

Abstract
Objective
With no effective therapies to attenuate cartilage degeneration in osteoarthritis (OA), the result is pain and disability. Activation of hedgehog (HH) signaling causes changes related to the progression of OA, with higher levels of Gli‐mediated transcriptional activation associated with increased disease severity. To elucidate the mechanism through which this occurs, this study sought to identify genes regulated by HH signaling in human OA chondrocytes.
Methods
Using human OA cartilage samples, microarray analyses were performed to detect changes in gene expression when the HH pathway was modulated. Results were analyzed for differentially expressed genes, grouped into functional networks, and validated in independent samples. To investigate the effects of chondrocyte‐specific sterol accumulation, we generated mice lacking Insig1 and Insig2, which are major negative regulators of cholesterol homeostasis, under Col2a1 regulatory elements.
Results
HH signaling was found to regulate genes that govern cholesterol homeostasis, and this led to alterations in cholesterol accumulation in chondrocytes. A higher level of Gli‐mediated transcription resulted in accumulation of intracellular cholesterol. In genetically modified mice, chondrocyte‐specific cholesterol accumulation was associated with an OA phenotype. Reducing cholesterol accumulation attenuated the severity of OA in mice in vivo and decreased the expression of proteases in human OA cartilage in vitro.
Conclusion
HH signaling regulates cholesterol homeostasis in chondrocytes, and intracellular cholesterol accumulation contributes to the severity of OA. Our findings have therapeutic implications, since reduction of HH signaling reversed cholesterol accumulation and statin treatment attenuated cartilage degeneration.
Osteoarthritis (OA) is characterized by changes to the articular joint, including progressive degeneration of the articular cartilage. The pathogenesis of OA is complex, because it can result from multiple etiologies involving mechanical, genetic, traumatic, and metabolic factors 1, 2. Regardless of the cause, the changes that occur to the articular chondrocytes during OA, such as chondrocyte hypertrophy, recapitulate some of the changes that occur in growth plate chondrocytes during elongation of the long bones 3, 4. Hedgehog (HH) signaling is among the pathways that regulate chondrocyte differentiation in the growth plate of the long bones, including the changes that lead to chondrocyte hypertrophy 5, 6. In vertebrates, HH signaling is mediated by Gli transcription factors, with Gli2 acting as a transcriptional activator of the HH target genes Gli1, Ptch1, and Hhip 7. Previous studies have demonstrated that genetically modified mice with higher levels of Gli‐mediated transcription in chondrocytes develop more severe OA, and that inhibition of Gli‐mediated transcription reduces disease severity, but the mechanism through which this occurs remains unclear 4, 8.
To elucidate the role of HH signaling in OA severity, we identified targets of Gli‐mediated transcription in human OA cartilage. Using an unsupervised approach to analyze microarray results, we identified several pathways that potentially mediate the effect of HH signaling in the pathogenesis of OA. Among these, genes that are involved in cholesterol homeostasis were significantly represented, including the major negative regulator INSIG1, and the transcriptional regulator SREBF2. Because cholesterol is vital for cellular processes and has been previously implicated in OA 9, 10, 11, 12, we focused on exploring the role of HH signaling in regulating intracellular cholesterol biosynthesis in chondrocytes. We demonstrate that HH signaling positively regulates cholesterol accumulation in chondrocytes, and that modulation of the intracellular cholesterol level alters the severity of OA.
MATERIALS AND METHODS
Human OA cartilage
Human cartilage samples were obtained from patients undergoing total knee replacement surgery for clinically diagnosed OA. Articular cartilage was dissected from the subchondral bone of the lateral femoral condyle and incubated with either a pharmacologic HH antagonist (10 μM C31H42N4O5), an HH ligand (5 μg/ml Shh‐N; R&D Systems), lovastatin hydroxy acid (10 μM; Cayman Chemical), purmorphamine (10 μM; Sigma‐Aldrich), or a vehicle carrier as control for 48 hours. Explant processing and subsequent RNA extraction were conducted as previously described 13. Because each patient's articular cartilage sample was divided and used as its own control, extraneous variables were held constant and no additional patient history was required. All samples were obtained with the patients’ informed consent, and the study was performed under the approval of the Mount Sinai Hospital Research Ethics Board (Toronto, Ontario, Canada).
Gene expression analyses
Microarray was performed with human OA articular cartilage samples using the Affymetrix Human Gene 1.0 ST platform. The 3 samples included cartilage from 2 male patients and 1 female patient, whose mean age was 62.3 years. RNA was extracted from the cartilage using a previously described method 13, after the samples had been treated in vitro with either 10 μM C31H42N4O5 (HH antagonist) or a vehicle control. The microarray data can be accessed through the GEO database (GEO accession no. GSE54749). Results were analyzed independently for paired samples from each of the 3 patients (control 1 versus HH antagonist–treated 1, control 2 versus HH antagonist–treated 2, control 3 versus HH antagonist–treated 3), and differentially expressed genes were filtered using Partek Genomics Suite for those genes that were either up‐regulated or down‐regulated across all 3 samples.
Ingenuity Pathway Analysis was used to identify functional gene networks represented in the microarray data. Real‐time polymerase chain reaction (PCR) experiments were conducted using TaqMan assays (Applied Biosystems). For these experiments, human OA articular cartilage samples were obtained as described above, and mouse cartilage samples were obtained by isolation of cartilage from the knee joints, as described previously 14. Results were normalized to the expression of endogenous control genes (ASNS, ACTB, and GAPDH) and analyzed according to the comparative threshold cycle (Ct) method using the ΔΔCt formula.
For promoter analyses, primary human chondrocytes or ATDC5 cells were transfected with either an ADAMTS5 luciferase reporter construct or a negative control vector (GeneCopoeia), using the Neon Transfection System (Life Technologies) or Lipofectamine (Life Technologies). Site‐directed mutagenesis by inverse PCR was used to delete the sterol regulatory element (SRE) site, which was confirmed by sequencing.
Genetically modified mice
We crossed Col2a1‐Cre mice 15 with Insig1(fl/fl);Insig2(−/−) mice 16 to excise Insig1 in Col2a1‐expressing cells and generate mice with cartilage‐specific cholesterol accumulation. Mice expressing Cre are referred to as InsigDKO (Insig1[−/−];Insig2[−/−];Col2a1‐Cre). These mice were compared to their Cre‐negative (Insig1[fl/fl];Insig2[−/−]) littermates as controls. Cre‐mediated recombination and excision of Insig1 was confirmed by real‐time PCR and Western blot analysis.
To activate HH signaling by increasing Gli‐mediated transcription, InsigDKO mice were crossed with Col2a1‐Gli2 mice 17, the progeny for which were designated Col2a1‐Gli2;InsigDKO. To inhibit HH signaling by reducing Gli‐mediated transcription, InsigDKO mice were crossed with Gli2zfd mice (Gli2 +/−) 18, the progeny for which were designated Gli2 +/−;InsigDKO. To recapitulate OA in mice, we performed medial meniscectomy surgery, as described previously 4. Briefly, the medial meniscus was excised from the left knee of 8‐week‐old mice, and OA developed predictably 8 weeks postoperatively. Sham surgeries were used as the control, in which the entire surgical procedure, except excision of the medial meniscus, was followed.
To reduce cholesterol levels in vivo, mice were treated with a pharmacologic agent, lovastatin hydroxy acid, at a dosage of 3 mg/kg/day, administered via surgical implantation of drug pellets with slow release over 8 weeks. Within each genotype, mice were randomly selected to receive either statin treatment or placebo. Effective statin treatment was confirmed by increased expression of hydroxymethylglutaryl‐coenzyme A reductase (HMGCR) in the chondrocytes, as assessed by immunohistochemistry.
The knee joints of mice were evaluated for the development of OA by radiography, histology, and real‐time PCR. Male and female mice and their littermate controls were compared in all experiments. All animal studies were approved by the Toronto Centre for Phenogenomics (Ontario, Canada).
Western blot analysis
Primary chondrocytes were isolated from patients’ total knee arthroplasty samples and mouse knee joints using previously described methods 13, 14. Protein lysates from the chondrocytes were harvested using Reporter Lysis Buffer (Promega), according to the manufacturer's instructions. Antibodies against INSIG1 (1:100, sc‐25124‐R; Santa Cruz Biotechnology) and ACTIN (1:5,000, A5441; Sigma) were used. The signals were detected and quantified using the ChemiDoc MP Imaging System (Bio‐Rad).
Radiography and histology
Mouse knee joints were harvested and fixed in 10% phosphate‐buffered formalin. Radiographs of the joints were obtained using the Faxitron MX20 X‐ray system. Samples were decalcified in 20% EDTA (pH 8.0), dehydrated, and embedded in paraffin for sectioning, as previously described 4. Histologic analysis was performed using antibodies against COL10A1 (X53; Quartett) and HMGCR (ab174830; Abcam), and by staining with hematoxylin and eosin and Safranin O. Histomorphometry was performed to measure the osteophyte volume and the bone volume relative to total volume 19, as previously described 4.
Grading for OA severity was performed in a blinded manner using the International Cartilage Repair Society (ICRS) 20 and Osteoarthritis Research Society International (OARSI) 21 scoring systems, as previously described 4. For scoring with the ICRS system, images of the knee joints were assessed for 6 features commonly associated with OA, including changes to the subchondral bone, and categories were tallied into an overall score to represent disease severity (lower scores represent greater severity).
Sterol quantification
Primary mouse chondrocytes were isolated from the knee joints and cultured as previously described 14. To measure total sterol and lipid levels, these chondrocytes were fixed with 10% phosphate‐buffered formalin for 10 minutes. Cells were stained with oil red O, and the alcohol‐extracted stain was quantified by spectrophotometry (optical density [OD] 500 nm), with readings normalized to the values for crystal violet stain (OD 540 nm).
To measure cholesterol synthesis, chondrocytes were pooled and incubated with 50 μCi/ml 3H‐acetic acid sodium salt overnight. Lipid extracted from the cells was subjected to thin layer chromatography for separation of the components, including cholesterol. Incorporation of 3H‐acetic acid sodium salt was measured in triplicate, and results are reported as the change (in counts per minute) relative to the values in controls.
Chromatin immunoprecipitation (ChIP) assay
A ChIP assay was conducted using a SimpleChIP Plus Enzymatic Chromatin Immunoprecipitation Kit with magnetic beads (catalog no. 9005; Cell Signaling Technology) according to the manufacturer's protocol, with modifications. Cultured ATDC5 cells were treated with 10 μM lovastatin and fixed with 1% formaldehyde to maintain DNA–protein binding interactions. Following nuclei preparation and enzymatic chromatin digestion (in accordance with the manufacturer's protocol), sonication was performed to shear the chromatin and achieve a target size range of 200–700 bp. A SREBF2 antibody (catalog no. AF7119‐SP; R&D Systems) and a negative control IgG antibody (Active Motif) were used to immunoprecipitate the DNA–protein complexes. Quantification of DNA was performed by real‐time PCR using custom primers for Adamts5, in the proximal promoter region and in a distal control region ∼38 kb away.
Statistical analysis
Values are reported as the mean and 95% confidence interval (95% CI), unless stated otherwise. Power calculations were conducted based on the variation observed in our previous study 4. Statistically significant differences between the groups were determined by Mann‐Whitney U test for comparisons of histologic grading, and by t‐test for comparisons of average gene expression and spectrophotometric quantification. P values less than 0.05 were considered statistically significant.
RESULTS
Regulation of cholesterol homeostatic gene expression by HH signaling
To identify HH target genes, we performed microarray analysis of human OA cartilage samples obtained from patients undergoing total knee replacement surgery. Articular cartilage explants were incubated overnight with an antagonist of HH signaling, C31H42N4O5, or a vehicle carrier as control 22. RNA was extracted using an optimized protocol 13. Known HH target genes were down‐regulated in the treated groups (results not shown), thereby confirming the modulation of HH signaling in human OA articular cartilage.
Results from the Affymetrix Human Gene 1.0 ST microarray including 3 independent biologic replicates were analyzed. In comparing the HH antagonist–treated group to the control group, differentially expressed genes were filtered for those whose expression changed in the same direction across all 3 data sets. This method of analysis was chosen in an effort to control for the heterogeneity that is inherent to human samples. Criteria requiring arbitrary values for the fold change or statistical cutoff were not applied, in order to capture subtle changes across entire gene networks. This approach is biologically relevant for detecting small perturbations with effects that can accumulate over time to cause progressive diseases such as OA.
Ingenuity Pathway Analysis was used as an unbiased method for identifying signaling pathways, molecular networks, and biologic processes that were represented in the microarray gene list. After filtering for all validated direct and indirect relationships according to the Ingenuity Knowledge Base, we found that the cholesterol biosynthetic pathway was the most significantly dysregulated (P = 3.91 × 10−13) (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract). Several genes known to be involved in cholesterol homeostasis 23, 24 were found to be up‐regulated in the human OA cartilage tissue with HH inhibition (Figure 1A).
Figure 1.
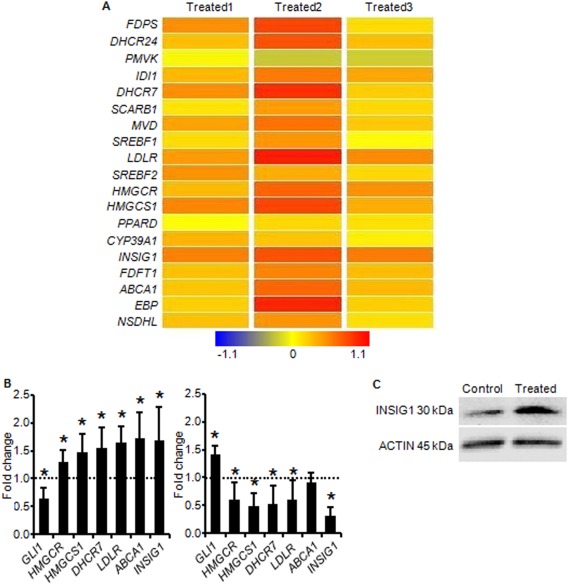
Hedgehog (HH) signaling regulates expression of cholesterol homeostatic genes. A, Results from Affymetrix Human Gene 1.0 ST microarray analysis of independent human osteoarthritic (OA) cartilage samples treated with an HH antagonist. Greater intensity of red represents increased gene expression with HH inhibition. B, Results from real‐time polymerase chain reaction analysis validate HH regulation of the cholesterol homeostatic genes identified by microarray. The human OA cartilage samples (n = 3) were treated with an HH antagonist (left) or an HH agonist (right). Values are the mean fold change in gene expression (with 95% confidence intervals) relative to that in untreated controls (set at 1.0 [broken horizontal line]). ∗ = P < 0.05 versus controls. C, Representative Western blot of INSIG1 protein expression in extracts from human OA cartilage explants treated with an HH antagonist or vehicle control. Actin was used as a loading control.
Although dysregulation of systemic cholesterol has been previously associated with OA 9, 10, 11, 12, intracellular cholesterol homeostasis in OA chondrocytes is not well understood. We sought to assess the role of HH signaling in regulating the expression of cholesterol homeostatic genes in chondrocytes.
Independent human OA cartilage samples were subjected to HH modulation and assayed for expression of genes that are known to be involved in cholesterol homeostasis 23, 24. The results of real‐time PCR validated the microarray findings by showing that HH inhibition, as evidenced by down‐regulation of GLI1, resulted in up‐regulation of the cholesterol homeostatic genes HMGCR, HMGCS1, DHCR7, LDLR, ABCA1, and INSIG1. Consistent with this inverse relationship, HH activation, as evidenced by up‐regulation of GLI1, resulted in a trend toward down‐regulation of these same genes (Figure 1B). The changes in expression were small but occurred across multiple genes in the pathway. This finding recapitulates the findings from microarray and is consistent with previously reported trends for changes in the expression of cholesterol homeostatic genes 16.
Transcription of cholesterol homeostatic genes is ultimately governed by SRE binding transcription factor 2 (SREBF2) in an end‐product feedback manner that is dependent on regulators such as insulin induced gene 1 (INSIG1) 16, 23. When the intracellular cholesterol level is high, the INSIG proteins prevent cholesterol biosynthesis, in part by tethering the SREBF proteins to the endoplasmic reticulum membrane and preventing transcription of target genes. When the intracellular cholesterol level is low, INSIG allows SREBF to be processed and to translocate to the nucleus, thereby activating transcription and restoring cholesterol homeostasis 16. In human OA cartilage, INSIG1 gene expression has been found to be significantly down‐regulated (fold change −2.38; P < 0.05) when compared to that in normal cartilage 25. In comparing more severely affected areas of human OA cartilage to less severely affected areas, expression of both INSIG1 and INSIG2 was found to be down‐regulated (fold change −7.09 and −3.89, respectively) (results not shown). Taken together, these data show that reduced INSIG expression correlates with increased OA severity.
These findings suggest that cholesterol homeostasis is perturbed, possibly as a result of the HH activation that is reported to accompany OA 4, 8. We found that when HH signaling was inhibited in OA cartilage, INSIG1 gene expression increased, and this translated to an increase in the level of INSIG1 protein (Figure 1C). As a critical negative regulator of cholesterol homeostasis, the inverse relationship between HH signaling and INSIG1 expression suggests that inhibition of HH signaling reduces cholesterol biosynthesis in chondrocytes. This was further supported by our observations of the overall increase in transcription of cholesterol homeostatic genes following HH inhibition, an increase that typically occurs in response to reduced intracellular cholesterol levels.
Regulation of cholesterol biosynthesis by HH signaling in chondrocytes
To investigate the net effect of HH signaling on cholesterol homeostasis in chondrocytes, we used genetically modified mice in which Gli‐mediated transcription was modulated. Previous studies have demonstrated that Gli‐mediated transcription is reduced in Gli2zfd mice (designated Gli2 +/−) by disrupting a single Gli2 allele 18, and Gli‐mediated transcription is increased in Col2a1‐Gli2 mice by overexpressing Gli2 17. Expression of both the Insig1 gene and the INSIG1 protein was increased in Gli2 +/− mouse cartilage and decreased in Col2a1‐Gli2 mouse cartilage (Figure 2A and Supplementary Figure 1A, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract). This finding confirmed that there is an inverse relationship between HH signaling and INSIG1 expression in chondrocytes.
Figure 2.
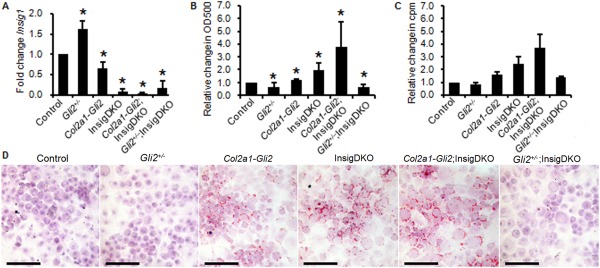
Hedgehog signaling regulates cholesterol biosynthesis in chondrocytes. Chondrocytes were obtained from mice with Gli transcriptional reduction (Gli2 +/−), Gli transcriptional activation (Col2a1‐Gli2), or Gli transcriptional reduction or activation and sterol accumulation (Insig1 double‐knockout [InsigDKO]) (n = 3 per group). Insig1(fl/fl);Insig2(−/−) mice were used as controls. A, Real‐time polymerase chain reaction analyses of change in Insig1 expression according to genotype. Results are the mean fold change (with 95% confidence intervals) relative to control. B, Spectrophotometric analyses for quantification of sterol and lipid accumulation in mouse chondrocytes. Results are the mean change (with 95% confidence intervals) in oil red O staining intensity relative to the values for crystal violet staining as a control. In A and B, ∗ = P < 0.05 versus controls. C, Cholesterol biosynthesis, as measured by 3H‐acetic acid sodium salt incorporation, in pooled primary mouse chondrocytes. Results, measured in triplicate, are shown as the mean ± SEM counts per minute relative to control. In A–C, control values were arbitrarily set at 1.0. D, Representative images of primary mouse chondrocytes stained with oil red O (same as those shown in B) to illustrate sterol and lipid accumulation. Bars = 100 μm.
To assess the effects of modulated HH signaling on total sterol and lipid levels, primary chondrocyte cultures were established using cartilage dissected from mouse knee joints, as previously described 14. Oil red O staining for sterol and lipid accumulation showed lower levels of sterol and lipid with Gli transcriptional reduction (represented in Gli2 +/− mice) and higher levels of sterol and lipid with Gli transcriptional activation (represented in Col2a1‐Gli2 mice) (Figure 2D). These differences in total sterol and lipid levels were quantified in the chondrocytes by spectrophotometry and found to be significant when compared to those in control chondrocytes (Figure 2B).
In primary human chondrocytes, stimulation of the HH pathway with the agonist purmorphamine resulted in a 3.70‐fold increase in oil red O staining as compared to that in untreated chondrocytes. This demonstrated that HH signaling regulates total sterol and lipid content in chondrocytes.
To determine whether Insig1 was mediating the effect of HH signaling on sterol homeostasis, we generated genetically modified mice in which Insig1 was removed from the chondrocytes. There are 2 mammalian Insig genes, Insig1 and Insig2, and these genes functionally compensate for each other in the regulation of sterol biosynthesis 16, 26. In the absence of these genes, mice accumulate cholesterol and triglycerides in a robust manner. To induce cholesterol accumulation in the chondrocytes, we crossed Insig1(fl/fl);Insig2(−/−) mice 16 with Col2a1‐Cre–transgenic mice 15, 27 (see Supplementary Figure 1B, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract). Analyses were performed by comparing Insig1 double‐knockout mice (designated InsigDKO) with control littermates (Insig1[fl/fl];Insig2[−/−]). To modulate HH signaling, InsigDKO mice were crossed with Gli2 +/− mice and Col2a1‐Gli2 mice, to generate Gli2 +/−;InsigDKO mice and Col2a1‐Gli2;InsigDKO mice, respectively (see Supplementary Figure 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract).
Using oil red O staining of primary mouse chondrocyte cultures, we confirmed that InsigDKO mouse chondrocytes had robust sterol and lipid accumulation. Despite the absence of Insig1, Gli‐mediated transcription still modulated sterol and lipid accumulation in the chondrocytes. Gli transcriptional activation (in Col2a1‐Gli2;InsigDKO mice) increased accumulation, and Gli transcriptional reduction (in Gli2 +/−;InsigDKO mice) decreased accumulation (Figures 2B and D).
To refine the potential sterol intermediates contributing to this accumulation, we took a candidate approach to determine changes to cholesterol. Radiotracer studies were conducted to measure cholesterol production from 3H‐acetic acid in primary mouse chondrocyte cultures. The results were consistent with those from spectrophotometric quantification with oil red O staining, showing that levels of cholesterol were positively modulated by HH signaling in primary mouse chondrocytes (Figure 2C). By examining changes to the cholesterol level in both Insig1‐expressing mice (Gli2 +/− and Col2a1‐Gli2 mice) and Insig1‐knockout mice (InsigDKO, Col2a1‐Gli2;InsigDKO, and Gli2 +/−;InsigDKO mice), we determined that HH signaling could alter the cholesterol level in the absence of Insig1, showing that additional regulators are involved in this process.
Exacerbation of OA by intracellular cholesterol accumulation in chondrocytes
Since InsigDKO mice exhibited robust sterol accumulation in the chondrocytes, we evaluated the knees of 6‐month‐old mice for markers of OA, to determine whether intracellular sterol accumulation contributes to disease development. Compared to control littermates, InsigDKO mice showed characteristic signs of OA. Histologic analyses revealed loss of proteoglycan and thinner articular cartilage in InsigDKO mice (Figure 3A). This was accompanied, and potentially mediated, by increased expression of the proteases Mmp13 and Adamts5 in the cartilage (Figure 3C). Expression of the hypertrophic marker Col10a1 was also increased and shown to be concentrated at the surface of the eroding cartilage in InsigDKO mice (Figures 3C and D).
Figure 3.

Intracellular cholesterol accumulation in mouse chondrocytes induces osteoarthritic (OA) changes. A, Representative histologic sections of the knees of Insig1 double‐knockout (InsigDKO) mice compared to Insig1(fl/fl);Insig2(−/−) control mice at age 6 months, showing Safranin O staining for proteoglycan. Arrows indicate loss of proteoglycan (red) and cartilage defects in InsigDKO mice compared to controls. Bars = 100 μm. B, Representative radiographic images (lateral views) of the knees of the mice at age 4 months. Arrows indicate areas of subchondral sclerosis (whitening) in InsigDKO mice compared to controls. C, Results of real‐time polymerase chain reaction analyses of OA markers in cartilage microdissected from the knees of InsigDKO mice (shaded bars) and control mice (solid bars) at age 6 months (n = 4 per group). Results are the mean fold change (with 95% confidence intervals) in InsigDKO mice relative to controls (set at 1.0). ∗ = P < 0.05 versus controls. D, Representative histologic sections of articular cartilage from the femur of mice at age 6 months, showing staining for Col10a1 (brown). Arrow indicates increased staining of hypertrophic chondrocytes in InsigDKO mice compared to controls. Bars = 100 μm.
Radiographic analyses revealed irregularity of the bone contour and increased sclerosis in the subchondral bone of InsigDKO mice (Figure 3B). Histomorphometry analyses indicated that the bone volume was reduced in both the tibia and the femur of InsigDKO mice (see Supplementary Figure 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract). Whether the subchondral bone changes preceded changes to the articular cartilage or were a consequence of changes to the articular cartilage remains unclear, but the contribution of subchondral bone to OA progression is well recognized 28, 29. Following surgical induction of OA, InsigDKO mice showed osteophyte‐like protrusions that were 15% larger by volume than those in the control littermates (see Supplementary Figure 4, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract).
Blinded grading of the mouse knee joints for OA severity was conducted using the ICRS Visual Histological Assessment Scale. This grading scale, in which lower scores represent more severe OA 20, was chosen because it accounts for changes to the subchondral bone, whereas other scoring systems may minimize this feature 21, 30. InsigDKO mice showed overall ICRS scores that were reduced in comparison to control mice (mean IRCS score 11.3 in InsigDKO mice versus 17.3 in control mice; P < 0.05) (see Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract). OARSI grading, in which higher scores represent more severe OA 21, confirmed this finding (mean OARSI score 2.1 in InsigDKO mice versus 1.4 in control mice; P < 0.05).
There were no sex‐dependent differences in the overall ICRS scores (results not shown), but for the grading of OA severity on the cartilage surface category, male mice showed a more severe phenotype than female mice (mean ± 95% CI ICRS score 1.5 ± 1.0 in male mice versus 3.0 ± 0.0 in female mice) at 6 months of age. This is consistent with the findings from previous studies indicating that male mice develop a more severe OA phenotype than female mice 31.
To determine whether the cholesterol accumulation in InsigDKO mice was exacerbating OA by activating HH signaling, we examined expression of HH target genes in the mouse cartilage. Because there was no change in expression of Gli1, Ptch1, and Hhip, this phenotype cannot be attributed to changes in Gli transcriptional activity in InsigDKO mouse chondrocytes (see Supplementary Figure 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract). These results demonstrate that intracellular sterol accumulation in chondrocytes exacerbates the severity of OA in mice.
Attenuation of OA by pharmacologic cholesterol inhibition in cartilage
To assess whether inhibition of cholesterol production could mitigate the progression of OA, mice were treated with 3 mg/kg/day lovastatin. Drug pellets were placed adjacent to the synovial membrane of the knees of Col2a1‐Gli2 mice in which HH signaling was activated, the knees of InsigDKO mice in which cholesterol accumulation occurred, and the knees of mice in which OA was induced by medial meniscectomy 4. Lovastatin treatment increased the expression of HMGCR by 25% in mouse chondrocytes (P < 0.05; n = 3) (see Supplementary Figure 5, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract), which is the typical response observed in relation to lowered intracellular cholesterol levels. In all cases, lovastatin treatment reduced cartilage fibrillation and thinning (Figure 4).
Figure 4.
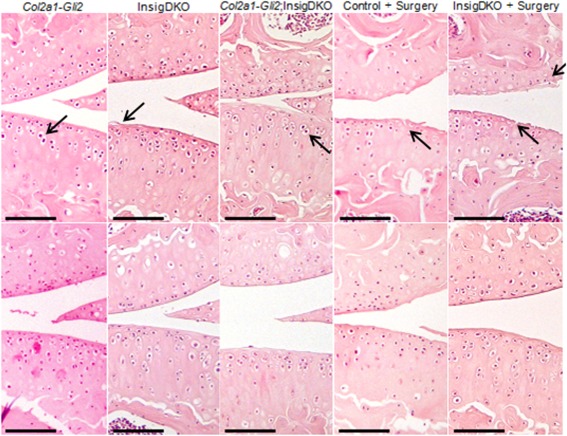
Pharmacologic cholesterol inhibition attenuates osteoarthritic (OA) changes in mice. Representative histologic sections show hematoxylin and eosin staining of the knee joints of 4‐month‐old mice after implantation with a slow‐release placebo (top panels) or statin pellet (bottom panels). Methods for inducing OA included Gli transcriptional activation by hedgehog signaling (Col2a1‐Gli2), cholesterol accumulation (Insig1 double‐knockout [InsigDKO]), or both (Col2a1‐Gli2;InsigDKO), as well as surgical excision of the medial meniscus (+ surgery) in either control or InsigDKO mice. Arrows indicate hypertrophic chondrocytes and cartilage fibrillation in placebo‐treated mice. Bars = 100 μm.
This finding was reflected by improvements in the overall ICRS scores for OA severity 20. For example, in InsigDKO mice with OA induced by medial meniscectomy surgery, the mean overall ICRS scores were 11.3 in the placebo group and 16.7 in the lovastatin group (P < 0.05) (see Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract). Furthermore, in Col2a1‐Gli2, InsigDKO, and Col2a1‐Gli2;InsigDKO mice, treatment with lovastatin attenuated chondrocyte hypertrophy, as indicated by changes to Col10a1 staining in the cartilage (see Supplementary Figure 6, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract).
To investigate the role of cholesterol inhibition in human OA, articular cartilage explants from patients with knee OA were treated with 10 μM lovastatin. Treatment with lovastatin induced increased HMGCR expression in the chondrocytes from human OA articular cartilage (see Supplementary Figure 7, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract), as expected in response to lowered intracellular cholesterol levels. Furthermore, lovastatin treatment significantly reduced the expression of MMP13 32, 33 and ADAMTS5, and also decreased ADAMTS5 promoter activity, in human OA chondrocytes (Figures 5A and B).
Figure 5.
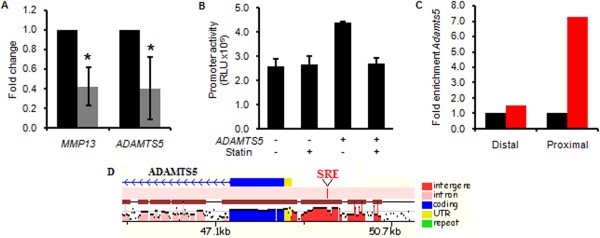
Pharmacologic cholesterol inhibition attenuates ADAMTS5 expression in human osteoarthritic (OA) cartilage. A, Results of real‐time polymerase chain reaction (PCR) analyses in human OA cartilage explants, showing a reduction in expression of the OA markers MMP13 and ADAMTS5 in samples treated with statin (shaded bars) as compared to vehicle control (solid bars) (n = 4 per group). Results are the mean fold change (with 95% confidence intervals) relative to controls (set at 1.0). ∗ = P < 0.05 versus controls. B, Luciferase activity from the ADAMTS5 promoter construct after transfection into primary human OA chondrocytes with or without statin treatment. Results, measured in triplicate, are the mean ± SEM relative light units (RLU). C, Enrichment of ADAMTS5 gene expression in ATDC5 cells. Chromatin immunoprecipitation was performed in ATDC5 cells with antibodies to SREBF2 (red bars) or IgG as a control (black bars). ADAMTS5 expression was assessed by real‐time PCR with primers designed in the proximal promoter region (Proximal) or a control region (Distal). Results are the fold change in ADAMTS5 expression with anti‐SREBF2 relative to IgG (set at 1.0), D, Schematic diagram of the ADAMTS5 promoter, showing a sterol regulatory element (SRE) consensus binding site (red) that is conserved in both mice and humans (results obtained from Mulan analysis; see http://mulan.dcode.org). UTR = untranslated region.
Results of the ChIP assay showed binding of SREBF2 to the proximal promoter of ADAMTS5, a region that contains a conserved SRE binding site (Figures 5C and D). Deletion of the SRE site diminished ADAMTS5 promoter activity (see Supplementary Figure 8, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39337/abstract), suggesting that ADAMTS5 is a target of the SREBF transcription factors. Since cholesterol levels regulate processing of the SREBF proteins 34, these data suggest that cholesterol accumulation can impact OA, at least in part, by modulating expression of ADAMTS5.
DISCUSSION
In this study, we show that HH signaling regulates the cholesterol homeostatic pathway in human and mouse cartilage. In genetically modified mice, the level of Gli‐mediated transcription positively correlates with the level of intracellular cholesterol accumulation and the severity of OA. Furthermore, cholesterol blockade reduces the severity of OA in Col2a1‐Gli2 mice with HH activation. This demonstrates that intracellular cholesterol biosynthesis at least partially mediates the effect of HH signaling in chondrocytes in OA 4.
Sterols have been shown to modulate HH signaling 35, 36, 37, 38, but this is the first study to demonstrate that HH signaling modulates cholesterol homeostasis. Cholesterol modifies HH signaling with the covalent addition of a cholesterol moiety to HH ligands 39. Key regulators of HH signaling contain conserved sterol‐sensing domains, suggesting that their behavior may be affected by sterols 40. Perturbations in either HH signaling or cholesterol homeostasis produce similar central nervous system abnormalities, facial dysmorphisms, and skeletal defects 41, 42. These parallels point to a mutual regulatory relationship between HH signaling and cholesterol homeostasis.
A previous study by Engelking et al noted the phenotypic similarity between total InsigDKO mouse embryos and mouse embryos with modulated HH signaling 26. Engelking et al hypothesized that the sterol accumulation in InsigDKO mice caused HH activation, but that study did not find differences in gene expression levels of Shh, Smo, Ptch1, or Gli1 in the palate tissues at 13.5 days postcoitum 26. Similarly, our InsigDKO mice did not show changes to HH target gene expression in the chondrocytes. Thus, the phenotypic changes observed in response to cholesterol accumulation are not likely attributable to perturbations in HH signaling in the cartilage. It is possible that HH signaling is modulated in surrounding tissues due to altered ligand trafficking, or is modulated in cartilage by sterol intermediates other than cholesterol 24. The potential mutual regulatory relationship between HH signaling and sterol homeostasis merits further investigation.
Given that OA is a degenerative disease, it conceivably results from the culmination of small changes over time. The magnitude of the changes to both gene expression and cholesterol accumulation, as reported herein, represent relatively small perturbations that accumulate over time, causing progressive cartilage degeneration and subchondral bone abnormalities. The resulting OA phenotype is mild, but reflective of early disease presentation in humans. Although it is possible that this phenotype could be attributed, in part, to developmental alterations or to structural changes to the joints, the rescue we observed with 8 weeks of statin treatment demonstrates at least a partial contribution of cholesterol dysregulation to the phenotype.
Studies examining the relationship between sterols and the occurrence of OA have been largely epidemiologic 9, 11, 43, 44. One limitation of those studies is the assumption that systemic sterol levels are representative of intraarticular sterol levels. Prete et al measured total cholesterol levels in patients with OA in comparison to control subjects, and showed that plasma levels of total cholesterol were comparable whereas the synovial fluid levels were markedly increased in patients with OA (range 4–169 mg/dl in OA patients versus 7–8 mg/dl in control subjects) 45. This finding suggests that any pharmacologic cholesterol inhibitor should target the synovial joint specifically 46.
Gierman et al reported that rescue of the OA phenotype could be achieved with statin treatment, but not with ezetimibe treatment 47, suggesting that reductions in intracellular cholesterol production (by statin), and not reductions in serum cholesterol (by ezetimibe), are required to attenuate OA progression. In support of this finding, the changes we observed in OA could be attributed to chondrocyte‐specific cholesterol accumulation. These changes were attenuated by statin treatment administered locally to the knee in vivo, which was effective in altering cholesterol homeostasis in the chondrocytes, thereby rescuing the OA phenotype.
We showed that statin treatment reduced the expression of proteases in human OA cartilage in vitro, including the promoter activity of ADAMTS5, the major protease responsible for cartilage degradation in OA 31. Our results indicate that activation of HH signaling in the chondrocytes induces cholesterol accumulation, the levels of which govern processing of SREBF2 transcription factors. We also show that ADAMTS5 contains a conserved SRE binding site to which SREBF2 binds, making it a putative target of SREBF2 and subject to regulation by intracellular cholesterol levels. This is one mechanism through which cholesterol levels can regulate OA progression.
Alternate mechanisms include induction of chondrocyte hypertrophy by cholesterol‐mediated activation of the lipid regulator retinoic acid receptor–related orphan nuclear receptor α 48, or cartilage degradation by proteases that are regulated by protein geranylgeranylation, which requires intermediates from the cholesterol biosynthetic pathway 32. Furthermore, statins have been shown to reduce inflammation 49 and induce anabolic effects in cartilage 33, 46, 50, and therefore additional studies are required to elucidate the exact mechanism through which sterol levels impact cartilage biology.
Identifying intracellular cholesterol as a mediator of OA reinforces the importance of chondrocyte homeostasis in this disease, and puts forth a mechanism by which HH signaling exacerbates OA. Cholesterol biosynthesis is identified as a novel downstream pathway that is regulated by HH signaling, a relationship that may be relevant to both the physiologic and pathologic processes of cartilage.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Alman had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Ali, Adeli, Alman.
Acquisition of data
Ali, Al‐Jazrawe, Ma, Whetstone, Poon, Farr, Naples, Adeli.
Analysis and interpretation of data
Ali, Al‐Jazrawe, Ma, Whetstone, Poon, Adeli, Alman.
Supporting information
Supplementary Figure 1. Hedgehog signaling modulates expression of Insig1, a major negative regulator of cholesterol biosynthesis. (A) Western blot analysis for INSIG1 in murine cartilage with Gli transcriptional reduction (Gli2+/‐) or Gli transcriptional activation (Col2a1‐Gli2). (B) Western blot analysis for INSIG1 in murine cartilage resulting from the cross between Insig1(fl/fl);Insig2(‐/‐) and Col2a1‐Cre. Reduction of INSIG1 was observed in Insig1(‐/‐) cartilage [Insig1(‐/‐);Insig2(‐/‐), subsequently designated InsigDKO] but not Insig1(‐/fl) cartilage [Insig1(‐/fl);Insig2(‐/‐)], so all analyses focused on Insig1(‐/‐) and Insig1(fl/fl) cartilage [Insig1(fl/fl);Insig2(‐/‐), subsequently designated Control]. ACTIN is shown as a loading control.
Supplementary Figure 2. Expression of Hedgehog target genes is unaltered in InsigDKO cartilage. Real‐time PCR for Hh target genes Gli1, Ptch1, and Hhip in the articular cartilage of mice with cholesterol accumulation (InsigDKO), with reduced Gli‐mediated transcription (Gli2+/‐;InsigDKO), or with increased Gli‐mediated transcription (Col2a1‐Gli2;InsigDKO). Expression in Control mice was arbitrarily defined as ‘1’ (dashed line) and data for each genotype given as the mean. Error bars are 95% confidence intervals (n = 3; *P < 0.05).
Supplementary Figure 3. InsigDKO mice show reduced bone volume compared to control mice. Histomorphometry analysis for bone volume in the tibia and femur of InsigDKO mice (gray bars) and control mice (black bars) at 6 months of age (*P < 0.05).
Supplementary Figure 4. Osteophyte‐like protrusions are more pronounced in InsigDKO mice compared to control mice. Representative histologic sections showing hematoxylin and eosin staining of the knee joints in 4‐month‐old mice following surgical induction of osteoarthritis (top panels). A magnified view of the joint space shows areas containing osteophyte‐like protrusions at the periphery of the joint (arrows; bottom panels).
Supplementary Figure 5. Effective statin treatment is shown by increased HMGCR in mouse cartilage. Representative histologic sections showing HMGCR immunohistochemistry (brown) in the articular cartilage at 4 months for InsigDKO mice treated with placebo or statin. Scale bar, 50 μm.
Supplementary Figure 6. Chondrocyte hypertrophy in the articular cartilage is altered by cholesterol accumulation. Representative histologic sections showing Col10a1 immunohistochemistry in the articular cartilage of the femur at 4 months of age for each genotype/treatment group shown in Figure 4. Scale bar, 100 μm.
Supplementary Figure 7. Effective statin treatment is shown by increased HMGCR in human cartilage. Real‐time PCR of HMGCR in human osteoarthritic articular cartilage explants treated with statin. Expression in the control group was arbitrarily defined as ‘1’ and data from the statin‐treated group given as the mean. Error bar is 95% confidence interval (n = 4; *P < 0.05).
Supplementary Figure 8. Deletion of the SRE binding site in the ADAMTS5 promoter diminishes reporter activity. Luciferase activity from a negative control vector (Negative), the ADAMTS5 promoter construct (ADAMTS5), and the ADAMTS5 promoter construct with deleted SRE site (ADAMTS5‐SRE), following transfection into ATDC5 cells. Measured in triplicate and reported as relative light units (RLU). Error bars are SEM.
Supplementary Table 1. Summary of Ingenuity Pathway Analysis. The gene list resulting from microarray analysis was subjected to Ingenuity Pathway Analysis. The settings were adjusted to consider all molecules and/or relationships, direct and indirect, according to the Ingenuity Knowledge Base reference set. This unsupervised analysis identified Cholesterol Biosynthesis to be the most significantly dysregulated pathway.
Supplementary Table 2. ICRS scores for osteoarthritis. The International Cartilage Repair Society (ICRS) score was used to grade histologic sections in a blinded manner. As previously reported4, the mean and 95% confidence interval are given for each criterion, and a summary score is provided with Mann‐Whitney U statistical analysis to determine significant differences. (A) Grading of knee joints from 6‐month‐old mice reveal more severe osteoarthritis in InsigDKO mice as compared to Control mice (P < 0.05). (B) Knee joints from 4‐month‐old mice were graded for genotypes (without surgery) shown in Figure 4. Statistical analyses compared placebo to statin treatment for each of Col2a1‐Gli2, InsigDKO, and Col2a1‐Gli2;InsigDKO. (C) Knee joints from 4‐month‐old mice were graded for genotypes (with surgery) shown in Figure 4. Statistical analyses compared placebo to statin treatment in mice that underwent excision of the medial meniscus. Statin treatment was found to have a statistically significant difference in all groups (P < 0.05).
ACKNOWLEDGMENT
We thank D. Backstein (Mount Sinai Hospital, Toronto, Ontario, Canada) for assistance in obtaining human cartilage specimens.
The content herein is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1. Hunter DJ, Eckstein F. Exercise and osteoarthritis. J Anat 2009;214:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Issa RI, Griffin TM. Pathobiology of obesity and osteoarthritis: integrating biomechanics and inflammation. Pathobiol Aging Age Relat Dis 2012;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aigner T, Soder S, Gebhard PM, McAlinden A, Haag J. Mechanisms of disease: role of chondrocytes in the pathogenesis of osteoarthritis—structure, chaos and senescence. Nat Clin Pract Rheumatol 2007;3:391–9. [DOI] [PubMed] [Google Scholar]
- 4. Lin AC, Seeto BL, Bartoszko JM, Khoury MA, Whetstone H, Ho L, et al. Modulating Hedgehog signaling can attenuate the severity of osteoarthritis. Nat Med 2009;15:1421–5. [DOI] [PubMed] [Google Scholar]
- 5. Mak KK, Kronenberg HM, Chuang PT, Mackem S, Yang Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 2008;135:1947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH‐related protein. Science 1996;273:613–22. [DOI] [PubMed] [Google Scholar]
- 7. Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell 2008;15:801–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou J, Chen Q, Lanske B, Fleming BC, Terek R, Wei X, et al. Disrupting the Indian hedgehog signaling pathway in vivo attenuates surgically induced osteoarthritis progression in Col2a1‐CreERT2; Ihhfl/fl mice. Arthritis Res Ther 2014;16:R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sturmer T, Sun Y, Sauerland S, Zeissig I, Gunther KP, Puhl W, et al. Serum cholesterol and osteoarthritis: the baseline examination of the Ulm Osteoarthritis Study. J Rheumatol 1998;25:1827–32. [PubMed] [Google Scholar]
- 10. Kostopoulou F, Gkretsi V, Malizos KN, Iliopoulos D, Oikonomou P, Poultsides L, et al. Central role of SREBP‐2 in the pathogenesis of osteoarthritis. PLoS One 2012;7:e35753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Al‐Arfaj AS. Radiographic osteoarthritis and serum cholesterol. Saudi Med J 2003;24:745–7. [PubMed] [Google Scholar]
- 12. Villalvilla A, Gomez R, Largo R, Herrero‐Beaumont G. Lipid transport and metabolism in healthy and osteoarthritic cartilage. Int J Mol Sci 2013;14:20793–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ali SA, Alman B. RNA extraction from human articular cartilage by chondrocyte isolation. Anal Biochem 2012;429:39–41. [DOI] [PubMed] [Google Scholar]
- 14. Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc 2008;3:1253–60. [DOI] [PubMed] [Google Scholar]
- 15. Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Col2a1‐directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 2000;26:145–6. [PubMed] [Google Scholar]
- 16. Engelking LJ, Liang G, Hammer RE, Takaishi K, Kuriyama H, Evers BM, et al. Schoenheimer effect explained: feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J Clin Invest 2005;115:2489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hopyan S, Gokgoz N, Poon R, Gensure RC, Yu C, Cole WG, et al. A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat Genet 2002;30:306–10. [DOI] [PubMed] [Google Scholar]
- 18. Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 1997;124:113–23. [DOI] [PubMed] [Google Scholar]
- 19. Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 1987;2:595–610. [DOI] [PubMed] [Google Scholar]
- 20. Mainil‐Varlet P, Aigner T, Brittberg M, Bullough P, Hollander A, Hunziker E, et al. Histological assessment of cartilage repair: a report by the Histology Endpoint Committee of the International Cartilage Repair Society (ICRS). J Bone Joint Surg Am 2003;85‐A Suppl 2:45–57. [PubMed] [Google Scholar]
- 21. Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative: recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 2010;18 Suppl 3:S17–23. [DOI] [PubMed] [Google Scholar]
- 22. Williams JA, Guicherit OM, Zaharian BI, Xu Y, Chai L, Wichterle H, et al. Identification of a small molecule inhibitor of the Hedgehog signaling pathway: effects on basal cell carcinoma‐like lesions [published erratum appears in Proc Natl Acad Sci U S A 2003;100:8607]. Proc Natl Acad Sci U S A 2003;100:4616–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A 2003;100:12027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gill S, Chow R, Brown AJ. Sterol regulators of cholesterol homeostasis and beyond: the oxysterol hypothesis revisited and revised. Prog Lipid Res 2008;47:391–404. [DOI] [PubMed] [Google Scholar]
- 25. Karlsson C, Dehne T, Lindahl A, Brittberg M, Pruss A, Sittinger M, et al. Genome‐wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage 2010;18:581–92. [DOI] [PubMed] [Google Scholar]
- 26. Engelking LJ, Evers BM, Richardson JA, Goldstein JL, Brown MS, Liang G. Severe facial clefting in Insig‐deficient mouse embryos caused by sterol accumulation and reversed by lovastatin. J Clin Invest 2006;116:2356–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grant TD, Cho J, Ariail KS, Weksler NB, Smith RW, Horton WA. Col2‐GFP reporter marks chondrocyte lineage and chondrogenesis during mouse skeletal development. Dev Dyn 2000;218:394–400. [DOI] [PubMed] [Google Scholar]
- 28. Mansell JP, Bailey AJ. Abnormal cancellous bone collagen metabolism in osteoarthritis. J Clin Invest 1998;101:1596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang R, Fang H, Chen Y, Shen J, Lu H, Zeng C, et al. Gene expression analyses of subchondral bone in early experimental osteoarthritis by microarray. PLoS One 2012;7:e32356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mankin HJ, Dorfman H, Lippiello L, Zarins A. Biochemical and metabolic abnormalities in articular cartilage from osteo‐arthritic human hips. II. Correlation of morphology with biochemical and metabolic data. J Bone Joint Surg Am 1971;53:523–37. [PubMed] [Google Scholar]
- 31. Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005;434:644–8. [DOI] [PubMed] [Google Scholar]
- 32. Barter MJ, Hui W, Lakey RL, Catterall JB, Cawston TE, Young DA. Lipophilic statins prevent matrix metalloproteinase‐mediated cartilage collagen breakdown by inhibiting protein geranylgeranylation. Ann Rheum Dis 2010;69:2189–98. [DOI] [PubMed] [Google Scholar]
- 33. Simopoulou T, Malizos KN, Poultsides L, Tsezou A. Protective effect of atorvastatin in cultured osteoarthritic chondrocytes. J Orthop Res 2010;28:110–5. [DOI] [PubMed] [Google Scholar]
- 34. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane‐bound transcription factor. Cell 1997;89:331–40. [DOI] [PubMed] [Google Scholar]
- 35. Corcoran RB, Scott MP. Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc Natl Acad Sci U S A 2006;103:8408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stottmann RW, Turbe‐Doan A, Tran P, Kratz LE, Moran JL, Kelley RI, et al. Cholesterol metabolism is required for intracellular Hedgehog signal transduction in vivo. PLoS Genet 2011;7:e1002224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eaton S. Multiple roles for lipids in the Hedgehog signalling pathway. Nat Rev Mol Cell Biol 2008;9:437–45. [DOI] [PubMed] [Google Scholar]
- 38. Dwyer JR, Sever N, Carlson M, Nelson SF, Beachy PA, Parhami F. Oxysterols are novel activators of the Hedgehog signaling pathway in pluripotent mesenchymal cells. J Biol Chem 2007;282:8959–68. [DOI] [PubMed] [Google Scholar]
- 39. Jeong J, McMahon AP. Cholesterol modification of Hedgehog family proteins. J Clin Invest 2002;110:591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuwabara PE, Labouesse M. The sterol‐sensing domain: multiple families, a unique role? Trends Genet 2002;18:193–201. [DOI] [PubMed] [Google Scholar]
- 41. Gofflot F, Hars C, Illien F, Chevy F, Wolf C, Picard JJ, et al. Molecular mechanisms underlying limb anomalies associated with cholesterol deficiency during gestation: implications of Hedgehog signaling. Hum Mol Genet 2003;12:1187–98. [DOI] [PubMed] [Google Scholar]
- 42. Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, et al. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996;383:407–13. [DOI] [PubMed] [Google Scholar]
- 43. Kadam UT, Blagojevic M, Belcher J. Statin use and clinical osteoarthritis in the general population: a longitudinal study. J Gen Intern Med 2013;28:943–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Clockaerts S, Van Osch GJ, Bastiaansen‐Jenniskens YM, Verhaar JA, Van Glabbeek F, Van Meurs JB, et al. Statin use is associated with reduced incidence and progression of knee osteoarthritis in the Rotterdam study [published erratum appears in Ann Rheum Dis 2012;71:1264]. Ann Rheum Dis 2012;71:642–7. [DOI] [PubMed] [Google Scholar]
- 45. Prete PE, Gurakar‐Osborne A, Kashyap ML. Synovial fluid lipoproteins: review of current concepts and new directions. Semin Arthritis Rheum 1993;23:79–89. [DOI] [PubMed] [Google Scholar]
- 46. Baker JF, Walsh P, Mulhall KJ. Statins: a potential role in the management of osteoarthritis? Joint Bone Spine 2011;78:31–4. [DOI] [PubMed] [Google Scholar]
- 47. Gierman LM, Kuhnast S, Koudijs A, Pieterman EJ, Kloppenburg M, van Osch GJ, et al. Osteoarthritis development is induced by increased dietary cholesterol and can be inhibited by atorvastatin in APOE*3Leiden.CETP mice—a translational model for atherosclerosis. Ann Rheum Dis 2014;73:921–7. [DOI] [PubMed] [Google Scholar]
- 48. Woods A, James CG, Wang G, Dupuis H, Beier F. Control of chondrocyte gene expression by actin dynamics: a novel role of cholesterol/Ror‐α signalling in endochondral bone growth. J Cell Mol Med 2009;13:3497–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leung BP, Sattar N, Crilly A, Prach M, McCarey DW, Payne H, et al. A novel anti‐inflammatory role for simvastatin in inflammatory arthritis. J Immunol 2003;170:1524–30. [DOI] [PubMed] [Google Scholar]
- 50. Yamashita A, Morioka M, Kishi H, Kimura T, Yahara Y, Okada M, et al. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature 2014;513:507–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Hedgehog signaling modulates expression of Insig1, a major negative regulator of cholesterol biosynthesis. (A) Western blot analysis for INSIG1 in murine cartilage with Gli transcriptional reduction (Gli2+/‐) or Gli transcriptional activation (Col2a1‐Gli2). (B) Western blot analysis for INSIG1 in murine cartilage resulting from the cross between Insig1(fl/fl);Insig2(‐/‐) and Col2a1‐Cre. Reduction of INSIG1 was observed in Insig1(‐/‐) cartilage [Insig1(‐/‐);Insig2(‐/‐), subsequently designated InsigDKO] but not Insig1(‐/fl) cartilage [Insig1(‐/fl);Insig2(‐/‐)], so all analyses focused on Insig1(‐/‐) and Insig1(fl/fl) cartilage [Insig1(fl/fl);Insig2(‐/‐), subsequently designated Control]. ACTIN is shown as a loading control.
Supplementary Figure 2. Expression of Hedgehog target genes is unaltered in InsigDKO cartilage. Real‐time PCR for Hh target genes Gli1, Ptch1, and Hhip in the articular cartilage of mice with cholesterol accumulation (InsigDKO), with reduced Gli‐mediated transcription (Gli2+/‐;InsigDKO), or with increased Gli‐mediated transcription (Col2a1‐Gli2;InsigDKO). Expression in Control mice was arbitrarily defined as ‘1’ (dashed line) and data for each genotype given as the mean. Error bars are 95% confidence intervals (n = 3; *P < 0.05).
Supplementary Figure 3. InsigDKO mice show reduced bone volume compared to control mice. Histomorphometry analysis for bone volume in the tibia and femur of InsigDKO mice (gray bars) and control mice (black bars) at 6 months of age (*P < 0.05).
Supplementary Figure 4. Osteophyte‐like protrusions are more pronounced in InsigDKO mice compared to control mice. Representative histologic sections showing hematoxylin and eosin staining of the knee joints in 4‐month‐old mice following surgical induction of osteoarthritis (top panels). A magnified view of the joint space shows areas containing osteophyte‐like protrusions at the periphery of the joint (arrows; bottom panels).
Supplementary Figure 5. Effective statin treatment is shown by increased HMGCR in mouse cartilage. Representative histologic sections showing HMGCR immunohistochemistry (brown) in the articular cartilage at 4 months for InsigDKO mice treated with placebo or statin. Scale bar, 50 μm.
Supplementary Figure 6. Chondrocyte hypertrophy in the articular cartilage is altered by cholesterol accumulation. Representative histologic sections showing Col10a1 immunohistochemistry in the articular cartilage of the femur at 4 months of age for each genotype/treatment group shown in Figure 4. Scale bar, 100 μm.
Supplementary Figure 7. Effective statin treatment is shown by increased HMGCR in human cartilage. Real‐time PCR of HMGCR in human osteoarthritic articular cartilage explants treated with statin. Expression in the control group was arbitrarily defined as ‘1’ and data from the statin‐treated group given as the mean. Error bar is 95% confidence interval (n = 4; *P < 0.05).
Supplementary Figure 8. Deletion of the SRE binding site in the ADAMTS5 promoter diminishes reporter activity. Luciferase activity from a negative control vector (Negative), the ADAMTS5 promoter construct (ADAMTS5), and the ADAMTS5 promoter construct with deleted SRE site (ADAMTS5‐SRE), following transfection into ATDC5 cells. Measured in triplicate and reported as relative light units (RLU). Error bars are SEM.
Supplementary Table 1. Summary of Ingenuity Pathway Analysis. The gene list resulting from microarray analysis was subjected to Ingenuity Pathway Analysis. The settings were adjusted to consider all molecules and/or relationships, direct and indirect, according to the Ingenuity Knowledge Base reference set. This unsupervised analysis identified Cholesterol Biosynthesis to be the most significantly dysregulated pathway.
Supplementary Table 2. ICRS scores for osteoarthritis. The International Cartilage Repair Society (ICRS) score was used to grade histologic sections in a blinded manner. As previously reported4, the mean and 95% confidence interval are given for each criterion, and a summary score is provided with Mann‐Whitney U statistical analysis to determine significant differences. (A) Grading of knee joints from 6‐month‐old mice reveal more severe osteoarthritis in InsigDKO mice as compared to Control mice (P < 0.05). (B) Knee joints from 4‐month‐old mice were graded for genotypes (without surgery) shown in Figure 4. Statistical analyses compared placebo to statin treatment for each of Col2a1‐Gli2, InsigDKO, and Col2a1‐Gli2;InsigDKO. (C) Knee joints from 4‐month‐old mice were graded for genotypes (with surgery) shown in Figure 4. Statistical analyses compared placebo to statin treatment in mice that underwent excision of the medial meniscus. Statin treatment was found to have a statistically significant difference in all groups (P < 0.05).