

Abstract
Background. Both endothelin receptor type B ([ETBR], a G protein-coupled receptor that mediates the vascular effects of the potent vasoconstrictor endothelin-1) and human cytomegalovirus ([HCMV], a ubiquitous herpesvirus) have been implicated in the pathogenesis of cardiovascular disease (CVD). The effects of HCMV infection on ETBR expression are unknown. We hypothesized that HCMV may contribute to the pathogenesis of CVD via ETBR modulation.
Methods. Human CMV effects on ETBR were studied in vitro in endothelial cells (ECs) and smooth muscle cells (SMCs) and ex vivo in human carotid plaque tissue specimens. Expression of ETBR and viral immediate-early were quantified using quantitative polymerase chain reaction. Functional consequences after ETBR blockade in ECs were examined by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide proliferation, wound healing, tube formation, and flow adhesion assays.
Results. Human CMV is capable of upregulating both ETBR mRNA and protein expression in ECs and SMCs. The ETBR was also abundantly expressed in ECs, foam cells, and SMCs, and, more importantly, in HCMV-positive cells in human carotid plaques. Endothelin receptor type B blockade led to decreased proliferation and reduced tumor necrosis factor α-mediated leukocyte recruitment in both uninfected and HCMV-infected ECs. Direct HCMV infection was antimigratory and antiangiogenic in ECs.
Conclusions. Human CMV may contribute to CVD via ETBR induction.
Keywords: angiogenesis, BQ788, cytomegalovirus, endothelial cells, endothelin receptor type B, leukocyte recruitment, vasculopathies
Human cytomegalovirus (HCMV) is a ubiquitous, enveloped virus with a 196–241 kilobase pair double-stranded DNA genome that encodes at least 166 gene products, most of which are nonessential for viral replication but are crucial for survival in host cells [1]. A recent study utilizing ribosomal profiling and transcript analysis demonstrates detection of potentially 751 unique RNAs being translated in CMV-infected fibroblast cells, which suggest a far more complex biology of this virus than previously thought [2]. Like other herpesviruses, primary infection of HCMV is followed by lifelong latency and reactivation of virus contributes to HCMV-associated diseases [1]. Viral replication occurs in the nucleus of infected cells and is regulated in a stepwise and coordinated manner with the prior expression of immediate-early (IE or α) genes, followed by delayed-early (E or β) and late (L or γ) genes in the permissive cells [1]. Human CMV infection in immunocompetent hosts is usually mild but can be fatal in immunocompromised individuals such as in acquired immune deficiency syndrome patients or organ transplant patients. It is also the main viral etiological agent responsible for congenital diseases in newborns [1]. Besides being a prominent opportunistic pathogen, and based on evidence accrued to date, HCMV may have a plausible active role in a number of different diseases, such as in cardiovascular diseases (CVDs), cancers, and autoimmune or chronic inflammatory diseases [3, 4].
For the probable role of HCMV in CVD, we and others have not only showed the high prevalence of HCMV in carotid plaques but also have begun to elucidate the possible multifaceted ways of HCMV in modulating atherogenesis [5–12]. For example, we recently proposed that HCMV may perturb nitric oxide (NO) production via up-regulation of arginase II, a metalloenzyme that shares the same substrate as endothelial NO synthase and with a potential to mediate endothelial dysfunction [8]. Endothelial cells (ECs) that line the blood vessels are crucial in angiogenesis, a complex process of forming new vessels from pre-existing blood vessels. As such, endothelial dysfunction is the cardinal feature of CVD, especially in ischemic CVD, and it is believed to be responsible for the failure of “therapeutic angiogenesis”, which is a promising strategy for the use of proangiogenic growth factors to promote the development of collateral blood vessels [13].
Endothelin receptor type B (ETBR) is a 7-transmembrane G protein-coupled receptor that mediates the vascular effects of endothelin-1 (ET-1), which is a potent vasoconstrictor [14]. It is predominantly expressed in ECs but can also be detected in vascular smooth muscle cells (SMCs) and other cell types [14, 15]. The ETBR is located in the plasma membrane, cytosol, nucleoplasm, and the nuclear envelope membrane [14], and its functions depend on the effector cell type. In ECs, ETBR primarily mediates vasodilation by releasing prostacyclin and NO. In SMCs, ETBR instead acts as a vasoconstrictor [14], similar to endothelin receptor type A (ETAR), which mediates vasoconstriction in vascular SMCs [16]. In addition, ETBR also functions as a “clearance receptor” for ET-1 [17].
Substantial evidence indicates a crucial role for ETBR in vascular homeostasis. The deregulation of ETBR is associated with CVDs such as coronary artery disease, cerebral ischemia, and atherosclerosis [14, 15]. In addition, mutations in the ETBR gene have been linked to certain genetic diseases [18, 19]. In cancer patients, overexpression of ETBR is associated with melanoma progression [20] and poor prognosis in breast cancer patients [21], and, consequently, its specific antagonist, BQ-788, has been evaluated as a potential cancer treatment [22].
Given the role of HCMV in atherogenesis and the importance of ETBR in vascular biology, we hypothesized that HCMV may promote CVD by inducing ETBR. To our knowledge, this is the first study to examine the effects of HCMV on ETBR and functional consequences after ETBR blockade.
METHODS
Virus and Cells
The virus stock, virus titration, and cells used in this study were as described previously [8]. In brief, a plaque-purified HCMV strain VR1814 propagated in human umbilical vein cells (HUVECs) was used as virus stock. The source and growth conditions for HUVEC, human pulmonary arterial SMCs (hPASMCs), and human aortic SMCs (hSMCs) were as described previously [8]. For the flow adhesion assay, HUVECs from 3 different donors were isolated and cultured in complete endothelial basal medium (EBM)-2 medium supplemented with EGM-2 SingleQuots (hereafter referred to as complete EBM-2 medium) instead of M199 as described in the protocol [23]. The use of HUVECs from a healthy donor was approved by the Karolinska Institutet Ethnical Committee (reference number 2012/654–31/3).
Virus Infection and Ganciclovir Treatment
At the indicated times, cells were infected with different multiplicity of infection (moi) of HCMVs, lysed, and RNA was isolated. Both ultraviolet (UV)-inactivation protocol and ganciclovir (GCV) treatment were performed as described previously [8].
Quantitative TaqMan Polymerase Chain Reaction
The RNA isolation, cDNA synthesis, and TaqMan polymerase chain reaction assay were performed as described previously [8]. The primers and probes (all obtained from Applied Biosystems; Life Technologies, Thermo Fisher Scientific Corporation) were as follows: ETAR (EDNRA, Hs00609865_m1), ETBR (EDNRB, Hs00240747_m1), and IE (custom made with sequence as described in [24]). The human β2-microglobulin (Hs00984230_m1) was used for normalization, and the 2−ΔΔCT method was used to quantify the relative expression.
Flow Cytometry Analysis
Flow cytometry was performed as described previously [8]. Single-color staining was performed using primary mouse anti-ETBR (1:200; a gift from Dr. Tomoko Doi, Kyoto University) [25] or mouse anti-IE (1:300) (MAB810R; Millipore). For dual staining, cells were incubated with rabbit anti-ETBR (1:200) (AER-002; Alomone Labs) for 30 minutes at room temperature and then washed and fixed with Reagent A followed by permeabilization with Reagent B (Molecular Probes; Invitrogen, Life Technologies) along with primary mouse anti-IE according to the manufacturer's protocol. The positivity was revealed using secondary antibody swine anti-rabbit (1:100) (F0054; Dako) conjugated to fluorescein isothiocyanate isomer 1 or goat anti-mouse (1:100) (R0480; Dako) conjugated to R-phycoerythrin (RPE).
Immunofluorescence and Immunohistochemical Staining
The immunofluorescence (IF) was performed on ice-cold absolute acetone/methanol (1:1) fixed cells in 8-chamber slides as previously described [8]. Primary antibodies used in this study were as follows: mouse anti-HCMV-IE (1:500), rabbit anti-ETBR (1:500), mouse anti-giantin (1:300) (clone G1/133, marker for Golgi; Enzo Life Sciences), and endoplasmic reticulum marker PDI (1:300) (clone 1D3; Enzo Life Sciences), with secondary antibody goat anti-mouse or goat anti-rabbit conjugated to Alexa Fluor 488 or 594 (both from the Molecular Probes and used at 1:500 dilution). For double-immunofluorescent staining, a mixture of indicated primary antibodies was incubated before incubation with corresponding secondary antibodies.
For the immunohistochemical (IHC) staining on frozen carotid plaque tissue sections, a previously used protocol was applied [7]. Primary antibodies were mouse monoclonal anti-HCMV-IE (1:500), mouse monoclonal anti-human Von Willebrand factor (1:50) (Clone F8/86; Dako), and rabbit anti-ETBR (1:250); immunoreactivity was revealed with chromogen 3,3’-Diaminobenzidine. The omission of primary antibody or corresponding isotype antibody served as negative control. Twenty carotid plaques were randomly selected from our previously described biobank, the Biobank of St. Petersburg Investigation of Carotid Endarterectomies [7]. The use of human materials in this study was approved by the Ethical Committee of St. Petersburg State Pavlov Medical University, the Russia Federation. The immunoreactivity was graded as previously described [7].
3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl Tetrazolium Bromide Cell Proliferation Assay
The assay was performed using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide ([MTT] Roche) according to the vendor's instruction. Approximately 1 × 104 viable cells were seeded in 6 replicate wells before treatment with BQ123, an ETAR blocker or BQ788, an ETBR blocker (both at a final concentration of 100 µM) either in the presence or absence of HCMV at moi = 1, and assayed for proliferation at indicated time points.
Wound Healing: In Vitro Scratch Assay
The wound healing assay was performed using a 6-well plate with 90% confluent HUVECs as described in the Wallert and Provost lab's Wound Assay Protocol (http://web.mnstate.edu/provost/woundAssayProtocol.pdf). In brief, cells were gently washed after scratch of HUVEC monolayers and replaced with 2 mL complete EBM-2 medium containing the ETBR blocker with or without HCMV. Pictures were acquired with Olympus CKX 41 inverted microscope using a 4× objective lens at an indicated time.
Angiogenesis: Tube Forming Assay
The assay was performed using angiogenesis µ-slides (lbidi, cat. no. 81506) according to the manufacturer's instructions. In brief, 10 µL Matrigel (BD Biosciences, cat. no. 354230) were dispensed onto each well and set at 37°C. Approximately 1 × 104 human umbilical vein cells/well in 50 µL complete EBM-2 medium were seeded in triplicate with or without the presence of ETBR blocker or with or without the HCMV in a final concentration of 100 µM. Tube formation was monitored at indicated times, and images were taken with Olympus CKX 41 inverted microscope.
Isolation of Human Leukocyte
The isolation was performed by a double gradient density centrifugation using Histopaque 1077 and 1119 (both from Sigma-Aldrich) with anticoagulated EDTA-whole blood, as described previously [26].
Flow Adhesion Assay
The assay was performed as described previously [26, 27] with the use of ibiTreat µ-slide VI0.4 (ibidi GmbH). The details of instrument setup, recording, and analysis was performed as described in [28]. In brief, approximately 4 × 104 human umbilical vein cells/chamber were seeded and treated with BQ788 (100 µM) in the presence or absence of HCMV for 16 hours before the assay. All HUVECs were pretreated with 2 ng/mL tumor necrosis factor α (TNF-α) (R&D Systems, cat.no. 210-TA-005) for 2 hours, except for the untreated cells before the assay. At least 6–10 images of each condition were taken for the analysis.
Statistical Analysis
Results were expressed as mean ± standard deviation and analyzed with a Student's t test (version 11.0 software; SigmaPlot) and compared with the uninfected or untreated group. A P < .05 was considered to be statistically significant.
RESULTS
Human Cytomegalovirus Induces Up-Regulation of Endothelin Receptor Type B in Both Endothelial Cells and Smooth Muscle Cells
We infected ECs (HUVEC) and SMCs (hPASMC and hSMC) with a different moi of HCMV and quantified the relative expression of ETBR mRNA at 1, 3, and 5 days of postinfection (dpi) for HUVEC and hPASMC and at 3 and 5 dpi for hSMC. We found that HCMV up-regulated the ETBR mRNA expression in both ECs and SMCs (Figure 1A.i–A.iii). A similar up-regulation of ETBR mRNA was observed in hPASMC, but to a lesser extend in hSMC (Figure 1A.ii and A.iii, respectively). As expected, viral IE mRNA was increased over time after infection and was moi-dependent (Figure 1A.iv–A.vi). The induction of ETBR mRNA required active viral replication because the UV-irradiated virus failed to exert any effect on ETBR in both ECs and SMCs (Supplementary Figure S1 and Figure 1A.vii–A.viii). As depicted in Figure 1B.i–B.iii, ETBR protein level was distinctly induced upon HCMV infection in both ECs and SMCs. Most but not all HCMV-infected cells expressed high levels of ETBR (Figure 1B.i–B.iii, white arrows), suggesting that a threshold of infection is needed for ETBR expression. Consistent with the mRNA levels, ETBR protein expression also appeared to be moi-dependent in HUVECs and hSMCs (Figure 1B.iv and B.vi, respectively); the same level of ETBR protein induction was observed with moi = 1 and 5 in the HCMV-infected hPASMC (Figure 1B.v). Hence, we conclude that active HCMV infection induces the expression of ETBR in both ECs and SMCs.
Figure 1.


Effects of human cytomegalovirus (HCMV) on vascular endothelial and smooth muscle cells. The graphic depicts relative expression of endothelin receptor type B (ETBR) with indicated multiplicity of infection (moi) of HCMV as assayed by quantitative TaqMan polymerase chain reaction (PCR) at indicated days postinfection (dpi) for endothelial cells (human umbilical vein cells [HUVECs]; A.i) or smooth muscle cells (SMCs) (human pulmonary arterial SMCs [hPASMCs], A.ii) and human aortic SMCs [hSMCs], A.iii). The corresponding infection levels in HCMV-infected HUVECs (A.iv), hPASMCs (A.v) and hSMCs (A.vi), as assessed by quantitative TaqMan PCR are shown. The effects of UV-irradiated HCMV on the ETBR mRNA level in hPASMC (A.vii) and hSMC (A.viii) are illustrated. The uninfected cells at the indicated time course were used as a calibrator for relative quantification using the comparative 2−ΔΔCt method with β2-microglobulin as an endogenous control (biological replicates for A.i-A.vi [n = 4] and A.vii and A.viii [n = 3] with technical duplicates for each PCR; values are mean ± standard deviation [SD]). Representative immunofluorescence staining on HCMV-infected endothelial cells (HUVECs; B.i) and SMCs (hPASMC, B.ii and hSMC, B.iii) with indicated MOI at 3 dpi. Arrows in B.i-B.iii depict HCMV-infected cells without up-regulation of ETBR. Nucleus was stained with 4′-6-diamidino-2-phenylindole ([DAPI], blue), and the immunoreactivity of ETBR or HCMV immediate-early (IE) was revealed with Alexa Fluor 594 (red) or Alexa Fluor 488 (green), respectively (biological replicates, n = 3). The corresponding quantitative results are shown for HCMV-infected HUVECs (B.iv), hPASMC (B.v) and hSMC (B.vi), by mean florescent intensity expressed as pixel sum as analyzed with Leica Application Suite Advanced Fluorescence software. At least 5 different areas of interest were quantified (values are mean ± SD; P < .05 was considered to be statistically significant; *P = .01–.05; **P = .001–.01; ***P < .001; n.s = not significant).
Human Cytomegalovirus Immediate-Early or Delayed-Early Protein Mediates Up-Regulation of Endothelin Receptor Type B
Because ETBR receptor expression was not induced in uninfected cells in close proximity to HCMV-infected ETBR-positive cells (see Figure 1B.i–B.iii), paracrine effects of cytokine(s) were excluded. To determine the viral gene mediating the ETBR up-regulation, we infected and treated HUVECs with GCV that inhibits CMV DNA replication and L gene expression but not expression of IE or E genes [29]. The GCV treatment did not abrogate the induction of ETBR transcripts but instead significantly induced higher expression of ETBR (Figure 2), suggesting that viral IE or E gene products are responsible for effect. We tested both siRNAs against the well-characterized IE1-72 or IE2-86, which we did not observe any effects on the ETBR mRNA expression (Supplementary Figure S2). Taken together, HCMV induced both ETBR mRNA and protein expression most likely via its IE or E proteins.
Figure 2.
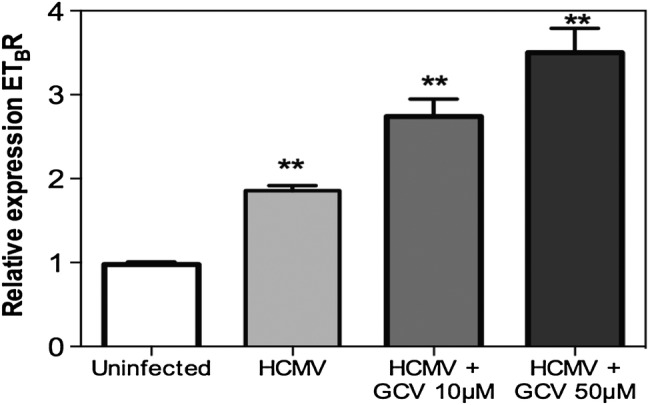
Human cytomegalovirus (HCMV) immediate-early or early gene is responsible for endothelin receptor type B (ETBR) up-regulation. Relative expression of ETBR in uninfected or HCMV-infected cells (human umbilical vein cells) treated with different concentrations of ganciclovir and analyzed at 5 days postinfection (values are mean ± standard deviation, biological replicates, n = 2 with technical duplicates for each polymerase chain reaction; **P = .001–.01; ***P < .001; Abbreviation: n.s = not significant).
Human Cytomegalovirus Alters the Cellular Distribution of Endothelin Receptor Type B (ETBR) and Induces ETBR Surface Expression
Endothelin receptor type B is known to be expressed at the plasma membrane, in the cytosol, at the nuclear envelope membrane, and in the nucleoplasm [14]. Consistent with this, we detected ETBR mainly close to the plasma membrane and/or in the cytoplasm but very little in the perinuclear membrane or in the nucleus of the uninfected HUVECs (Figure 3A, top row). We were surprised to find that HCMV infection resulted in strong expression of ETBR in the cytoplasm (Figure 3A, bottom row, panel ETBR, arrow) and colocalized with a Golgi marker (giantin) with little or no colocalization in the endoplasmic reticulum (Figure 3B).
Figure 3.


Human cytomegalovirus (HCMV) infection alters endothelin receptor type B (ETBR) cellular distribution, induces surface expression of ETBR and its effect on endothelin receptor type A (ETAR). (A) Representative immunofluorescence staining on HCMV-infected endothelial cells at 3 days postinfection to illustrate the subcellular distribution of ETBR in uninfected and infected cells. Arrow shows increased ETBR protein expression in the perinuclear region. Nucleus was stained with 4′-6-diamidino-2-phenylindole ([DAPI] blue), and the immunoreactivity of ETBR or HCMV immediate-early (IE) was revealed with Alexa Fluor 594 (red) or Alexa Fluor 488 (green), respectively (biological replicates, n = 3). (B) Representative immunofluorescence staining of ETBR and giantin, a Golgi marker (top panel), or PDI, an endoplasmic reticulum marker (bottom panel). Nucleus was stained with DAPI (blue). Flow cytometry analysis on the effects of HCMV-infected endothelial cells (human umbilical vein cells [HUVECs]) or smooth muscle cells (SMCs) (human pulmonary arterial SMCs [hPASMCs] and human aortic SMCs [hSMCs]) on ETBR at indicated days postinfection (dpi). (C) The effects of different multiplicity of infection (moi) of HCMV on ETBR surface expression at 1, 3, and 5 dpi (left panel) in HUVECs. The HCMV IE is shown on the right panel. Y-axis denotes log intensity of ETBR (left panel) or IE (right panel), whereas x-axis is side scatter in linear scale (SS Lin). (D) The effects of ultraviolet (UV)-irradiated HCMV on ETBR surface expression in HUVECs. Little or no IE was detected with the UV treatment (bottom row). (E) The effects of UV-irradiated HCMV (middle row) or HCMV (bottom row) on ETBR surface expression in SMCs, hPASMCs (left column) and hSMCs (right column) at 5 dpi. Y-axis denotes log intensity of ETBR (left panel), whereas x-axis is SS Lin. The effects of HCMV on ETAR mRNA level in smooth muscle cells, hPASMC (F) and hSMC (G) (MOI; biological replicates, n = 3 with technical duplicates for each polymerase chain reaction; values are mean ± standard deviation; P < .05 was considered to be statistically significant; *P = .01–.05; **P = .001–.01; ***P < .001).
To determine whether HCMV infection also induces surface expression of ETBR, we compared surface expression on uninfected and infected ECs and SMCs by flow cytometry analysis. As depicted in Figure 3C (left panel), the ETBR surface expression was increased over time after HCMV infection in HUVECs (Figure 3C, column panel of moi = 0.5 and 5); this effect was moi-dependent, which was consistent with the data for induced mRNA expression (Figure 3C, right panel). Very little or no induction of ETBR surface expression was noted in HUVEC treated with UV-irradiated virus, suggesting that viral replication is required for this effect (Figure 3D, top row of the left panel). Ultraviolet treatment was considered effective because little or no IE protein expression was detected (Figure 3D, bottom row). Similar ETBR induction was noted in both hPASMC and hSMC (Figure 3E, bottom row) but not in cells treated with UV-irradiated virus (Figure 3E, middle row).
We did not detect any ETAR expression with the commercial HUVECs (unpublished data), which is in line with previous studies [30]. However, we did detect low ETAR mRNA expression (threshold of cycle [Ct] = 34) in freshly isolated primary HUVEC from healthy donor, which is in line with what has been reported previously [31]. This allowed us the opportunity to investigate both ETAR and ETBR expression upon HCMV infection in HUVECs. We detected a balanced effect between ETAR and ETBR upon HCMV infection, at least in the early stage of infection (1 dpi to 3 dpi); when there was a reduction of ETBR expression, ETAR was induced at 3 dpi (Supplementary Figure S3). It is interesting to note that both receptors were up-regulated at 5 dpi when infected with HCMV (MOI = 10). In contrast to the ETBR induction, HCMV caused significantly reduced levels of ETAR expression in both hPASMC and hSMC (Figure 3F–G).
Human Cytomegalovirus and Endothelin Receptor Type B Expression Colocalizes in Atherosclerotic Plaques
To examine whether ETBR was present in carotid atherosclerotic plaques ex vivo and whether the virus was colocalized with ETBR, we performed IHC and IF on 20 frozen sections from carotid endarterectomy biopsies obtained from patients. We found that 80% (16 of 20) of the plaques showed immunoreactivity to HCMV-IE antigens; the majority of them, 62.5%, were grade I and 37.5% were grade II (see Supplementary Table S1). Endothelin receptor type B was expressed in 80% of the examined plaques and localized to ECs, SMCs, and macrophages/foam cells. We observed no apparent correlation between percentage of IE and ETBR-positive cells (Supplementary Figure S4), likely due to the low infectivity or other factors modulating ETBR expression in carotid plaques. More importantly, we detected colocalization of both ETBR and HCMV IE in ECs and SMCs-like cells in carotid plaques (n = 3; Figure 4A and B). Using IF, we observed distinctly enhanced ETBR expression levels in the nucleus and cytoplasm primarily in HCMV-infected cells ex vivo (Figure 4B).
Figure 4.

Human cytomegalovirus (HCMV) antigens are closely associated with endothelin receptor type B (ETBR) in human carotid atherosclerotic plaques. Representative photomicrographs of immunohistochemistry and immunofluorescence (IF) staining on frozen human carotid atherosclerotic plaques. (A) Immunohistochemistry staining to show colocalization of HCMV immediate-early (IE) and ETBR in Von Willebrand factor (vWF)-positive cells as revealed by chromogen diaminobenzidine, brown products in a boxed area with consecutive sections (left column; right column is a higher magnification of the boxed areas). (B) The IF staining on a human carotid atherosclerotic plaque to illustrate the colocalization of HCMV-IE with ETBR in fibroblast-like cells. Nucleus was stained with 4′-6-diamidino-2-phenylindole ([DAPI] blue), and the immunoreactivity of HCMV-IE or ETBR was revealed with Alexa Fluor 594 (red) or Alexa Fluor 488 (green), respectively. Twenty human carotid atherosclerotic plaques were examined.
Endothelin Receptor Type B Blockade Reduces Cell Growth and Impairs Tumor Necrosis Factor-α-Mediated Leukocyte Recruitment
Because we observed that HCMV is capable of modulating ETAR or ETBR expression in vitro and observed colocalization of HCMV and ETBR in atherosclerotic plaques, we wondered about the physiological relevance of ETBR on ECs functions. Hitherto, the role of ETBR in ECs, besides the well known cardiovascular effects, remains obscure. Endothelial cells were chosen for further studies due to their critical role in HCMV pathogenesis [32] and because ETBR, but not ETAR, was expressed on the HUVECs used in this study. We first examined the effects of an ETBR blocker, BQ788, on ECs proliferation, migration, and angiogenesis by MTT, wound healing, and tube formation assays, respectively. BQ788 did not affect ECs proliferation, migration, or angiogenesis at 1-day treatment (Figure 5A–C), but it resulted in slightly retarded cell growth at 4 days compared with that of untreated cells (Figure 5A).
Figure 5.

The effects of an endothelin receptor type B (ETBR) blocker, BQ788, on endothelial cells proliferation, migration, vasculogenecity, and leukocyte recruitment. (A) Cell proliferation assay by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) was performed in human umbilical vein cells (HUVECs) untreated or treated with BQ788 at the indicated time points after treatment. Optical density was read at 590 nm with reference wavelength at 670 nm. (B) Light microscopic images (×4) of the wound healing assay were taken at different times after the “scratch” of HUVEC monolayers untreated or treated with BQ788. (C) Light microscopic images (×4) of tube formation by HUVECs untreated or treated with BQ788. (D) Light microscopic images of flow adhesion assay in cells stimulated with tumor necrosis factor-α (TNFα) and untreated or treated with BQ788 (left panel). The number of adherent cells was quantified for all the conditions (right panel). Values are mean ± standard deviation; P < .05 was considered to be statistically significant; *P = .01–.05; **P = .001–.01; ***P < .001).
Given that the crucial role of ECs in leukocyte recruitment is fundamental during inflammatory responses [33], we further examined the effect of BQ788 on leukocyte recruitment to ECs using a flow adhesion assay. To do this, we seeded ECs and activated them with TNF-α in the presence or absence of BQ788. As expected, little or no leukocyte recruitment was recorded in the “nonactivated” HUVECs, whereas leukocytes were recruited upon TNF-α activation (Figure 5D). Treatment with BQ788 significantly reduced TNF-α-mediated leukocyte recruitment (Figure 5D), implying a role of ETBR in leukocyte recruitment.
The Effects of Human Cytomegalovirus Infection on Endothelial Cells Proliferation, Migration, and Angiogenesis
Having shown a potential “physiological” role of ETBR in ECs, we next addressed the question of how HCMV modulates these processes. To this end, we infected HUVECs with or without BQ788 and assayed its effects on cellular proliferation, migration, or tube formation. Treatment of BQ788 significantly reduced ECs proliferation at 3 dpi, compared with that of untreated cells (Figure 6A); HCMV infection enhanced the inhibitory effect by BQ788 on cellular proliferation (Figure 6A). The viral infection (moi = 1) was not cytotoxic to the cells at 3 dpi (Figure 6A). Human CMV infection alone or in the presence of BQ788 abolished the migratory properties of ECs in the wound healing assay (Figure 6B) as well as its capacity to form tubes in the tube formation assay (Figure 6C). The antimigratory and antiangiogenic effect of the HCMV was dependent on the virus load, and, once executed, the effect sustained even after 7 dpi (unpublished observations). In the flow adhesion assay, BQ788 reduced leukocyte recruitment to infected and TNF-α-stimulated HUVECs (Figure 6D). Taken together, these findings suggest that HCMV blunts the migratory and angiogenic properties of ECs.
Figure 6.

The effects of human cytomegalovirus (HCMV) and endothelin receptor type B (ETBR) blocker, BQ788, on endothelial cells proliferation, migration, vasculogenecity, and leukocyte recruitment. (A) Cell proliferation assay by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) was performed in human umbilical vein cells (HUVECs) uninfected or HCMV-infected, untreated or treated with BQ788 at the indicated time points after infection. Optical density was read at 590 nm with reference wavelength at 670 nm. (B) Light microscopic images (×4) of the wound healing assay were taken at different times after the “scratch” of HUVEC monolayers uninfected or HCMV-infected treated with BQ788. (C) Light microscopic images (×4) of tube formation by HUVEC uninfected or HCMV-infected and treated with BQ788. (D) Light microscopic images of flow adhesion assay in cells stimulated with tumor necrosis factor-α (TNFα) and uninfected or HCMV-infected (left panel). The number of adherent cells was quantified for all the conditions (right panel). Values are mean ± standard deviation; P < .05 was considered to be statistically significant; *P = .01–.05; **P = .001–.01; ***P < .001).
DISCUSSION
It is well accepted that increased ETBR expression in ECs denotes vasodilation via release of NO and prostaglandins, whereas up-regulation of ETBR in SMCs is expected to mediate vasoconstriction [14, 34]. In the present study, we found that HCMV affects the expression of both ETBR and ETAR. On one hand, our finding that HCMV increases ETBR in ECs suggests a possible vasoprotective role or the prevention of ECs apoptosis [35]. The antiapoptotic effects mediated by ETBR may favor viral survival in the early stage of viral replication [36]. Thus, in view of current knowledge, up-regulation of ETBR in ECs would promote vasodilation and also enhanced depletion of ET-1 due to its clearance properties [17]. Human CMV also appears to reduce leukocyte recruitment and prevents angiogenesis and migration, which may impair the repair functions after myocardial infarction and result in increased heart damage [37]. On the other hand, up-regulation of ETBR in SMCs might lead to vasoconstriction, proliferation, inflammation, and increased oxidative stress that underlies the pathogenesis of many CVD [14, 34]. Furthermore, the ETBR-induced vascular SMCs proliferation would favor viral growth and survival and contribute to vascular pathogenesis; eg, intima hyperplasia. However, because we also observed a down-regulation of ETAR in SMCs, the net functional effect attributed by HCMV infection via altered ET receptors expression remains unclear. Therefore, further studies are needed to understand the biological net effects of HCMV infection on both ECs and SMCs in terms of vasoconstriction or vasodilation in vivo, but this will be complicated due to the lack of a suitable animal model for HCMV.
It is interesting to note that blockade of ETBR by BQ788 alone or in the presence of HCMV significantly reduced TNF-α-mediated leukocyte recruitment, suggesting a plausible role of ETBR in leukocyte recruitment and inflammation. Leukocyte recruitment includes a cascade of stepwise events that culminates in leukocyte transmigration, with the aid and concrete effort of certain chemoattractants, selectins, and integrins on the ECs (reviewed in [33]). BQ788 has been shown to suppress up-regulation of TNF-α and rescued retinal ganglion cells from optic nerve injury by attenuating inflammation [38]. The mechanism by which ETBR regulates leukocyte recruitment and whether this affects inflammation remains to be elucidated. Endothelin-1 has been shown to promote neutrophil recruitment via TNF-α and CXCL1/CXCR2 in mice, which is mediated by both ETAR and ETBR [39]. Because ECs are known to be the major source of ET-1 under physiological conditions [40], it is conceivable that ET-1 may be responsible for the influence of ETBR on leukocyte recruitment [41]. However, a major role of ET-1 in leukocyte recruitment in HCMV-infected HUVECs is unlikely since we observed that HCMV down-regulates both ET-1 mRNA and protein levels (unpublished data), whereas HCMV may use ETBR for other purposes during infection.
We detected ETBR in ECs, SMCs, and macrophages/foam cells, which is consistent with previously reported data of increased ETBR immunoreactivity in human atherosclerotic lesions [42, 43]. Consistent with these studies and the new data presented here, we observed that HCMV-positive cells in atherosclerotic plaques exhibited intense ETBR staining, mainly in ECs and SMCs. Although all of the examined carotid plaques showed high prevalence of both HCMV-IE antigens (80%) and ETBR (80%), the majority of them was graded I (62.5% for both antigens in their positive staining, respectively). These observations demonstrate a close association between HCMV and ETBR in human atherosclerotic plaques, but the functional relevance of this observation will require further studies. In this study, only 1 case had abundant ETBR (graded III) expression (Supplementary Table S1); this makes the direct correlation between HCMV-IE antigens and ETBR difficult to assess. Moreover, HCMV infection is known to produce a myriad of indirect effects such as enhanced expression of adhesion molecules, induced production of proinflammatory cytokines, and activation of T cells, which may also contribute to atherosclerosis [3].
In this study, we also found that although the expression of ETBR in vitro is predominantly cytoplasmic or perinuclear upon HCMV infection, its expression is localized to nucleus in ex vivo carotid plaques. The similar dominant nuclear localization in cardiac myocytes has been reported previously [44], and recently it was proposed that the endothelin receptors of the nuclear membrane may play an important role in ET-1 action by receiving the ET-1 receptor-mediated cytosolic signaling [45]. In the context of virus infection, we speculate that the nuclear localization of ETBR that occurs upon HCMV infection may facilitate virus replication, and this hypothesis is under investigation in our laboratory.
We found that HCMV infection renders ECs refractory to migration and blunts their capacity to form tubes. Our finding is in line with Yamamoto-Tabata et al [46], who used the same virus strain and cells but in contrast with another study that used different virus strain appended with enhanced green fluorescent protein and microvascular ECs [47]. Endothelial cells from different vascular beds are known to respond differently to HCMV infection [48]. In a recent study, we provided multifaceted evidence that direct infection with HCMV strain VR1814 induces an antimigratory and antiangiogenic ECs phenotype that could have detrimental effects on the vasculature development (unpublished data). The antiangiogenic properties conferred by HCMV may also pose a threat to therapeutic angiogenesis. On the other hand, both HCMV-encoded pUL7 protein and/or its major secretome, interleukin-6, were shown to promote angiogenesis [49, 50]. Thus, it appears that HCMV-infected cells are antiangiogenic whereas its secretome is proangiogenic.
Our present study is limited by lack of an appropriate in vivo model because HCMV is highly species-specific and does not efficiently infect or cause any apparent disease in animals. Therefore, it is difficult to delineate the net effects of HCMV infection on endothelin receptors and thereby provide a better understanding of the molecular mechanisms by which HCMV may promote atherosclerosis through regulation of ETBR expression.
CONCLUSIONS
In conclusion, our data suggest that HCMV is capable of inducing ETBR expression, and close association between HCMV and ETBR in human atherosclerotic plaques may suggest a role in atherogenesis that requires further investigation. Moreover, our finding that direct HCMV infection is antiangiogenic highlights the need to consider the presence of this pathogen in therapeutic angiogenesis.
Supplementary Data
Supplementary material is available online at Open Forum Infectious Diseases online (http://OpenForumInfectiousDiseases.oxfordjournals.org/).
Acknowledgments
We are indebted to Drs. Vladimir V. Shlomin, Michail L. Gordeev, and Vera A. Ovchinnikova for the help during collection of carotid plaques. We also thank the late Maria Ekström and Jessica Patterson for excellent technical assistance and members of Cecilia Söderberg-Naucler’s laboratory for valuable comments and suggestions.
Financial support. This work was supported by Karolinska Institutet Foundation (2012fobi35508 and 2013fobi38188; to K.-C. Y.); Swedish Heart and Lung Foundation (grant nos. 20100614 and 20100259; to C. S.-N.); the Swedish Medical Research Council (K2010-56X-12615-13-3, K2012-64X-10857-19-5; to C. S.-N.); and Cardiovascular Programme and Stockholm County Council.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.
References
- 1.Mocarski ES, Courcelle CT. Cytomegaloviruses and their replication. In: Knipe D, Howley PM, ed. Fields Virology, Volume 2, 4th ed Philadelphia, PA: Lippincott Williams & Wilkins; 2001: pp 2629–73. [Google Scholar]
- 2.Stern-Ginossar N, Weisburd B, Michalski A et al. . Decoding human cytomegalovirus. Science 2012; 338:1088–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popovic M, Smiljanic K, Dobutovic B et al. . Human cytomegalovirus infection and atherothrombosis. J Thromb Thrombolysis 2012; 33:160–72. [DOI] [PubMed] [Google Scholar]
- 4.Soderberg-Naucler C. HCMV microinfections in inflammatory diseases and cancer. J Clin Virol 2008; 41:218–23. [DOI] [PubMed] [Google Scholar]
- 5.Chiu B, Viira E, Tucker W, Fong IW. Chlamydia pneumoniae, cytomegalovirus, and herpes simplex virus in atherosclerosis of the carotid artery. Circulation 1997; 96:2144–8. [DOI] [PubMed] [Google Scholar]
- 6.Yi L, Wang DX, Feng ZJ. Detection of human cytomegalovirus in atherosclerotic carotid arteries in humans. J Formos Med Assoc 2008; 107:774–81. [DOI] [PubMed] [Google Scholar]
- 7.Yaiw KC, Ovchinnikova O, Taher C et al. . High prevalence of human cytomegalovirus in carotid atherosclerotic plaques obtained from Russian patients undergoing carotid endarterectomy. Herpesviridae 2013; 4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yaiw KC, Mohammad AA, Taher C et al. . Human cytomegalovirus induces upregulation of arginase II: possible implications for vasculopathies. Basic Res Cardiol 2014; 109:401. [DOI] [PubMed] [Google Scholar]
- 9.Assinger A, Kral JB, Yaiw KC et al. . Human cytomegalovirus-platelet interaction triggers Toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arterioscler Thromb Vasc Biol 2014; 34:801–9. [DOI] [PubMed] [Google Scholar]
- 10.Rahbar A, Soderberg-Naucler C. Human cytomegalovirus infection of endothelial cells triggers platelet adhesion and aggregation. J Virol 2005; 79:2211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen YH, Zhang L, Utama B et al. . Human cytomegalovirus inhibits Akt-mediated eNOS activation through upregulating PTEN (phosphatase and tensin homolog deleted on chromosome 10). Cardiovasc Res 2006; 69:502–11. [DOI] [PubMed] [Google Scholar]
- 12.Weis M, Kledal TN, Lin KY et al. . Cytomegalovirus infection impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine in transplant arteriosclerosis. Circulation 2004; 109:500–5. [DOI] [PubMed] [Google Scholar]
- 13.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature 2005; 438:967–74. [DOI] [PubMed] [Google Scholar]
- 14.Mazzuca MQ, Khalil RA. Vascular endothelin receptor type B: structure, function and dysregulation in vascular disease. Biochem Pharmacol 2012; 84:147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Orleans-Juste P, Labonte J, Bkaily G et al. . Function of the endothelin(B) receptor in cardiovascular physiology and pathophysiology. Pharmacol Ther 2002; 95:221–38. [DOI] [PubMed] [Google Scholar]
- 16.Watts SW. Endothelin receptors: what's new and what do we need to know? Am J Physiol Regul Integr Comp Physiol 2010; 298:R254–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukuroda T, Fujikawa T, Ozaki S et al. . Clearance of circulating endothelin-1 by ETB receptors in rats. Biochem Biophys Res Commun 1994; 199:1461–5. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka H, Moroi K, Iwai J et al. . Novel mutations of the endothelin B receptor gene in patients with Hirschsprung's disease and their characterization. J Biol Chem 1998; 273:11378–83. [DOI] [PubMed] [Google Scholar]
- 19.Attie T, Till M, Pelet A et al. . Mutation of the endothelin-receptor B gene in Waardenburg-Hirschsprung disease. Hum Mol Genet 1995; 4:2407–9. [DOI] [PubMed] [Google Scholar]
- 20.Demunter A, De Wolf-Peeters C, Degreef H et al. . Expression of the endothelin-B receptor in pigment cell lesions of the skin. Evidence for its role as tumor progression marker in malignant melanoma. Virchows Arch 2001; 438:485–91. [DOI] [PubMed] [Google Scholar]
- 21.Wulfing P, Diallo R, Kersting C et al. . Expression of endothelin-1, endothelin-A, and endothelin-B receptor in human breast cancer and correlation with long-term follow-up. Clin Cancer Res 2003; 9:4125–31. [PubMed] [Google Scholar]
- 22.Lahav R, Heffner G, Patterson PH. An endothelin receptor B antagonist inhibits growth and induces cell death in human melanoma cells in vitro and in vivo. Proc Natl Acad Sci U S A 1999; 96:11496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badrnya S, Schrottmaier WC, Kral JB et al. . Platelets mediate oxidized low-density lipoprotein-induced monocyte extravasation and foam cell formation. Arterioscler Thromb Vasc Biol 2014; 34:571–80. [DOI] [PubMed] [Google Scholar]
- 24.Dzabic M, Rahbar A, Yaiw KC et al. . Intragraft cytomegalovirus protein expression is associated with reduced renal allograft survival. Clin Infect Dis 2011; 53:969–76. [DOI] [PubMed] [Google Scholar]
- 25.Yamaguchi T, Arimoto-Tahara I, Fujiyoshi Y, Doi T. Characterization and application of monoclonal antibodies against human endothelin B receptor expressed in insect cells. Biotechnol Lett 2004; 26:293–9. [DOI] [PubMed] [Google Scholar]
- 26.Bahra P, Rainger GE, Wautier JL et al. . Each step during transendothelial migration of flowing neutrophils is regulated by the stimulatory concentration of tumour necrosis factor-alpha. Cell Adhes Commun 1998; 6:491–501. [DOI] [PubMed] [Google Scholar]
- 27.Rainger GE, Fisher A, Shearman C, Nash GB. Adhesion of flowing neutrophils to cultured endothelial cells after hypoxia and reoxygenation in vitro. Am J Physiol 1995; 269:H1398–406. [DOI] [PubMed] [Google Scholar]
- 28.Shetty S, Weston CJ, Adams DH, Lalor PF. A flow adhesion assay to study leucocyte recruitment to human hepatic sinusoidal endothelium under conditions of shear stress. J Vis Exp 2014; 85:51330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McSharry JJ, Lurain NS, Drusano GL et al. . Rapid ganciclovir susceptibility assay using flow cytometry for human cytomegalovirus clinical isolates. Antimicrob Agents Chemother 1998; 42:2326–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh P, Horner T, Witkiewicz H, Schnitzer JE. Endothelin induces rapid, dynamin-mediated budding of endothelial caveolae rich in ET-B. J Biol Chem 2012; 287:17353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sek AC, Xie Z, Terai K et al. . Endothelial expression of endothelin receptor a in the systemic capillary leak syndrome. PLoS One 2015; 10:e0133266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerna G, Baldanti F, Revello MG. Pathogenesis of human cytomegalovirus infection and cellular targets. Hum Immunol 2004; 65:381–6. [DOI] [PubMed] [Google Scholar]
- 33.Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res 2007; 101:234–47. [DOI] [PubMed] [Google Scholar]
- 34.Pernow J, Shemyakin A, Bohm F. New perspectives on endothelin-1 in atherosclerosis and diabetes mellitus. Life Sci 2012; 91:507–16. [DOI] [PubMed] [Google Scholar]
- 35.Shichiri M, Marumo F, Hirata Y. Endothelin-B receptor-mediated suppression of endothelial apoptosis. J Cardiovasc Pharmacol 1998; 31:S138–41. [DOI] [PubMed] [Google Scholar]
- 36.Roulston A, Marcellus RC, Branton PE. Viruses and apoptosis. Annu Rev Microbiol 1999; 53:577–628. [DOI] [PubMed] [Google Scholar]
- 37.Jeffery HC, Soderberg-Naucler C, Butler LM. Human cytomegalovirus induces a biphasic inflammatory response in primary endothelial cells. J Virol 2013; 87:6530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tonari M, Kurimoto T, Horie T et al. . Blocking endothelin-B receptors rescues retinal ganglion cells from optic nerve injury through suppression of neuroinflammation. Invest Ophth Vis Sci 2012; 53:3490–500. [DOI] [PubMed] [Google Scholar]
- 39.Zarpelon AC, Pinto LG, Cunha TM et al. . Endothelin-1 induces neutrophil recruitment in adaptive inflammation via TNFalpha and CXCL1/CXCR2 in mice. Can J Physiol Pharmacol 2012; 90:187–99. [DOI] [PubMed] [Google Scholar]
- 40.Bohm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res 2007; 76:8–18. [DOI] [PubMed] [Google Scholar]
- 41.Piechota-Polanczyk A, Goraca A. Influence of specific endothelin-1 receptor blockers on hemodynamic parameters and antioxidant status of plasma in LPS-induced endotoxemia. Pharmacol Rep 2012; 64:1434–41. [DOI] [PubMed] [Google Scholar]
- 42.Iwasa S, Fan J, Shimokama T et al. . Increased immunoreactivity of endothelin-1 and endothelin B receptor in human atherosclerotic lesions. A possible role in atherogenesis . Atherosclerosis 1999; 146:93–100. [DOI] [PubMed] [Google Scholar]
- 43.Dagassan PH, Breu V, Clozel M et al. . Up-regulation of endothelin-B receptors in atherosclerotic human coronary arteries. J Cardiovasc Pharmacol 1996; 27:147–53. [DOI] [PubMed] [Google Scholar]
- 44.Boivin B, Chevalier D, Villeneuve LR et al. . Functional endothelin receptors are present on nuclei in cardiac ventricular myocytes. J Biol Chem 2003; 278:29153–63. [DOI] [PubMed] [Google Scholar]
- 45.Bkaily G, Avedanian L, Al-Khoury J et al. . Nuclear membrane receptors for ET-1 in cardiovascular function. Am J Physiol Regul Integr Comp Physiol 2011; 300:R251–63. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto-Tabata T, McDonagh S, Chang HT et al. . Human cytomegalovirus interleukin-10 downregulates metalloproteinase activity and impairs endothelial cell migration and placental cytotrophoblast invasiveness in vitro. J Virol 2004; 78:2831–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bentz GL, Yurochko AD. Human CMV infection of endothelial cells induces an angiogenic response through viral binding to EGF receptor and beta(1) and beta(3) integrins. Proc Natl Acad Sci U S A 2008; 105:5531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bentz GL, Jarquin-Pardo M, Chan G et al. . Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol 2006; 80:11539–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Botto S, Streblow DN, DeFilippis V et al. . IL-6 in human cytomegalovirus secretome promotes angiogenesis and survival of endothelial cells through the stimulation of survivin. Blood 2011; 117:352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacManiman JD, Meuser A, Botto S et al. . Human cytomegalovirus-encoded pUL7 is a novel CEACAM1-like molecule responsible for promotion of angiogenesis. MBio 2014; 5:e02035. [DOI] [PMC free article] [PubMed] [Google Scholar]