

SUMMARY
The MLL1 gene is a frequent target for recurrent chromosomal translocations, resulting in transformation of hematopoietic precursors into leukemia stem cells. Here, we report on structure-function studies that elucidate molecular events in MLL1 binding of histone H3K4me3/2 marks and recruitment of the cyclophilin CyP33. CyP33 contains a PPIase and a RRM domain and regulates MLL1 function through HDAC recruitment. We find that the PPIase domain of CyP33 regulates the conformation of MLL1 through proline isomerization within the PHD3-Bromo linker, thereby disrupting the PHD3-Bromo interface and facilitating binding of the MLL1-PHD3 domain to the CyP33-RRM domain. H3K4me3/2 and CyP33-RRM target different surfaces of MLL1-PHD3 and can bind simultaneously to form a ternary complex. Furthermore, the MLL1-CyP33 interaction is required for repression of HOXA9 and HOXC8 genes in vivo. Our results highlight the role of PHD3-Bromo cassette as a regulatory platform, orchestrating MLL1 binding of H3K4me3/2 marks and cyclophilin-mediated repression through HDAC recruitment.
INTRODUCTION
The mixed lineage leukemia 1 (MLL1) gene is essential for embryonic development and hematopoiesis (Hess, 2004). The encoded protein belongs to a family of H3K4 methyltransferases (Milne et al., 2002; Nakamura et al., 2002), with a domain architecture that includes three AT-hook motifs, a transcriptional repression region containing a CXXC zinc-finger domain, four PHD fingers and one PHD-flanking bromodomain (Bromo), FYRN and FYRC domains, and a C-terminal methyltransferase SET domain (Figure 1A). The MLL1 gene is a frequent target for chromosomal translocations in human acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) (Ayton and Cleary, 2001). More than 70% of infant leukemias and approximately 10% of adult AMLs involve MLL1 rearrangements, which are often associated with a poor outcome. In most MLL1 translocations, ~1400 residues from the N terminus of MLL1 are fused in-frame to one of more than 50 translocation partners, a process that displaces the segment starting at the PHD finger cluster and ending at the C-terminal SET domain. Many MLL1 fusion partners belong to a protein network associated with transcriptional regulation through chromatin remodeling, thereby implying that MLL1 fusions may correlate with aberrant histone modifications at target promoters, such as HOX gene clusters (Krivtsov and Armstrong, 2007; Linggi et al., 2005).
Figure 1. Construct Design and Crystal Structure of MLL1 PHD3-Bromo Cassette in the Free State.

(A) Domain architecture of MLL1 is shown in the top panel. Two coexpression constructs of MLL1 PHD3-Bromo cassette used for crystallization are shown in the bottom panel.
(B) A ribbon view of the crystal structure of MLL1 PHD3-Bromo cassette with labeling of secondary structure elements. Pocket residues within PHD3 involved in Kme recognition are shown in stick representation, together with residues involved in direct interaction at the interface of the PHD3-Bromo cassette.
(C) An expanded view highlighting the cis peptide bond at Pro1629 in the linker segment connecting MLL1 PHD3 and Bromo domains. Residues 1628–1630 are shown in stick representation, with the corresponding 2Fo-Fc electron density contoured at 1σ.
(D) An expanded view of the interdomain interaction between Glu1605 from PHD3 and the Val1723-Leu1724 backbone amides from Bromo.
(E) An expanded view highlighting the interfacial hydrophobic and hydrogen bonding interactions between PHD3 (in blue) and Bromo (in salmon).
(F) The partially formed Kme recognition pocket on the surface of PHD3. The Tyr1581 residue, which is not part of the aromatic-lined pocket, is indicated by a red asterisk.
See also Figure S1.
MLL1 is a member of the trithorax group of proteins that function in opposition to the polycomb group proteins in maintaining ‘‘cellular memory’’ (Jenuwein and Allis, 2001). MLL1 is critical for maintaining HOX gene expression during embryonic development (Yu et al., 1995) and in hematopoiesis (Ernst et al., 2004). MLL1 globally colocalizes with RNA polymerase II at the 5′ end of actively transcribed genes (Guenther et al., 2005; Milne et al., 2005), which are also enrichment sites for histone H3 lysine 4 trimethylation (H3K4me3) marks (Guenther et al., 2005). This coincidence of MLL1 target sites and H3K4me3 mark enrichment at the genomic level implies that MLL1 recruitment to such H3K4me3-enriched loci could be mediated in part by an interaction involving one of its PHD fingers, a well-established reader of methylated lysine marks (Ruthenburg et al., 2007a; Baker et al., 2008).
MLL1 is a component of large nuclear complexes (Nakamura et al., 2002; Dou et al., 2005) and is regulated through extensive interactions with multiple proteins that either activate or suppress gene expression. Among suppressor factors, CyP33 is quite unique in that it is neither a polycomb group protein nor a histone deacetylase (HDAC). Rather, CyP33 is a cyclophilin that contains an RNA recognition motif (RRM) within its N terminus, separated by a long linker from its C-terminally positioned peptidyl prolyl isomerase (PPIase) domain. Moreover, CyP33 is found in nuclear fractions of cultured cells exhibiting overlapping patterns of nuclear localization with MLL1 (Mi et al., 1996; Fair et al., 2001). CyP33 has been shown to specifically interact with the third PHD finger (PHD3) of MLL1 (Fair et al., 2001), and in the process facilitates an increase in the level of HDAC1 binding to the MLL1 repression domain (Xia et al., 2003). Both PPIase activity and RRM binding are required to facilitate this switch in MLL1 function from transcriptional activation to repression (Fair et al., 2001; Xia et al., 2003). Despite these advances, the molecular principles governing the interplay between MLL1 and CyP33 remain elusive, given that there is no understanding of the roles of the RRM and PPIase domains of CyP33 in regulating the activity of MLL1. Further, since MLL1 PHD3 is directly involved in binding CyP33 and is also predicted to be a potential reader of lysine methylation marks, it remains unresolved as to how these two distinct molecular events can be integrated together to provide comprehensive mechanistic insights into MLL1 function. To address these questions, we undertook systematic structure-function studies of the MLL1 PHD3-Bromo cassette in the absence and presence of bound histone peptides and CyP33.
In this study, we present X-ray and nuclear magnetic resonance (NMR)-based structures of MLL1 PHD3-Bromo cassette in the free state, as well in complexes with H3K4me3/2 peptides and the RRM domain of CyP33. We also performed isothermal titration calorimetry (ITC)-based binding and NMR-based titration studies to establish that the H3K4me3/2 marks and the RRM domain target distinct surfaces of PHD3 and can coexist as a ternary complex. Significantly, we demonstrate that cis-trans isomerization of a linker proline within the MLL1 PHD3-Bromo cassette by the CyP33 PPIase domain dramatically disrupts the interdomain contacts within the PHD3-Bromo cassette, thereby topologically altering the relative alignments of the PHD3 and Bromo domains and facilitating access of an otherwise occluded PHD3 binding surface to the RRM domain. Further, in vivo studies establish that CyP33 can be recruited to the HOXA9 and HOXC8 genes independently of MLL1 but that the MLL1-CyP33 interaction is required for histone deacetylase-mediated repression of these target genes in vivo. Collectively, our studies establish a role for the MLL1 PHD3-Bromo cassette as a functional platform connecting histone targeting and CyP33-mediated regulation. These observations provide insights into mechanisms that regulate MLL1 in its normal physiological state and shed light on how MLL1 fusions are mis-regulated as a result of dysfunction of this disease-associated regulatory platform.
RESULTS
Crystal Structure of MLL1 PHD3-Bromo Cassette in the Free State
To begin to understand the physical basis of chromatin interactions of the PHD3-Bromo cassette of the MLL1 protein, we sought to structurally characterize this region both alone and in complex with relevant binding partners. Initially, we were unable to express (in E. coli) and purify soluble constructs of the PHD3-Bromo cassette, as a result of premature cleavage within the ZA loop of Bromo. This problem was overcome by deletion of residues 1666–1702 within the extra long ZA loop (Figure 1A and Figure S1A available online) and coexpression of the resulting fragments in a duet vector. The coexpressed MLL1 PHD3-Bromo cassette expressed well in E.coli and could be purified to homogeneity.
The 1.72 Å crystal structure of the coexpressed MLL1 PHD3-Bromo cassette (residues 1566–1784, with deletion of 1666–1702 ZA loop segment) in the free state is shown in Figure 1B (X-ray statistics in Table S1). Both PHD3 and Bromo adopt characteristic folds of their family members. The linker segment (1625–1631) connecting PHD3 and Bromo adopts a turn segment, which leads into the extended α helix of Bromo. Significantly, Pro1629 in the linker segment adopts a cis conformation (Figure 1C), facilitating an overall compact globular fold involving the two domains, mediated in part by a pair of salt bridges between a carboxylate group and backbone amide protons (Figure 1D). The dominant interaction between PHD3 and Bromo is mediated by the extended αZ helix of Bromo, which is anchored in place by a network of hydrogen-bonding and hydrophobic interactions (Figure 1E; buried surface area 660 Å2). Residues Tyr1576, Met1585, and Trp1594 form a partial aromatic cage on the surface of PHD3, with Tyr1581 positioned nearby, but not part of, the cage in the free state (red asterisk in Figure 1F).
Crystal Structure of MLL1 PHD3-Bromo Cassette Bound to H3(1-9)K4me3/2
By peptide pulldown assays, we first identified that the PHD3-Bromo cassette is a reader of higher-order lysine methylation states in the H3K4me context (Figure S1B). We then monitored the binding energetics for complex formation between the coex-pressed PHD3-Bromo cassette and H3(1-15)K4me-containing peptides by ITC. PHD3 of the dual domain binds H3K4me3 and H3K4me2 peptides with dissociation constants (KD) of 4.3 μM and 6.9 μM, respectively, while it binds H3K4me1 peptide with an approximately 10-fold weaker affinity (KD = 53 μM) (Figure 2A). By comparison, MLL1 PHD3 alone showed much weaker binding affinity for H3K4me-containing peptides as measured by ITC (Table S2), which provided an early indication that the adjacent Bromo may contribute to recognition of histone methylation marks by MLL1 PHD3.
Figure 2. Crystal Structure of MLL1 PHD3-Bromo Cassette Bound to H3K4me3/2-Containing Peptides, Together with Binding Data on Wild-Type and Mutant PHD3.

(A) ITC binding curves of MLL1 PHD3-Bromo cassette with H3K4me-containing peptides.
(B) A ribbon view of the crystal structure of MLL1 PHD3-Bromo cassette in complex with H3(1-9)K4me3 peptide. cis-configured Pro1629 and pocket residues for trimethyl-lysine recognition are labeled accordingly.
(C) A view of the hydrogen bonding interactions between MLL1 PHD3 (blue) and bound H3K4me3 peptide (yellow) in the crystal structure of the complex.
(D) ITC profiles for the binding of H3(1-15)K4me3 peptide to wild-type (WT) MLL1 PHD3-Bromo and its W1594E, Y1581A, and Q1587A mutants.
(E) A view of the superimposed structures of the K4me3 binding pocket of MLL1 PHD3 in the free (silver) and H3K4me3-bound (blue) states. The blue and red arrows highlight the conformational changes in the loop spanning Tyr1576-Met1585 segment and involving Tyr1581, respectively, on complex formation.
(F) A view of the superimposed structures of the peptide N terminus binding pocket of MLL1 PHD3 in the free (silver) and H3K4me3-bound (blue) states. The Pro1614-Val1617 segment adopts an α helix (silver) in the free state but changes into a turn fold (blue) in the bound state.
(G) Surface electrostatic view of MLL1 PHD3 with bound H3(1-6)K4me3 peptide in a space-filling representation. Note that access is blocked to both the trime-thylammonium group of H3K4me3 (indicated by red asterisk) and the N terminus (indicated by red pound symbol) in this complex.
(H) ITC profiles for the binding of MLL1 PHD3-Bromo to wild-type and two modified H3K4me3 peptides containing one or three additional Ala residues added to the N terminus.
See also Figure S2.
Bromodomains are well documented as acetyllysine-reading modules (Zeng and Zhou, 2002; Mujtaba et al., 2007). The MLL1 Bromo exhibits the typical bromodomain fold, with a main chain root-mean-square deviation (rmsd) of 1.5 Å when compared to BPTF Bromo (Li et al., 2006) (Figure S1C). To test whether MLL1 Bromo also binds acetylated lysines, we expressed and purified 15N-labeled MLL1 PHD3-Bromo cassette (1566–1784) and MLL1 Bromo alone (1639–1784) without deleting the ZA loop, using a modified expression protocol outlined in the Extended Experimental Procedures. We did not observe any complexation shifts in the 1H,15N-HSQC spectra of either the PHD3-Bromo cassette or Bromo alone after addition of five equivalents of H4(7-17)K12ac or H4(14-19)K16ac (Figure S1D) peptides, as well as peptides containing H3K23ac and H3K27ac modifications (data not shown). The inability of MLL1 Bromo to recognize acetyllysine in a range of histone sequence contexts may be due to replacement of a conserved Asn in the BC loop of acetyllysine-recognizing Bromos, such as Asn407 in GCN5 (Owen et al., 2000) and Asn3007 in BPTF, by an Asp in the corresponding position of MLL1 (Figure S1A).
Guided by the above binding studies, we next solved the crystal structure of the coexpressed MLL1 PHD3-Bromo cassette with bound H3(1-9)K4me3 peptide at 1.90 Å resolution (Figure 2B; X-ray statistics in Table S1; electron density of bound peptide shown in Figure S2A). The details of the molecular recognition between residues 1 to 6 of H3K4me3 and MLL1 PHD3 in the complex are shown in Figure 2C, with the corresponding stereo view shown in Figure S2B. The bound peptide forms a β turn at the K4me3-Q5 step and is stabilized by a set of intermolecular hydrogen bonds. The trimethylammonium group of K4 is positioned within an aromatic cage composed of Tyr1576, Tyr1581, Met1585, and Trp1594 side chains (Figure 2B), where it is stabilized by methyl-π and cation-π interactions (Ma and Dougherty, 1997). The guanidinium group of R2 of the bound peptide is directed away from the surface of PHD3, and its ε guanidinium proton is hydrogen bonded to the side chain of Gln1587 (Figure 2C).
We also solved the 1.92 Å crystal structure of the complex of the coexpressed MLL1 PHD3-Bromo cassette with H3(1-9) K4me2 peptide (electron density of bound peptide shown in Figure S2C; X-ray statistics in Table S1) and observed similar intermolecular contacts as in the H3K4me3 complex (stereo view in Figure S2D), with the ammonium proton of K4me2 forming an intermolecular hydrogen bond with the peptide carbonyl of Asp1580.
We mutated Tyr1581 and Trp1594 aromatic residues, which line the aromatic pocket, to Ala and Glu, respectively, and also mutated Gln1587, which forms a salt bridge with R2, to Ala so as to measure the impact of these mutations on complex formation between H3K4me3/2 and PHD3-Bromo cassette by ITC. All the above mutants, which are associated with key intermolecular contacts, are destabilizing and exhibit increased dissociation constants (Table S2).
Conformational Changes in MLL1 PHD3 upon Complex Formation
Quite unexpectedly, we observed that the aromatic ring of Tyr1581 is looped out from the aromatic cage of PHD3 in MLL1 PHD3-Bromo in the free state (Figure 1F) but flips inwards by 3.1 Å to complete the aromatic cage upon formation of the H3K4me3 complex (red arrow, Figure 2E). In addition to this change, the loop segment between residues Tyr1576 and Met1585 also undergoes a remarkable and previously unrecognized conformational change by shifting (3.6 Å) toward the central β strands (β1 and β2) on complex formation (blue arrow, Figure 2E), thus contributing to closing an additional face of the aromatic cage. Strikingly, the conformational transition buries the K4me3 side chain bound to MLL1 PHD3 (Figure 2G), a feature not observed previously for binding of the K4me3 side chain to the preformed binding pocket of BPTF PHD2 (Li et al., 2006).
We also observed a conformational change within the Asn1612-Ala1618 segment of PHD3 on formation of the H3K4me3 complex (rmsd between states for this segment is 3.0 Å). The Pro1614-Val1617 segment switches from an α-helical conformer for the PHD3-Bromo cassette in the free state (ribbon, silver color) to a turn fold on complex formation (stick, blue color) as shown in Figure 2F. This transition tends to widen the channel between segment 1612–1618 and the central β sheet, such that A1 can be positioned snugly in the newly created pocket, with its methyl group surrounded by a cluster of hydrophobic residues on complex formation. The turn fold in the bound state of PHD3 is stabilized by a set of direct and water-mediated intermolecular hydrogen bonds involving the N terminus of the bound peptide (Figure 2F).
Concomitant with the conformational changes, the N terminus of bound H3K4me3/2 becomes less accessible to solvent on complex formation (Figure 2G). We designed experiments to probe the accessibility of the N terminus of the bound H3K4me3 peptide in the MLL1 PHD3-Bromo cassette complex by measuring the impact on dissociation constants after addition of either a single Ala or an Ala-Ala-Ala tripeptide at the N terminus. The dissociation constants increased by 6-fold on addition of a single Ala (KD = 26 μM) and by 15-fold on addition of an Ala-Ala-Ala tri-peptide (KD = 64 μM) (Figure 2H).
In all previously solved structures of H3K4me3/2-PHD finger complexes, there were no evident conformational changes, other than minor adjustments, in the K4me3 binding pocket on the surface of the PHD finger (Taverna et al., 2007). In striking contrast to such static recognition pockets, the conformational changes within H3K4me3/2-MLL1 PHD3 complexes involved both significant side-chain and main-chain movements (Figures 2E and 2F), with such conformational transitions within recognition pockets also exhibiting qualitative differences between recognition of H3K4me3 and H3K4me2 marks (stereo view in Figure S2E). These conformational rearrangements contribute to the binding specificity of MLL1 between methylation states in the H3K4me context and also explain why a free N terminus of the H3K4me-containing peptide is favored by the MLL1 PHD3 finger.
MLL1 PHD3 Is Important for Stable Target Gene Binding In Vivo
To test the contribution of H3K4me3 binding to MLL1 function in vivo, we examined the colocalization of MLL1 and H3K4me3 marks on well-established MLL1 target genes in vivo. Chromatin immunoprecipitation (ChIP) assays were performed for four regions on HOXA9 and HOXC8 loci and two regions on the p27KIP1 locus (Figure 3A). Using an antibody against MLLC in the ChIP assay, endogenous MLL1 binds to all the loci (Figure 3B) in a pattern that correlates reasonably well with the relative level of H3K4me3 marks at these loci (Figure 3C). To test the effect of H3K4me3-binding activity of MLL1, we established four individual stable cell lines that express FLAG and HA double-tagged MLLN fragments that were either wild-type (fh-MLLCXXC-PHD3, lines 2 and 3) or mutant (fh-MLLCXXC-PHD3 (W1594E), lines 4 and 5) for H3K4me3-binding activity (Figure 3D). Lines 4 and 5 contain the W1594E mutation in MLL1 PHD3 that specifically disrupts the binding between MLL1 PHD3 and the H3K4me3 mark. Anti-FLAG western blots indicate that expression levels were equal in all four cell lines (Figure 3D). Using an anti-HA antibody, ChIP assays show that the average combined signal for binding of the two mutant lines (Figure 3E, green bars) is consistently 30% to 50% lower than the two wild-type lines (Figure 3E, red bars), at primer positions B, C, and D for HOXA9, C for HOXC8, and A for p27KIP1. These are all primer positions in regions with relatively high levels of H3K4me3 (Figure 3C). For less abundant H3K4me3 regions, both fragments showed low and comparable binding. These results verified that binding of H3K4me3 marks by MLL1 PHD3 is important, although not solely responsible, for MLL1 localization at target genes (Milne et al., 2010).
Figure 3. MLL1 PHD3 Is Important for Stable Target Gene Binding In Vivo.

(A) Positions of primer sets used for real-time PCR. (B and C) ChIP analysis with antibodies for MLLC
(B) or H3K4me3 (normalized to H3 levels)
(C) at the target genes HOXA9, HOXC8, and p27KIP1. ChIP results shown are typical for at least three independent experiments. Error bars represent standard deviation of three separate PCR reactions.
(D) Four separate stable cell lines were generated expressing the MLLN fragments fh-MLLCXXC-PHD3 and fh-MLLCXXC-PHD3 (W1594E) (W1594E point mutation in MLL1 PHD3).
(E) ChIP signal with an HA antibody for the stable cell lines in (D) at MLL1 target genes. ChIP signal was calculated for each cell line individually, and then the results for 2 and 3 were averaged together, as were the results for 4 and 5. Averages were the results of three independent experiments for each cell line. Error bars represent standard deviation across the averaged values.
PPIase-Mediated Complex Formation between MLL1 PHD3 and CyP33 RRM Domains
Cyclophilin CyP33, containing an independently folded N-terminal RRM and a C-terminal PPIase, has been previously shown to target MLL1 through recognition of PHD3 (Fair et al., 2001). We tested for complex formation between intact CyP33 (1–301), as well as its individual RRM (1–82) and PPIase (136–301) domains, with either PHD3 alone or PHD3-Bromo cassette of MLL1. We observed from gel filtration profiles that CyP33 RRM formed a complex with the PHD3 domain alone (Figure 4, line 1, and Figure S3A), but not with the PHD3-Bromo cassette (Figure 4, line 3, and Figure S3B), and that complex formation in the latter case occurred only in the context of full-length CyP33 (Figure 4, line 4, and Figure S3C) or in the presence of coexpressed CyP33 PPIase domain (Figure 4, line 5, and Figure S3D). A more detailed analysis is outlined in the Extended Experimental Procedures. Thus, the CyP33 PPIase domain appears to regulate the interaction between CyP33 RRM and MLL1 PHD3 in the presence of linked MLL1 Bromo, presumably via its PPIase activity for peptidyl prolyl isomerization. Indeed, the linker segment connecting PHD3 and Bromo of MLL1 contains a single Pro at position 1629, with the His1628-Pro1629 peptide bond adopting a cis alignment both for PHD3-Bromo in the free state (Figure 1C) and when bound to H3K4me3/2 peptides (Figure 2B). In this cis Pro1629 conformation, a segment of the surface of PHD3 is occluded through interaction with Bromo (Figure 1E), suggesting that this buried surface in the PHD3-Bromo cassette, were it to become accessible, could be involved in interaction with the CyP33 RRM domain.
Figure 4. Complex Formation Results between Different Combinations of Constructs of MLL1/2 PHD3-Bromo Cassette and CyP33.
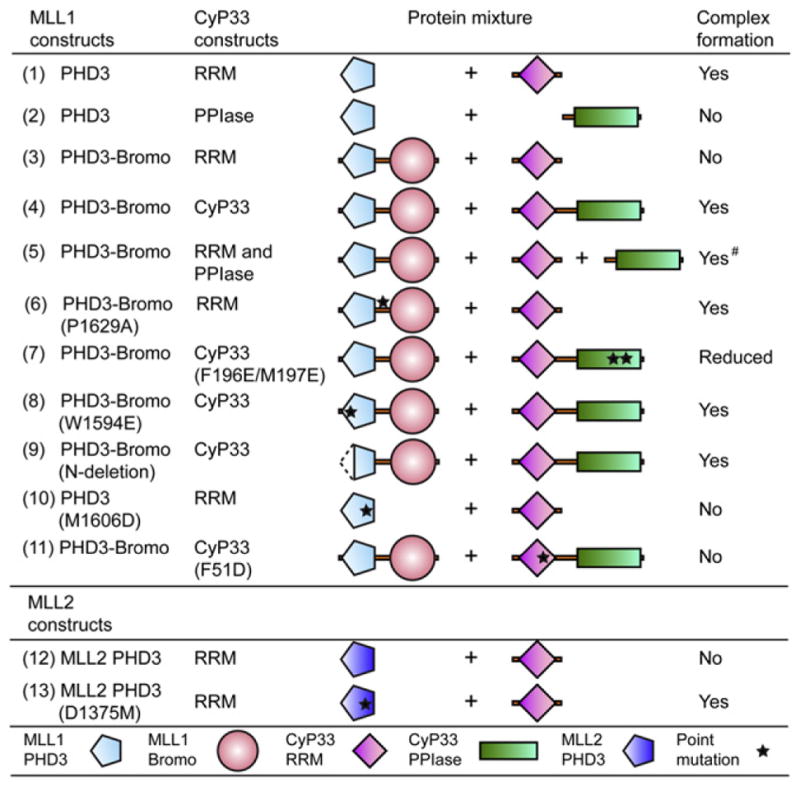
The # symbol indicates that complex was formed between PHD3-Bromo and RRM by coexpression of vectors of PHD3-Bromo, RRM, and PPIase together in E. coli. See also Figure S3.
We next examined the impact of cis-trans conformational transition of Pro1629 and the requirement for CyP33 PPIase activity in terms of the interaction between CyP33 and the MLL1 PHD3-Bromo cassette. First, the CyP33 RRM domain interacted with a PHD3-Bromo mutant, where Pro1629 was replaced by Ala, so as to force a trans peptide bond at this position, even in the absence of CyP33 PPIase domain (Figure 4, line 6, and Figure S3E). Second, the double mutation F196E/M197E incorporated within the CyP33 PPIase domain, which disrupts the hydrophobic propensity of the proline-binding pocket (Wang et al., 2005), reduced the binding between CyP33 and PHD3-Bromo (Figure 4, line 7, and Figure S3F). Taking these data together, we infer that the CyP33 PPIase acts on the Pro1629 peptide bond to regulate, through cis-trans isomerization, the interaction between CyP33 RRM and PHD3 in the context of PHD3-Bromo cassette.
Intermolecular Interface between MLL1 PHD3 and CyP33 RRM on Complex Formation
We have solved the 2.0 Å crystal structure of the RRM domain of CyP33 (5–82), whose topology is comprised of a four-stranded antiparallel β sheet packed against two α helices (stereo view in Figure S4A; X-ray statistics in Table S3). We next turned our structural efforts to the NMR-based characterization of the complex between MLL1 PHD3 and CyP33 RRM in solution. We mapped NMR complexation shifts for amide resonances of uniformly 13C,15N-labeled MLL1 PHD3 C1575S mutant upon addition of unlabeled CyP33 RRM (Figure S4B), with the largest complexation shifts (Δδave > 0.15 ppm, highlighted in yellow) restricted to residues from the long α helix (α2) toward the C-terminal end of the PHD3 domain (Figure 5A). In parallel, we mapped complexation shifts for amide resonances of uniformly 13C,15N-labeled CyP33 RRM domain upon addition of unlabeled MLL1 PHD3 C1575S mutant (Figure S4C), with the largest complexation shifts (Δδave > 0.15 ppm, highlighted in yellow) restricted to one face of the β sheet of the RRM domain, as well as the loop connecting the second and third β strands (Figure 5B). Despite these insights, the NMR spectra of MLL1 PHD3 bound to the RRM domain of CyP33 exhibited significant signal broadening and spectral overlap, precluding unambiguous structural characterization of the complex.
Figure 5. NMR-Based Mapping of Interacting Surfaces and Solution Structure Defining the Interface between MLL1 PHD3 and CyP33 RRM.

(A) Crystal structure of MLL1 PHD3 (1566–1628) with labeling of secondary structure elements. Residues exhibiting large chemical shift perturbations (Δδave > 0.15 ppm) upon interacting with unlabeled CyP33 RRM are colored in yellow. Δδave = [(Δδ2HN+(ΔδN/5)2)/2]1/2.
(B) Crystal structure of CyP33 RRM (5–82) with labeling of secondary structure elements. Residues exhibiting large chemical shift perturbations (Δδave > 0.15 ppm) upon interacting with unlabeled MLL1 PHD3 are colored in yellow. br>(C) The backbone trace of 20 NMR refined structures of the complex containing the MLL1 PHD3 fragment (1603–1619, blue) and CyP33 RRM (2–82, magenta).
(D) An alternate view of the NMR solution structure of the complex between a fragment of MLL1 PHD3 (1603–1619, blue) and the CyP33 RRM (2–82, magenta).
(E) A ribbon representation of the interaction between MLL1 PHD3 fragment (1603–1619, blue) and CyP33 RRM (2–82, magenta) based on the NMR structure of the complex. Residues involved in the interaction between MLL1 PHD3 and CyP33 RRM are colored in green and yellow, respectively.
(F) Spectral overlap of the selected regions of 1H,13C-HSQC of MLL1 (1566–1639; segment contains entire PHD3 finger, the linker, and a short segment of the αZ helix of Bromo) in the free state (black) and bound to CyP33 RRM (1–82) (red).
(G) Model of the complex between MLL1 PHD3 (1566–1628, blue) and the CyP33 RRM (2–82, magenta) based on the structure in (D) after superimposing of their respective PHD3 α2 helices.
(H) ITC curves for binding of H3(1-15)K4me3/2 peptide to the complex of MLL1 PHD3-Bromo and full-length CyP33.
(I) Complexation shifts in Trp1594 indole resonance (black cross peak) in the 1H,15N-HSQC spectrum of MLL1 PHD3 on sequential addition of H3K4me3 (green cross peak), and CyP33 RRM (red cross peak).
See also Figure S4.
To overcome the above limitations, and guided by the information on interacting elements associated with complex formation (Figures 5A and 5B), we next designed a construct in which the interacting segment of MLL1 PHD3 (1585–1628) was fused to the N terminus of CyP33 RRM (2–82) through a VD(GGS)4 peptide linker, so as to facilitate intramolecular complex formation. The 13C,15N-labeled fusion construct gave an exceptionally well-resolved 1H,15N-HSQC NMR spectrum (Figure S4D), facilitating resonance assignment and structure determination of the complex. The defined segments of the complex are composed of a fragment (1603–1619) of MLL1 PHD3 bound to intact CyP33 RRM (20 superimposed structures shown in Figure 5C; NMR statistics in Table S4). The long α helix α2 (1603–1611) of MLL1 PHD3, along with its immediate downstream segment (1612–1619) traverses along the exposed face of the β sheet of the CyP33 RRM domain (Figures 5C and 5D), with this association being primarily mediated by hydrophobic contacts involving the side chains of Met1606, Ile1609, and Leu1610 along one face of the α helix of PHD3, projecting into the groove formed by Val7, Gln36, Phe51, Glu53, and Ala82 on the β sheet face of RRM (Figure 5E). In addition, hydrophobic contacts between Leu1613, Ala1618, and Tyr1619 residues C-terminal to the α helix of PHD3 and Tyr9, Pro38, and Phe49 of RRM further strengthen the association.
CyP33-Mediated cis-to-trans Transition of Linker Pro1629 in MLL1 PHD3-Bromo Cassette
The structure of the complex (Figure 5D) confirms our deduction that the surface of PHD3 that is buried because of its interaction with Bromo within the PHD3-Bromo cassette is involved in the interaction with CyP33 RRM. To expose this PHD3 surface for targeting by the CyP33 RRM, a dramatic conformational change has to occur, which necessitates repositioning of the PHD3 and Bromo domains within the cassette. We anticipate that such a transition requires Pro1629, located within the linker segment connecting domains, to undergo a PPIase-mediated cis-to-trans conformational transition. To directly monitor this transition, we first characterized the configuration of Pro1629 in the MLL1 PHD3-Bromo cassette by NMR. The sequence specific chemical shift assignments of Pro1629-Ala1630 were achieved using a uniformly 13C,15N-labeled sample, as well as a sample containing selectively 13C-labeled Pro and 15N-labeled Ala. The results revealed that the His1628-Pro1629 peptide bond adopts a cis configuration in the MLL1 PHD3-Bromo cassette in the free state in solution, as reflected in the characteristic cis-Pro Cγ chemical shift of Pro1629 at 25.4 ppm (Figure S4E).
To facilitate monitoring the cis-to-trans transition of Pro1629 following MLL1 PHD3 binding to CyP33 RRM, we resorted to a simpler system. We designed a short construct (residues 1566–1639) containing PHD3, Pro1629-containing linker, and a part of helix αZ that is involved in the interaction with PHD3 (Figure 1E). The His1628-Pro1629 peptide bond predominantly adopts a cis configuration in this construct in the free state, as reflected in its major cross peak exhibiting a Cγ chemical shift of 25.3 ppm (Figure 5F, black cross peak P1629 cis). Upon addition of CyP33 RRM, the Cγ resonance of Pro1629 shifted to 27.5 ppm (Figure 5F, red cross peak labeled P1629 trans), a value characteristic of a trans Pro configuration. This establishes that the His1628-Pro1629 peptide bond in MLL1 PHD3-Bromo transits from cis-to-trans configuration upon interaction with CyP33 RRM.
MLL1 PHD3 Can Simultaneously Bind H3K4me3 and CyP33 RRM
We next aligned the crystal structure of full-length H3K4me3-bound PHD3 (1566–1628) onto the NMR-based structure of CyP33 RRM-bound MLL1 PHD3 fragment (1603–1619; Figure 5D), to yield a model of the H3K4me3-PHD3-RRM ternary complex (Figure 5G). Notably, MLL1 PHD3 uses different surfaces/pockets to bind H3K4me3 and the RRM domain of CyP33. This was further confirmed from ITC-based binding studies, where H3K4me3 and H3K4me2 peptides bound to the complex of MLL1 PHD3-Bromo cassette and full-length CyP33 with KD values of 11.6 μM and 15.2 μM, respectively (Figure 5H). These dissociation constants are increased by a factor of around 2 when compared to binary complex formation in the absence of full-length CyP33 (Figure 2A).
Further, we used the well-resolved Trp1594 indole resonance in the 1H,15N-HSQC spectrum of MLL1 PHD3 to monitor complex formation on sequential addition of H3K4me3 and CyP33 RRM. We observed a large downfield shift of the Trp indole cross-peak on addition of H3K4me3 to form a binary complex, followed by an upfield shift on addition of CyP33 RRM to form a ternary complex (Figure 5I). Similarly, addition of the components in the reverse order resulted in the same shift on ternary complex formation (Figure S4F). Together, these results unequivocally confirm that MLL1 PHD3 uses different surfaces to simultaneously bind H3K4me3 and CyP33 RRM.
Unique Features of MLL1 PHD3 among MLL Family Members
Four members of the MLL SET family of histone methyl transferases, designated MLL1 to MLL4, contain one or more clusters of PHD fingers (Ruthenburg et al., 2007a). We compared the sequence of MLL1 PHD3 with other potential H3K4me-binding PHD fingers from MLL SET family members. Sequence alignments show that the PHD3 domains of MLL1 and MLL2 (sometimes called MLL4 in the literature) exhibit the highest similarity (70% identity) (Figure S3I). Unexpectedly, our gel filtration data demonstrate that MLL2 PHD3 does not bind CyP33 RRM (Figure 4, line 12, and Figure S3J). The loss of binding can be attributed to replacement of CyP33 RRM-interacting residue Met1606 on helix α2 of MLL1 PHD3 by corresponding residue Asp1375 in MLL2 PHD3 (Figure S3I, residues labeled with green background). In support of this inference, introduction of a M1606D mutation into MLL1 PHD3 completely disrupted its complex formation with CyP33 RRM (Figure 4, line 10, and Figure S3K), thereby verifying the importance of hydrophobic interactions at the interface of the PHD3-RRM complex (Figure 5E). Conversely, introduction of a D1375M mutation in MLL2 PHD3 converted MLL2 PHD3 to a CyP33 RRM binding partner (Figure 4, line 13, and Figure S3L). Similarly, the corresponding residues in other potential H3K4me-binding PHD fingers within MLL family members are either Asp or Glu (Figure S3I), which indicates that regulation by CyP33 is specific for MLL1 among MLL family members.
CyP33-Mediated Histone Deacetylation and Repression of HOX Genes
To test whether the interaction between MLL1 and CyP33 was necessary for target gene regulation in vivo, we made mutations that disrupt the PPIase domain of CyP33 (F196E/M197E) (Wang et al., 2005), as well as a mutation in the RRM domain (F51D) that should disrupt the MLL1-CyP33 interaction. In vitro, a single mutation F51D in the RRM domain that disrupts the hydrophobic interaction between CyP33 RRM and MLL1 PHD3 was sufficient to abolish the binding between CyP33 and MLL1 PHD3-Bromo (Figure 4, line 11, and Figure S3M).
To test the impact of these mutants on CyP33-regulated HOX gene expression, we transfected plasmids containing FLAG-HA-tagged wild-type CyP33, CyP33 RRM mutant (F51D), or CyP33 PPIase-dead mutant (F196E/M197E) into 293T cells along with an empty vector as the control. All three constructs expressed well in 293T cells, and the expression levels were verified by western blots with an anti-FLAG antibody (Figure 6A). In K562 cells, CyP33 has been shown to be a repressor of the HOXC8 locus (Fair et al., 2001). Similarly, for 293T cells, we found that overexpression of wild-type CyP33 resulted in an approximately 30%–40% downregulation of both HOXA9 and HOXC8 expression (Figure 6B), which is comparable to disruptions in HOX gene expression that were seen with MLL1 complex component knockdowns (Dou et al., 2006). As a comparison, overexpression of the mutants F51D or F196E/M197E did not cause any repression of either HOX gene (Figure 6B).
Figure 6. Mutations in Either RRM or PPIase Domains Affect CyP33-Regulated Gene Expression and Histone Deacetylation.

(A) 293T cells were transiently transfected with an empty vector (1, blue), wild-type full-length FLAG and HA tagged CyP33 (2, red) or full-length CyP33 that has either the F51D (3, green) or F196E/M197E (4, purple) point mutations. A western blot with an anti-FLAG antibody indicates that all three constructs were expressed at high levels.
(B) Real-time PCR quantification of gene expression levels from the transfected cells in (A) shows that HOXA9 and HOXC8 expression were reduced in the presence of wild-type CyP33 (red bars) but not in cells expressing the F51D (green bars)or F196E/M197E (purple bars) point mutations or the empty vector control (blue bars). GAPDH expression levels were unaffected by expression of any constructs. Error bars represent the standard deviation of two independent experiments.
(C and D) ChIP experiments with an anti-H3 pan acetyl (AcH3) antibody (C), and an anti-HA antibody (D) at HOXA9, HOXC8, and at a control region. PCR primer/probe sets are as indicated in Figure 3. ChIP results shown are typical for at least three independent experiments. Error bars represent standard deviation of three separate PCR reactions.
(C) ChIP shows decreased H3 acetylation levels at HOXA9 and HOXC8 in wild-type CyP33-expressing cells (red bars) but not in cells expressing the F51D (green bars) and F196E/M197E (purple bars) point mutations or the empty vector control (blue bars)
(D) Anti-HA ChIP shows that wild-type (red bars) and F196E/M197E (purple bars) versions of CyP33 bound to HOXA9 and HOXC8, while the F51D point mutation exhibited variably reduced CyP33 binding.
See also Figure S5.
Consistent with the gene-expression results, ChIP with an anti-H3 pan acetyl antibody (AcH3) showed that HOXA9 and HOXC8 loci exhibited decreased acetylation levels when wild-type CyP33 but not its mutants were overexpressed (Figure 6C), suggesting a role for histone deacetylation in repression (Xia et al., 2003). In addition, CyP33 has no effect on MLL1 binding at HOXA9 and HOXC8 (Figure S5), suggesting that its mechanism of action does not involve modulating MLL1 binding to target genes.
The CyP33 F51D mutant is only partially disrupted in its ability to bind to HOXA9 and HOXC8 (Figure 6D). This suggests that CyP33 recruitment to target genes in vivo is not dependent on interacting with MLL1. However, since this mutant is unable to deacetylate and repress target genes, this shows that the CyP33 MLL1 interaction is essential for MLL1-mediated repression at these target genes. Thus, CyP33 is recruited to HOX genes by a mechanism independent of MLL1 (perhaps through other protein interactions or by interacting with a specific RNA), but once it is bound to a target gene, its repressor function requires a direct interaction with MLL1. These results establish that the interaction between CyP33 and MLL1 is necessary for CyP33-mediated histone deacetylation and repression of HOXA9 and HOXC8 genes.
DISCUSSION
Cis-trans Proline Isomerization within MLL1 PHD3-Bromo Controls Access to CyP33 RRM
Here, we present a detailed model of a PPIase-mediated switch modulating the interaction between MLL1 PHD3-Bromo and CyP33. PPIases belong to a ubiquitous protein family that catalyzes the peptidyl prolyl cis-trans isomerization reaction (Andreotti, 2003; Fanghänel and Fischer, 2004). PPIases play an important role in protein folding/refolding by catalyzing a rate-limiting step, as well as in protein transport and signaling (Galat and Metcalfe, 1995). Our studies reveal that the independently folded CyP33 RRM and PPIase domains, which are separated by a long flexible linker without any hint of direct interactions, have distinct and separable interaction surfaces on the MLL1 PHD3-Bromo cassette. Based on our observations that CyP33 PPIase is required for the interaction between CyP33 RRM and MLL1 PHD3-Bromo cassette but is not associated with formation of a stable complex (Figure 4, line 5, and Figure S3D), the main function of PPIase in this process is as a chaperone that accelerates the cis-to-trans conformational transition of the His1628-Pro1629 peptide bond within the linker segment. This transition in turn releases the otherwise occluded surface of MLL1 PHD3, in the context of the PHD3-Bromo cassette, for recognition by the RRM domain of CyP33.
By comparison, an earlier functional study established proline isomerization as a novel noncovalent histone modification that regulates transcription by showing that the conformational state of Pro38 on histone 3 is governed by the proline isomerase Fpr4, which in turn controls methylation of H3K36 by Set2 (Nelson et al., 2006). The above complementary examples at the reader level (this study) and the histone peptide level (Nelson et al., 2006) reinforce the concept that proline isomerization could function on both sides of a regulatory switch controlling both the relative topology of a single peptide or individual domains and their downstream biological activities.
MLL1 PHD3-Bromo as a Regulatory Platform Controlling Transcriptional Regulation
Previous biochemical studies, together with the molecular details presented in this paper, enable us to generate a model describing CyP33-mediated regulation of MLL1 (Figure 7A). In the native state, MLL1 (as a complex of MLLN and MLLC) (Hsieh et al., 2003) is targeted by corepressors and coactivators, while exhibiting a net activation role in transcriptional regulation. MLL1 accumulates at H3K4me3-abundant regions of promoters through PHD3-specific targeting, with the binding surface on PHD3 for CyP33 RRM recognition blocked by Bromo because of a cis alignment at Pro1629 (Figure 7A, top segment). CyP33 binding catalyzes a cis-to-trans peptide bond conversion at linker Pro1629, allowing MLL1 to transit to the CyP33 bound state, stabilized by interaction between the RRM domain of CyP33 and its target site on PHD3, while retaining the ability of MLL1 to bind nucleosomes through H3K4me3-based PHD3 recognition. As CyP33-mediated and/or HDAC1-related repression components accumulate at the MLL1 repression sites (Xia et al., 2003), the overall functional balance of MLL1 switches to repression (Figure 7A, bottom segment). AU-rich RNAs may compete for binding to CyP33 RRM (Figure 7A, middle segment) (Mi et al., 1996), thereby releasing CyP33 from the MLL1 complex, resulting in MLL1 cycling back to the native conformation.
Figure 7. Schematic Outlining the Proposed Regulation of MLL1 Function by Cyclophilin-Mediated Proline Isomerization within PHD3-Bromo Cassette.

(A) A scheme showing transitions between two states of the MLL1 complex mediated by CyP33. CyP33 binding to MLL1 increases the recruitment of core-pressors to the repression domain, switching the balance of MLL1 functions from activation to repression of target genes. It should be noted that involvement of the RNA component in the scheme needs validation.
(B) Structure of MLL1 PHD3-Bromo cassette with cis-configured Pro1629 (left) and model of MLL1 PHD3-Bromo-CyP33 RRM complex with trans-configured Pro1629 (right).
In this model, H3K4me3/2 peptide and CyP33 RRM target distinct surfaces on MLL1 PHD3 (Figure 5G) and thus can bind simultaneously to form a ternary complex (Figures 5H and 5I). The ability of a PHD finger to simultaneously bind histone peptides and a range of interacting partners extends our understanding of PHD fingers, which function not only as readers of histone marks, but also as mediators that interact with other regulatory proteins. This concept is reinforced from a related study of the Pygo1 PHD finger, which also uses two distinct surfaces to simultaneously engage H3K4me3/2 peptide and the HD1 domain of BCL9 (Fiedler et al., 2008). This property of simultaneous binding also indicates that regulation of MLL1 by CyP33 may also occur at the chromatin level, especially at H3K4me3/2 enriched loci.
Our results also imply that after the recruitment of CyP33 to the MLL1 PHD3-Bromo cassette, a large conformational rearrangement between PHD3 and Bromo associated with the cis-trans isomerization of the linker proline (Figure 7B) would result in a dramatic reorientation of the remaining MLL1 domains relative to each other, thereby potentially facilitating the recruitment of other repression components.
Further, as the canonical RNA-binding surface of RRM (Cléry et al., 2008) in CyP33 is involved in MLL1 PHD3 recognition (Figure 5C) and CyP33 RRM has been shown to exhibit some preference for AU-rich RNAs (Mi et al., 1996; Wang et al., 2008), one anticipates competition between RNA and MLL1 PHD3 for binding to CyP33 RRM, thereby providing a mechanism for releasing CyP33 from the MLL1 complex.
These results establish that the MLL1 PHD3-Bromo cassette serves as a critical regulatory platform, connecting the downstream regulator CyP33 to the H3K4me3/2 enriched nucleosomal platform. Loss of this regulatory platform could contribute to the leukemogenic potential of MLL1 fusion proteins. Supporting this concept, experiments with MLL1-ENL fusions have shown that loss of the MLL1 PHD3 was necessary for MLL1-ENL-induced immortalization of hematopoietic stem cells, while insertion of MLL1 PHD3 into MLL1 suppressed such transformations (Chen et al., 2008). Similarly, insertion of the PHD3-containing region of MLL1 back into MLL1-AF9 fusions also blocked immortalization of hematopoietic progenitors (Muntean et al., 2008). An improved understanding of the molecular mechanisms regulating interactions between MLL1 and CyP33 could provide therapeutic insights into treatment of MLL1 aberrant-expression related leukemias.
Concluding Remarks
Our structure-function studies have put forward the general concept that the PHD3-Bromo cassette is key for both nucleosomal recruitment and CyP33 targeting of MLL1, adding new insights into the potential function of multimodule reading regions observed in many chromatin remodelers and histone-modifying enzymes (Ruthenburg et al., 2007a). We envision that linked multiple modules, such as the PHD3-Bromo cassette of MLL1 studied here, are used not only to increase the binding affinity and specificity of the writers and/or erasers that contain them by engaging multiple marks in histone and/or DNA components (Ruthenburg et al., 2007b), but also to modulate critical associations with other components in the multisubunit architecture that they reside in. Deletion of this region of MLL1 will not only destabilize its recruitment to target promoter regions, but also impair the CyP33 related repression pathway, disrupting the balance between MLL1-mediated gene activation and repression, thereby resulting in constant activation leading to the onset of leukemogenesis.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Detailed methods for vector construction, GST-tagged or His-Sumo-tagged protein expression and purification were outlined in the Extended Experimental Procedures.
Crystallization and X-Ray Structure Determination
All the crystals were obtained by the hanging-drop method at 20°C. Data sets for crystals of MLL1 PHD3-Bromo cassette in the free form or in the complex form with H3K4me3/2 peptides were collected at APS NE-CAT 24ID-C or 24ID-E beam lines. The free-form MLL1 PHD3-Bromo structure was solved by SAD method with zinc anomalous signals. The subsequent complex structures of MLL1 PHD3-Bromo with H3K4me3/2 peptides were solved by molecular replacement method using the free-form structure as the model. The data set for CyP33 RRM crystals was collected on our in-house X-ray diffractometer. The structure was solved by molecular replacement using the NMR model of the structure in PDB (PDB ID: 2CQB). For detailed method of crystallization and structure calculation, see the Extended Experimental Procedures.
NMR Sample Preparation and Structure Determination
Uniformly 15N,13C- or 15N-labeled proteins were expressed from Rosetta2 (DE3) E. coli cells in M9 minimum medium supplemented with 13C-labeled glucose and/or 15N-labeled NH4Cl. For (U-15N Ala, U-13C Pro)-selective labeling, the medium was supplemented with 15N-labeled alanine and 13C-labeled proline. The proteins were then subject to column purification before NMR data collection. Sequence-specific chemical shift assignments were achieved through a set of multidimensional heteronuclear NMR spectra. In addition, NOE restraints for structural calculation were derived from a 3D 15N-edited NOESY-HSQC, a 3D aliphatic 13C-edited NOESY-HSQC, and a 3D aromatic 13C-edited NOESY-HSQC. For details of structural calculation and experimental procedures, see the Extended Experimental Procedures.
Isothermal Titration Calorimetry Measurements
Calorimetric experiments were conducted at 25°C with a MicroCal iTC200 instrument. MLL1 PHD3-Bromo and its mutants purified by coexpression method, or MLL1 PHD3 alone, were dialyzed overnight against buffer containing 20 mM Tris (pH 7.5), 100 mM NaCl, and 2 mM β-mercaptoethanol. The sample of the MLL1 PHD3-Bromo-CyP33 complex was prepared by dialyzing MLL1 PHD3-Bromo and CyP33 separately in above buffer first, then by mixing together at 1:1.05 ratio for 2 hr on ice. Aliquots of lyophilized peptides were dissolved in the same buffer before use. Calorimetric titration was performed by injection of synthetic peptide into protein samples at various concentrations. Calorimetric titration data were fitted with software Origin 7.0 on the basis of a 1:1 binding stoichiometry.
Peptide Pulldown Assays
All biotin-labeled histone peptides were synthesized by the Peptide Core Facilities of Sloan-Kettering and Rockefeller University. Peptide pulldown experiments were performed as described previously (Wysocka et al., 2006).
293T Transient Transfections
Constructs were transfected into 10 cm dishes of 293T cells with 1–2 μ of DNA and 6 μl FuGene reagent according to the manufacturer’s instructions. Forty-eight hours later, cells were collected and divided into two aliquots: (1) whole-cell extracts were made with 300 mM KCl + 0.05% NP40 and used for western blotting, and (2) RNA was purified with QIAGEN RNeasy kits and complementary DNA (cDNA) was made with Invitrogen SuperScript II. cDNA was then quantified in real-time PCR reactions with an ABI 7300 machine and SYBR green with GAPDH-, HOXC8-, or HOXA9-specific primers.
293 Stable Cell Lines
293 cells were transfected with linearized vectors expressing either fh-MLLCXXC-PHD3 or mutant fh-MLLCXXC-PHD3 (W1594E) under the control of a CMV promoter and with an IRES/neo selection gene. Stably expressing cells were selected for G418 resistance over a 1 month period, and then expression levels and ChIP experiments were done.
Chromatin Immunoprecipitation Experiments
ChIP experiments were performed as described previously (Milne et al., 2009). Antibodies to H3K4me3 are from Active Motif (catalog# 39159), pan acetyl of H3 from Upstate (catalog# 06-599), unmodified H3 from Abcam (catalog# 1791), and HA from Roche (clone 12CA5, catalog# 11583816001). Antibodies to endogenous MLLC were a generous gift of the Robert Roeder laboratory.
Supplementary Material
Table S1. X-ray statistics of MLL1 PHD3-Bromo cassette in the free and H3K4me3/2 bound states. Related to Figures 1 and 2.
Table S2. ITC-based dissociation constants for complex formation between H3K4me-containing peptides and wild-type or PHD3 mutants of MLL1 PHD3-Bromo cassette. Related to Figures 2 and 4.
Table S3. X-ray statistics of CyP33 RRM structure. Related to Figure 4.
Table S4. NMR statistics for the 20 energy-minimized conformers of MLL1 PHD3-CyP33 RRM fusion protein. Related to Figure 4.
Acknowledgments
This research was funded by the Leukemia and Lymphoma Society Program Project grant LLS-SCOR 7132-08 and a Starr Foundation grant, both of them jointly to D.J.P. and C.D.A. D.J.P. is a consultant in GlaxoSmithKline’s epigenetic programs.
Footnotes
ACCESSION NUMBERS
The X-ray coordinates of MLL1 PHD3-Bromo cassette in the free state (accession number 3LQH) and when bound to H3K4me3 (3LQJ) and H3K4me2 (3LQI) peptides have been deposited in the Protein Data Bank (PDB). The X-ray coordinates of CyP33 RRM domain in the free state (3LPY) and the NMR-based coordinates for MLL1 PHD3-CyP33 RRM fusion protein (2KU7) have also been deposited in the PDB. The NMR chemical shift assignments (16725) have been deposited in Biological Magnetic Resonance Bank .
Supplemental Information includes Extended Experimental Procedures, five figures, and four tables and can be found with this article online at doi:10. 1016/j.cell.2010.05.016.
References
- Andreotti AH. Native state proline isomerization: an intrinsic molecular switch. Biochemistry. 2003;42:9515–9524. doi: 10.1021/bi0350710. [DOI] [PubMed] [Google Scholar]
- Ayton PM, Cleary ML. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene. 2001;20:5695–5707. doi: 10.1038/sj.onc.1204639. [DOI] [PubMed] [Google Scholar]
- Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks. Mutat Res. 2008;647:3–12. doi: 10.1016/j.mrfmmm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Santillan DA, Koonce M, Wei W, Luo R, Thirman MJ, Zeleznik-Le NJ, Diaz MO. Loss of MLL PHD finger 3 is necessary for MLL-ENL-induced hematopoietic stem cell immortalization. Cancer Res. 2008;68:6199–6207. doi: 10.1158/0008-5472.CAN-07-6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cléry A, Blatter M, Allain FH. RNA recognition motifs: boring? Not quite. Curr Opin Struct Biol. 2008;18:290–298. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- Ernst P, Mabon M, Davidson AJ, Zon LI, Korsmeyer SJ. An Mll-dependent Hox program drives hematopoietic progenitor expansion. Curr Biol. 2004;14:2063–2069. doi: 10.1016/j.cub.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Fair K, Anderson M, Bulanova E, Mi H, Tropschug M, Diaz MO. Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol Cell Biol. 2001;21:3589–3597. doi: 10.1128/MCB.21.10.3589-3597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanghänel J, Fischer G. Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front Biosci. 2004;9:3453–3478. doi: 10.2741/1494. [DOI] [PubMed] [Google Scholar]
- Fiedler M, Sánchez-Barrena MJ, Nekrasov M, Mieszczanek J, Rybin V, Müller J, Evans P, Bienz M. Decoding of methylated histone H3 tail by the Pygo-BCL9 Wnt signaling complex. Mol Cell. 2008;30:507–518. doi: 10.1016/j.molcel.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galat A, Metcalfe SM. Peptidylproline cis/trans isomerases. Prog Biophys Mol Biol. 1995;63:67–118. doi: 10.1016/0079-6107(94)00009-x. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, Young RA. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci USA. 2005;102:8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess JL. MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med. 2004;10:500–507. doi: 10.1016/j.molmed.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Hsieh JJ, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23:186–194. doi: 10.1128/MCB.23.1.186-194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linggi BE, Brandt SJ, Sun ZW, Hiebert SW. Translating the histone code into leukemia. J Cell Biochem. 2005;96:938–950. doi: 10.1002/jcb.20604. [DOI] [PubMed] [Google Scholar]
- Ma JC, Dougherty DA. The cation-π Interaction. Chem Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- Mi H, Kops O, Zimmermann E, Jäschke A, Tropschug M. A nuclear RNA-binding cyclophilin in human T cells. FEBS Lett. 1996;398:201–205. doi: 10.1016/s0014-5793(96)01248-3. [DOI] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- Milne TA, Dou Y, Martin ME, Brock HW, Roeder RG, Hess JL. MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci USA. 2005;102:14765–14770. doi: 10.1073/pnas.0503630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Zhao K, Hess JL. Chromatin immunoprecipitation (ChIP) for analysis of histone modifications and chromatin-associated proteins. Methods Mol Biol. 2009;538:409–423. doi: 10.1007/978-1-59745-418-6_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Kim J, Wang GG, Stadler SC, Basrur V, Whitcomb S, Wang Z, Ruthenburg A, Elenitoba-Johnson K, Roeder RG, Allis CD. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HoxA9 locus in leukemogenesis. Mol Cell. 2010;38:853–863. doi: 10.1016/j.molcel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–5527. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- Muntean AG, Giannola D, Udager AM, Hess JL. The PHD fingers of MLL block MLL fusion protein-mediated transformation. Blood. 2008;112:4690–4693. doi: 10.1182/blood-2008-01-134056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- Nelson CJ, Santos-Rosa H, Kouzarides T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell. 2006;126:905–916. doi: 10.1016/j.cell.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Owen DJ, Ornaghi P, Yang JC, Lowe N, Evans PR, Ballario P, Neuhaus D, Filetici P, Travers AA. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyl-transferase gcn5p. EMBO J. 2000;19:6141–6149. doi: 10.1093/emboj/19.22.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007a;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007b;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yun CH, Gu SY, Chang WR, Liang DC. 1.88 Å crystal structure of the C domain of hCyP33: a novel domain of peptidyl-prolyl cis-trans isomerase. Biochem Biophys Res Commun. 2005;333:845–849. doi: 10.1016/j.bbrc.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Wang Y, Han R, Zhang W, Yuan Y, Zhang X, Long Y, Mi H. Human CyP33 binds specifically to mRNA and binding stimulates PPIase activity of hCyP33. FEBS Lett. 2008;582:835–839. doi: 10.1016/j.febslet.2008.01.055. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci USA. 2003;100:8342–8347. doi: 10.1073/pnas.1436338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002;513:124–128. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. X-ray statistics of MLL1 PHD3-Bromo cassette in the free and H3K4me3/2 bound states. Related to Figures 1 and 2.
Table S2. ITC-based dissociation constants for complex formation between H3K4me-containing peptides and wild-type or PHD3 mutants of MLL1 PHD3-Bromo cassette. Related to Figures 2 and 4.
Table S3. X-ray statistics of CyP33 RRM structure. Related to Figure 4.
Table S4. NMR statistics for the 20 energy-minimized conformers of MLL1 PHD3-CyP33 RRM fusion protein. Related to Figure 4.