

Abstract
Various chemical modifications on histones and regions of associated DNA play crucial roles in genome management by binding specific factors that, in turn, serve to alter the structural properties of chromatin. These so-called effector proteins have typically been studied with the biochemist's paring knife — the capacity to recognize specific chromatin modifications has been mapped to an increasing number of domains that frequently appear in the nuclear subset of the proteome, often present in large, multisubunit complexes that bristle with modification-dependent binding potential. We propose that multivalent interactions on a single histone tail and beyond may have a significant, if not dominant, role in chromatin transactions.
The eukaryotic genome is assembled into chromatin, and the nucleosome serves as its fundamental organizational unit. This unit is composed of an octamer of core histone proteins (two copies of H2A, H2B, H3 and H4) encircled by ∼146 bp of DNA. Histones project unstructured N-terminal ‘tails’ from the α-helical protein core of the nucleosome through the superhelical turns of DNA that enshroud the radial surface of the histone octamer. The majority of known histone post-translational modifications (PTMs) localize to residues in the unstructured tails, particularly at the N termini, yet a burgeoning number of modifications also appear to reside within the helical secondary structure and loops of folded histones1. Further diversifying the nucleosome core particle is a set of histone isoforms known as histone variants, some of which appear to have essential roles in various stages of DNA management2–5.
The lowest order of chromatin structure is the nucleosomal unit iterated in extended conformation to resemble ‘beads on a string’, which can be consolidated into higher-order structures through the intermediacy of attendant proteins, RNA and cations. Physiological chromatin structure is a vital arbiter of DNA function, in that structural variation appears to regulate the accessibility of underlying DNA, ranging from condensed heterochromatin to more ‘open’ euchromatin6,7. Rather than mere static packaging of the genome, the spatial arrangement of chromatin serves as an information carrier that may help to preserve cell identity through mitotic division8, and yet the local structure is sufficiently dynamic that it may be rapidly modulated by signalling cascades in response to external stimuli9–11.
Phenotypic traits that are not encoded in the Watson– Crick base pairing of the genome are collectively referred to as epigenetic phenomena and appear to manifest physically as the faithful heritability of chromatin states by daughter cells12–14. The precise mechanisms of epigenetic phenomena are poorly understood, but causal connections between chemical modifications to DNA15 and histone proteins16–18, as well as other non-histone proteins19–21 and resultant local chromatin structure, are increasingly recognized as crucial intermediaries. It is becoming clear that chromatin modifications rarely occur in isolation — rather, new patterns of covalent modifications are emerging rapidly from mass spectrometry and large-scale ‘epigenomics’ efforts1,3,22–27.
How are these patterns interpreted? There are several non-mutually exclusive mechanisms by which DNA methylation and histone PTMs may produce crucial structural transformations in the chromatin polymer. First, direct nucleosome-intrinsic effects alter the physical properties of individual nucleosomes, particularly by neutralization or addition of charge, which enhances nucleosome mobility by abrogating individual histone–DNA contacts within a given nucleosome28–30. Second, direct nucleosome-extrinsic effects of chromatin modifications toggle the ability of nucleosomes to form higher-order structures through the modulation of internucleosomal contacts31. Third, effector-mediated consequences are changes that are elicited in the chromatin fibre due to specific binding events that couple a particular histone modification with a cognate non-histone binding partner, termed an effector32. effector proteins may alter the properties of chromatin by crosslinking two or more nucleosomes33–35, by enhancing the occupancy of the RNA polymerase complex and related factors36 or by recruiting active structure remodelling or further chemical modification activities32.
Since the landmark discovery that bromodomains may specifically engage acetylated Lys residues, particularly in histone sequence contexts37, the chromatin field has focused heavily on elucidating additional effector-mediated pathways. Subsequent studies have revealed a wealth of protein folds that bind various histone PTMs17,38,39. The focus of most of these efforts has been pairing a single PTM with a cognate effector module, followed by examination of the functional significance of this association event. Cellular processes as varied as transcription40, replication23, stem-cell pluripotency26, gene silencing40,41, X-chromosome inactivation24, DNA repair42, apoptosis43,44, certain cancers45,46, epigenetic inheritance47 and gene-expression programmes during development48,49 all appear to require effector–chromatin-modification interactions in their course or causation.
Early formulations of the histone/epigenetic code hypothesis suggested that distinct functional consequences result from histone PTMs and that a given outcome is encoded in the precise nature and pattern of marks18,32,50. In subsequent years, the discovery of many novel histone-binding modules has fuelled much attention and considerable interest in this general area. For example, certain PHD finger domains have recently been identified as ‘readers’ of the trimethylated Lys4 mark on histone H3 (H3K4me3)51–54. However, despite these advances, some have concluded that there is an inherent flaw in the logic underlying the histone code hypothesis55–57. one criticism is that multiple binding partners have been reported for a single histone PTM55. Furthermore, some bromodomains are somewhat promiscuous with regard to the sequence context of sub-strate acetylation marks37,58, which plausibly accounts for the functional redundancy observed for some acetylation marks in yeast56. This apparent redundancy confounds a simple one-mark-to-one-module type of decoding. others have questioned the functional significance of molecular interactions that are individually weak in nature14. These valid concerns prompt us to re-evaluate the original hypothesis by asking: is there a theoretical framework that accommodates these issues without abandoning the core of the original histone code hypothesis? It is our contention that the phenomenon of multivalency59,60 — that is, the cooperative engagement of several linked substrates by a species with more than one discrete interaction surface — may be widespread in chromatin transactions and that this biophysical effect may allay some of the above concerns.
Here, we seek to apply to chromatin biology the general concepts of multivalency, the biophysics of which is amply developed in other areas of biology and chemistry57,59–62. Indeed, there are hints of multi-valency littered throughout the vast body of chromatin literature: certain nuclear proteins are replete with predicted effector domains, and many coincident predicted binding modules occur within the subunits of chromatin-associated macromolecular complexes. However, direct proof in the form of a systematically characterized example is still lacking. This review examines the literature that establishes the coexistence of certain chromatin modifications, briefly describes the thermodynamics of multivalency and marshals the available evidence to support the proposed hypothesis, with the hope of stimulating further efforts to study the extent and magnitude of these putative effects. In trying to describe these concepts, we seek to advance a theoretical framework for what may be thought of as a ‘nucleosome code’; we note that variations of this theme have been proposed by others32,50,63. We hope that the biophysics of multivalency will provide valuable new mechanistic insights into the signalling potential of chromatin modification patterns.
Coexistence of chromatin modifications
Proximal modifications that constitute a putative ‘code’ need not be restricted to a single histone tail as originally anticipated18, but may span two or more tails on a given nucleosome, adjacent nucleosomes, or nucleosomes that are discontinuous in primary DNA sequence but spatially colocalized in a chromatin territory64. Patterns of native chromatin modifications are studied principally through mass spectrometry1,27 and chromatin immunoprecipitation (ChIP) methods65. New genome-scale extensions of ChIP, such as ChIP-chip66,67 and ChIP-seq68, have enabled a transition from investigating single marks at discrete loci to correlating complex epigenetic signatures, expression patterns and other physical features at the megabase level and beyond with spatial resolution well below a single nucleosome footprint22,23,25,68,69.
The first well-studied histone modification class, Lys acetylation, is correlated with transcriptional activation through direct alteration of the physical properties of chromatin70 and recruitment of bromodomain-containing effectors37. Histone hyperacetylation is correlated positively with actively transcribed chromatin71,72, and biophysical studies reveal a more translationally mobile octamer73–75, as well as inhibition of 30-nm packing contacts in the case of acetylated Lys16 of histone H4 (H4K16ac)31. Alternatively, there are numerous bromodomain effector-mediated pathways that also generally serve to enhance transcriptional activation37,76–80.
Conversely, Lys methylation and Arg methylation events are more varied in their correlated functions. Some are generally associated with gene activation, and others are involved with repression and silencing — all known examples of which are effector mediated (for recent reviews that catalogue individual marks and associated functions, see REFS 1,17). However, the greater the resolution and percentage of the genome that is covered by epigenomics, the more these canonical associations between a given mark and gene expression become nuanced and idiosyncratic16,25,81. For example, H3K4me3 is traditionally associated with the promoter-proximate regions of genes undergoing active transcription, yet in embryonic stem cells, there is a significant colocalization of this mark with a canonical heterochromatic PTM, H3K27me3, at developmentally regulated loci22,26. These so-called bivalent domains are largely transcriptionally repressed. Moreover, the H3K4me3 modification is found at nearly all gene start sites in embryonic stem cells, only a subset of which are producing full-length transcripts, so it may now be thought of as principally associated with transcription initiation25. A collection of spatially juxtaposed chromatin marks is presented in TABLE 1.
Table 1. Notable patterns of coexisting histone marks.
| Histone marks | Locus/chromatin state | Method |
|---|---|---|
| H3K4me2/3 + H4K16ac | Transcriptionally active homeotic genes | ChIP122 |
| H3K4me2/3 + H3K9/14/18/23ac | Transcriptionally active chromatin | MS125 |
| H3S10ph + H3K9/14ac | Mitogen-stimulated transcription | Ab9,10 and MS126 |
| H3R17me1/2a + H3K18ac | Oestrogen-stimulated transcription | Ab127 |
| H4K5ac + H4K12ac | Pre-deposition | Ab and MS128 |
| H3K4me3 + H3K27me3 | ‘Bivalent domains’ at key developmental genes | ChIP-chip, reChIP22 |
| H3K9me3 + H3K27me3 + 5-MeC | Silent loci | ChIP, bisulphite sequencing91,129, MS |
| H3K27me3 + H2AK119ub1 | Silent homeotic genes | ChIP, ChIP-chip41,69 |
| H3K9me3 + H4K20me3 + CpG 5-MeC | Heterochromatin | IF and nucleosomal CoIP130, ChIP-seq26 and bisulphite sequencing |
| H3K9me2/3 + H4K20me1 + H3K27me3 + CpG 5-MeC | Inactive X-chromosome | ChIP, ChIP-chip, reChIP24, bisulphite sequencing |
5-MeC, 5-methylcytosine; Ab, specific antibody in western blot; ac, acetyl; ChIP, chromatin immunoprecipitation; ChIP-chip, ChIP followed by amplification and microarray hybridization; ChIP-seq, ChIP followed by massively multiplexed sequencing; CoIP, co-immunoprecipitation; CpG, the DNA sequence that is often targeted for epigenetic 5-cytosine methylation; IF, immunofluorescence; me, methyl; MS, mass spectrometry; ph, phosphoryl; reChIP, ChIP with two sequential immunoprecipitations with different antibodies; Rme2a, asymmetric dimethylated Arg; ub, ubiquitin.
Although epigenomics-level efforts strongly imply mark coexistence, most do not definitively prove it because the ChIP method yields relative ensemble measurements. That is, heterogeneity in the cell population and cell cycle may cause a mixture of different chromatin states at a locus that might be averaged together inappropriately. (When collections of cells with uncertain degrees of homogeneity are used, the resulting net signal may be susceptible to large fluctuations in a small subpopulation.) Further complicating analyses in diploid genomes is the mixture of allele-specific marks, an issue that may be partially resolved by allele-specific ChIP-seq26. Furthermore, because of variable immunoprecipitation efficiencies and inconsistent antibody quality, these measurements must be normalized in such a way that makes them relative measurements without clear relation to an actual number of modifications over a given chromatin span. Without measuring an absolute number of modifications per nucleosome, it is unclear whether two marks actually simultaneously exist, or on average are both biased towards a particular region but occur at a frequency well below one mark per locus. BOX 1 lists methods for studying this coincidence of marks and presents ways of addressing these problems that have been recently reported in the literature.
Box 1. Experimental approaches to characterize epigenetic signatures.
Epigenomics
High-resolution chromatin immunoprecipitation (ChIP) technologies (ChIP-chip and ChIP-seq) imply the coexistence of various marks but do not simultaneously and directly establish the residence of two or more marks within the same nucleosome. Furthermore, because the information gleaned from epigenomics experiments crucially depends on the quality of the antibody used in ChIP experiments, more robust reagents that are specific for given modifications are needed. There are many identified modifications for which there are no ChIP-grade antibodies; consequently, the patterns of these modifications have not been spatially correlated with genomic elements. Thus, in addition to continued progress in epigenomics approaches, parallel efforts are necessary to implicate directly the concurrence of patterns at the nucleosome level.
Mass spectrometry
Although several proximate post-translational modifications within particular histones have been identified from tryptic digest mass spectrometry (MS)/MS1, new forms of high-resolution MS/MS (such as electron transfer and electron capture dissociation) permit the examination of modifications present on residues that are distal in primary sequence on fragment sizes approaching whole histones27,111,112. More whole-histone MS/MS from staged or select cell (that is, stem cell) populations should dramatically enhance the current set of coincident post-translational modifications within a given histone113.
Acid-urea gels
Various forms of acid-urea gels are sensitive enough to provide single-charge difference resolution of modified histones. When coupled with western blot analysis, this method can effectively discern multiple modifications on a given histone9,10,114.
Sequential IP and rechIP
Sequential immunoprecipitation (IP) methodologies permit the investigation of chromatin modifications that occur within a single nucleosome22,24,115,116. This technique, which is often performed on mononucleosomes, entails a second IP after elution from an initial IP with an orthogonal antibody, such that two marks can be simultaneously investigated116. More widespread application of this technically demanding technique, in concert with PCR using locus-specific probes, should reveal more precise patterns of nucleosome modification mark coexistence22,115,116.The systematic combination of all of the above approaches with synchronized and staged cell populations should reveal more histone modification patterns with higher resolution.
Many questions remain about the patterns of modification discovered thus far. To what extent does the appearance of proximate ‘opposing’ marks coexist for extended periods of time? Do some apparent bivalent domains represent transient intermediate chromatin states that are in transit from one limiting state to another in a developmental context (simplistically, off-to-on)? The finding that the jumonji domain-containing protein-2A (JMJD2A) enzyme demethylates H3K9me3 and is recruited by binding the H3K4me3 mark suggests that such incipient states exist82,83. Mediators and intermediaries in this process have also been implicated in the particulars of establishing, maintaining or modulating different chromatin structural states — the collections of marks themselves may serve as signalling platforms, integrate inputs (embodied by individual marks), and recruit chromatin-modulating or -stabilizing factors. Indeed, ample circumstantial evidence links distinct patterns of chromatin modifications to different chromatin structural states. How specific congregations of marks might physically collaborate to achieve these outcomes is the subject of the remainder of this article.
A physical basis for the histone code?
Mainly ignored in the feverish elucidation of chromatin modifications and their correlation to genome function are the details by which the physical processes stimulated by the modifications actually transpire. often, the problem of quantitative attribution of the individual binding contributions may not be experimentally tractable. But frequently, when one specific interaction is discovered that has some role in locus targeting, it is deemed sufficient even though the studied interaction is too weak and/or too non-specific to explain the observed phenomenon adequately. We propose that the multivalent nature of many complexes that manage chromatin may explain the magnitude of the isolated binding affinities and their individually modest substrate specificities, as well as serving a crucial role in chromatin fibre dynamics. As we have recently outlined the theoretical argument for widespread multivalency in chromatin biology in some detail38, only a brief treatment will be undertaken here.
In well-characterized systems outside of chromatin biology, multivalent binding results in dramatic affinity enhancements and additional specificity while remaining much more dynamic and susceptible to competition than a correspondingly tight monovalent interaction59,60. The physical basis of the multivalency phenomenon is attributable to both thermodynamic and kinetic effects. In thermodynamic terms, binding enhancement is caused by roughly additive enthalpies of each binding event (assuming no strain is introduced) with the concomitant reduction of entropy loss59 (FIG. 1). This reduction of the entropy term relative to a similar number of uncoupled interactions is the result of a sacrifice of rotational and translational degrees of freedom in binding that occurs as a group (FIG. 1b). under optimal conditions of little conformational flexibility and no introduced strain, much of the entropy loss on binding is ‘prepaid’ by the initial complex assembly and is not levied against the enthalpy during association61. Thus, the free energies for multiple individual effector–substrate binding events can achieve levels of significant synergy that are dependent on the degree of valence and the spatial organization of effectors relative to the arrangement of substrates61.
Figure 1. Thermodynamics of multivalent binding.

a | Monovalent association of a hypothetical effector module (purple) to a chromatin substrate (yellow tail with green diamond) is simplistically compared to a bivalent association of the same effector in a complex, representing the lowest order of multivalent interactions. The change in free energy ΔGi for the monovalent system undergoing binding is indicated by the change in enthalpy ΔHi minus the change in entropy ΔSi, scaled by the temperature (T). b | By tethering the two effector modules, the entropy term may be, to a first approximation, similar for each of the binding equilibria in panels a and b (TΔScomplex ≈ TΔSi). For our purposes, this example assumes that hydrogen-bonding electrostatic interactions dominate and desolvation is negligible, so that ΔS for the system will be negative. Thus, the entropic penalty to binding is lessened approximately twofold by pre-organizing the effector domains in a complex (TΔScomplex ≈ TΔSi), while the enthalpy of the bivalent domain interaction is effectively double that of the monovalent case (∼2ΔHi, if enthalpic penalties due to the strain induced by bivalent binding are negligible). In this manner, the reduced net entropy loss for the binding process can be a significant determinant of free energy, especially in low-binding enthalpy regimes. Losses of entropy on the substrate side would be expected to be minimal due to the low intrinsic rotational and translational freedom of chromatin; however, conformational entropy losses here are assumed to be negligible for simplicity.
The kinetic explanation arises from the significant enhancement of individual rates of association, not by altering individual rate constants, but by enhancing the local concentration of a given substrate and cognate effector module. local concentration increases are caused by effective tethering of an effector complex to a locus through other similar interactions. This sort of cooperativity in binding has been widely observed in DNA annealing, gene activation by sequence-specific DNA-binding factors57 and other chemical and biological systems59. When multiple association events between chromatin-modification complexes and chemically modified chromatin substrates are coupled and elaborated by the degree of valence, substantial additive or super-additive binding may ensue. Several unusual aspects of this phenomenon are worth noting because they are likely to be crucial for the functional mechanisms of chromatin transactions.
First, not all contacts need to be modification dependent; for example, contacts with DNA84 (sequence dependent and independent) or contacts with a region of the histone octamer surface that is not subject to modification may have significant roles in dictating ensemble association85. If the overall binding energetics are distributed among several non-specific and specific interactions, the non-specific interactions may not be sufficient to drive binding on their own, so that the crucial free-energy balance may be dictated by one or more specific contacts.
Second, the higher the degree of valence, the more profound the effect; for example, moving from bivalent to trivalent oligosaccharide ligand binding to mammalian hepatic GalNAc lectin produces apparent affinity enhancement over monovalent galactose of 103-fold and 106-fold, respectively62. Similar energetic consequences might be expected in multivalent interactions with the chromatin fibre38.
Third, composite specificity, which is greater than the intrinsic specificity of any of the discrete binding interactions, may also arise from multivalent interactions. In principle, the apposition of effector modules in an optimal alignment creates surfaces that are complementary to the spatial positions of each of the substrate elements when they are displayed in chromatin, so that the distances between discrete interactions become additional specificity determinants. Thereby, greater net specificity may be imparted by constraining productive binding to the specific spatial relationships among each substrate element in its native context (or the ability to attain an ideal conformation). This phenomenon is how type IIP restriction enzymes build up huge sequence specificity from a symmetric bivalent interaction with two half-sites86.
Finally, a system of recognition and localization that results from a collection of individually weak interactions may also be more readily competed because the individual dissociation rates are high relative to comparably strong monovalent binding interactions59,87. During development, the plasticity of chromatin structure and attendant modifications requires dynamics: with multivalent interactions, high affinity can be coupled with a susceptibility to competition, as well as a greater potential specificity through the synchronous recognition of several marks. However, to our knowledge, no PTM-dependent multivalent effect has been reported that systematically and quantitatively measures the isolated affinities of each effector–substrate interaction as well as the net affinity. Thus, the magnitude of the effect, even in the form of a proof-of-principle experiment, still remains obscure for chromatin transactions. BOX 2 details empirical approaches to rectify this dearth of empirical evidence.
Box 2. Methods for examining nucleosome engagement.
‘Designer chromatin’ from semi-synthetic histones
Any of the known histone modifications may be introduced semisynthetically by native chemical ligation of a recombinant histone fragment and the appropriate peptide117–119. A simpler route to methyl-Lys analogues is through direct alkylation of the appropriate Cys mutant in a given histone background120. Histones with chemically incorporated modifications may then be assembled with recombinant histones and DNA to form mononucleosomes or arrays of ‘designer chromatin’31 that bear multiple specific modifications at once. The advantage of chemical methods over the enzymatic production of modified histones lies primarily in the 90–100% homogeneity of the resultant species. By contrast, enzymatic installation of particular modifications often suffers from poor control of the extent and degree (in the case of methylation) of modification, with additional concerns regarding the specificity of a given modification enzyme under the forcing in vitro conditions. In the latter case, resultant heterogeneity often makes qualitative analysis difficult and quantitative analysis impossible. Quantitative mono- and dinucleosome binding assays, nucleosomal array compaction assays121, electron microscopy33 and even transcription assays with multiply modified designer chromatin can be profitably deployed to understand the physical basis of a given mark in the presence of multivalent effector modules.
Structures of multi-effector unit fragments
Although it is often more convenient to solve individual domain structures in complex with their respective substrates, this level of structural analysis reveals little about the crucial spatial relationships between modification-dependent binding pockets of multi-module protein fragments or multi-effector subunit complexes. Moving structural analysis away from this reductionist routine will invariably enhance our understanding of chromatin engagement mechanisms by these complexes.
In vivo approaches
Once the particulars of a multivalent interaction have been well characterized in vitro, appropriate mutations (which are preferable to domain deletions) may be introduced, either in a null genetic background or in a tagged form (especially in dominant-negative cases). Spatial requirements for effector-module juxtaposition can be probed by insertion or deletion of rigid protein spacer modules, for example, creating a longer helix between two effectors. Because putative multivalent interactions are implicated in recruitment or stabilization of chromatin-modifying complexes to particular genomic regions, chromatin immunoprecipitation experiments with mutants should recapitulate the in vitro quantitative binding experiments. Combinatorial assemblies of effectors can also be probed by swapping analogous portions or domains with different binding specificities84, or by serial duplication of a putative effector domain, and examining locus residence times at a related inducible gene target following synchronized activation.
Linked effectors engaging chromatin
Examination of the predicted domains in most enzymatic complexes that operate on chromatin (many of which contain several putative modification-dependent binding modules) shows that the combinatorial engagement of chromatin through numerous modalities may indeed be the rule rather than the exception (FIG. 2a,c). Structural details of the molecular discrimination of individual effector domains have been extensively reviewed38,39,80,88 and will not be addressed here. Distinguishing among several possible modes of synchronous chromatin mark binding is crucial to understanding the actual mechanisms of multivalent chromatin mark ‘readers’. In particular, simultaneous tail binding may occur at the level of a single tail or at two tails of the same nucleosome, adjacent nucleosomes or discontinuous nucleosomes (FIG. 3).
Figure 2. Polypeptides with many putative effector modules and representative complexes.
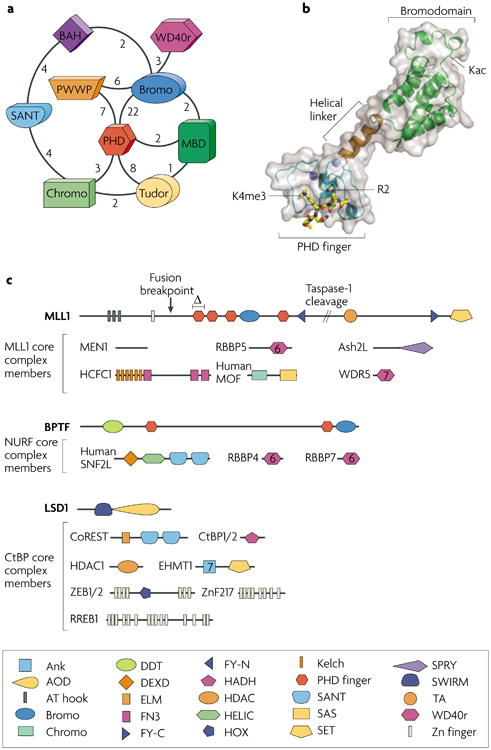
a | The coexistence of possible effector module domains within single polypeptides is depicted schematically, with the number of instances of linkage for any two domains within the human proteome listed near the line connecting them. The SMART database was used as the source of these linkages, and redundant entries were removed. b | A structurally characterized example of two linked effector domains is provided by the structure of a BPTF module that comprises a PHD finger, a helical linker and a bromodomain, with a trimethylated Lys4 of histone H3 (H3K4me3) peptide bound to the PHD finger85. The acetyl-Lys (Kac)-binding pocket on the bromodomain is shown, as well as residues R2 and K4me3 of the H3 peptide. c | Chromatin metabolism complexes, exemplified by the MLL1 (ref. 122), NURF103,123 and CtBP11 core complexes, have multiple putative effector domains. The predicted domain structure of subunits of the complex members are shown as a linear arrangement from N to C terminus. Chromatin-associated domains, most of which are modification sensitive, are coloured as in panel a, and are shown with additional predicted domains given in the key. The portion of the MLL1 protein that is cleaved by taspase-1 to yield two functional fragments (MLL1-N and MLL1-C) is shown. A frequent breakpoint at which fusion partners are appended and a domain deletion (Δ) that causes certain leukaemias are also depicted on the MLL1 domain structure. Ash2L, Set1–Ash2 histone methyltransferase complex subunit; BAH, bromo-adjacent homology domain; BPTF, bromodomain PHD finger transcription factor; Bromo, bromodomain; Chromo, chromodomain; CoREST, corepressor to the RE1 silencing transcription factor; CtBP, C-terminal binding protein; EHMT1, euchromatic histone-Lys N-methyltransferase-1; HCFC1, host cell factor C1; HDAC1, histone deacetylase-1; LSD1, Lys-specific demethylase-1; MBD, methyl-CpG binding domain; MEN1, multiple endocrine neoplasia-1; MLL1, mixed lineage leukaemia; MOF, males absent on first histone acetyltransferase; NURF, nucleosome remodelling factor; PHD, plant homeodomain; PWWP, PWWP motif protein of the Royal superfamily; RBBP, retinoblastoma binding protein; RREB1, Ras responsive element binding protein-1; SNF2L, sucrose non-fermenting-2-like ATPase; WD40r, WD40 repeat; WDR5, WD repeat domain-5; ZEB1/2, zinc finger E-box binding homeobox-1/2; ZnF217, zinc finger protein-217.
Figure 3. Modes of multivalent chromatin engagement.

To distinguish among several potential mechanisms of multivalent association, we propose the following nomenclature. a | Intranucleosomal association can be subdivided into two distinct classes124: cis-histone, when more than one discrete binding contact is made to a single histone, in particular the same tail; and trans-histone, whereby contacts are made to different histone protomers or attendant DNA within the same nucleosome. b | By contrast, internucleosomal binding modes crosslink two nucleosomes that are either adjacent or discontinuous in DNA sequence. Most of these crucial interactions are envisioned as modification dependent; however, DNA interactions and modification-independent contacts may have a vital energetic role. BPTF, bromodomain PHD finger transcription factor; HP1, heterochromatin protein-1; TAF1, TATA-binding protein-associated factor-1.
Additional recognition of covalent modifications on other surfaces of the folded core of the histone octamer and DNA contacts (including sequence-specific recognition, 5-methylcytosine recognition and general DNA affinity) can presumably occur in an intra- or internucleosomal manner. These multivalent mode distinctions are important because they are thought to yield different functional consequences — ranging from gripping a particular histone tail in a multidentate fashion while ratcheting on DNA to enhance nucleosomal mobility, to crosslinking two nucleosomes in disparate chromosomal positions so that a heterochromatic region is bridged and compacted34,35. examples could include effector modules with conserved PTM recognition elements that coexist within a given polypeptide or complex, but at present there is little direct evidence to support these claims (FIG. 2a,b). Nevertheless, the sheer number of such associations tempts speculation that this phenomenon is general. The remainder of this section will examine numerous cases in which multivalency may be inferred, organized according to the putative mode of the interaction.
Cooperative binding of PTMs on a single histone tail
Elegant work by the Tjian laboratory first discerned modest cooperativity in binding multiply acetylated histone peptides in tandem bromodomain proteins, human TAF1 (TATA-binding protein-associated factor-1) and a partial yeast homologue, bromodomain-containing factor-1 (Bdf1)76,78. In particular, the TAF1 tandem bromodomains display moderate affinity increases for multi-acetylated histone H4 peptides that may arise from the synergistic binding energetics that are typical of multivalent systems. The initial work centred on the structural characterization of the tandem bromodomain module of TAF1, the largest subunit of the TFIID basal transcription factor (FIG. 3a; cis-histone, intranucleosomal). The two bromodomains appear to be rigidly confined in a relative orientation that positions each acetyl-Lys-binding pocket on roughly the same face, such that a single peptide with two acetylated Lys residues positioned ∼25 Å apart could bridge the two pockets76. Typical single bromodomain–acetylated-peptide interactions are so weak (with a dissociation constant (Kd) of 100–350 µM37,79) that their in vitro significance comes into question, whereas the Kd for double bromodomains that bind doubly acetylated counterparts is considerably lower (∼1–20 µM).
There is some apparent specificity for acetylated H3 and H4 tails, and in the case of TAF1, a potential preference for a certain spacing of acetylated Lys residues; however, the extent to which this preference is truly restricted to two particular acetyl-Lys residues remains unclear58,76,78. It is possible that the magnitude of cooperativity of this bivalent interaction may be even greater with other intranucleosomal substrates. Regardless of the magnitude of the effect, cooperative binding was deemed vital to the function of these two tandem bromodomain proteins. Bromodomain pocket mutations and mutation of potential H4 acetylated Lys residues also negatively influence the efficacy of TFIID in promoting transcription, particularly when the TATA box is obscured by nucleosome wrapping89,90. Although the precise role, specificity and molecular detail for each of the two bromodomains involved in composite binding have not been systematically detailed, the current data attest to significant cooperativity in the binding.
CMT3, a homodimeric Arabidopsis thaliana DNA methyltransferase that is implicated in gene silencing, bears two copies of a chromodomain that together bind well only to the combination of H3K9me3 and H3K27me3 in the same polypeptide91. Although neither individual mark is bound tightly enough to be measured, nor is sufficient to recruit CMT3 in vivo, it seems that the combination of the two trimethyl-Lys residues within a single polypeptide satisfies this requirement91.
A third example of the cis-histone phenomenon involves negative cooperativity of adjacent modifications within the histone H3 tail in the binding and expulsion of the chromodomain of heterochromatin protein-1 (HP1)92,93. The affinity of HP1 for H3K9me2 or H3K9me3 is in the low micromolar range; however, when this modification is paired with a subsequent phosphorylation of H3S10 mediated by Aurora B kinase, the affinity of the chromodomain is diminished by several orders of magnitude (an effect referred to as phosphomethyl switching)92. Repulsive forces in biology can easily achieve much greater magnitude than the corresponding attractive forces. HP1 binding to H3K9me3 is thought to be crucial for the formation of pericentric heterochromatin, and its subsequent ejection triggered by phosphorylation appears to be necessary for mitotic progression92,93.
Harnessing the nucleosome unit
There are several potential modification-dependent binding modules that paradoxically associate with complexes that have opposing enzymatic activities. one such example is provided by the Eaf3 protein that resides in both the histone deacetylase Rpd3S and the histone acetyltransferase NuA4 complexes. The chromodomain of Eaf3 alone displays a modest preference for several methyl forms of H3K36 in vitro (H3K36me3 binds approximately two-fold tighter than the unmodified peptide in a pull-down assay, although no quantitative binding experiments have yet been reported)94,95. In the Rpd3S complex, this chromodomain appears to engage enzymatically modified nucleosomes as a function of methylation at H3K36, whereas no such nucleosome binding was observed with the NuA4 complex under the same conditions84.
Closer examination reveals that several additional contacts are made by the Rpd3S complex with the nucleosome that appear to tip the balance of binding energetics through multivalency. The complex has overall affinity for linker DNA and histone hyperacetylation that is not yet traceable to a particular subunit, and a PHD finger in the Rco1 subunit has an as-yet-undefined role in enhancing binding84. The PHD finger of Rco1 and the chromodomain of Eaf3 have significant roles in targeting the complex to genes or maintaining complex stability in vivo, because individual domain deletions phenocopy Set2 (the K36 methyltransferase) null strains in their inability to suppress aberrant transcripts as a downstream consequence of localized histone deacetylase activity84. Despite the incomplete characterization of each module's preferred portion of the modified nucleosomal substrate, this work is a pioneering demonstration that the product of a series of individually weak contacts, when assembled into a multivalent complex, can be important for in vivo chromatin function (FIG. 3a; oligovalent complex).
What is the role of Eaf3 in NuA4 binding to nucleosomes? A plausible interpretation is that a suboptimal nucleosomal substrate methylated only at H3K36 may explain the lack of affinity under the conditions described. The absence of the H3K4me3 mark in this nucleosomal species precludes a potential second modification-dependent interaction with Yng2, a known H3K4me2/3 effector and NuA4 subunit52, from productive participation. Furthermore, it would be interesting to examine whether the PHD finger within the Pho23 subunit of the related Rpd3L complex (which also has demonstrated intrinsic H3K4me3-binding capacity52,96) has an analogous role to Eaf3, supporting binding for H3K4me3-modified nucleosomes rather than H3K36me3-modified ones. This study demonstrates that it is not always an individual constituent of a complex that dictates the overall binding capacity; rather, it is the ensemble of all different binding elements in varying complexes that may give rise to differing specificities. other examples of common subunits shared by oligovalent protein complexes with opposing activities have been documented in the literature41,97,98, which is suggestive of a more general theme operating in chromatin biology.
Several models for multivalent engagement of a single nucleosome have been suggested, yet still none has been tested for actual cooperativity. A striking example is provided by the Lys-specific demethylase-1 (LSD1)–corepressor to the Re1 silencing transcription factor (CoREST) complex. Structural analysis reveals that LSD1 projects an extended helical stalk from the amine oxidase–histone H3 recognition centre that binds to CoREST, thereby positioning a second SANT domain at the apex of the stalk >100 Å away99 (FIG. 4a). Given the intrinsic DNA-binding capacity of this SANT domain99 and the H3 Lys demethylase function of LSD1, a model has been presented that is consistent with the noted requirement for DNA binding by CoREST in the processing of nucleosomal substrates100. This situation appears to reflect the composite recognition of a particular histone modification and sequence non-specific binding of DNA, which together collaborate to yield favourable nucleosomal-binding free energy.
Figure 4. Models of nucleosomal engagement.
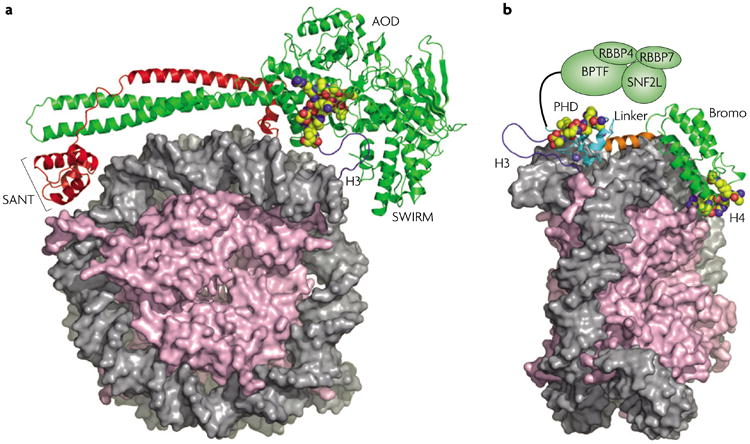
a | A model of the LSD1–CoREST complex docked with the nucleosome99 in a bipartite manner with the second SANT domain binding to DNA and the AOD and SWIRM domains binding an H3 tail bearing Lys methylation. LSD1 is shown in green and CoREST in red. Adapted from REF. 99. b | A model of the PHD–bromodomain module of BPTF (green) binding a nucleosome with modifications in the tails of two different histones, H3K4me3 and H4ac (the precise site is unknown but H4K16ac is modelled here). The remaining portion of the NURF complex is shown as green ovals, including the N terminus of BPTF, SNF2L and RBBP4 and -7. In both panels, core histones are pink with tail cartoons in dark blue. The modification recognition epitope is shown in space-filling spheres (carbon, yellow; nitrogen, blue; oxygen, red) with DNA in grey. AOD, amine oxidase domain; BPTF, bromodomain PHD finger transcription factor; Bromo, bromodomain; CoREST, corepressor to the RE1 silencing transcription factor; LSD1, Lys-specific demethylase-1; NURF, nucleosome remodelling factor; PHD, plant homeodomain; RBBP, retinoblastoma binding protein; SNF2L, sucrose non-fermenting-2-like ATPase.
Furthermore, BHC80, another subunit of some LSD1 complexes, has recently been demonstrated to bind specifically to non-methylated H3 tails, providing yet another point of contact with substrate nucleosomes101. Interestingly, a similar unmodified H3 tail interaction was uncovered within a Cys-rich (PHD finger-like) domain of the DNA methyltransferase subunit DNMT3l, providing a link between a lack of methylation at H3K4 and de novo methylation of DNA in mammals102. In addition, pull-down assays with an epitope-tagged version of DNMT3L indicate an association with all four core histones, foreshadowing possible nucleosomal recognition. The energetics of these and other similar modules have been individually investigated, but how these discrete interactions assemble into presumptive multivalent associations with nucleosomal units remains to be elucidated (see below and BOX 2).
A final example of intranucleosomal engagement is provided by the bromodomain PHD finger transcription factor (BPTF) component of the nucleosome remodelling factor (NURF) complex. In particular, the structurally characterized PHD–bromodomain module can bind H3K4me3 (REFS 51,54) and shows acetylation-dependent H4 recognition (H.L., A.J.R., C.D.A and D.J.P., unpublished observations). The structure suggests that these two effector domains are rigidly spanned by an α-helix that enforces a particular spatial disposition of the binding pockets (FIG. 4b). Interestingly, modelling of this bivalent module by superimposing the two histone tail substrates provides a plausible model for bivalent nucleosomal engagement that predicts additional DNA contacts due to close proximity of the helical linker to the DNA major groove (FIG. 4b) (H.L., A.J.R., C.D.A and D.J.P., unpublished observations). In this case, we predict a clear bivalent enhancement that is well beyond the sum of the individual affinities. Further experiments are currently underway to measure precisely the exact energetic contributions and spatial requirements of the interaction through perturbations to both the substrate and the dual-effector module.
It should be noted that there are likely to be two copies of BPTF within the NURF complex103; in addition, there are further putative effector modules within the BPTF polypeptide and in other subunits of the complex (FIG. 2b). Moreover, two other complex members, the WD40-repeat proteins RBBP4 and RBBP7 (formerly known as RbAp48 and RbAp46, respectively), can bind histones through the first helix of the histone fold98 (FIG. 4b). An intriguing facet of this minimally bivalent interaction is the pre-organized rigidity of the α-helical linker between effector modules. This fixed linkage appears to reduce the conformational entropy of the module before binding so that less entropy is lost in the act. Nevertheless, by inspection of polypeptides with several putative binding modules, one can envision that flexible linkers are also likely to be used and, in such cases, the accompanying induced fit during complex formation on the nucleosome must be energetically offset by greater enthalpies of binding.
Beyond the nucleosome
The formation and stabilization of higher-order chromatin organization requires several architectural chromatin proteins. The archetypal example is HP1, which has been implicated in numerous protein–protein associations, including the H3K9me-mediated histone binding described above. Several studies have suggested that HP1 crosslinks H3K9me-rich chromatin domains by virtue of dimerization — a known property of its chromoshadow domain34,35. Whether this phenomenon occurs at adjacent nucleosomes along the same chromatin fibre or is able to span large sections of unconnected chromosomal arrays remains an open question (FIG. 3b; compare the panels labelled ‘adjacent bridging’ and ‘discontinuous bridging’), as does the actual importance and energetic consequences of this bridging.
A related chromodomain complex is similarly associated with chromatin crosslinking; the Polycomb repressive complex-1 (PRC1) appears to bind approximately three nucleosomes simultaneously and, consequently, compacts arrays of nucleosomes in a histone-tail-independent manner (as shown by electron microscopy)33. Intriguingly, the Polycomb protein (Pc) of PRC1 is known to specifically engage K27me3 in the H3 tail104, although this interaction is dispensable in this in vitro experiment. The precise molecular contacts that underlie this essential heterochromatin complex that engages chromatin remain enigmatic.
Beyond the well-studied chromatin regulators described above, other examples of potential multivalency may easily be proposed. For example, 8 out of the 15 annotated bromodomains in yeast reside in RSC, a chromatin remodelling complex of the SWI/SNF family. Many of these bromodomains bear the canonical residues that are implicated in acetyl-Lys binding. One subunit, Rsc4, has a tandem bromodomain architecture that is similar to human TAF1 (FIG. 3a) and exhibits a preference for binding H3K14ac77. Remarkably, mutants in both bromodomains are required to expose synthetic lethality with mutations of H3K14 (REF. 77). This paradox is resolved by a recent study that revealed a surprising autoinhibitory role for the first bromodomain — it binds an N-terminal peptidic extension of Rsc4, but only when this substrate has been acetylated at a conserved Lys by the histone acetyltransferase GCN5 (REF. 105). It is suggested that autoinhibition of Rsc4 binding to H3K14ac is involved in destabilizing the binding of the RSC complex to attenuate its residence time at promoters. In the RSC complex, it is likely that there is synergistic binding with other affinity determinants outside the Rsc4 subunit; in particular, it is tantalizing to speculate that other bromodomains in the complex also have roles in a multivalent system.
By no means does the above discussion provide a comprehensive examination of prospective multivalent chromatin associations. Undoubtedly, as more modules in multivalent chromatin complexes succumb to detailed structural and biophysical analyses, a clearer picture of the importance of cooperativity and putative combinatorial readout of nucleosomal units will emerge. We look forward to experimental tests of these and other theories of multivalent behaviour that potentially govern the association of key regulators (proteins or complexes) in chromatin biology, some of which are outlined in BOX 2.
Conclusion
Large-scale experiments that have examined chromatin modification patterns through evolutionary space suggest a strong conservation of chromatin signatures and nucleosome positioning biases69,106. Chromatin modifications serve as an effective and economic way to regulate gene expression through the alteration of modification patterns, which, in turn, modulate the higher-order structure of the fibre and/or govern recruitment of effector modules. local patterns of epigenetic marks appear to dictate transcriptional activity at particular regulons and chromatin territories as well as sequential firing of developmental loci69,107. More long-lived and self-reinforcing patterns are involved in silencing phenomena — DNA methylation is heritable, and certain histone marks may be effectively heritable108. Histone modifications do not operate alone; rather, they appear to act in concert with other putative epigenetic information carriers and a host of DNA-sequence-specific factors to constitute another level of regulation that governs DNA-templated processes. More complex histone or nucleosomal code combinations may provide an important distinction between lower and higher eukaryotes. Indeed, there is evidence for more sophisticated patterns of histone modification and histone variant utilization in metazoans3,27.
We propose that combinations of effectors may greatly increase the reading specificity, affinity and dynamics of chromatin-associated macromolecular assemblages. The promiscuity of certain marks with regard to binding partners (there are now >10 effector proteins that are known to bind H3K4me3) suggests that, in many cases, one histone modification is not sufficient to recruit a given complex. Instead, we suspect that it is the collusion of chromatin marks and possible sequence-specific DNA-binding factors that all contribute significantly to recruitment and stabilization. The ‘one mark, many partners’ promiscuity paradox55,56 may be resolved by the addition of binding constraints and free energies that are imparted by other domains within each distinct complex. The converse of this idea, that a single effector protein may serve as a subunit in several different complexes, even those with opposing enzymatic activity, has recently been examined84 and we expect more examples of this phenomenon to be discovered in the near future. There is some evolutionary economy to both phenomena in that they represent combinatorial diversification from a smaller set of interaction elements that maximizes the signalling potential of a given mark or module capable of binding it.
When the overall affinity and specificity are divided into a modular set of interactions, the individual importance of any one interaction can either be vital or dispensable for binding. It has been suggested that the ‘code’ of covalent chromatin modifications is highly redundant109. Some of this apparent redundancy may actually be instances of multivalent recognition where not every constituent interaction is necessary to transduce an apparent effect, while no single interaction is individually sufficient. Perhaps there are even instances of multivalent chromatin association when only subsets of competent binding modules are engaged at a given time — this would allow readout of different combinations of marks in different temporal and tissue contexts. An example of broadened specificity as a consequence of distinct combinations of interaction modules is provided by the CCCTC-binding factor (CTCF): when CTCF binds to sequence-heterogeneous insulator elements, different combinations of its 11 zinc-finger binding modules are used depending on the precise sequence and DNA methylation pattern of the substrate110.
In some cases, one mark may be sufficient to elicit a specific biologic output42; in other cases, it appears that multiple marks are required9. How do we begin to think about these multiple marks with the goal of providing crucial experimental tests of the multivalency-based theories provided here? We must venture beyond the reductionist conventional wisdom that largely restricts studies to a single mark considered in isolation. Rather, we should begin to explore how the coexistence of marks within a given tail, within a given nucleosome and within a given chromatin domain may serve to dictate functional outcomes in combination. Here, we hope to emphasize that multivalent assemblages of effector domains are emerging as crucial interpreters of chromatin modification patterns. Their study will undoubtedly be central to enhancing our understanding of genome management mechanisms. Although direct evidence may still be lacking, assembled genetic and biochemical data suggests the widespread use of multivalent recognition that governs many interactions with chromatin. We suspect that the nucleosomal unit, with all its rich binding surfaces, promises to have many more secrets left to share with far-reaching implications for human biology and disease.
Acknowledgments
We apologize to all authors whose important contributions could not be acknowledged due to space limitations. We thank members of the Patel and Allis laboratories for reading the manuscript and providing constructive criticism. We are especially grateful to H. Dormann, A. Goldberg, P. Lewis, S. Taverna, J. Wysocka and the anonymous reviewers. D.J.P. is supported by funds from the Abby Rockefeller Mauze Trust and the Dewitt Wallace and Maloris Foundations, and C.D.A. is supported by National Institutes of Health grants and funds from The Rockefeller University. A.J.R. is supported by an Irvington postdoctoral fellowship from the Cancer Research Institute.
Glossary
- PTM
A post-translational modification or chemical alteration of an amino acid residue (or residues) that occurs subsequent to its translation, introducing a new functional group into a given protein.
- Histone variant
An isoform of a particular histone type that is encoded as a distinct gene and denoted by an additional designation (for example, the bar body-deficient histone H2A gene product is called H2A.bbd). Some variants have specialized functions: notably, H2A.X is involved in DNA repair and H2A.Z is involved in gene regulation.
- Heterochromatin
A highly condensed and transcriptionally less active form of chromatin that occurs at defined sites, such as centromeres, silencer DNA elements or telomeres.
- Euchromatin
Chromatin that appears to be less compact than condensed mitotic chromosomes. Active genes are contained within euchromatin.
- DNA methylation
A covalent modification of DNA at the 5-position of the cytosine nucleobase that is coupled to transcriptional repression. CpG sequence elements in gene regulatory regions are often modified in this manner to attenuate gene transcription.
- Direct nucleosome-intrinsic
A histone modification-dependent process that alters the physical properties of the nucleosome by modulation of interactions of the core histone octamer with DNA.
- Direct nucleosome-extrinsic
A histone modification-dependent process that alters the physical properties of chromatin by modulation of interactions between nucleosomes.
- Effector mediated
A chromatin modification-dependent process that changes or stabilizes the chromatin architecture through modification-dependent recruitment of accessory factors.
- Effector
A protein domain that binds a particular histone modification to transduce a downstream function. effector proteins themselves or other subunits of complexes in which they reside can be histone-modifying enzymes or chromatin-remodelling factors, or they can be involved in stabilization of heterochromatin or have some other gene regulatory role.
- Bromodomain
A domain with sequence conservation that is found in several transcriptional regulatory proteins involved in gene activation, and that has acetyl-Lys-binding activity.
- PHD finger domain
The plant homeodomain (PHD) zinc finger is found in many nuclear proteins that are thought to be involved in chromatin transactions.
- ChIP-chip
Chromatin immunoprecipitation followed by amplification and microarray hybridization.
- ChIP-seq
Chromatin immunoprecipitation followed by massively multiplexed sequencing.
- Lys acetylation
Acetylation of the ζ-amine of the Lys side chain, a PTM that is catalysed by histone acetyltransferases.
- Lys methylation
A PTM at the ζ-amine of Lys that adds one, two or three methyl groups.
- Arg methylation
A PTM of the η-nitrogens of Arg to introduce one or two methyl groups.
- Cooperativity
Binding enhancement caused by one or more discrete binding interactions that further assist such interactions, although not necessarily as a consequence of a conformational change.
- Chromodomain
The canonical methyl-Lys-binding protein fold that was initially characterized in HP1 and Polycomb proteins. A member of the royal superfamily of folds.
- Negative cooperativity
Antagonism in binding that leads to sub-additive or repulsive binding energies.
- WD40 repeat
A protein motif that is composed of a 40-amino-acid repeat that forms a four-stranded antiparallel β-propeller sheet. WD40 proteins that contain 5–7 WD40 repeats may form β-propeller structures that participate in many cellular functions, including G-protein-mediated signal transduction, transcriptional regulation, rNA processing and regulation of vesicle metabolism.
References
- 1.Cosgrove MS. Histone proteomics and the epigenetic regulation of nucleosome mobility. Expert Rev Proteomics. 2007;4:465–478. doi: 10.1586/14789450.4.4.465. A recent overview of known histone PTMs as identified by mass spectrometry It focuses on direct nucleosome-intrinsic effects of histone modifications and represents a nice catalogue of marks. [DOI] [PubMed] [Google Scholar]
- 2.Clarkson MJ, Wells JR, Gibson F, Saint R, Tremethick DJ. Regions of variant histone His2AvD required for Drosophila development. Nature. 1999;399:694–697. doi: 10.1038/21436. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein E, Hake SB. The nucleosome: a little variation goes a long way. Biochem Cell Biol. 2006;84:505–517. doi: 10.1139/o06-085. [DOI] [PubMed] [Google Scholar]
- 4.Henikoff S, Ahmad K. Assembly of variant histones into chromatin. Annu Rev Cell Dev Biol. 2005;21:133–153. doi: 10.1146/annurev.cellbio.21.012704.133518. A definitive review on histone variants that describes the functional associations of variants with various stages of chromatin metabolism. [DOI] [PubMed] [Google Scholar]
- 5.Pidoux AL, Allshire RC. The role of heterochromatin in centromere function. Philos Trans R Soc Lond B Biol Sci. 2005;360:569–579. doi: 10.1098/rstb.2004.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allis CD, Jenuwein T, Reinberg D. Epigenetics. Cold Spring Harbor Laboratory Press; 2006. [Google Scholar]
- 7.Sproul D, Gilbert N, Bickmore WA. The role of chromatin structure in regulating the expression of clustered genes. Nature Rev Genet. 2005;6:775–781. doi: 10.1038/nrg1688. [DOI] [PubMed] [Google Scholar]
- 8.Patterton D, Wolffe AP. Developmental roles for chromatin and chromosomal structure. Dev Biol. 1996;173:2–13. doi: 10.1006/dbio.1996.0002. [DOI] [PubMed] [Google Scholar]
- 9.Barratt MJ, Hazzalin CA, Cano E, Mahadevan LC. Mitogen-stimulated phosphorylation of histone H3 is targeted to a small hyperacetylation-sensitive fraction. Proc Natl Acad Sci USA. 1994;91:4781–4785. doi: 10.1073/pnas.91.11.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheung P, et al. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol Cell. 2000;5:905–915. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- 11.Shi Y, et al. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 12.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. A thoughtful examination of the meaning of the term epigenetics. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Ptashne M. On the use of the word ‘epigenetic’. Curr Biol. 2007;17:R233–R236. doi: 10.1016/j.cub.2007.02.030. [DOI] [PubMed] [Google Scholar]
- 15.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 17.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 19.Huang J, et al. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 20.Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains. Nature Rev Mol Cell Biol. 2006;7:473–483. doi: 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- 21.Yang XJ. Multisite protein modification and intramolecular signaling. Oncogene. 2005;24:1653–1662. doi: 10.1038/sj.onc.1208173. [DOI] [PubMed] [Google Scholar]
- 22.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 23.Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brinkman AB, et al. Histone modification patterns associated with the human X chromosome. EMBO Rep. 2006;7:628–634. doi: 10.1038/sj.embor.7400686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. This study reports widespread abortive transcriptional initiation and the appearance of H3K4me3 at nearly every coding gene in ES cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. A genome-wide profiling of chromatin states in stem cells as they differentiate, describing the first use of allele-specific ChIP. This work also represents the first direct comparison of ChIP-chip and ChIP-seq approaches. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia BA, Shabanowitz J, Hunt DF. Characterization of histones and their post-translational modifications by mass spectrometry. Curr Opin Chem Biol. 2007;11:66–73. doi: 10.1016/j.cbpa.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 28.Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code. Nature Struct Mol Biol. 2004;11:1037–1043. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Ramirez M, Rocchini C, Ausio J. Modulation of chromatin folding by histone acetylation. J Biol Chem. 1995;270:17923–17928. doi: 10.1074/jbc.270.30.17923. [DOI] [PubMed] [Google Scholar]
- 30.Tse C, Sera T, Wolffe AP, Hansen JC. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol Cell Biol. 1998;18:4629–4638. doi: 10.1128/mcb.18.8.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shogren-Knaak M, et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 32.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 33.Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science. 2004;306:1574–1577. doi: 10.1126/science.1100576. [DOI] [PubMed] [Google Scholar]
- 34.Zhao T, Heyduk T, Allis CD, Eissenberg JC. Heterochromatin protein 1 binds to nucleosomes and DNA in vitro. J Biol Chem. 2000;275:28332–28338. doi: 10.1074/jbc.M003493200. [DOI] [PubMed] [Google Scholar]
- 35.Nielsen AL, et al. Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol Cell. 2001;7:729–739. doi: 10.1016/s1097-2765(01)00218-0. [DOI] [PubMed] [Google Scholar]
- 36.Vermeulen M, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 37.Dhalluin C, et al. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 38.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. A review that evaluates the biology and biophysics of H3K4 methylation and recognition, with an emphasis on structure and function. [DOI] [PubMed] [Google Scholar]
- 39.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nature Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. A recent and comprehensive enumeration of chromatin-binding module structures in complex with modified histones, coupled to the attendant biology of these interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 41.Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 42.van Attikum H, Gasser SM. The histone code at DNA breaks: a guide to repair? Nature Rev Mol Cell Biol. 2005;6:757–765. doi: 10.1038/nrm1737. [DOI] [PubMed] [Google Scholar]
- 43.Ahn SH, et al. Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in Scerevisiae. Cell. 2005;120:25–36. doi: 10.1016/j.cell.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 44.Cheung WL, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003;113:507–517. doi: 10.1016/s0092-8674(03)00355-6. [DOI] [PubMed] [Google Scholar]
- 45.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer — a mechanism for early oncogenic pathway addiction? Nature Rev Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 46.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, part I: covalent histone modifications. Trends Mol Med. 2007;13:363–372. doi: 10.1016/j.molmed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genet. 2003;33 Suppl:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 48.Lan F, et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449:689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- 49.Swigut T, Wysocka J. H3K27 demethylases, at long last. Cell. 2007;131:29–32. doi: 10.1016/j.cell.2007.09.026. [DOI] [PubMed] [Google Scholar]
- 50.Turner BM. Histone acetylation and an epigenetic code. Bioessays. 2000;22:836–845. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 51.Li H, et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. Reports the first structure of a mixed two-effector module (one of each binding-domain type), as well as the first structures of the PHD fingers in complex with H3K4me3-binding modules. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pena PV, et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi X, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wysocka J, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 55.Becker PB. Gene regulation: a finger on the mark. Nature. 2006;442:31–32. doi: 10.1038/442031a. [DOI] [PubMed] [Google Scholar]
- 56.Henikoff S. Histone modifications: combinatorial complexity or cumulative simplicity? Proc Natl Acad Sci USA. 2005;102:5308–5309. doi: 10.1073/pnas.0501853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ptashne M, Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- 58.Hassan AH, et al. Selective recognition of acetylated histones by bromodomains in transcriptional co-activators. Biochem J. 2007;402:125–133. doi: 10.1042/BJ20060907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krishnamurthy VM, Estroff LM, Whitesides GM. In: Fragment-based Approaches in Drug Discovery. Jahnke W, Erlanson DA, editors. Wiley-VCH; Weinheim: 2006. pp. 11–53. A comprehensive review of the thermodynamics of multivalency. [Google Scholar]
- 60.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed Engl. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. This review is an exhaustive enumeration of examples of multivalency from the literature of chemistry and biology. [DOI] [PubMed] [Google Scholar]
- 61.Jencks WP. On the attribution and additivity of binding energies. Proc Natl Acad Sci USA. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee YC, et al. Binding of synthetic oligosaccharides to the hepatic Gal/GalNAc lectin. Dependence on fine structural features J Biol Chem. 1983;258:199–202. [PubMed] [Google Scholar]
- 63.Turner BM. Defining an epigenetic code. Nature Cell Biol. 2007;9:2–6. doi: 10.1038/ncb0107-2. [DOI] [PubMed] [Google Scholar]
- 64.Fraser P, Bickmore W. Nuclear organization of the genome and the potential for gene regulation. Nature. 2007;447:413–417. doi: 10.1038/nature05916. A wonderful review of chromatin territories by two of the key experts on in vivo chromatin structural organization. [DOI] [PubMed] [Google Scholar]
- 65.Hecht A, Grunstein M. Mapping DNA interaction sites of chromosomal proteins using immunoprecipitation and polymerase chain reaction. Methods Enzymol. 1999;304:399–414. doi: 10.1016/s0076-6879(99)04024-0. [DOI] [PubMed] [Google Scholar]
- 66.Bernstein BE, et al. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci USA. 2002;99:8695–8700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robyr D, et al. Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell. 2002;109:437–446. doi: 10.1016/s0092-8674(02)00746-8. [DOI] [PubMed] [Google Scholar]
- 68.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. The first systematic application of ChIP-seq methodology to histone PTMs reports numerous histone modifications and their correlations to chromatin states. A comparison of different methylation states at the same Lys residue is also described. [DOI] [PubMed] [Google Scholar]
- 69.Bernstein BE, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 70.Wade PA, Pruss D, Wolffe AP. Histone acetylation: chromatin in action. Trends Biochem Sci. 1997;22:128–132. doi: 10.1016/s0968-0004(97)01016-5. [DOI] [PubMed] [Google Scholar]
- 71.Kurdistani SK, Tavazoie S, Grunstein M. Mapping global histone acetylation patterns to gene expression. Cell. 2004;117:721–733. doi: 10.1016/j.cell.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 72.Schubeler D, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18:1263–1271. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnson EM, Sterner R, Allfrey VG. Altered nucleosomes of active nucleolar chromatin contain accessible histone H3 in its hyperacetylated forms. J Biol Chem. 1987;262:6943–6946. [PubMed] [Google Scholar]
- 74.Ura K, Kurumizaka H, Dimitrov S, Almouzni G, Wolffe AP. Histone acetylation: influence on transcription, nucleosome mobility and positioning, and linker histone-dependent transcriptional repression. EMBO J. 1997;16:2096–2107. doi: 10.1093/emboj/16.8.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X, Hayes JJ. Site-specific binding affinities within the H2B tail domain indicate specific effects of lysine acetylation. J Biol Chem. 2007;282:23867–32876. doi: 10.1074/jbc.M706035200. [DOI] [PubMed] [Google Scholar]
- 76.Jacobson RH, Ladurner AG, King DS, Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–1425. doi: 10.1126/science.288.5470.1422. Reports the first dual histone-binding module structure, and represents a landmark study indicating cooperative binding by two bromodomains of multiply acetylated histone substrates. [DOI] [PubMed] [Google Scholar]
- 77.Kasten M, et al. Tandem bromodomains in the chromatin remodeler RSC recognize acetylated histone H3 Lys14. EMBO J. 2004;23:1348–1359. doi: 10.1038/sj.emboj.7600143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ladurner AG, Inouye C, Jain R, Tjian R. Bromodomains mediate an acetyl-histone encoded antisilencing function at heterochromatin boundaries. Mol Cell. 2003;11:365–376. doi: 10.1016/s1097-2765(03)00035-2. [DOI] [PubMed] [Google Scholar]
- 79.Mujtaba S, et al. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol Cell. 2004;13:251–263. doi: 10.1016/s1097-2765(03)00528-8. [DOI] [PubMed] [Google Scholar]
- 80.Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–5527. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- 81.Mellor J. Dynamic nucleosomes and gene transcription. Trends Genet. 2006;22:320–329. doi: 10.1016/j.tig.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 82.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 83.Klose RJ, et al. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 84.Li B, et al. Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science. 2007;316:1050–1054. doi: 10.1126/science.1139004. An important example of multivalent binding capacity that is combinatorially assembled into chromatin-modification complexes. [DOI] [PubMed] [Google Scholar]
- 85.Berndsen CE, et al. Nucleosome recognition by the Piccolo NuA4 histone acetyltransferase complex. Biochemistry. 2007;46:2091–2099. doi: 10.1021/bi602366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim YC, Grable JC, Love R, Greene PJ, Rosenberg JM. Refinement of EcoRI endonuclease crystal structure: a revised protein chain tracing. Science. 1990;249:1307–1309. doi: 10.1126/science.2399465. [DOI] [PubMed] [Google Scholar]
- 87.Rao J, Lahiri J, Isaacs L, Weis RM, Whitesides GM. A trivalent system from vancomycin.D-ala-D-Ala with higher affinity than avidin.biotin. Science. 1998;280:708–711. doi: 10.1126/science.280.5364.708. A landmark study in multivalency that demonstrates the enormous gain in overall affinity by systematic comparison of monovalent and trivalent vancomycin molecules. [DOI] [PubMed] [Google Scholar]
- 88.Khorasanizadeh S. The nucleosome: from genomic organization to genomic regulation. Cell. 2004;116:259–272. doi: 10.1016/s0092-8674(04)00044-3. [DOI] [PubMed] [Google Scholar]
- 89.Martinez-Campa C, et al. Precise nucleosome positioning and the TATA box dictate requirements for the histone H4 tail and the bromodomain factor Bdf1. Mol Cell. 2004;15:69–81. doi: 10.1016/j.molcel.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 90.Matangkasombut O, Buratowski S. Different sensitivities of bromodomain factors 1 and 2 to histone H4 acetylation. Mol Cell. 2003;11:353–363. doi: 10.1016/s1097-2765(03)00033-9. [DOI] [PubMed] [Google Scholar]
- 91.Lindroth AM, et al. Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. EMBO J. 2004;23:4286–4296. doi: 10.1038/sj.emboj.7600430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fischle W, et al. Regulation of HP1–chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 93.Hirota T, Lipp JJ, Toh BH, Peters JM. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- 94.Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol Cell. 2005;20:971–978. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 95.Carrozza MJ, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 96.Shi X, et al. Proteome-wide analysis in Saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J Biol Chem. 2007;282:2450–2455. doi: 10.1074/jbc.C600286200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST– human histone deacetylase complex. Proc Natl Acad Sci USA. 2001;98:1454–1458. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosomal DNA regulates the core-histone-binding subunit of the human Hat1 acetyltransferase. Curr Biol. 1998;8:96–108. doi: 10.1016/s0960-9822(98)70040-5. [DOI] [PubMed] [Google Scholar]
- 99.Yang M, et al. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell. 2006;23:377–387. doi: 10.1016/j.molcel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 100.Shi YJ, et al. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 101.Lan F, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ooi SK, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Barak O, et al. Isolation of human NURF: a regulator of Engrailed gene expression. EMBO J. 2003;22:6089–6100. doi: 10.1093/emboj/cdg582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fischle W, et al. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vandemark AP, et al. Autoregulation of the RSC4 tandem bromodomain by GCN5 acetylation. Mol Cell. 2007;27:817–828. doi: 10.1016/j.molcel.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Segal E, et al. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Milne TA, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 108.Dodd IB, Micheelsen MA, Sneppen K, Thon G. Theoretical analysis of epigenetic cell memory by nucleosome modification. Cell. 2007;129:813–822. doi: 10.1016/j.cell.2007.02.053. [DOI] [PubMed] [Google Scholar]
- 109.Schreiber SL, Bernstein BE. Signaling network model of chromatin. Cell. 2002;111:771–778. doi: 10.1016/s0092-8674(02)01196-0. [DOI] [PubMed] [Google Scholar]
- 110.Filippova GN, et al. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. 1996;16:2802–2813. doi: 10.1128/mcb.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zubarev RA, et al. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 112.Mikesh LM, et al. The utility of ETD mass spectrometry in proteomic analysis. Biochim Biophys Acta. 2006;1764:1811–1822. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chicoine LG, Schulman IG, Richman R, Cook RG, Allis CD. Nonrandom utilization of acetylation sites in histones isolated from Tetrahymena. Evidence for functionally distinct H4 acetylation sites. J Biol Chem. 1986;261:1071–1076. [PubMed] [Google Scholar]
- 114.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nature Protoc. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- 115.Metivier R, et al. Transcriptional complexes engaged by apo-estrogen receptor-α isoforms have divergent outcomes. EMBO J. 2004;23:3653–3666. doi: 10.1038/sj.emboj.7600377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 117.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 118.Shogren-Knaak MA, Peterson CL. Creating designer histones by native chemical ligation. Methods Enzymol. 2004;375:62–76. doi: 10.1016/s0076-6879(03)75004-6. [DOI] [PubMed] [Google Scholar]
- 119.Chatterjee C, McGinty RK, Pellois JP, Muir TW. Auxiliary-mediated site-specific peptide ubiquitylation. Angew Chem Int Ed Engl. 2007;46:2814–2818. doi: 10.1002/anie.200605155. [DOI] [PubMed] [Google Scholar]
- 120.Simon MD, et al. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dorigo B, Schalch T, Bystricky K, Richmond TJ. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J Mol Biol. 2003;327:85–96. doi: 10.1016/s0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 122.Dou Y, et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 123.Xiao H, et al. Dual functions of largest NURF subunit NURF301 in nucleosome sliding and transcription factor interactions. Mol Cell. 2001;8:531–543. doi: 10.1016/s1097-2765(01)00345-8. [DOI] [PubMed] [Google Scholar]
- 124.Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- 125.Taverna SD, et al. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc Natl Acad Sci USA. 2007;104:2086–2091. doi: 10.1073/pnas.0610993104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Garcia BA, et al. Modifications of human histone H3 variants during mitosis. Biochemistry. 2005;44:13202–13213. doi: 10.1021/bi050906n. [DOI] [PubMed] [Google Scholar]
- 127.Daujat S, et al. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr Biol. 2002;12:2090–2097. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- 128.Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol Cell. 2006;24:309–316. doi: 10.1016/j.molcel.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 129.Lindroth AM, et al. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science. 2001;292:2077–2080. doi: 10.1126/science.1059745. [DOI] [PubMed] [Google Scholar]
- 130.Sims JK, Houston SI, Magazinnik T, Rice JC. A trans-tail histone code defined by monomethylated H4 Lys-20 and H3 Lys-9 demarcates distinct regions of silent chromatin. J Biol Chem. 2006;281:12760–12766. doi: 10.1074/jbc.M513462200. [DOI] [PubMed] [Google Scholar]