martin P., Comeau M.E., Sajuthi S., Andrews R. et al. (2013) Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat. Genet., 45, 664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Genetics of Ankylosing Spondylitis Consortium Cortes A., Hadler J., Pointon J.P., Robinson P.C., Karaderi T., Leo P., Cremin K., Pryce K., Harris J. et al. (2013) Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet., 45, 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.International Multiple Sclerosis Genetics Consortium Beecham A.H., Patsopoulos N.A., Xifara D.K., Davis M.F., Kemppinen A., Cotsapas C., Shah T.S., Spencer C., Booth D. et al. (2013) Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet., 45, 1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J.Z., Almarri M.A., Gaffney D.J., Mells G.F., Jostins L., Cordell H.J., Ducker S.J., Day D.B., Heneghan M.A., Neuberger J.M. et al. (2012) Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat. Genet., 44, 1137–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Onengut-Gumuscu S., Chen W.M., Burren O., Cooper N.J., Quinlan A.R., Mychaleckyj J.C., Farber E., Bonnie J.K., Szpak M., Schofield E. et al. (2015) Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet., 47, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trynka G., Hunt K.A., Bockett N.A., Romanos J., Mistry V., Szperl A., Bakker S.F., Bardella M.T., Bhaw-Rosun L., Castillejo G. et al. (2011) Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet., 43, 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsoi L.C., Spain S.L., Knight J., Ellinghaus E., Stuart P.E., Capon F., Ding J., Li Y., Tejasvi T., Gudjonsson J.E. et al. (2012) Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet., 44, 1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gutierrez-Achury J., Zhernakova A., Pulit S.L., Trynka G., Hunt K.A., Romanos J., Raychaudhuri S., van Heel D.A., Wijmenga C., de Bakker P.I. (2015) Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat. Genet., 47, 577–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raychaudhuri S., Sandor C., Stahl E.A., Freudenberg J., Lee H.S., Jia X., Alfredsson L., Padyukov L., Klareskog L., Worthington J. et al. (2012) Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet., 44, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stahl E.A., Wegmann D., Trynka G., Gutierrez-Achury J., Do R., Voight B.F., Kraft P., Chen R., Kallberg H.J., Kurreeman F.A. et al. (2012) Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat. Genet., 44, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhernakova A., Stahl E.A., Trynka G., Raychaudhuri S., Festen E.A., Franke L., Westra H.J., Fehrmann R.S., Kurreeman F.A., Thomson B. et al. (2011) Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet., 7, e1002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eyre S., Bowes J., Diogo D., Lee A., Barton A., Martin P., Zhernakova A., Stahl E., Viatte S., McAllister K. et al. (2012) High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet., 44, 1336–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fortune M.D., Guo H., Burren O., Schofield E., Walker N.M., Ban M., Sawcer S.J., Bowes J., Worthington J., Barton A. et al. (2015) Statistical colocalization of genetic risk variants for related autoimmune diseases in the context of common controls. Nat. Genet., 47, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lauret E., Rodrigo L. (2013) Celiac disease and autoimmune-associated conditions. Biomed. Res. Int., 2013, 127589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cortes A., Brown M.A. (2011) Promise and pitfalls of the immunochip. Arthritis Res. Ther., 13, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tigchelaar E.F., Zhernakova A., Dekens J.A., Hermes G., Baranska A., Mujagic Z., Swertz M.A., Muñoz A.M., Deelen P., Cénit M.C. et al. (2014) Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open, 25;58:e006772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westra H.J., Peters M.J., Esko T., Yaghootkar H., Schurmann C., Kettunen J., Christiansen M.W., Fairfax B.P., Schramm K., Powell J.E. et al. (2013) Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet., 45, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lappalainen T., Sammeth M., Friedlander M.R., t Hoen P.A., Monlong J., Rivas M.A., Gonzalez-Porta M., Kurbatova N., Griebel T., Ferreira P.G. et al. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toms T.E., Panoulas V.F., Smith J.P., Douglas K.M., Metsios G.S., Stavropoulos-Kalinoglou A., Kitas G.D. (2011) Rheumatoid arthritis susceptibility genes associate with lipid levels in patients with rheumatoid arthritis. Ann. Rheum. Dis., 70, 1025–1032. [DOI] [PubMed] [Google Scholar]
- 22.Stahl E.A., Raychaudhuri S., Remmers E.F., Xie G., Eyre S., Thomson B.P., Li Y., Kurreeman F.A., Zhernakova A., Hinks A. et al. (2010) Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet., 42, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levels J.H., Marquart J.A., Abraham P.R., van den Ende A.E., Molhuizen H.O., van Deventer S.J., Meijers J.C. (2005) Lipopolysaccharide is transferred from high-density to low-density lipoproteins by lipopolysaccharide-binding protein and phospholipid transfer protein. Infect. Immun., 73, 2321–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zannis V.I., Fotakis P., Koukos G., Kardassis D., Ehnholm C., Jauhiainen M., Chroni A. (2015) HDL biogenesis, remodeling, and catabolism. Handb. Exp. Pharmacol., 224, 53–111. [DOI] [PubMed] [Google Scholar]
- 25.Kaji H. (2013) High-density lipoproteins and the immune system. J. Lipids, 2013, 684903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norata G.D., Catapano A.L. (2012) HDL and adaptive immunity: a tale of lipid rafts. Atherosclerosis, 225, 34–35. [DOI] [PubMed] [Google Scholar]
- 27.Gruaz L., Delucinge-Vivier C., Descombes P., Dayer J.M., Burger D. (2010) Blockade of T cell contact-activation of human monocytes by high-density lipoproteins reveals a new pattern of cytokine and inflammatory genes. PLoS One, 5, e9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta N., DeFranco A.L. (2007) Lipid rafts and B cell signaling. Semin. Cell. Dev. Biol., 18, 616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cruz W., Fialho S., Morato E., Castro G., Zimmermann A., Ribeiro G., Neves F., Pereira I. (2010) Is there a link between inflammation and abnormal lipoprotein profile in Sjogren's syndrome? Joint Bone Spine, 77, 229–231. [DOI] [PubMed] [Google Scholar]
- 30.Mathieu S., Gossec L., Dougados M., Soubrier M. (2011) Cardiovascular profile in ankylosing spondylitis: a systematic review and meta-analysis. Arthritis Care. Res., 63, 557–563. [DOI] [PubMed] [Google Scholar]
- 31.van Leuven S.I., Hezemans R., Levels J.H., Snoek S., Stokkers P.C., Hovingh G.K., Kastelein J.J., Stroes E.S., de Groot E., Hommes D.W. (2007) Enhanced atherogenesis and altered high density lipoprotein in patients with Crohn's disease. J. Lipid. Res., 48, 2640–2646. [DOI] [PubMed] [Google Scholar]
- 32.Hahn B.H., Grossman J., Ansell B.J., Skaggs B.J., McMahon M. (2008) Altered lipoprotein metabolism in chronic inflammatory states: proinflammatory high-density lipoprotein and accelerated atherosclerosis in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res. Ther., 10, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andreassen O.A., Desikan R.S., Wang Y., Thompson W.K., Schork A.J., Zuber V., Doncheva N.T., Ellinghaus E., Albrecht M., Mattingsdal M. et al. (2015) Abundant genetic overlap between blood lipids and immune-mediated diseases indicates shared molecular genetic mechanisms. PLoS One, 10, e0123057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung C.P., Oeser A., Raggi P., Sokka T., Pincus T., Solus J.F., Linton M.F., Fazio S., Stein C.M. (2010) Lipoprotein subclasses determined by nuclear magnetic resonance spectroscopy and coronary atherosclerosis in patients with rheumatoid arthritis. J. Rheumatol., 37, 1633–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alonso-Ramirez R., Loisel S., Buors C., Pers J.O., Montero E., Youinou P., Renaudineau Y. (2010) Rationale for targeting CD6 as a treatment for autoimmune diseases. Arthritis, 2010, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez P.C., Torres-Moya R., Reyes G., Molinero C., Prada D., Lopez A.M., Hernandez I.M., Hernandez M.V., Martinez J.P., Hernandez X. et al. (2012) A clinical exploratory study with itolizumab, an anti-CD6 monoclonal antibody, in patients with rheumatoid arthritis. Results Immunol., 2, 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sollid L.M., Iversen R., Steinsbo O., Qiao S.W., Bergseng E., Dorum S., du Pre M.F., Stamnaes J., Christophersen A., Cardoso I. et al. (2015) Small bowel, celiac disease and adaptive immunity. Dig. Dis., 33, 115–121. [DOI] [PubMed] [Google Scholar]
- 38.Paulissen S.M., van Hamburg J.P., Dankers W., Lubberts E. (2015) The role and modulation of CCR6+ Th17 cell populations in rheumatoid arthritis. Cytokine, 74, 43–53. [DOI] [PubMed] [Google Scholar]
- 39.Husby S., Koletzko S., Korponay-Szabo I.R., Mearin M.L., Phillips A., Shamir R., Troncone R., Giersiepen K., Branski D., Catassi C. et al. (2012) European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J. Pediatr. Gastroenterol. Nutr., 54, 136–160. [DOI] [PubMed] [Google Scholar]
- 40.Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.M. (2010) Robust relationship inference in genome-wide association studies. Bioinformatics, 26, 2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deelen P., Bonder M.J., van der Velde K.J., Westra H.J., Winder E., Hendriksen D. (2014) Genotype harmonizer: automatic strand alignment and format conversion for genotype data integration. BMC Res. Notes, 7, 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trynka G., Sandor C., Han B., Xu H., Stranger B.E., Liu X.S., Raychaudhuri S. (2013) Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet., 45, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R. et al. (2010) The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol., 28, 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trynka G., Westra H.J., Slowikowski K., Hu X., Xu H., Stranger B.E., Klein R.J., Han B., Raychaudhuri S. (2015) Disentangling the effects of colocalizing genomic annotations to functionally prioritize non-coding variants within complex-trait loci. Am. J. Hum. Genet., 97, 139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.