

Summary
During mitosis, the macromolecular kinetochore complex assembles on the centromere to orchestrate chromosome segregation. The properties and architecture of the 16-subunit Constitutive Centromere-Associated Network (CCAN) that allow it to build a robust platform for kinetochore assembly are poorly understood. Here, we use inducible CRISPR knockouts and biochemical reconstitutions to define the interactions between the human CCAN proteins. We find that the CCAN does not assemble as a linear hierarchy, and instead, each sub-complex requires multiple non-redundant interactions for its localization to centromeres and the structural integrity of the overall assembly. We demonstrate that the CENP-L-N complex plays a crucial role at the core of this assembly through interactions with CENP-C and CENP-H-I-K-M. Finally, we show that the CCAN is remodeled over the cell cycle such that sub-complexes depend on their interactions differentially. Thus, an interdependent meshwork within the CCAN underlies the centromere specificity and stability of the kinetochore.
Keywords: kinetochore, centromere, mitosis, CENP-A, CenH3
Graphical Abstract
Introduction
The transmission of the genome to daughter cells during mitosis and meiosis requires the attachment of spindle microtubules to a specialized region of each chromosome, termed the centromere (McKinley and Cheeseman, 2016, Fukagawa and Earnshaw, 2014). The histone H3-variant, centromere protein A, CENP-A, epigenetically marks the centromere (Black and Cleveland, 2011), and is recognized by the macromolecular kinetochore structure to orchestrate chromosome segregation. The centromere-kinetochore interface must provide specificity to ensure that the kinetochore is recruited to a single region on each chromosome, as well as forming a robust and stable platform for kinetochore assembly. Defining the molecular basis for these functions is a critical outstanding goal.
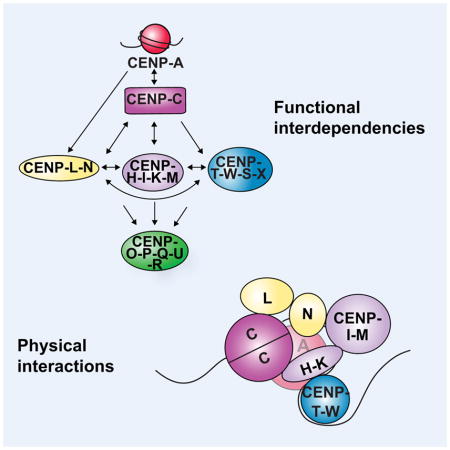
The 16-subunit Constitutive Centromere-Associated Network (CCAN) localizes to centromeres throughout the cell cycle and provides the foundation for outer kinetochore assembly on CENP-A-containing chromatin (Cheeseman and Desai, 2008). The CCAN proteins can be grouped into five sub-complexes: CENP-C, CENP-L-N, CENP-H-I-K-M, CENP-T-W-S-X and CENP-O-P-Q-U-R (Fig. 1A; Amano et al., 2009; Basilico et al., 2014; Carroll et al., 2009; Earnshaw and Rothfield, 1985; Foltz et al., 2006; Hori et al., 2008b; Izuta et al., 2006; Nishino et al., 2012; Okada et al., 2006; Saitoh et al., 1992). Amongst these proteins, CENP-C and CENP-N bind to CENP-A nucleosomes directly (Carroll et al., 2010; Carroll et al., 2009; Falk et al., 2015; Guse et al., 2011; Kato et al., 2013), and CENP-C and CENP-T associate with the proteins that comprise the kinetochore microtubule-binding interface (Gascoigne et al., 2011; Nishino et al., 2013; Przewloka et al., 2011; Screpanti et al., 2011). Thus, the CCAN proteins mediate the connection between the centromere and the outer kinetochore. Previous work has analyzed the relationships between CCAN components, with varying results between studies and differences in the architecture of this assembly between organisms (Amano et al., 2009; Basilico et al., 2014; Carroll et al., 2010; Carroll et al., 2009; Eskat et al., 2012; Folco et al., 2015; Foltz et al., 2006; Gascoigne et al., 2011; Hori et al., 2008a; Hori et al., 2008b; Hori et al., 2013; Klare et al., 2015; Kwon et al., 2007; Liu et al., 2006; Logsdon et al., 2015; McClelland et al., 2007; Nagpal et al., 2015; Okada et al., 2006; Tachiwana et al., 2015). However, it remains unclear how the interactions of the CCAN subcomplexes are integrated to achieve the critical functions and properties of the centromere-kinetochore interface.
Figure 1. Inducible CRISPR knockouts define the contributions of the CCAN to chromosome segregation.

A) Diagram of the CCAN components in their sub-complexes. B) Strategy for the generation of inducible knockouts of CCAN genes. spCas9 under control of a tet-on promoter is stably integrated into HeLa cells and sgRNAs targeting early exons of the gene of interest are stably integrated by lentiviral transduction. C) Representative immunofluorescence images of mitotic phenotypes following CENP-N inducible knockout. D) Quantification of mitotic phenotypes following inducible knockout of a component of each CCAN subcomplex for four days. Cells were classified as having an alignment defect if they displayed bipolar-like spindles with > 4 off-axis chromosomes. n = 100 cells per condition. E) Representative immunofluorescence images of interphase phenotypes following CENP-C inducible knockout. F) Quantification of interphase phenotypes following inducible knockout of a component of each CCAN subcomplex for four days. n = 100 cells per condition. Scale bars, 5 μm.
Here, we define extensive functional and physical interactions between CCAN components in human cells. Instead of representing a linear hierarchy of pairwise interactions, our results demonstrate that each CCAN sub-complex forms multiple independent interactions with other sub-complexes, centromeric nucleosomes, and/or DNA. Importantly, each sub-complex critically depends on the combination of these interactions for its centromere localization, such that no single interaction is sufficient for its recruitment. For example, we find that CENP-L-N complex localization requires direct interactions with CENP-A nucleosomes, CENP-C, and the CENP-H-I-K-M complex, and that the contributions of these interactions vary during the cell cycle. Our data present a coherent model for the organization of the centromere-kinetochore interface and reveal that the extensive network of interactions formed between sub-complexes plays a critical role in providing a specific and robust foundation for the chromosome segregation machinery.
Results
Inducible CRISPR-based knockouts robustly eliminate CCAN components
Previous work has analyzed the consequences of depleting CCAN components in human cells, but in some cases has resulted in conflicting results, possibly due to variable efficiencies of protein depletion. To define the functions and relationships of the CCAN sub-complexes, we adapted the CRISPR-Cas9 knockout strategy recently employed for genome-wide knockout screens (Shalem et al., 2014; Wang et al., 2014b). For these experiments, we generated a clonal cell line expressing doxycycline-inducible Streptococcus pyogenes Cas9 (spCas9) in human HeLa cells and then stably integrated a single guide RNA (sgRNA) targeting an early exon for each gene of interest (Fig. 1B, S1A). Upon spCas9 induction, double stranded DNA breaks are generated in the targeted exon such that repair of these cuts can generate indels that disrupt the open reading frame and abolish protein synthesis. Previous work demonstrated that the subunits within a given CCAN sub-complex are interdependent for their centromere localization (Basilico et al., 2014; Hori et al., 2008b; Okada et al., 2006), with the exception of the CENP-T-W-S-X complex in which CENP-T and CENP-W are upstream of CENP-S and CENP-X (Amano et al., 2009). Therefore, we employed this inducible knockout strategy to target the following core components of each CCAN sub-complex (the components targeted are indicated by bold and underlined letters): CENP-C, CENP-H-I-K-M, CENP-L-N, CENP-O-P-Q-U-R, and CENP-T-W-S-X.
We first defined the phenotypes resulting from the inducible knockouts of CCAN components after four days of spCas9 induction. We observed severe mitotic defects in each CCAN knockout, with the exception of CENP-O, which did not display a detectable phenotype (Fig. 1C and D). The phenotypes were qualitatively similar across the CENP-C, CENP-N, CENP-I and CENP-T knockouts in mitotic cells (Fig. 1C and D). However, the CENP-C knockout also exhibited a dramatic accumulation of interphase cells with micronuclei (Fig. 1E and F), suggesting a bypass of the spindle-assembly checkpoint as reported previously (Kwon et al., 2007). In most cases, similar phenotypes have been reported in previous RNAi studies in human cells and knockout analyses in chicken DT40 cells. However, in contrast to the relatively mild phenotypes observed following treatment with CENP-N siRNAs (Foltz et al., 2006; McClelland et al., 2007; our unpublished data), induction of the CENP-N knockout resulted in severely disrupted chromosome alignment and mitotic arrest (Fig. 1C and D, Fig. S1B). The defects observed in the inducible CENP-N knockout could be rescued by the expression of a GFP-CENP-N construct with the sequence altered to disrupt sgRNA targeting (Fig. S1C). Thus, the inducible CRISPR-based knockout system allows for the on-target elimination of a protein-of-interest, in some cases resulting in a more potent phenotype than standard RNAi approaches.
Inducible knockouts define the functional interdependencies between CCAN sub-complexes
We next employed the inducible knockouts to define the functional requirements for the centromere localization of each CCAN sub-complex. For these experiments, we induced the knockout of each CCAN sub-complex and performed quantitative immunofluorescence for representative CCAN components in mitotic cells (Fig. 2A, S2A). Upon induction of the knockout, the protein corresponding to the target gene was dramatically reduced in the majority of mitotic cells (Fig. S2B and C), although a subset of cells escaped the cutting or repaired cuts in at least one allele without error. We sought to define the effects on CCAN assembly when one sub-complex was completely eliminated. Although a reduction in protein levels was observed 2 days after Cas9 induction (Fig. S2D), complete elimination of the target subcomplex based on quantitative immunofluorescence was not detected in some cases until 5 days after Cas9 induction (Fig. S2D). Therefore, we analyzed cells 5 days following induction. We note that this strategy presents the potential for secondary effects due to the depletion of these proteins over several mitoses, as we address below with an inducible degron system.
Figure 2. Inducible knockouts reveal interdependencies between CCAN components.

A) Representative immunofluorescence images of control mitotic cells and CENP-N knockout mitotic cells stained for selected CCAN components. Images are deconvolved and scaled equivalently for each antibody. The centromeres of unprocessed images of this type were quantified to generate the data in panels B–F. See Fig. S2A for corresponding images from these cells showing DNA staining. B–F) Mean centromeric fluorescence intensity of a component of each CCAN subcomplex in mitotic cells following knockout of the indicated CCAN subunit for five days, normalized to cells in which spCas9 is not induced. n = 20 cells per condition per antibody per knockout, error bars represent s.e.m. For CENP-C, -N, -I, and –T knockouts, cells exhibiting mitotic errors were quantified. For the CENP-O knockout, which does not exhibit a mitotic phenotype, mitotic cells were selected at random. G) Summary of interdependencies of CCAN components determined by the inducible knockout strategy (Figures 2B–F). “+” indicates fluorescence intensity following knockout > 20 % of control; “−” indicates fluorescence intensity following knockout < 20 % of control. H) Summary of functional relationships between CCAN components determined using the inducible knockout strategy. Scale bar, 5 μm.
Using this information, we analyzed the centromeric levels of each CCAN sub-complex in each knockout 5 days after induction to test the functional relationships between CCAN sub-complexes (Fig. 2B–G). CENP-C has been implicated in recruiting the CENP-T-W-S-X and CENP-H-I-K-M complexes (Basilico et al., 2014; Klare et al., 2015). Expanding on this, we found that the localization of all CCAN sub-complexes was disrupted in the inducible CENP-C knockout (Fig. 2B). In addition to recruiting downstream components, previous work found that CENP-C is required for the incorporation and stabilization of CENP-A nucleosomes (Dambacher et al., 2012; Falk et al., 2015; McKinley and Cheeseman, 2014; Moree et al., 2011). Indeed, in the CENP-C knockout we found that CENP-A levels were significantly reduced (Fig. S2E). As CENP-A is required for the localization of all CCAN components to centromere chromatin (Fachinetti et al., 2013; Liu et al., 2006), the severe defects in the localization of other CCAN sub-complexes following CENP-C knockout likely reflect a combination of the contributions of CENP-C to recruiting CCAN components via direct protein interactions (see below) plus indirect contributions to overall centromere integrity via CENP-A.
In reciprocal experiments, we found that CENP-C localization to mitotic kinetochores was largely maintained following knockout of the remaining CCAN components (Fig. 2C–G), although we note that there was some reduction in CENP-C levels in the CENP-N, CENP-I, and CENP-T knockouts (Fig. 2C–E). Importantly, we found that CENP-N, CENP-I and CENP-T were all interdependent, such that the centromere localization of all three sub-complexes was severely disrupted in each inducible knockout (Fig. 2C–E). In these depletions, we additionally found a modest reduction in CENP-A levels at centromeres (Fig. S2E). Finally, we found that the inducible knockout of CENP-O did not affect the localization of any other CCAN sub-complexes (Fig. 2F), indicating that it occupies a downstream position within the CCAN hierarchy (Fig. 2H).
A central function of the CCAN is to recruit the microtubule binding proteins of the kinetochore, the KNL1/Mis12 complex/Ndc80 complex (KMN) network (Cheeseman et al., 2006). Therefore, we analyzed the contributions of the CCAN components to KMN recruitment using the inducible knockouts. We found that induction of the CENP-C knockout resulted in a severe decrease in the levels of KNL1, the Mis12 complex subunit Dsn1, and the Ndc80 complex subunit Hec1 (Fig. S2F–H). These defects were more severe than those observed in the CENP-N, CENP-I and CENP-T knockouts (Fig. S2F–H). These data are consistent with the relationships between CCAN components that we defined above and existing models for vertebrate KMN recruitment through direct interactions with CENP-C and CENP-T (Gascoigne et al., 2011; Nishino et al., 2013; Przewloka et al., 2011; Screpanti et al., 2011). In addition, we found that induction of the CENP-O knockout did not affect KMN recruitment (Fig. S2F–H), consistent with the absence of a discernible mitotic phenotype (Fig. 1D). As the CENP-O-P-Q-U-R complex does not play an essential role in the centromere-kinetochore interface (Fig. 2F, H), we did not analyze its associations further in this study. Defining the contributions of the CENP-O-P-Q-U-R complex at the kinetochore remains an important outstanding goal.
Together, our analysis of the inducible CCAN knockouts establishes a three-level hierarchy of CCAN recruitment to the kinetochore, with all proteins depending on CENP-C, an interdependence of the CENP-L-N, CENP-H-I-K-M, and CENP-T-W-S-X complexes, and the CENP-O-P-Q-U-R complex downstream of the other CCAN sub-complexes (Fig. 2H).
An integrated assembly of CENP-C, CENP-H-I-K-M and CENP-L-N is formed by pairwise interactions between all three components
Our analysis of the inducible knockouts demonstrated that there are numerous functional interdependencies between each essential CCAN sub-complex (Fig. 2H), such that multiple other CCAN sub-complexes fail to localize when a single sub-complex is eliminated (Fig. 2G). Therefore, we next sought to dissect the molecular basis for these interdependencies. To define the direct physical relationships between CCAN sub-complexes, we reconstituted all of the human CCAN components by co-expression in insect cells as five discrete sub-complexes (Fig. 3A). We tested the interactions between CCAN sub-complexes by tagging each CCAN sub-complex with either a hepta-histidine (His) tag or a glutathione-S-transferase (GST) tag and determining whether co-expressed CCAN sub-complexes co-purified from insect cells over Nickel-NTA agarose and subsequently over glutathione agarose (Fig. 3B).
Figure 3. CCAN reconstitution facilitates the analysis of the interactions of CENP-C, the CENP-H-I-K-M complex and the CENP-L-N complex.

A) SDS-PAGE gel showing reconstitution and purification of all 16 proteins of the human CCAN as five subcomplexes. B) Schematic for the identification of biochemical interactions by co-infection and sequential His- and GST-purifications. C) SDS-PAGE gel showing co-purification of CENP-L-N-His and GST-CENP-C. D) SDS-PAGE gel showing co-purification of GST-CENP-C with His-CENP-I-H-K-M. E) SDS-PAGE gel showing co-purification of GST-CENP-I-H-K-M with CENP-L-N-His. F) SDS-PAGE gel showing co-purification of CENP-L-N-His, GST-CENP-C and untagged CENP-H-I-K-M. The results of the sequential purification (Nickel purification followed by glutathione purification) are shown for each lane, such that only those complexes that co-purify over both Nickel and glutathione are detected. G) Schematic of the direct pairwise interactions between CENP-C, the CENP-H-I-K-M complex and the CENP-L-N complex. All gels were stained with Coomassie Brilliant Blue. *: Contaminant.
We first defined the physical interactions of CENP-C, which is required for the localization of all other CCAN sub-complexes (Fig. 2B, G, H). We found that both the CENP-L-N and CENP-H-I-K-M complexes interacted independently with CENP-C (Fig. 2C and D). These interactions are consistent with a CENP-L and CENP-C interaction reported in Schizosaccharomyces pombe (Tanaka et al., 2009), between CENP-L-N and CENP-C in chicken (Nagpal et al., 2015) and between human CENP-C and CENP-H-I-K-M (Klare et al., 2015). The CENP-C-CENP-L-N and CENP-C-CENP-H-I-K-M interactions were both mediated by a middle region within CENP-C (amino acids 235–509 out of 943; Fig. S3A and B). Within the CENP-H-I-K-M complex, we found that CENP-H-K interacted with CENP-C (Fig. S3C).
Intriguingly, we additionally identified an interaction between CENP-H-I-K-M and CENP-L-N (Figure 3E). Thus, the hierarchy of CCAN recruitment downstream of CENP-C defined by our functional analyses does not consist of linear set of physical connections. Instead, CENP-L-N, CENP-H-I-K-M and CENP-C are intimately interrelated by pairwise interactions between all three components. In addition, we found that CENP-L-N, CENP-H-I-K-M, and CENP-C were able to interact simultaneously to form a higher-order complex (Figure 3F and 3G).
CENP-C requires the CENP-H-I-K-M and CENP-L-N complexes for its robust interphase localization
Our biochemical analysis identified extensive interactions between CENP-C and the CENP-L-N and CENP-H-I-K-M complexes. We sought to dissect the contributions of these interrelated interactions to the integrity of the CCAN with temporal resolution. Strategies such as the inducible knockout system or RNAi provide powerful tools for the progressive depletion of a target protein, but do not allow the analysis of protein function during specific windows of the cell cycle. For example, the depletion of most CCAN proteins results in a potent mitotic arrest (Fig. 1D), preventing the analysis of their contributions to interphase CCAN assembly. As noted above, such strategies also carry the potential for additional effects due to the extended period of depletion. To overcome these challenges, we employed auxin-inducible degrons (AID) to rapidly eliminate CCAN components (Holland et al., 2012; Nishimura et al., 2009). We tagged the endogenous alleles of CENP-N and CENP-I by modifying our previous approach for C-terminal tagging by CRISPR/Cas9 (McKinley and Cheeseman, 2014) in pseudo-diploid DLD-1 cells (Fig. 4A). This system results in the potent elimination of the AID-EGFP-tagged protein following addition of the auxin-family hormone indole-3-acetic acid (IAA), and the delocalization of associated proteins from centromeres. For example, addition of IAA to CENP-N-AID-EGFP cells results in the loss of CENP-N-AID-EGFP and its associated protein CENP-L from centromeres (Fig. 4B).
Figure 4. Interphase stabilization of CENP-C by the CENP-L-N and CENP-H-I-K-M complexes revealed by inducible degron analysis.

A) Gene-targeting strategy for introduction of an auxin-inducible degron (AID) tag at the 3′ end of the coding sequence of the endogenous locus. The PAM (NGG) was mutated (NTT) to resist re-cutting by the spCas9 after repair with the template. B) Representative immunofluorescence images of an interphase cell expressing CENP-N-AID-EGFP from the endogenous locus following treatment with the auxin-class hormone indole-3-acetic acid (IAA) and stained with anti-CENP-L and anti-CENP-A to mark centromeres. C) Strategy for cell synchronization to analyze protein requirements in G1/S phase or mitosis. D) Mean centromeric fluorescence intensity of a component of each essential CCAN subcomplex in CENP-N-AID-expressing cells (left) and CENP-I-AID-expressing cells (right) in mitosis. Numbers represent centromeric fluorescence intensity as percent of untreated cells, +/− s.e.m., n = 20 cells per condition. E) Mean centromeric fluorescence intensity of a component of each essential CCAN subcomplex in CENP-N-AID-expressing cells (left) and CENP-I-AID-expressing cells (right) in G1/S. Numbers represent centromeric fluorescence intensity as percent of untreated cells, +/− s.e.m., n = 20 cells per condition. F) Schematic of interactions between CENP-C, the CENP-L-N complex, and the CENP-H-I-K-M complex in mitosis (top) and interphase (bottom). Scale bar, 5 μm.
We used the auxin degron alleles to define the relationships between CCAN proteins during either G1/S phase or mitosis. For these experiments, we synchronized cells such that they were arrested at a specific cell cycle stage while the AID-tagged protein was being degraded, and did not transit through the cell cycle during this time (Fig. 4C). Consistent with our analyses of mitotic cells using the inducible knockout strategy (Fig. 2C and D), CENP-L, CENP-I and CENP-T de-localized from centromeres following the degradation of CENP-N or CENP-I in interphase or mitosis (Fig. 4D). In addition, CENP-C localization was only mildly affected by elimination of these proteins in mitosis (Fig. 4D, Fig. 2C and D). In contrast, following the degradation of CENP-I or CENP-N during G1/S we found that CENP-C localization was severely diminished (Fig. 4E). Similarly, CENP-C localization was greatly reduced in asynchronous interphase cells following CENP-I or CENP-N degradation, but was not affected by IAA addition to control cells lacking AID-tagged alleles (Fig. S4). These data indicate that CENP-I and CENP-N stabilize CENP-C much more significantly in G1/S than in mitosis in human cells, as previously proposed in chicken cells (Kwon et al., 2007; Nagpal et al., 2015). Thus, although CENP-C occupies the top position of the CCAN hierarchy during mitosis and recruits the complete CCAN via direct interactions with CENP-N and CENP-H-I-K-M, in interphase these interactions reciprocally stabilize the centromere recruitment of CENP-C itself (Fig. 4F).
The localization of the CENP-T-W-S-X complex requires separable interactions with both the CENP-H-I-K-M complex and DNA
Our functional data from both the inducible knockouts and auxin degron system indicate that the CENP-T-W-S-X complex depends on CENP-C, the CENP-L-N complex, and the CENP-H-I-K-M complex for its centromere localization (Fig. 2 and 4). In addition, we found that CENP-L-N and CENP-H-I-K-M complex localization reciprocally depended on the CENP-T-W-S-X complex (Fig. 2). We next assessed the physical basis for these functional relationships. Consistent with previous work (Basilico et al., 2014), we found that the CENP-H-I-K-M complex interacted with the CENP-T-W complex (Fig. 5A). We found that this CENP-T-W-CENP-H-I-K-M interaction was mediated by CENP-H-K (Fig. 5B). In contrast, we did not detect interactions between CENP-T-W and either CENP-C (Fig. 5C) or the CENP-L-N complex (Fig. 5D). Thus, the functional requirements of CENP-L-N and CENP-C for CENP-T-W-S-X localization arise from a direct interaction between CENP-T-W and CENP-H-I-K-M, and contributions of CENP-C and CENP-L-N to promoting CENP-H-I-K-M localization as described above.
Figure 5. The CENP-T-W complex requires interactions with CENP-H-I-K-M and DNA for its centromeric localization.

A) SDS-PAGE gel showing co-purification of His-CENP-T-W and GST-CENP-I-H-K-M. CENP-W was not retained on the gel due to its small size. B) SDS-PAGE gel showing co-purification of His-CENP-T-W with untagged CENP-H-K, and GST-I-H-K-M as a positive control. C) SDS-PAGE gel indicating that His-CENP-T-W does not interact with GST-CENP-C. The sequential purification confirms that the bands co-purifying with His-CENP-T are contaminants and not GST-CENP-C. A parallel glutathione purification was performed (far right lane) to confirm the presence of GST-CENP-C in the extract. D) SDS-PAGE gel indicating the absence of an interaction of GST-CENP-T-W with CENP-L-N-His. E) Localization of transiently transfected GFP-CENP-W constructs in interphase cells. CENP-WDNA contains 5 mutations to disrupt its DNA binding (Nishino et al., 2012). Numbers represent percent of GFP-positive transfected cells showing the indicated phenotype, n = 100 cells. F) SDS-PAGE gel showing co-purification of GST-CENP-I-H-K-M with both His-CENP-T-W and His-CENP-T-WDNA. F) Schematic of the interactions underlying CENP-T-W localization. Gels stained were with Coomassie Brilliant Blue unless otherwise indicated. *: Contaminant. Scale bar, 5 μm.
The work described above (Fig. 2D, Fig. 4D and E), and work from others (Basilico et al., 2014), indicates that the interaction between the CENP-H-I-K-M complex and CENP-T-W plays a crucial role in CENP-T-W localization to centromeres. However, our previous work also demonstrated that the CENP-T-W-S-X proteins contain histone fold domains that assemble as a nucleosome-like structure that binds to and wraps DNA (Hori et al., 2008a; Nishino et al., 2012). To test the relative contributions of the CENP-H-I-K-M interactions and DNA binding to CENP-T-W-S-X complex localization, we employed a mutant of CENP-W in which five residues that contribute to DNA binding are disrupted (CENP-WDNA; Nishino et al., 2012). As reported previously, this CENP-WDNA mutant protein is severely compromised for its centromere localization (Fig. 5E; Nishino et al., 2012). However, we found that the CENP-T-WDNA mutant complex maintained its interaction with the CENP-H-I-K-M complex (Fig. 5F). This indicates that the defective localization of the CENP-WDNA mutant is a consequence of its effects on DNA binding, not an indirect consequence of disrupting its interaction with the CENP-H-I-K-M complex. Together with the functional analyses described above, these data indicate that the centromere localization of the CENP-T-W-S-X complex requires both its interaction with the CENP-H-I-K-M complex and its intrinsic DNA binding activity (Fig. 5G).
Dual pathways recruit the CENP-L-N complex to centromeres
In addition to the extensive network of contacts between the CCAN sub-complexes described above, components of the CCAN interact directly with CENP-A nucleosomes to anchor this network at centromeres. For example, previous work demonstrated that CENP-N binds to a region of CENP-A nucleosomes known as the CENP-A targeting domain (CATD) (Carroll et al., 2009; Fang et al., 2015), and that this interaction is sufficient for the initial recruitment of CENP-N (Logsdon et al., 2015). However, we found that CENP-C is also required for CENP-L-N localization (Fig. 2B), and that the CENP-L-N complex interacts with CENP-C directly (Fig. 3C). We therefore dissected the interactions of the CENP-L-N complex with CENP-A nucleosomes and with CENP-C to determine their relative contributions to CENP-L-N localization and CCAN integrity.
We first analyzed the interactions between CENP-L and CENP-N. We found that truncation of CENP-N at amino acid 240 was critical for the generation of biochemically tractable N- and C-terminal CENP-N fragments. Expression of an N-terminal region of CENP-N (amino acids 1–240; CENP-NNT) on its own resulted in well-behaved protein for biochemical analysis (Fig. S5A). In contrast, we found that the CENP-N C-terminal domain (CENP-NCT) (amino acids 241–339) required co-expression with CENP-L (Fig. S5A). This interaction between the C terminus of CENP-N and CENP-L is consistent with previous data using in vitro translated human proteins (Carroll et al., 2009) or proteins from budding yeast (Hinshaw and Harrison, 2013).
We next analyzed the interactions of the CENP-L-N complex with CENP-A nucleosomes using purified complexes separated by native gel. We found that the full-length CENP-L-N complex bound robustly to CENP-A nucleosomes (Fig. 6A), but not to histone H3-containing nucleosomes (Fig. S5B). In addition, the CENP-L-N complex bound to H3CATD nucleosomes in which the loop 1 and alpha 2 structures of H3 are replaced with the CATD of CENP-A (Fig. S5C), indicating that this region of CENP-A is sufficient for the CENP-N-CENP-A interaction. Previous reports suggested that the N terminal domain of CENP-N (residues 1–289) contains its CENP-A nucleosome binding activity (Carroll et al., 2009; Fang et al., 2015). Indeed, we found that our CENP-NNT protein (residues 1–240) was sufficient to bind to CENP-A nucleosomes (Fig. 6B, S5D). In contrast, we found the CENP-L-NCT complex interacted with CENP-C (Fig. 6C), but was unable to bind to CENP-A nucleosomes (Fig. S5E). Thus, different domains of the CENP-L-N complex mediate its interactions with CENP-A nucleosomes and CENP-C.
Figure 6. Interactions with CENP-A nucleosomes and CENP-C contribute differentially to CENP-N recruitment in interphase and mitosis.

A) Native gel showing binding of full-length CENP-L-N complex to CENP-A nucleosomes assembled on 145 bp alpha-satellite DNA, stained with ethidium bromide (EtBr) to detect DNA and subsequently with Coomassie to detect protein. B) Native gel showing binding of CENP-NNT to CENP-A nucleosomes assembled on 145 bp alpha-satellite DNA. C) SDS-PAGE gel testing the interactions of fragments of CENP-L-N with CENP-C. Gel stained with Coomassie Brilliant Blue. D) Live cell imaging of cells stably expressing either GFP-CENP-NFL or CENP-NNT-GFP in interphase and mitosis, either in control cells or following induction of the CENP-N knockout. Corresponding control and CENP-N knockout pairs are scaled equivalently. E) Live cell imaging of cells stably expressing either GFP-CENP-NFL or CENP-NNT-GFP in interphase and mitosis following 72 h RNAi of CENP-L. Numbers represent fraction of cells showing the observed phenotype. F) Quantification of mitotic phenotypes following inducible knockout of CENP-N for four days in cells stably expressing CENP-NNT-GFP, n = 100 cells. G) Schematic of the interactions underlying CENP-L-N localization in interphase (top), and mitosis (bottom). CENP-N N- and C-termini are labeled NT and CT, respectively. Scale bars, 5 μm.
To dissect the contributions of CENP-A and CENP-C to the localization of the CENP-L-N complex in vivo, we expressed GFP-tagged versions of CENP-N in HeLa cells. We found that CENP-NNT-GFP localized to centromeres in interphase, similar to full-length GFP-CENP-N (CENP-NFL) (Fig. 6D, S5F, S5G). This suggests that the direct binding between the CENP-NNT and CENP-A nucleosomes is sufficient for its interphase localization. Consistent with this, CENP-NNT-GFP did not require CENP-L or CENP-C in interphase (Fig. 6E, S5H and data not shown). In contrast, we found that CENP-NNT localization to centromeres was barely detectable during mitosis in either the presence or absence of endogenous CENP-N (Fig. 6D and S5G). Instead, we found that CENP-NNT localized diffusely across chromosome arms (Fig. 6D), indicating that centromere-specificity is disrupted in this mutant. These data indicate that the robust localization of CENP-N to mitotic centromeres requires the CENP-N C-terminus and CENP-L, which bind to CENP-C (Fig. 6C). However, CENP-NCT did not localize to centromeres in either interphase or mitosis (Fig. S5I), indicating that CENP-C binding alone is not sufficient to target CENP-N to centromeres. Instead, CENP-N localization to mitotic kinetochores requires direct CENP-A nucleosome binding via its N terminus that is stabilized by the CENP-C interaction with its C terminus and CENP-L. The centromere localization and interactions conferred by the C terminus and CENP-L are critical for kinetochore assembly, as CENP-NNT was unable to rescue the mitotic defects in the CENP-N inducible knockout (Fig. 6D and F).
Collectively, our data support a model in which CENP-N is recruited to interphase centromeres through direct binding to the CATD of CENP-A nucleosomes. However, this binding is disrupted in mitosis, potentially as a result of chromatin compaction (Fang et al., 2015) or loss of an additional interaction partner that is dynamic in mitosis. The CENP-L-N complex is stabilized in mitosis and directed specifically to the centromere by its additional interaction with CENP-C (Fig. 6G) to maintain its central position within the CCAN. These data reveal that the interactions within the CCAN and between CCAN components and CENP-A nucleosomes can make differential contributions over the course of the cell cycle.
Discussion
Here we analyzed the physical and functional interactions of the CCAN and the contributions of this organization to the critical properties of the centromere-kinetochore interface. Our work reveals that the CCAN is an integrated meshwork that relies on a multiplicity of interactions to generate its requisite specificity and robustness (Fig. 7). We demonstrate that each essential CCAN sub-complex interacts with multiple other elements, but that no individual interaction is sufficient for the localization of a given sub-complex to centromeres. Instead, each sub-complex relies on multiple interactions for its centromere localization and the integrity of the overall network, such that perturbing a single node can disrupt the entire structure even if other contacts remain. For example, CENP-I localization is eliminated during mitosis following depletion of its binding partner, the CENP-L-N complex, even though the localization of a second CENP-H-I-K-M complex binding partner, CENP-C, remains unaffected (Figs. 2 and 4). Similarly, our work demonstrates that DNA and CENP-H-I-K-M interactions both play crucial roles for the localization of the CENP-T-W-S-X complex. Thus, this assembly is not a simple linear connector between CENP-A and the proteins of the outer kinetochore, but in fact forms an interconnected network of interactions more extensive than previously appreciated. This meshwork paradigm may extend beyond the interactions that we have identified and refined here, as weak or highly dynamic interactions that are not detectable in solution may also contribute to this interface when these interactions are combined at the kinetochore.
Figure 7. Model for the architecture of the CCAN.

Left: the connections of the CCAN to the kinetochore. Right: The direct interactions between CCAN sub-complexes as viewed from above the CENP-A nucleosome, either occurring on a single nucleosome (top) or between two different nucleosomes (bottom).
Defining the role of the CCAN in kinetochore function has been a central goal since the discovery of these proteins. The network of integrated interactions that we defined here has the potential to contribute several key functions to this assembly. In particular, the multiplicity of contacts at this interface provides a robust and stable platform for kinetochore assembly. Previous work proposed that the CCAN provides force resistance at kinetochores (Ribeiro et al., 2010; Suzuki et al., 2014) and the numerous interaction interfaces that we identified provide an attractive model for the molecular architecture underlying this property.
Although the CCAN proteins localize to centromeres throughout the cell cycle, our work reveals that the CCAN is not a static assembly, but is instead formed and reformed from different interactions over time. Recent work proposed that CENP-A accessibility is reduced during mitotic chromosome compaction, disrupting the interaction between CENP-N and CENP-A (Fang et al., 2015). Our analysis of the CENP-NNT mutant provides in vivo support for this model, as this mutant localizes to interphase, but not mitotic kinetochores. However, our work also reveals that CENP-N localization and the overall CCAN assembly remains intact throughout the cell cycle despite this chromosome compaction, as the CENP-L-N complex is stabilized by the association between the CENP-L-N complex and CENP-C. Reciprocally, although CENP-C does not require other CCAN proteins for its mitotic centromere localization, it depends on CENP-N and the CENP-H-I-K-M complex for its robust interphase localization, as also reported in chicken cells (Kwon et al., 2007; Nagpal et al., 2015). Thus, the multiplicity of interactions between CCAN sub-complexes maintains stable contacts with the centromere throughout the cell cycle and in the face of a changing foundation of CENP-A chromatin.
Finally, and perhaps most intriguingly, the multiplicity of interactions that we defined allows the CCAN to potentially bridge CENP-A nucleosomes that are in close spatial proximity (Fig. 7). This could theoretically occur between CENP-A molecules that occupy consecutive positions along DNA, as immunofluorescence of stretched chromatin fibers indicates that segments of centromeric DNA may be locally enriched for CENP-A molecules (Blower et al., 2002; Sullivan and Karpen, 2004). Alternatively, the higher order organization of centromeric chromatin could bring non-adjacent CENP-A molecules together in three dimensions (Ribeiro et al., 2010). The connections between CCAN components of adjacent CENP-A nucleosomes may contribute to the formation of active centromeres only in the presence of a high local concentration of CENP-A nucleosomes, and not at the individual CENP-A molecules that are found frequently throughout bulk chromatin (Athwal et al., 2015; Bodor et al., 2014; Shang et al., 2013). The possibility that the CCAN contributes to constraining kinetochore assembly to true centromeres is particularly appealing in light of the fact that organisms such as Drosophila melanogaster, which have a minimal CCAN containing only CENP-C, are permissive for ectopic kinetochore assembly, such that they assemble functional kinetochores at non-centromeric sites following CENP-A overexpression (Heun et al., 2006). In contrast, human cells anchor their kinetochores through the complete network of interactions that we defined here, and do not form ectopic kinetochores when CENP-A is diffusely incorporated along chromosome arms by overexpression (Gascoigne et al., 2011; Sullivan et al., 1994; Van Hooser et al., 2001). Instead, the formation of kinetochores at ectopic sites in vertebrates requires a high local concentration of CENP-A generated by tethering the CENP-A chaperone, HJURP (Barnhart et al., 2011), or CENP-A itself (Logsdon et al., 2015; Tachiwana et al., 2015; Westhorpe et al., 2015) to a targeted site. Thus, the requirement for a multiplicity of interactions within the CCAN can achieve both stability and specificity for the kinetochore structure.
Experimental Procedures
Cell culture
The cell lines used in this study are described in Table S1. All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% tetracycline-free fetal bovine serum (FBS), penicillin/streptomycin and 2 mM L-glutamine. Clonal cell lines stably expressing GFPLAP fusions were generated in HeLa cells as described previously (Cheeseman and Desai, 2005). The generation of the inducible knockout cell lines and auxin degron cell lines are described below.
Pooled siRNAs against CENP-L (GGACAUUUCUUUCGCAAUA; AAGAUUAGUUCGUGUUUCA; GCAAUCAAUGCAUUUAAUC; UUAUUGGAGUGUUAGCAUA) and a non-targeting control were obtained from Dharmacon. RNAi experiments were conducted using Lipofectamine RNAi MAX and serum-free OptiMEM (Life Technologies). DMEM + 10% FBS was added 5–6 h after incubation. Cells were assayed 72 h after transfection. Transient transfections were performed using Lipofectamine 2000 and OptiMEM (Life Technologies) according to manufacturer’s instructions.
Generation of inducible knockouts
A HeLa cell line containing doxycycline-inducible human codon-optimized spCas9 was generated by co-transfecting 2 μg of HP138-neo (a piggyBac transposon expressing: spCas9 under control of the tetOn promoter, the reverse tetracycline activator, and neomycin resistance; Fig. 1B), a derivative of the transposon described in (Wang et al., 2014a) with 1 μg of HP137 (the transposase under control of the CAGGS promoter). Both plasmids were gifts from Chikdu Shivalila and Rudolf Jaenisch (Whitehead/MIT). Cells were selected with 800 μg/ml G418 (Life Technologies) for 2 weeks. Clonal cell lines were isolated by single-cell sorting and screened by Western blot for expression of Cas9.
The plasmid used to express sgRNAs under control of the hU6 promoter (pLenti-sgRNA) was a generous gift from Tim Wang, David Sabatini and Eric Lander (Whitehead/Broad/MIT). The sgRNAs used in this study are listed in Table S3. sgRNAs were designed against 5′ exons using crispr.mit.edu. The sgRNAs were introduced by lentiviral infection as described (Wang et al. 2015), and clonal populations isolated by single-cell sorting. For each of the targeted genes, we tested 2–3 independent sgRNA sequences, each of which generated identical results (data not shown). spCas9 expression was induced with 1 μM doxycycline hyclate (Sigma).
Generation of auxin-inducible-degron-tagged cell lines
The CENP-I and CENP-N loci were tagged with EGFP-AID at the C-terminus using CRISPR/Cas-mediated genome engineering in DLD-1-TIR1 cells (a gift from Andrew Holland, Johns Hopkins University) (Holland et al., 2012) as described previously (McKinley and Cheeseman, 2014). To induce degradation, Indole-3-acetic acid (Sigma) was prepared in water and added to cells at a concentration of 500 μM for 12 hr (synchronized cells) or 5 h (asynchronous cells). The design of the repair template is described in the Supplemental Experimental Procedures. sgRNAs designed to target the 3′ UTR of the target gene were expressed in pX330 (Cong et al., 2013) and are listed in Table S3. The repair and guide plasmids were co-transfected as described previously (McKinley and Cheeseman, 2014) and selected with 300 μg/ml G418 for 2 weeks before single-cell sorting. Clones were visually inspected for correct localization of the EGFP fusion, loss of the EGFP upon addition of IAA, and loss of all endogenous protein upon addition of IAA as determined by immunofluorescence. A small subset of cells in the clonal population failed to degrade the EGFP tagged protein upon IAA addition, potentially due to loss of the TIR1 construct. Cells that retained EGFP upon IAA addition were not included in the analyses described.
Immunofluorescence, microscopy, and flow cytometry analysis
Immunofluorescence was performed using the antibodies listed in Table S2. In general, cells were pre-extracted in 0.5% Triton-X-100 for 7 min before fixation in PBS + 3.8% formaldehyde. The pre-extraction step was excluded for the visualization of microtubules. The CENP-L antibody was generated against His-CENP-L expressed in Sf9 cells. The CENP-O-P antibody was generated against CENP-O-P-His expressed in BL21 (DE3) Escherichia coli. The CENP-K antibody was generated against GST-CENP-K expressed in BL21 (DE3) E. coli. Cy2-, Cy3-, and Cy5-conjugated secondary antibodies were obtained from Jackson Laboratories. DNA was visualized using 10 μg/ml Hoechst. Immunofluorescence and live cell images were acquired on a DeltaVision Core deconvolution microscope (Applied Precision) equipped with a CoolSnap HQ2 CCD camera and deconvolved where appropriate. For immunofluorescence, approximately 10–20 Z-sections were acquired at 0.2 μm steps using a 100×, 1.4 Numerical Aperture (NA) Olympus U-PlanApo objective. Live cell imaging was performed using a 60×/1.42 NA Olympus U-PlanApo objective.
For analysis of DNA content by flow cytometry, cells were fixed in 70% ethanol on ice for > 15 minutes, washed in PBS and incubated in PBS + 0.1% BSA + 0.5% Tween for 30 minutes on ice. Cells were then incubated at 37° C for 40 minutes in 600 μl PBS + 0.1% FBS + 0.25 mg/ml RNAseA (Sigma) + 10μg/ml propidium iodide (Life Technologies) before analysis using the BD FACSCanto II (BD Biosciences). Data were analyzed using FlowJo.
CCAN protein expression and purification
Genes encoding the CCAN components were cloned into pACEBac1, combined into their respective sub-complexes using the MultiBac system (ATG:biosynthetics) and electroporated into E. coli DH10EmBacY cells to generate bacmids. Bacmids used in this study are listed in Table S4. Sf9 cells were maintained in Sf900 III SFM (Life Technologies) between 500,000 and 4,000,000 cells/ml at 27 °C with shaking at 150 rpm. V0 and V1 virus were generated as described (Fitzgerald et al., 2006). CENP-NNT-His was cloned into pET3aTr and expressed in BL21 (DE3) E. coli for 6 h at 18 °C. For co-expression, 200 ml of Sf9 cells at 500,000 cells/ml were infected with 3.5 ml each V1 virus and incubated at 27 °C with shaking for 48h.
Buffers used in this study are listed in Table S5. All buffers were supplemented with 10 mM β-mercaptoethanol (Sigma). Details of the purifications are described in the Supplemental Experimental Procedures. For sequential purifications, complexes co-expressing GST- and His-tagged subunits were first purified on Ni-agarose and the elution was bound to glutathione agarose, washed three times and eluted.
Nucleosome purification and native gel assays
Nucleosomes were assembled as described previously (Falk et al., 2015) using purified histones and 145bp alpha-satellite DNA (Falk et al., 2015). For binding assays, nucleosomes were incubated with the proteins of interest in the indicated molar ratio at room temperature for ~30 min in a final NaCl concentration of 200 mM. Complexes were analyzed on 5% native gel and stained with ethidium bromide to visualize DNA and Coomassie Brilliant Blue to visualize protein components.
Supplementary Material
Acknowledgments
We are grateful to members of the Cheeseman and Black laboratories for helpful discussions and critical reading of the manuscript. We thank Tim Wang, Kevin Krupczak and David Sabatini for generously teaching us their CRISPR/Cas9 knockout strategy, and Chikdu Shivalila and Rudolf Jaenisch for reagents for the inducible Cas9 transposon system. We thank David Kern for plasmids, help with CENP-L-N expression, and troubleshooting Sf9 expression systems in the Cheeseman laboratory. We thank Andy Holland for the DLD-1-TIR1 cell line and advice on the AID system. I.M.C. and K.L.M. are inventors on a patent application for the use of the CRISPR/Cas9 system for degron tagging. This work was supported by a Scholar award to IMC from the Leukemia & Lymphoma Society, grants from the NIH/National Institute of General Medical Sciences to IMC (GM088313 and GM108718) and BEB (GM082989), a Research Scholar Grant to IMC (121776) from the American Cancer Society, and an NRSA to LYG (CA186430).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amano M, Suzuki A, Hori T, Backer C, Okawa K, Cheeseman IM, Fukagawa T. The CENP-S complex is essential for the stable assembly of outer kinetochore structure. J Cell Biol. 2009;186:173–182. doi: 10.1083/jcb.200903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athwal RK, Walkiewicz MP, Baek S, Fu S, Bui M, Camps J, Ried T, Sung MH, Dalal Y. CENP-A nucleosomes localize to transcription factor hotspots and subtelomeric sites in human cancer cells. Epigenetics Chromatin. 2015;8:2. doi: 10.1186/1756-8935-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart MC, Kuich PH, Stellfox ME, Ward JA, Bassett EA, Black BE, Foltz DR. HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J Cell Biol. 2011;194:229–243. doi: 10.1083/jcb.201012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basilico F, Maffini S, Weir JR, Prumbaum D, Rojas AM, Zimniak T, De Antoni A, Jeganathan S, Voss B, van Gerwen S, et al. The pseudo GTPase CENP-M drives human kinetochore assembly. Elife. 2014;3:e02978. doi: 10.7554/eLife.02978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BE, Cleveland DW. Epigenetic centromere propagation and the nature of CENP-a nucleosomes. Cell. 2011;144:471–479. doi: 10.1016/j.cell.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower MD, Sullivan BA, Karpen GH. Conserved organization of centromeric chromatin in flies and humans. Dev Cell. 2002;2:319–330. doi: 10.1016/s1534-5807(02)00135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor DL, Mata JF, Sergeev M, David AF, Salimian KJ, Panchenko T, Cleveland DW, Black BE, Shah JV, Jansen LE. The quantitative architecture of centromeric chromatin. Elife. 2014;3:e02137. doi: 10.7554/eLife.02137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Milks KJ, Straight AF. Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol. 2010;189:1143–1155. doi: 10.1083/jcb.201001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Silva MC, Godek KM, Jansen LE, Straight AF. Centromere assembly requires the direct recognition of CENP-A nucleosomes by CENP-N. Nat Cell Biol. 2009;11:896–902. doi: 10.1038/ncb1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 2006;127:983–997. doi: 10.1016/j.cell.2006.09.039. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Desai A. A combined approach for the localization and tandem affinity purification of protein complexes from metazoans. Sci STKE. 2005;2005:pl1. doi: 10.1126/stke.2662005pl1. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Desai A. Molecular architecture of the kinetochore-microtubule interface. Nat Rev Mol Cell Biol. 2008;9:33–46. doi: 10.1038/nrm2310. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambacher S, Deng W, Hahn M, Sadic D, Frohlich J, Nuber A, Hoischen C, Diekmann S, Leonhardt H, Schotta G. CENP-C facilitates the recruitment of M18BP1 to centromeric chromatin. Nucleus. 2012;3:101–110. doi: 10.4161/nucl.18955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Rothfield N. Identification of a family of human centromere proteins using autoimmune sera from patients with scleroderma. Chromosoma. 1985;91:313–321. doi: 10.1007/BF00328227. [DOI] [PubMed] [Google Scholar]
- Eskat A, Deng W, Hofmeister A, Rudolphi S, Emmerth S, Hellwig D, Ulbricht T, Doring V, Bancroft JM, McAinsh AD, et al. Step-wise assembly, maturation and dynamic behavior of the human CENP-P/O/R/Q/U kinetochore sub-complex. PLoS One. 2012;7:e44717. doi: 10.1371/journal.pone.0044717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinetti D, Folco HD, Nechemia-Arbely Y, Valente LP, Nguyen K, Wong AJ, Zhu Q, Holland AJ, Desai A, Jansen LE, et al. A two-step mechanism for epigenetic specification of centromere identity and function. Nat Cell Biol. 2013;15:1056–1066. doi: 10.1038/ncb2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk SJ, Guo LY, Sekulic N, Smoak EM, Mani T, Logsdon GA, Gupta K, Jansen LE, Van Duyne GD, Vinogradov SA, et al. Chromosomes. CENP-C reshapes and stabilizes CENP-A nucleosomes at the centromere. Science. 2015;348:699–703. doi: 10.1126/science.1259308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Liu Y, Wei Y, Deng W, Yu Z, Huang L, Teng Y, Yao T, You Q, Ruan H, et al. Structural transitions of centromeric chromatin regulate the cell cycle-dependent recruitment of CENP-N. Genes Dev. 2015;29:1058–1073. doi: 10.1101/gad.259432.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald DJ, Berger P, Schaffitzel C, Yamada K, Richmond TJ, Berger I. Protein complex expression by using multigene baculoviral vectors. Nat Methods. 2006;3:1021–1032. doi: 10.1038/nmeth983. [DOI] [PubMed] [Google Scholar]
- Folco HD, Campbell CS, May KM, Espinoza CA, Oegema K, Hardwick KG, Grewal SI, Desai A. The CENP-A N-tail confers epigenetic stability to centromeres via the CENP-T branch of the CCAN in fission yeast. Curr Biol. 2015;25:348–356. doi: 10.1016/j.cub.2014.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR, 3rd, Cleveland DW. The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol. 2006;8:458–469. doi: 10.1038/ncb1397. [DOI] [PubMed] [Google Scholar]
- Fukagawa T, Earnshaw WC. The centromere: chromatin foundation for the kinetochore machinery. Dev Cell. 2014;30:496–508. doi: 10.1016/j.devcel.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Takeuchi K, Suzuki A, Hori T, Fukagawa T, Cheeseman IM. Induced ectopic kinetochore assembly bypasses the requirement for CENP-A nucleosomes. Cell. 2011;145:410–422. doi: 10.1016/j.cell.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature. 2011;477:354–358. doi: 10.1038/nature10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heun P, Erhardt S, Blower MD, Weiss S, Skora AD, Karpen GH. Mislocalization of the Drosophila centromere-specific histone CID promotes formation of functional ectopic kinetochores. Dev Cell. 2006;10:303–315. doi: 10.1016/j.devcel.2006.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw SM, Harrison SC. An Iml3-Chl4 heterodimer links the core centromere to factors required for accurate chromosome segregation. Cell Rep. 2013;5:29–36. doi: 10.1016/j.celrep.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Fachinetti D, Han JS, Cleveland DW. Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc Natl Acad Sci U S A. 2012;109:E3350–3357. doi: 10.1073/pnas.1216880109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Amano M, Suzuki A, Backer CB, Welburn JP, Dong Y, McEwen BF, Shang WH, Suzuki E, Okawa K, et al. CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. Cell. 2008a;135:1039–1052. doi: 10.1016/j.cell.2008.10.019. [DOI] [PubMed] [Google Scholar]
- Hori T, Okada M, Maenaka K, Fukagawa T. CENP-O class proteins form a stable complex and are required for proper kinetochore function. Mol Biol Cell. 2008b;19:843–854. doi: 10.1091/mbc.E07-06-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Shang WH, Takeuchi K, Fukagawa T. The CCAN recruits CENP-A to the centromere and forms the structural core for kinetochore assembly. J Cell Biol. 2013;200:45–60. doi: 10.1083/jcb.201210106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izuta H, Ikeno M, Suzuki N, Tomonaga T, Nozaki N, Obuse C, Kisu Y, Goshima N, Nomura F, Nomura N, et al. Comprehensive analysis of the ICEN (Interphase Centromere Complex) components enriched in the CENP-A chromatin of human cells. Genes Cells. 2006;11:673–684. doi: 10.1111/j.1365-2443.2006.00969.x. [DOI] [PubMed] [Google Scholar]
- Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, Bai Y. A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science. 2013;340:1110–1113. doi: 10.1126/science.1235532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klare K, Weir JR, Basilico F, Zimniak T, Massimiliano L, Ludwigs N, Herzog F, Musacchio A. CENP-C is a blueprint for constitutive centromere-associated network assembly within human kinetochores. J Cell Biol. 2015;210:11–22. doi: 10.1083/jcb.201412028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon MS, Hori T, Okada M, Fukagawa T. CENP-C is involved in chromosome segregation, mitotic checkpoint function, and kinetochore assembly. Mol Biol Cell. 2007;18:2155–2168. doi: 10.1091/mbc.E07-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ST, Rattner JB, Jablonski SA, Yen TJ. Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J Cell Biol. 2006;175:41–53. doi: 10.1083/jcb.200606020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon GA, Barrey EJ, Bassett EA, DeNizio JE, Guo LY, Panchenko T, Dawicki-McKenna JM, Heun P, Black BE. Both tails and the centromere targeting domain of CENP-A are required for centromere establishment. J Cell Biol. 2015;208:521–531. doi: 10.1083/jcb.201412011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland SE, Borusu S, Amaro AC, Winter JR, Belwal M, McAinsh AD, Meraldi P. The CENP-A NAC/CAD kinetochore complex controls chromosome congression and spindle bipolarity. EMBO J. 2007;26:5033–5047. doi: 10.1038/sj.emboj.7601927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, Cheeseman IM. Polo-like kinase 1 licenses CENP-A deposition at centromeres. Cell. 2014;158:397–411. doi: 10.1016/j.cell.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, Cheeseman IM. The molecular basis for centromere identity and function. Nature Reviews Molecular Cell Biology. 2016 doi: 10.1038/nrm.2015.5. in press. http://dx.doi.org/10.1038/nrm.2015.5. Online November 25, 2015. [DOI] [PMC free article] [PubMed]
- Moree B, Meyer CB, Fuller CJ, Straight AF. CENP-C recruits M18BP1 to centromeres to promote CENP-A chromatin assembly. J Cell Biol. 2011;194:855–871. doi: 10.1083/jcb.201106079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal H, Hori T, Furukawa A, Sugase K, Osakabe A, Kurumizaka H, Fukagawa T. Dynamic changes in the CCAN organization through CENP-C during cell-cycle progression. Mol Biol Cell. 2015 doi: 10.1091/mbc.E15-07-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Nishino T, Rago F, Hori T, Tomii K, Cheeseman IM, Fukagawa T. CENP-T provides a structural platform for outer kinetochore assembly. EMBO J. 2013;32:424–436. doi: 10.1038/emboj.2012.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino T, Takeuchi K, Gascoigne KE, Suzuki A, Hori T, Oyama T, Morikawa K, Cheeseman IM, Fukagawa T. CENP-T-W-S-X forms a unique centromeric chromatin structure with a histone-like fold. Cell. 2012;148:487–501. doi: 10.1016/j.cell.2011.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Cheeseman IM, Hori T, Okawa K, McLeod IX, Yates JR, 3rd, Desai A, Fukagawa T. The CENP-H-I complex is required for the efficient incorporation of newly synthesized CENP-A into centromeres. Nat Cell Biol. 2006;8:446–457. doi: 10.1038/ncb1396. [DOI] [PubMed] [Google Scholar]
- Przewloka MR, Venkei Z, Bolanos-Garcia VM, Debski J, Dadlez M, Glover DM. CENP-C is a structural platform for kinetochore assembly. Curr Biol. 2011;21:399–405. doi: 10.1016/j.cub.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Ribeiro SA, Vagnarelli P, Dong Y, Hori T, McEwen BF, Fukagawa T, Flors C, Earnshaw WC. A super-resolution map of the vertebrate kinetochore. Proc Natl Acad Sci U S A. 2010;107:10484–10489. doi: 10.1073/pnas.1002325107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh H, Tomkiel J, Cooke CA, Ratrie H, 3rd, Maurer M, Rothfield NF, Earnshaw WC. CENP-C, an autoantigen in scleroderma, is a component of the human inner kinetochore plate. Cell. 1992;70:115–125. doi: 10.1016/0092-8674(92)90538-n. [DOI] [PubMed] [Google Scholar]
- Screpanti E, De Antoni A, Alushin GM, Petrovic A, Melis T, Nogales E, Musacchio A. Direct binding of Cenp-C to the Mis12 complex joins the inner and outer kinetochore. Curr Biol. 2011;21:391–398. doi: 10.1016/j.cub.2010.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang WH, Hori T, Martins NM, Toyoda A, Misu S, Monma N, Hiratani I, Maeshima K, Ikeo K, Fujiyama A, et al. Chromosome engineering allows the efficient isolation of vertebrate neocentromeres. Dev Cell. 2013;24:635–648. doi: 10.1016/j.devcel.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BA, Karpen GH. Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat Struct Mol Biol. 2004;11:1076–1083. doi: 10.1038/nsmb845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KF, Hechenberger M, Masri K. Human CENP-A contains a histone H3 related histone fold domain that is required for targeting to the centromere. J Cell Biol. 1994;127:581–592. doi: 10.1083/jcb.127.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Badger BL, Wan X, DeLuca JG, Salmon ED. The architecture of CCAN proteins creates a structural integrity to resist spindle forces and achieve proper Intrakinetochore stretch. Dev Cell. 2014;30:717–730. doi: 10.1016/j.devcel.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachiwana H, Muller S, Blumer J, Klare K, Musacchio A, Almouzni G. HJURP involvement in de novo CenH3(CENP-A) and CENP-C recruitment. Cell Rep. 2015;11:22–32. doi: 10.1016/j.celrep.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Chang HL, Kagami A, Watanabe Y. CENP-C functions as a scaffold for effectors with essential kinetochore functions in mitosis and meiosis. Dev Cell. 2009;17:334–343. doi: 10.1016/j.devcel.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Van Hooser AA, Ouspenski II, Gregson HC, Starr DA, Yen TJ, Goldberg ML, Yokomori K, Earnshaw WC, Sullivan KF, Brinkley BR. Specification of kinetochore-forming chromatin by the histone H3 variant CENP-A. J Cell Sci. 2001;114:3529–3542. doi: 10.1242/jcs.114.19.3529. [DOI] [PubMed] [Google Scholar]
- Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014a;20:616–623. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014b;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. Identification and characterization of essential genes in the human genome. Science. 2015 Oct 15; doi: 10.1126/science.aac7041. pii: aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhorpe FG, Fuller CJ, Straight AF. A cell-free CENP-A assembly system defines the chromatin requirements for centromere maintenance. J Cell Biol. 2015;209:789–801. doi: 10.1083/jcb.201503132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.