

Abstract
The circadian clock synchronizes behavioral and physiological processes on a daily basis in anticipation of the light–dark cycle. In mammals, molecular clocks are present in both the central pacemaker neurons and in nearly all peripheral tissues. Clock transcription factors in metabolic tissues coordinate metabolic fuel utilization and storage with alternating periods of feeding and fasting corresponding to the rest–activity cycle. In vitro and in vivo biochemical approaches have led to the discovery of mechanisms underlying the interplay between the molecular clock and the metabolic networks. For example, recent studies have demonstrated that the circadian clock controls rhythmic synthesis of the cofactor nicotinamide adenine dinucleotide (NAD+) and activity of NAD+-dependent sirtuin deacetylase enzymes to regulate mitochondrial function across the circadian cycle. In this chapter, we review current state-of-the-art methods to analyze circadian cycles in mitochondrial bioenergetics, glycolysis, and nucleotide metabolism in both cell-based and animal models.
1. INTRODUCTION
All photosensitive organisms possess intrinsic self-sustained circadian clocks that oscillate with a frequency matching the rotation of the Earth on its axis. The universality of 24 h biologic rhythms is evident in the Metazoa, where central endocrine processes involved in metabolism, growth, and reproduction exhibit daily circadian rhythms, giving rise to oscillations in levels of peptidergic and steroidal ligands and their signaling pathways across the day and night cycle. At the organismal level, circadian timing is an emergent property of a complex network of cellular clocks, with the primary signal derived from the master pacemaker neurons of the suprachiasmatic nucleus (SCN). Projections from the SCN terminate in hypothalamic neuroendocrine centers critical in feeding and glucose homeostasis. At the molecular level, the core mechanism of the oscillator in both central and peripheral cells is a transcription–translation feedback loop involving members of the basic helix-loop-helix Period-Arnt-Sim superfamily. The forward limb is encoded by a heterodimeric complex of activators (CLOCK/BMAL1) that induce transcription of their own repressors (PER/CRY) in a cycle that repeats itself every 24 h (reviewed in Lowrey & Takahashi, 2004). A secondary stabilizing loop is embedded within the activator limb and involves the REV-ERB/ ROR proteins that control Bmal1 transcription (Lowrey & Takahashi, 2004).
Genomic and proteomic profiling experiments have demonstrated that the circadian clock governs a wide array of physiological functions through circadian control of approximately 10–20% of the transcriptome and proteome, including numerous metabolic genes and proteins in peripheral tissues (Koike et al., 2012; Panda et al., 2002). However, a major challenge remains in determining the functional impact of circadian oscillators in cell metabolism and organismal physiology. Our recent studies have shown that the clock produces rhythmic cycles of nicotinamide adenine dinucleotide (NAD+), an electron carrier in oxidoreductase reactions and a cofactor for class III deacetylase and poly-ADP ribosylase enzymes important in nutrient and stress response pathways. Clock control of NAD+ arises through the transcriptional regulation of the gene encoding the rate-limiting enzyme in NAD+ biosynthesis, nicotinamide phosphoribosyltransferase (NAMPT) (Imai & Guarente, 2014; Peek, Ramsey, Marcheva, & Bass, 2012; Ramsey et al., 2009). We have also demonstrated that the clock-NAD+ biosynthetic cycle generates daily cycles of mitochondrial respiration through rhythmic modulation of the NAD+-dependent mitochondrial deacetylase SIRT3 (Peek et al., 2013). Together, these findings highlight the multitiered integration of transcriptional, translational, and posttranslational pathways underlying circadian regulation of metabolism.
The tight interconnection between circadian and metabolic processes has direct implications for experimental design in studies of metabolism and bioenergetics in the whole animal and in cells. For instance, consideration of biological time of the organism is required in order to capture significant differences in activity of oscillating transcription factors in liver such as the D-albumin-binding protein (Schibler, 2009). Studies in humans also highlight the need to control for time-of-day in experimental design. For example, humans exhibit greater glucose tolerance, insulin sensitivity, and glucose-stimulated insulin secretion in the morning compared to evening (Boden, Ruiz, Urbain, & Chen, 1996; Carroll & Nestel, 1973; Jarrett & Keen, 1969; la Fleur, Kalsbeek, Wortel, Fekkes, & Buijs, 2001; Lee, Ader, Bray, & Bergman, 1992). Therefore, it is important to consider the following when characterizing metabolic function in animal models: (1) how metabolic systems may vary over the course of the day in relation to rhythmic behaviors such as feeding and sleeping, (2) whether the appropriate time of day has been chosen to test molecular hypotheses in each experiment, and (3) what additional insight can be gained by analyzing multiple time points throughout the day for a given metabolic or bioenergetic output.
Here, we highlight both in vitro and in vivo experimental approaches that integrate circadian and biochemical analyses with a specific focus on the rhythmicity of mitochondrial bioenergetics. Briefly, Section 3.1 outlines in vitro bioenergetics studies in synchronized cells, including assays of fuel selection using the Seahorse Bioanalyzer to measure oxidative and glycolytic pathways, while Section 3.2 outlines in vivo-based methods to analyze fatty acid oxidation and bioenergetics in live animals across the circadian cycle. Such integration of circadian and bioenergetics analyses into the design of both in vitro and in vivo experiments will yield novel insight into how peripheral tissues coordinate fuel utilization and metabolism and will shed light on the role of clock disruption in metabolic disorders.
2. MATERIALS
2.1. Circadian in vitro studies (in synchronized C2C12 myotubes)
C2C12 cells (ATCC# CRL-1772) (Yaffe & Saxel, 1977) infected with Period2-luciferase-expressing lentivirus (Liu et al., 2007, 2008) (see Note 1, Section 4.1)
C2C12 myoblast culture medium: Dulbecco’s Modified Eagle Medium (DMEM) with 4.5 g/l glucose, 10% FBS, 4 mM glutamine, 1 mM sodium pyruvate, and 100 U/ml penicillin/streptomycin
C2C12 myotube differentiation medium: DMEM with 4.5 g/l glucose, 2% horse serum, 4 mM glutamine, 1 mM sodium pyruvate, and 100 U/ml penicillin/streptomycin (Yaffe & Saxel, 1977)
C2C12 myotube synchronization medium: DMEM with 4.5 g/l glucose, 50% horse serum, 4 mM glutamine, 1 mM sodium pyruvate, and 100 U/ml penicillin/streptomycin (Zhang et al., 2012)
Cell scrapers
10-cm culture dishes and 96-well Seahorse cell culture plates
1×MAS buffer: 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1.0 mM EGTA, and 0.2% (w/v) fatty acid-free BSA, pH 7.2, at 37 °C
Seahorse medium: XF Base medium: Dulbecco’s Modified Eagle Medium (DMEM) with 4.5 g/l glucose, 4 mM glutamine (adjust pH to 7.4 at 37 °C)
Saponin
Mitochondrial substrates: e.g., acyl-carnitines (short-, medium-, or long-chain), malate, pyruvate, glutamate, and succinate
Mitochondrial inhibitors: antimycin A (complex III), rotenone (complex I), oligomycin (complex V), FCCP (uncoupler)
10% perchloric acid
3 M K2CO3
HPLC mobile phase: Buffer A (50 mM K2HPO4/50 mM KH2PO4) and Buffer B (methanol)
HPLC machine (e.g., Shimadzu LC-20AT), C18 HPLC column (e.g., Supelcosil LC-18-T HPLC column), HPLC guard column
2.2. Circadian in vivo studies (48 h constant darkness experiments in mice)
C57BL/6J mice, aged 12 weeks (Jackson Laboratories)
Light-tight circadian cabinets with computer-controlled light–dark cycles
Red light, red light-equipped room, and/or infrared night vision goggles
Standard housing mouse cages
Dissection equipment (scissors, forceps, etc.)
Eppendorf tubes for tissue collection
Liquid nitrogen
SETH buffer: 250 mM sucrose, 1 mM EDTA, 10 mM Tris-Cl (pH 7.4)
Dounce homogenizer
24-well plates
FAO reaction mixture: 100 mM sucrose, 10 mM Tris-Cl (pH 7.4), 5 mM K2PO4, 80 mM KCl, 1 mM MgCl2, 2 mM L-carnitine, 0.1 mM malate, 2 mM ATP, 0.05 mM coenzyme A, 1 mM dithiothreitol (DTT), 0.2 mM EDTA, 0.3% bovine serum albumin (BSA), 0.5% fatty acid-free BSA, 1 μCi (0.125 mM) [1-14C] oleic acid-BSA
35% perchloric acid
100% phenylethylamine
Whatman filter paper
Scintillation vials, scintillation fluid, scintillation counter
3. METHODS
3.1. Circadian in vitro cell-based methods (in synchronized C2C12 myotubes)
3.1.1 Advantages of cell-based models to test circadian control of metabolism
Studies of the circadian control of metabolism have been aided by the discovery that in the presence of entraining stimuli, cultured cells exhibit synchronized ~24-h rhythms of circadian clock gene expression (Balsalobre, Damiola, & Schibler, 1998). Remarkably, oscillation of the core transcriptional clock gives rise to rhythmic 24 h oscillations in downstream metabolic pathways even in cells maintained in constant nutrient conditions. Several methods of cell synchronization have been developed, including high serum treatment, heat shock, temperature cycling, and treatment with pharmacological modulators of cell-signaling pathways, including the cyclic AMP pathway (forskolin, butyryl cAMP), the glucocorticoid pathway (dexamethasone), and G protein-coupled receptor pathway (endothelin) (Balsalobre, Brown, et al., 2000; Balsalobre, Marcacci, & Schibler, 2000; Buhr, Yoo, & Takahashi, 2010; Saini, Morf, Stratmann, Gos, & Schibler, 2012). An advantage of cell-based studies of circadian metabolism is that such approaches are reductionist in that they eliminate the many complex upstream factors that impact metabolism, such as feeding and hormonal rhythms. Nonetheless, the ultimate goal of physiologic genomic studies of the circadian clock is to gain insight into cellular dynamics at the tissue and organismal level (described in Section 3.2). Ultimately, the integration of cell-based findings with studies in the intact animal provides a “holistic” temporal portrait of physiology and metabolism. In this regard, the following sections will provide detailed methodological parameters for circadian analysis of mitochondrial function, with a focus on applying such approaches to dissect the role of NAD+ metabolism and sirtuin deacetylase activity in rhythmic oxidative and glycolytic metabolism. A special feature of our approach has been to develop techniques to analyze immortalized skeletal muscle cells, and we provide details on our experience with this system as a powerful platform for cell-based studies of circadian physiology. Below, we provide a brief overview of the role of the clock-NAD+-sirtuin pathway in the circadian control of mitochondrial bioenergetics (Section 3.1.2) and describe methods to analyze mitochondrial metabolism and circadian function in the immortalized mouse muscle-derived cell type C2C12 (Section 3.1.3).
3.1.2 Circadian control of mitochondrial bioenergetics
Numerous studies have demonstrated that intact function of the autonomous circadian clock in peripheral tissues such as liver, muscle, and adipose is necessary for normal metabolic physiology. Yet a gap remains in our understanding of the link between the core clock transcription loop and the downstream cellular metabolism. As noted above, recent studies have elucidated one node coupling the clock to energetics with the discovery that the circadian program drives rhythmic synthesis of the metabolic cofactor NAD+ through transcriptional control of the rate-limiting enzyme in the NAD+ salvage pathway, NAMPT. Circadian control of NAD+ levels in liver has been shown to control rhythmic oscillations in the activity of NAD+-dependent sirtuin deacetylase enzymes (SIRTs), which are critical regulators of metabolic fuel switching in response to altered fuel availability. SIRT1 is responsible for inducing mitochondrial oxidative capacity via deacetylation and activation of transcription pathways for mitochondrial biogenesis either directly (e.g., histone modification) or indirectly through deacetylation of metabolic regulators (e.g., PCG1alpha, NRF1–2) (Rodgers, Lerin, Gerhart-Hines, & Puigserver, 2008; Rodgers & Puigserver, 2007), while the mitochondrial-localized sirtuins SIRT3–5 directly regulate oxidative enzymes in mitochondria during nutrient flux (He, Newman, Wang, Ho, & Verdin, 2012; Pirinen, Lo Sasso, & Auwerx, 2012). Thus, circadian control of sirtuin activity represents a central node in the circadian control of metabolic function throughout the 24-h circadian cycle.
3.1.3 In vitro bioenergetics measurements in synchronized cells
Here, we outline experimental approaches to study oxidative cycles in vitro using mouse muscle-derived C2C12 cells. The first step in these experiments is to differentiate the replication-competent C2C12 myoblast monolayer into myotubes (a more physiological state), thereby gaining access to a cell type that recapitulates features of intact muscle, including high rates of metabolic fuel flux and mitochondrial respiration (Yaffe & Saxel, 1977) (see Note 1, Section 4.1). Importantly, C2C12 myotubes display robust circadian oscillations lasting approximately 1 week after synchronization with high serum (Fig. 1). Below, we describe how C2C12 myotubes are serially synchronized over two circadian periods (every 4 h for 44 h) and collected simultaneously to assay for circadian rhythms of cellular NAD+, ATP, and metabolic fuel utilization (Fig. 2; Table 1). The techniques described below include HPLC analyses of the nucleotide metabolites NAD+, ATP, ADP, and AMP. We also describe methods to monitor mitochondrial oxygen consumption in response to various metabolic fuels, including fatty acids (acyl-carnitines), amino acids (glutamate), and glucose (pyruvate), using a Seahorse Biosciences XF96 Bioanalyzer, which directly measures the oxygen consumption rate (OCR) in small populations of cells following injection of fuel substrates, as well as pharmacological mitochondrial respiratory modulators which enable dissection of respiratory function in detail (see Note 2, Section 4.1) (Fig. 3). Below, we describe two methods to analyze circadian oscillations in cellular bioenergetics: (1) OCR measurements in permeabilized cells to analyze mitochondrial fuel utilization and respiratory chain function (Step 7), and (2) simultaneous measurement of respiratory (OCR) and nonrespiratory (extracellular medium acidification rate (ECAR) fuel utilization, which reflects lactate production in response to anaerobic utilization of glucose) in intact cells (Step 8).
Figure 1.
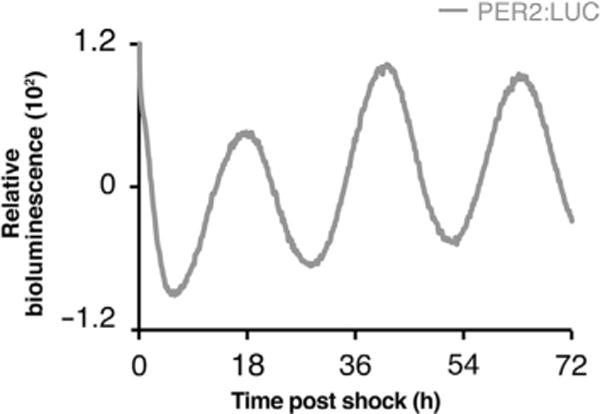
Robust in vitro circadian oscillations in synchronized C2C12 cells. Real-time bioluminescence recordings from PER2:LUC-expressing C2C12 myotubes. Bioluminescence was monitored for 3 days in a Lumicycler apparatus following synchronization of the cells with 50% horse serum.
Figure 2.
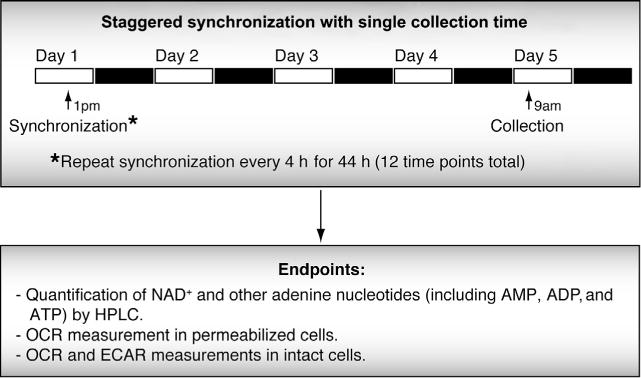
Serial synchronization for 48 h with single collection time point for C2C12 cells. A staggered synchronization every 4 h for a total of 48 h enables a single collection time point, which is necessary for assays that require simultaneous comparison of samples in live cells.
Table 1.
Example experimental setup for synchronization and 48 h collection in cells
| Time point | Synchronization time | Collection time | ||
|---|---|---|---|---|
| 44 | Day 1 | 1 pm | Day 5 | 9 am |
| 40 | 5 pm | |||
| 36 | 9 pm | |||
|
|
||||
| 32 | Day 2 | 1 am | ||
| 28 | 5 am | |||
| 24 | 9 am | |||
| 20 | 1 pm | |||
| 16 | 5 pm | |||
| 12 | 9 pm | |||
|
|
||||
| 8 | Day 3 | 1 am | ||
| 4 | 5 am | |||
| 0 | 9 am | |||
Figure 3.
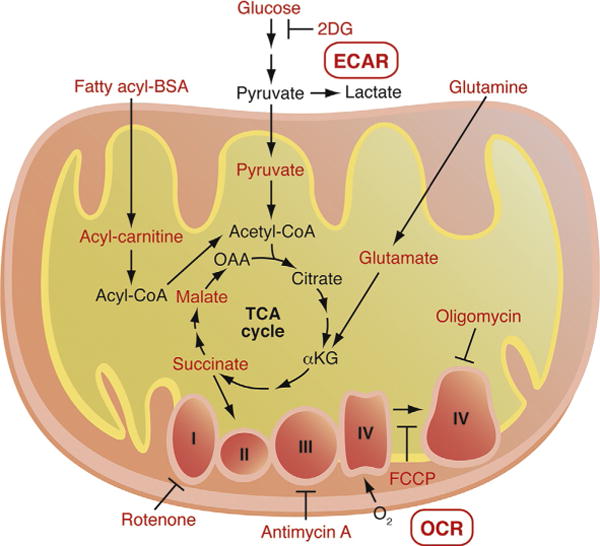
Model of fuel substrates and pharmacological mitochondrial respiratory modulators used in the in vitro bioenergetics measurements. Using a Seahorse Biosciences XF96 Bioanalyzer, small volumes of intact and permeabilized cells can be analyzed for both mitochondrial respiration (i.e., oxygen consumption rate—OCR) and lactate production via anaerobic glycolysis (i.e., extracellular medium acidification rate—ECAR). To compare utilization of fuel substrates, several oxidative substrates can be added to permeabilized cells, including acyl-carnitines to measure FAO, pyruvate/malate to measure flux from glucose into the mitochondria, glutamate/malate to measure flux from glutamine, and succinate plus rotenone (a complex I inhibitor) to measure respiratory chain from complex II. Respiratory function can also be analyzed in detail using pharmacological modulators of the electron transport chain, including oligomycin (inhibits ATP synthase and measures proton leak), FCCP (dissipates inner membrane proton gradient and measures maximal respiratory capacity), and antimycin A (inhibits complex III and measures nonrespiratory OCR).
Passage and maintain C2C12 myoblast cultures in 10-cm dishes in C2C12 myoblast medium (described above) according to ATCC instructions. For these studies, it may be advantageous to utilize C2C12 cells that have been stably infected with virus expressing the Period2-luciferase reporter gene (Liu et al., 2007, 2008), as parallel monitoring of PER2 reporter activity during the experiment will enable confirmation of proper synchronization of cells (see Note 3, Section 4.1).
Day 1: Plate C2C12myoblasts at a highcell density(~90%)on 12×10-cm dishes for HPLC and protein analysis and on the inner 60 wells of a Seahorse 96-well cell culture plate (i.e., rows B–G; columns 2–11) for bioenergetics measurements (see Note 4, Section 4.1).
Days 2–5: Replace culture medium with C2C12 myotube differentiation medium. Change the medium every 1–2 days and monitor cultures for myotube formation (Yaffe & Saxel, 1977). Myotube formation will happen in approximately 4–6 days in differentiation medium, and the presence of multinucleate fibers can be visualized using an inverted light microscope.
Days 6–8: Replace medium in all dishes with C2C12 differentiation medium containing 0.5% horse serum (instead of normal 2% horse serum). Synchronize cells according to Table 1 by replacing the medium with C2C12 synchronization medium (which contains 50% horse serum) and incubating for 2 h. Wash cells 1×with sterile PBS and replace the medium with C2C12 differentiation medium containing 0.5% horse serum (instead of normal 2% horse serum) (see Note 5, Section 4.1).
Day 9: Collect all cells at one time point (Table 1) and perform metabolic analyses as described in Steps 6–8 below.
For HPLC and protein collections: Wash 10-cm dishes 1×in sterile PBS, aspirate, and add 1 ml of 1×PBS. Scrape cells from dish and collect in 1.5-ml Eppendorf tubes. Centrifuge cells for 1 min at 200×g and aspirate supernatant. Resuspend cells in 1 ml of 1×PBS and aliquot 300 μl (for protein) and 700 μl (for HPLC) of cell suspensions into fresh 1.5-ml tubes. Pellet cells for 1 min at 200×g and freeze cell pellets at −80 ° C. Nucleotide measurements by HPLC can be performed at a later time. Protein quantifications are used to normalize HPLC peak areas to cell number. Method for nucleotide extraction and HPLC analysis are described in references Peek et al. (2013) and Klaidman, Leung, and Adams (1995).
For OCR measurement in permeabilized myotubes: Here, cells are permeabilized using detergent (saponin) to preserve mitochondrial inner membranes and respiratory function while permitting entry of most mitochondrial fuel substrates (Jamur & Oliver, 2010). Prepare all solutions of desired mitochondrial fuel substrates within 1–2 h of assay and adjust to pH 7.0. For measurements of fatty acid oxidation, use 80 μM acyl-carnitine plus 0.5 mM malate. For measurements of oxygen consumption after pyruvate entry into the TCA cycle, use 10 mM pyruvate plus 10 mM malate. For measurements of oxygen consumption after amino acid entry into the TCA cycle, use 10 mM glutamate plus 10 mM malate. Finally, to measure oxygen consumption directly starting at complex II, use 10 mM succinate plus 1 μM rotenone (a complex I inhibitor). See Fig. 3 for overview of biochemical pathways. 10–15 min prior to the start of the assay, replace wells with 175 μl of 1× MAS buffer containing fuel substrates and store in CO2-free 37 °C incubator. Prepare injection ports on XF96 assay cartridges with appropriate concentrations of injection compounds (outlined in Table 2). Place plate in Seahorse XF96 Bioanalyzer and run program according to Table 2.
For OCR/ECAR measurements in intact myotubes: To assess glycolytic metabolism (ECAR), oxidative metabolism (OCR), and the interplay between the two (OCR/ECAR ratio), cell membranes must remain intact, and thus saponin treatment is not used. Wash cells with 1×PBS and replace media with 200-μl XF Base media. Store in a 37 ° C incubator without CO2 for 2 h to remove any residual ions that can alter ECAR. Prepare desired fuels and inhibitors (as follows) within 1–2 h of assay. It is critical to adjust all solutions to pH 7.0. Glucose fuel (5 mM) can be used to assess conditions that primarily alter glycolytic efficiency, while glutamine (10 mM) with either galactose (5 mM) or fatty acyl-BSA (100 μM) (vs. unconjugated BSA control) can be used to assess oxidative metabolic fuel utilization. Immediately prior to loading the Seahorse plate into the machine, replace media with 175 μl of prewarmed XF Base media. Prepare injection ports on XF96 assay cartridges with appropriate concentrations of injection compounds (outlined in Table 2). Place plate in Seahorse XF96 Bioanalyzer and run program according to Table 2.
Table 2.
Seahorse Bioanalyzer program
| Steps | Whole cells | Permeabilized cells | ||
|---|---|---|---|---|
| 1 | Mix 3 min | Loop (2X) | START
|
|
| Mix 1 min
| ||||
| Wait 3 min | ||||
|
| ||||
| 2 | Loop (3X) | START
|
Loop (3X) | START
|
| Mix 1 min
|
Mix 1 min
|
|||
| Measure 3 min
|
Measure 3 min
|
|||
| Mix 1 min | Mix 30 s | |||
|
| ||||
| 3 | Loop END | Loop END | ||
|
| ||||
| 4 | Inject Port A: Substrate | Inject Port A: 100 μg/ml saponin | ||
|
| ||||
| 5 | Loop (3X) | START
|
Loop (5X) | START
|
| Mix 1 min
|
Mix 1 min
|
|||
| Measure 3 min
|
Measure 3 min
|
|||
| Mix 1 min | Mix 1 min | |||
|
| ||||
| 6 | Loop END | Loop END | ||
|
| ||||
| 7 | Inject Port B: 1 μM oligomycin | Inject Port B: Substrate | ||
|
| ||||
| 8 | Loop (3X) | START
|
Loop (2X) | START
|
| Mix 1 min
|
Mix 1 min
|
|||
| Measure 3 min
|
Measure 3 min
|
|||
| Mix 1 min | Mix 1 min | |||
|
| ||||
| 9 | Loop END | Loop END | ||
|
| ||||
| 10 | Inject Port C: 1 μM FCCP | Inject Port C: 1 μM FCCP | ||
|
| ||||
| 11 | Loop (3X) | START
|
Loop (2X) | START
|
| Mix 1 min
|
Mix 1 min
|
|||
| Measure 3 min
|
Measure 3 min
|
|||
| Mix 1 min | Mix 1 min | |||
|
| ||||
| 12 | Loop END | Loop END | ||
|
| ||||
| 13 | Inject Port D: 5 μM antimycin A | Inject Port D: 5 μM antimycin A | ||
|
| ||||
| 14 | Loop (3X) | START
|
Loop (2X) | START
|
| Mix 1 min
|
Mix 1 min
|
|||
| Measure 3 min
|
Measure 3 min
|
|||
| Mix 1 min | Mix 1 min | |||
|
| ||||
| 15 | Loop END | Loop END | ||
3.2. In vivo bioenergetics measurements—48 h DD mouse collection
In addition to in vitro cell-based studies, in vivo circadian measurements are critical to demonstrate that circadian processes occur in the physiological setting. A circadian process is defined by the presence of temperature-stable rhythms with a period length of approximately 24 h that persists in the absence of extrinsic temporal cues (e.g., light). Therefore, to determine whether metabolic processes are under circadian control in vivo, animals are sacrificed at regular intervals over the course of multiple circadian time periods while maintained in constant darkness. However, when measuring circadian rhythms of tissue-specific metabolic outputs such as glucose regulation and mitochondrial metabolism, it is important to equalize the nutrient state of the tissue around-the-clock. To do so, animals are analyzed under constant nutritional conditions by fasting all animals prior to tissue collection, although admittedly such an approach still does not control for the nutritional prehistory of the organism prior to the initiation of the fast (see Note 1, Section 4.2). By studying animals subjected to serial fasting, our laboratory has established that the clock drives self-sustained 24-h cycles of NAD+ turnover, sirtuin lysine deacetylase activity, and respiration. Importantly, in combination with cell-based studies, we demonstrate that the clock-NAD+-oxidative cycle is self-sustained independently of nutrient or feeding (Peek et al., 2013). Here, we outline an experimental approach to monitor mitochondrial oxidative metabolism in mouse liver tissue. Specifically, 18-h fasted mice are sacrificed every 4 h over the course of 44 h (i.e., two full circadian periods), and liver tissue is analyzed for NAD+ synthesis and the oxidation of radiolabeled fatty acid in CO2 (Bennett, 2007; Saudubray et al., 1982; Yoshino & Imai, 2013) (Fig. 4).
Figure 4.
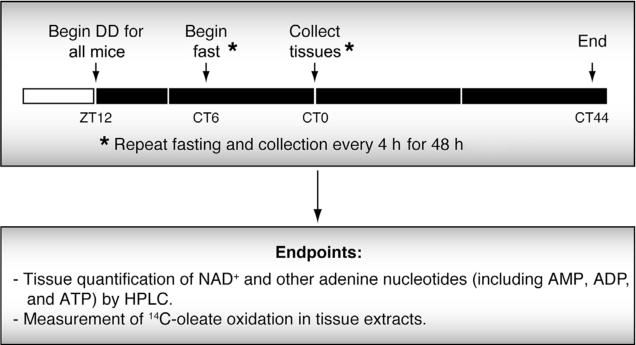
Experimental timeline for in vivo 48 h tissue collection in constant darkness. Mice are released into DD at ZT12 in order to coordinate with the start of the normal dark period. Food is removed from the first set of mice at CT6, which is 18 h prior to the first tissue collection (indicated by CT0). The fasting and tissue collection is then repeated every 4 h for 44 h in order to get a full set of samples to be analyzed for rhythms of the process of interest.
Place 48 wild-type C57Bl/6J mice in light-sealed boxes set to a 12 h:12 h light–dark cycle. It is important to keep mouse age and sex consistent between replicate animals (i.e., all male 8–10 week old mice). Place four mice per cage in 12 cages (12 collection groups—every 4 h for 48 h) (see Note 2, Section 4.2). Mice should be maintained in boxes for 1–2 weeks prior the beginning of the experiment to allow for acclimation to new environment. Change bedding, food, and water as needed without disrupting light cycle.
Day 1: Change light conditions to constant darkness, starting at ZT12 (ZT=Zeitgeber Time).
Day 2: All mice will be fasted for 18 h prior to tissue collection in order to uncouple the effects of rhythmic feeding behavior from the tissue autonomous circadian control of the metabolic processes being measured (i.e., oxygen consumption, ATP, NAD+) (Peek et al., 2013) (see Note 1, Section 4.2). Remove food from first collection group (CT0, CT=Circadian Time) at CT6 (18 h prior to collection at CT0) and begin serial fasting of mouse groups every 4 h (according to Table 3), taking care not to expose mice to light.
Days 3–5: Sacrifice mice every 4 h for 44 h (i.e., two full circadian periods, CT0–CT44) by CO2 inhalation in complete darkness using infrared goggles or under very dim red light. For HPLC and FAO analysis of metabolic tissues, lights can be turned on after sacrifice.
Tissue collection for HPLC: Rapidly excise ~10–20 mg of tissue (e.g., liver, skeletal muscle, heart, and adipose) and freeze in liquid nitrogen. It is important to freeze quickly and not weigh the tissue prior to freezing due to the rapid loss of nucleotides (weights can be taken later on the frozen tissue). Store tissue samples at −80 °C and perform nucleotide extraction and HPLC analysis at a later date using published methods (Yoshino & Imai, 2013).
Tissue collection for fatty acid oxidation (FAO): Prior to tissue collection, prepare a large batch of FAO reaction mixture containing [1-14C]oleic acid [enough for 60 reactions—((four mice per time point+one control reaction)×12 time points=60)]. Rapidly excise ~100–200 mg liver tissue (see Note 3, Section 4.2) and place in 1.5-ml Eppendorf tube containing 1-ml SETH buffer on ice. Weigh tissue and homogenize in 2 μl SETH per milligram tissue using a 2-ml dounce homogenizer. Mix 40-μl lysate with 160-μl reaction mixture and incubate for 1 h at 37 °C (set up duplicate reactions). Also prepare control reactions containing only SETH buffer and no lysate (to control for auto-oxidation of the radiolabeled fat). To capture 14C-CO2 produced from FAO, place reaction mixture in well of a 24-well plate. Under fume hood, add 100 μl of 35% perchloric acid and quickly cover well with Whatman filter paper soaked in 100% phenylethylamine. Incubate plates with slight agitation at room temperature for 1 h. To measure released radiolabeled CO2, place filter paper in scintillation fluid and measure counts per minute (CPM) using a scintillation counter (see Note 4, Section 4.2). Final CPM value is obtained by subtracting average CPM value of control wells from the average CPM value of test wells.
Table 3.
Example experimental setup for 48 h tissue collection in constant darkness
| Time point | Remove food | Tissue collection | ||
|---|---|---|---|---|
| CT0 | Day 2 | 12 pm | Day 3 | 6 am |
| CT4 | 4 pm | 10 am | ||
| CT8 | 8 pm | 2 pm | ||
|
|
||||
| CT12 | Day 3 | 12 am | 6 pm | |
| CT16 | 4 am | 10 pm | ||
|
|
||||
| CT20 | 8 am | Day 4 | 2 am | |
| CT24 | 12 pm | 6 am | ||
| CT28 | 4 pm | 10 am | ||
| CT32 | 8 pm | 2 pm | ||
|
|
||||
| CT36 | Day 4 | 12 am | 6 pm | |
| CT40 | 4 am | 10 pm | ||
|
|
||||
| CT44 | 8 am | Day 5 | 2 am | |
4. NOTES
4.1. Circadian in vitro cell-based methods
C2C12 cells are used in this experiment because they display both robust circadian rhythmicity after synchronization as well as high levels of oxidative phosphorylation. Similar experiments can be performed using other cell types. For example, mouse embryonic fibroblasts and the human osteosarcoma cell line U2OS have been used frequently in circadian studies (Hughes et al., 2009; Zhang et al., 2009); however, as is the case for many immortalized cells, these latter cell types are predominately glycolytic. For each cell type, synchronization methods and cell density must be optimized.
Pharmacological modulators of mitochondrial respiration can be useful for determining specific points of mitochondrial dysfunction. A common set of pharmacological drugs include (1) Oligomycin: inhibits the ATP synthase complex (complex V) and allows for the measurement of uncoupled oxygen consumption (i.e., electron flux that does not require proton gradient dissipation by ATP synthase). (2) Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP): dissipates inner membrane proton gradient and allows for maximal flux through the electron transport chain, enabling measurement of maximal (or spare) respiratory capacity. (3) Antimycin A: inhibits complex III and blocks respiratory oxygen consumption; thus any residual oxygen consumption is due to other oxygen-consuming processes (e.g., fatty acid synthesis). (4) Rotenone: inhibits complex I. When used in combination with succinate, OCRs reflect respiration from complex II specifically.
To ensure proper synchronization of cells to be collected for metabolic analysis, it is recommended to monitor expression of circadian gene expression. This can be done most easily using a well-developed reporter construct expressing the gene promoter for the circadian repressor PERIOD2 fused to the firefly luciferase gene reporter (Per2-luc) (Liu et al., 2007, 2008). To monitor PER2-LUC activity, C2C12 myoblast cells infected with PER2:LUC can be plated in 3.5-cm dishes in parallel to the main experiment and differentiated and synchronized as described in Section 3.1, Steps 2–4. After synchronization, cells are placed in C2C12 myotube differentiation medium containing 0.5% horse serum, 10 mM HEPES buffer, 0.035% (w/v) sodium bicarbonate, and 0.1 mM luciferin (light-sensitive—add immediately before use). Plates are placed in a 37 °C incubator (CO2-free) with a LumiCycle apparatus (Actimetrics) for continuous monitoring of luciferase activity. Data from these plates will allow for determination of circadian period length and amplitude, as well as the best length of time after synchronization to begin assaying for metabolic outputs (e.g., when oscillation appears “stable” with approximately 24-h period length).
Studies performed in the Bass laboratory using the XF96 Seahorse Bioanalyzer have determined that the most consistent data are obtained from the inner 60 wells of the plate. This is particularly apparent when performing studies using whole cells, as we suspect culture conditions to be different in these wells to the rest, potentially due to increased rates of medium evaporation. Other studies not outlined in this chapter, including studies using isolated tissue mitochondria, should be unaffected by the outer wells since they are plated immediately prior to the start of the assay.
Our laboratory and others have observed increased amplitude and persistence of C2C12 myotube circadian oscillations when synchronized cells are maintained in low serum conditions (e.g., 0.5% serum) (Peek et al., 2013; Zhang et al., 2012). It is important to make sure all culture dishes are incubated in lowered serum (0.5%) for the same amount of time as metabolic outputs may be affected. For this reason, all dishes are switched to 0.5% medium at the same time (Section 3.1, Step 4). Serum conditions following synchronization should be optimized for every cell type.
4.2. In vivo bioenergetics measurements—48 h DD mouse collection
Mice eat primarily during the dark period and fast during the light period; thus, it can be difficult to uncouple effects of centrally derived feeding rhythms from endogenous peripheral clock-driven rhythms. To circumvent this problem, mice can be fasted prior to collection, so that feeding does not affect metabolic outputs being measured. However, a significant caveat to this is that some mice will be deprived of nutrients longer than others due to the fact that they will be fasted during their normal rest or fasting phase (e.g., during the light period). To address this issue, it is important to first perform a time course of fasting in mice and assess the dynamics of the metabolic output over time. Fasting length should be chosen in which the output has “plateaued.” For example, fatty acid oxidation peaks after 14 h of fasting and remains at similar levels with longer fasting durations. Thus, 18 h of fasting was chosen to monitor endogenous rhythms of hepatic fatty acid oxidation (Peek et al., 2013). Another complementary approach to address whether a metabolic process is driven by peripheral clocks is the use of tissue-specific genetic mutant mice.
Sampling every 4 h allows for detection of six different phases of the circadian cycle. Sampling less frequently than 4 h results in a dramatic decrease in the ability to detect 24 h rhythms (Hughes et al., 2009). Due to the tedious nature of a continuous 48 h experiment, it is also possible to break the experiment into two separate 24 h experiments (CT0–CT20 and CT24–CT44), in which the second set of mice (CT24–CT44) should be placed into constant darkness 60 h prior to the first collection (CT24) to achieve equivalent free-running conditions.
The hepatic FAO assay is presented here as an example of a radiolabeled substrate-based experiment that can be performed using the 48-h DD in vivo circadian time course method. Other tissues in addition to liver, including skeletal muscle, heart, and white and brown adipose, are highly oxidative and can be also used to measure fuel substrate oxidation (Kim, Hickner, Cortright, Dohm, & Houmard, 2000). In addition, similar methods have been developed to measure oxidation of a wide array of radiolabeled oxidative fuel substrates including 14C-glucose,14C-pyruvate, and 14C-glutamate (Note: Attention should be paid to the particular carbon that is radiolabeled and which oxidation step is being measured).
In addition to measurement of complete 14C-oleate oxidation (i.e., 14C-CO2 production), measurements can also be made of incomplete oxidation products that remain in solution after addition of perchloric acid to the reaction mixture (i.e., acid-soluble metabolites, ASMs). These products include partially oxidized fatty acyl molecules of chain lengths smaller than C6–C8 and organic acids (Hirschey & Verdin, 2010; Koves et al., 2005). Measurement of ASMs can be useful if partial oxidation of substrate is suspected. To measure ASMs, remaining liquid in reaction wells is transferred to 1.5-ml Eppendorf tubes and centrifuged for 10 min at 10,000×g. Supernatants are transferred to scintillation vials containing scintillation fluid and CPM is measured using a scintillation counter.
Acknowledgments
We thank members of the Bass laboratory for helpful discussions. This work was supported by NIH grants R01DK090625 and P01AG011412, the Juvenile Diabetes Research Foundation, the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust, and the University of Chicago Diabetes Research and Training Center DK-20595 (J. B.), T32HL07909 (D. L. and M. P.).
References
- Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, et al. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science. 2000;289:2344–2347. doi: 10.1126/science.289.5488.2344. [DOI] [PubMed] [Google Scholar]
- Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–937. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- Balsalobre A, Marcacci L, Schibler U. Multiple signaling pathways elicit circadian gene expression in cultured Rat-1 fibroblasts. Current Biology. 2000;10:1291–1294. doi: 10.1016/s0960-9822(00)00758-2. [DOI] [PubMed] [Google Scholar]
- Bennett MJ. Assays of fatty acid beta-oxidation activity. Methods in Cell Biology. 2007;80:179–197. doi: 10.1016/S0091-679X(06)80009-9. [DOI] [PubMed] [Google Scholar]
- Boden G, Ruiz J, Urbain JL, Chen X. Evidence for a circadian rhythm of insulin secretion. The American Journal of Physiology. 1996;271:E246–E252. doi: 10.1152/ajpendo.1996.271.2.E246. [DOI] [PubMed] [Google Scholar]
- Buhr ED, Yoo SH, Takahashi JS. Temperature as a universal resetting cue for mammalian circadian oscillators. Science. 2010;330:379–385. doi: 10.1126/science.1195262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KF, Nestel PJ. Diurnal variation in glucose tolerance and in insulin secretion in man. Diabetes. 1973;22:333–348. doi: 10.2337/diab.22.5.333. [DOI] [PubMed] [Google Scholar]
- He W, Newman JC, Wang MZ, Ho L, Verdin E. Mitochondrial sirtuins: Regulators of protein acylation and metabolism. Trends in Endocrinology and Metabolism. 2012;23:467–476. doi: 10.1016/j.tem.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Hirschey MD, Verdin E. Measuring fatty acid oxidation in tissue homogenates. Protocol Exchange. 2010 http://dx.doi.org/10.1038/nprot.2010.92.
- Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, et al. Harmonics of circadian gene transcription in mammals. PLoS Genetics. 2009;5:e1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai SI, Guarente L. NAD and sirtuins in aging and disease. Trends in Cell Biology. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamur MC, Oliver C. Permeabilization of cell membranes. Methods in Molecular Biology. 2010;588:63–66. doi: 10.1007/978-1-59745-324-0_9. [DOI] [PubMed] [Google Scholar]
- Jarrett RJ, Keen H. Diurnal variation of oral glucose tolerance: A possible pointer to the evolution of diabetes mellitus. British Medical Journal. 1969;2:341–344. doi: 10.1136/bmj.2.5653.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. American Journal of Physiology, Endocrinology and Metabolism. 2000;279:E1039–E1044. doi: 10.1152/ajpendo.2000.279.5.E1039. [DOI] [PubMed] [Google Scholar]
- Klaidman LK, Leung AC, Adams JD., Jr High-performance liquid chromatography analysis of oxidized and reduced pyridine dinucleotides in specific brain regions. Analytical Biochemistry. 1995;228:312–317. doi: 10.1006/abio.1995.1356. [DOI] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, et al. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves TR, Li P, An J, Akimoto T, Slentz D, Ilkayeva O, et al. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. The Journal of Biological Chemistry. 2005;280:33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- la Fleur SE, Kalsbeek A, Wortel J, Fekkes ML, Buijs RM. A daily rhythm in glucose tolerance: A role for the suprachiasmatic nucleus. Diabetes. 2001;50:1237–1243. doi: 10.2337/diabetes.50.6.1237. [DOI] [PubMed] [Google Scholar]
- Lee A, Ader M, Bray GA, Bergman RN. Diurnal variation in glucose tolerance. Cyclic suppression of insulin action and insulin secretion in normal-weight, but not obese, subjects. Diabetes. 1992;41:742–749. doi: 10.2337/diab.41.6.750. [DOI] [PubMed] [Google Scholar]
- Liu AC, Tran HG, Zhang EE, Priest AA, Welsh DK, Kay SA. Redundant function of REV-ERBalpha and beta and non-essential role for Bmal1 cycling in transcriptional regulation of intracellular circadian rhythms. PLoS Genetics. 2008;4:e1000023. doi: 10.1371/journal.pgen.1000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AC, Welsh DK, Ko CH, Tran HG, Zhang EE, Priest AA, et al. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell. 2007;129:605–616. doi: 10.1016/j.cell.2007.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Takahashi JS. Mammalian circadian biology: Elucidating genome-wide levels of temporal organization. Annual Review of Genomics and Human Genetics. 2004;5:407–441. doi: 10.1146/annurev.genom.5.061903.175925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, et al. Circadian clock NAD+cycle drives mitochondrial oxidative metabolism in mice. Science. 2013;342:6158. doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek CB, Ramsey KM, Marcheva B, Bass J. Nutrient sensing and the circadian clock. Trends in Endocrinology and Metabolism. 2012;23:312–318. doi: 10.1016/j.tem.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirinen E, Lo Sasso G, Auwerx J. Mitochondrial sirtuins and metabolic homeostasis. Best Practice & Research Clinical Endocrinology & Metabolism. 2012;26:759–770. doi: 10.1016/j.beem.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Letters. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini C, Morf J, Stratmann M, Gos P, Schibler U. Simulated body temperature rhythms reveal the phase-shifting behavior and plasticity of mammalian circadian oscillators. Genes & Development. 2012;26:567–580. doi: 10.1101/gad.183251.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudubray JM, Coude FX, Demaugre F, Johnson C, Gibson KM, Nyhan WL. Oxidation of fatty acids in cultured fibroblasts: A model system for the detection and study of defects in oxidation. Pediatric Research. 1982;16:877–881. doi: 10.1203/00006450-198210000-00015. [DOI] [PubMed] [Google Scholar]
- Schibler U. The 2008 Pittendrigh/Aschoff lecture: Peripheral phase coordination in the mammalian circadian timing system. Journal of Biological Rhythms. 2009;24:3–15. doi: 10.1177/0748730408329383. [DOI] [PubMed] [Google Scholar]
- Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Imai S. Accurate measurement of nicotinamide adenine dinucleotide (NAD(+)) with high-performance liquid chromatography. Methods in Molecular Biology. 2013;1077:203–215. doi: 10.1007/978-1-62703-637-5_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EE, Liu AC, Hirota T, Miraglia LJ, Welch G, Pongsawakul PY, et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 2009;139:199–210. doi: 10.1016/j.cell.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Patel SP, McCarthy JJ, Rabchevsky AG, Goldhamer DJ, Esser KA. A non-canonical E-box within the MyoD core enhancer is necessary for circadian expression in skeletal muscle. Nucleic Acids Research. 2012;40:3419–3430. doi: 10.1093/nar/gkr1297. [DOI] [PMC free article] [PubMed] [Google Scholar]