

Abstract
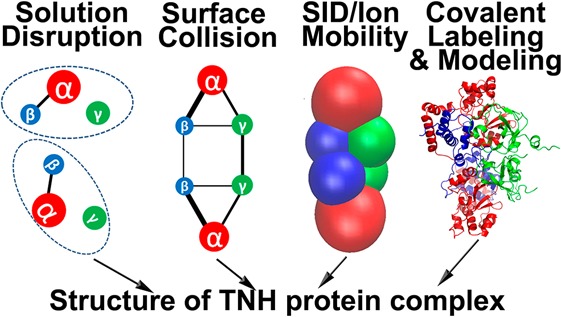
Toyocamycin nitrile hydratase (TNH) is a protein hexamer that catalyzes the hydration of toyocamycin to produce sangivamycin. The structure of hexameric TNH and the arrangement of subunits within the complex, however, have not been solved by NMR or X-ray crystallography. Native mass spectrometry (MS) clearly shows that TNH is composed of two copies each of the α, β, and γ subunits. Previous surface induced dissociation (SID) tandem mass spectrometry on a quadrupole time-of-flight (QTOF) platform suggests that the TNH hexamer is a dimer composed of two αβγ trimers; furthermore, the results suggest that α–β interact most strongly (Blackwell et al. Anal. Chem. 2011, 83, 2862–2865). Here, multiple complementary MS based approaches and homology modeling have been applied to refine the structure of TNH. Solution-phase organic solvent disruption coupled with native MS agrees with the previous SID results. By coupling surface induced dissociation with ion mobility mass spectrometry (SID/IM), further information on the intersubunit contacts and relative interfacial strengths are obtained. The results show that TNH is a dimer of αβγ trimers, that within the trimer the α, β subunits bind most strongly, and that the primary contact between the two trimers is through a γ–γ interface. Collisional cross sections (CCSs) measured from IM experiments are used as constraints for postulating the arrangement of the subunits represented by coarse-grained spheres. Covalent labeling (surface mapping) together with protein complex homology modeling and docking of trimers to form hexamer are utilized with all the above information to propose the likely quaternary structure of TNH, with chemical cross-linking providing cross-links consistent with the proposed structure. The novel feature of this approach is the use of SID-MS with ion mobility to define complete connectivity and relative interfacial areas of a heterohexameric protein complex, providing much more information than is available from solution disruption. That information, when combined with CCS-guided coarse-grained modeling and covalent labeling restraints for homology modeling and trimer–trimer docking, provides atomic models of a previously uncharacterized heterohexameric protein complex.
Short abstract
Multiple complementary mass spectrometry based approaches and homology modeling were applied to refine a structure of toyocamycin nitrile hydratase consistent with additional chemical crosslinking experiments.
Introduction
Mass spectrometry (MS) has become an indispensable tool for characterizing proteins. A frequent use of MS lies in the field of proteomics, which mainly involves protein identification by digestion to peptides, followed by analysis by liquid chromatography coupled tandem mass spectrometry (LC-MS/MS).1−3 More recently, MS has begun to play an influential role in structural biology,4 as illustrated by a number of sophisticated studies of protein conformations and dynamics.5−9 Many proteins exist and function as multimeric complexes, in which subunits noncovalently interact with each other. In fact, more than 60% of entries in the Protein Data Bank are dimers or larger assemblies.10 The characterization of protein complexes, including their composition and subunit interactions, is crucial to understanding how these protein complexes function, and MS based tools are often appropriate even when the sample is too complex for other tools, when the complex will not crystallize, or when the complex is too large for NMR or too small for cryo-EM.
Information provided through different MS-based experiments can be complementary, and thus once combined, can contribute to a better understanding of the structure of a specific protein complex.11 In native MS experiments, protein samples, prepared at neutral pH in aqueous buffers, are ionized, desolvated or partially desolvated, and introduced into the gas phase using nanoelectrospray ionization (nano-ESI). In this way, noncovalent interactions can often be preserved, and the mass of the intact complex can be obtained. (The measured mass may be higher than the sequence mass, because water, salts, and/or buffer molecules may remain attached to the complex.) In order to probe the constituents and also to gain information about the architecture of the complexes, gas phase disassembly methods can be applied. The most widely applied dissociation method is collision induced dissociation (CID). In CID, the dominant dissociation pathway is ejection of a single highly charged subunit, leaving behind its complementary (n–1)mer, and therefore CID alone generally does not provide information on substructure connectivity. Substructure is sought by solution disruption of the complexes as described below. A few cases have been reported in which CID can provide dissociation products reflective of the initial complex; however, this remains the atypical pathway.12 In contrast, surface induced dissociation (SID) has been shown to selectively disrupt the smaller interfaces in protein complexes with known crystal structures to release subcomplexes that reflect the native structures of protein complexes.13−18 Quintyn et al. have shown, for example, that for three D2 homotetramers, transthyretin, streptavidin and neutravidin, a dimer–dimer interface is cleaved, as would be expected based on known structure. Ma et al.19 have shown that SID of glutamate dehydrogenase, a dimer of trimers protein complex, results in trimer products. SID of 20S proteasome with α7β7β7α7 symmetry showed the generation of α7β7 products, which is consistent with the stacked ring topology of the complex.20 Similarly, GroEL, which is composed of two stacked heptamer rings, was dissociated to heptamers upon SID.21 On the basis of these results, SID is a useful tool in the study of unknown structures as it gives fragmentation reflective of the connectivity of the native structure.
Given a high purity of protein complex, once the complex is detected by native MS, a solution disruption approach can also provide useful connectivity information. In this approach, the complex is destabilized by adding organic solvent or increasing ionic strength of the sample solution. The subcomplexes produced by solution disruption can be analyzed by MS and provide subunit connectivity in addition to information on the relative binding strength of different subunits.22 In a study by Levy et al.,23 subcomplexes containing the larger interface upon solution disruption were observed in 13 out of 16 protein homooligomers. In an extensive study by Marsh et al.,24 23 out of 27 heteromeric protein complexes showed excellent agreement between solution disassembly and interface sizes.
Ion mobility (IM) coupled to mass spectrometry (IM/MS) provides an additional dimension of information, enabling determination of the size and shape of the analytes.25−27 IM/MS is based on measurement of the time that analytes take to travel through an ion mobility cell, with analytes driven by an electric field gradient and retarded by collisions with a bath gas. The mobility measurement can be converted to a rotationally averaged collisional cross-section (CCS),28,29 and it has been reported that the CCS measured for numerous protein complexes is in excellent agreement (RMSD of 3%) with theoretical CCS calculated from atomic coordinates via a corrected projection approximation (PA) algorithm.30 Furthermore, the CCSs of monomers or subcomplexes generated by solution disruption can be measured and assembled back into the intact complex based on the CCS constraints from IM/MS experiments, providing an overall topology of the complex.31,32 All of these discoveries motivate the use of IM/MS for structural studies of protein complexes.
Another powerful MS-based approach in structural biology is covalent labeling (surface mapping).4 This approach involves changing the mass of different parts of a protein in a conformation-dependent manner.33 Covalent labeling is performed either specifically or nonspecifically on amino acids. Nonspecific covalent labeling is usually carried out with hydroxyl radicals.34,35 A laser or synchrotron is normally required to generate the hydroxyl radicals. Amino acid-specific labeling, in contrast, has the advantages of readily accessible reagents and ease of use.36 The reactions usually take place in solution with more solvent-exposed amino acids labeled preferentially; the mass shift is detected by MS of peptides formed by enzymatic digestion. Covalent modification on amino acids is usually stable during analysis, unlike in hydrogen/deuterium exchange where back exchange or scrambling of mass labels is known to occur. The use of covalent labeling approaches requires consideration of whether the introduction of modifications alter the structure of the analytes;36 therefore, checking the structural integrity of the protein subjected to covalent labeling is essential.
Nitrile hydratases are an important class of industrially relevant biocatalysts37 that convert nitriles to their respective amides. Toyocamycin nitrile hydratase (TNH) catalyzes the formation of the antibiotic sangivamycin from its corresponding nitrile, toyocamycin.38 TNH is homologous to the industrially useful nitrile hydratase family of enzymes, with structures of some members known (47 PDB entries). The understanding of the detailed structure of TNH, which we seek in this work, can guide protein engineering, such as tailoring substrate specificity.39 The structure of TNH is particularly attractive for this goal as it is unique among nitrile hydratases in acting upon the relatively large substrate toyocamycin. All known nitrile hydratases (NHase) exist as dimers of two subunits, α and β. The α subunit of NHase is homologous to the α subunit of TNH. Interestingly, the N- and C-terminal halves of the β subunit of NHases are homologous to the β and γ subunits of TNH, respectively, suggesting that the β and γ subunits likely form a dimer that is structurally analogous to the prototypical β subunit of NHase. TNH has not been crystallized and falls in a size range that is too small for cryo-EM and too large for typical NMR characterization, making MS based structural biology approaches highly appealing.
In a previous study on TNH we utilized MS to confirm that TNH complex is composed of two α, two β, and two γ subunits (MW α 21190 Da, β 9974 Da, γ 11444 Da). CID applied on the +19 charge state of the hexamer resulted mainly in the ejection of α and β monomers. In contrast, the SID spectra were dominated by the αβγ trimer and led to the suggestion that TNH is a dimer of αβγ trimers.39 In this study, we extend the mass spectrometric methods to probe subunit–subunit interactions and combine the SID/IM data with covalent labeling to provide amino acid level constraints to assist homology modeling and refine the structure of TNH. Finally, chemical cross-linking is used as a validation of the proposed structures.
Results and Discussion
The overall approach used for this research, shown in Scheme 1, is a general approach that can be applied to unknown protein complexes. Each method of the scheme has been validated for known structures. This paper describes how these multiple complementary methods were combined to define the structure of the heterohexameric TNH protein complex. The approach hinges on the formation of surface collision products separated and characterized by ion mobility to provide connectivity and relative interface strength.
Scheme 1. Workflow for Characterizing the TNH Structure by Complementary Mass Spectrometric Tools.
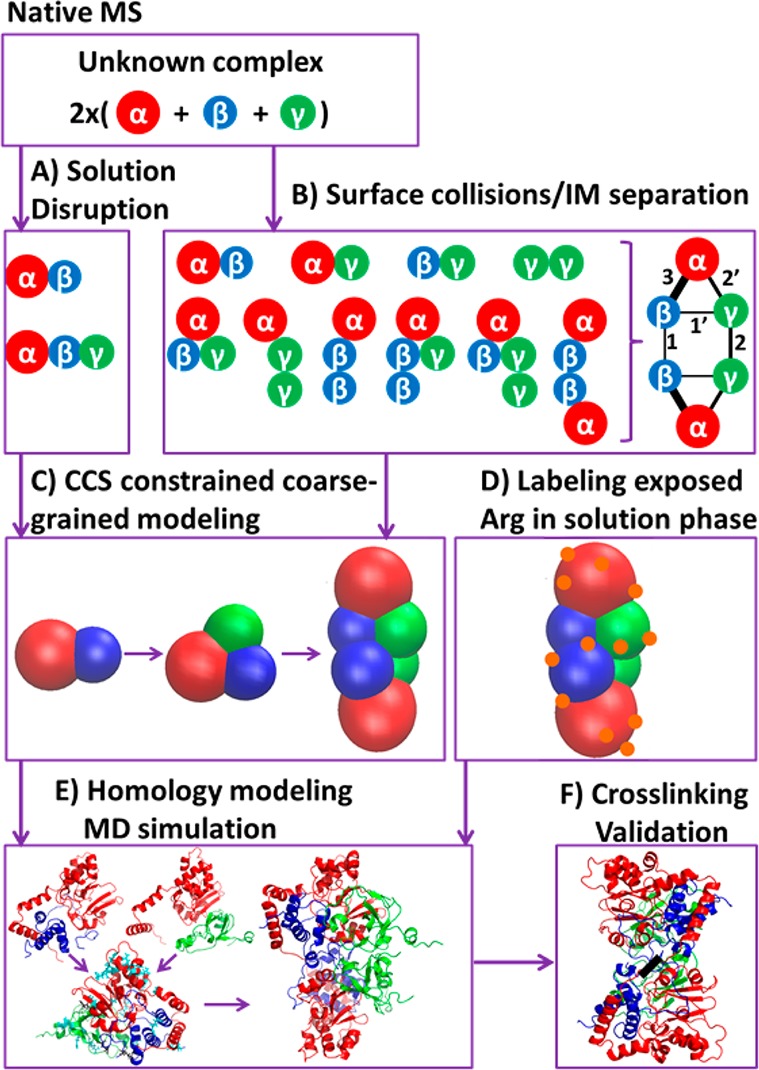
By native mass spectrometry, the stoichiometry of different subunits is obtained. (A) Performing solution disruption results in partial subunit connectivity. (B) Surface induced dissociation (SID) with ion mobility (IM) separation provides a complete connectivity map with relative interfacial strengths. (C) Collisional cross sections (CCSs) for TNH complex and subcomplexes, which are generated by solution disruption and SID gas phase disruption are used as constraints to directly build a coarse-grained model for the TNH complex. (D) Surface mapping experiments covalently label solvent accessible Arg residues, thus providing residue level buried/exposed constraints. (E) Homology modeling generates trimer atomic-level structural candidates, with RMSD during molecular dynamics (MD) simulations, CCSs, and residue buried/exposed constraints to evaluate candidates. Finally, the best trimer model is docked to provide possible hexamer structures. (F) The hexamer structures are partially validated by BS2G and BS3 chemical crosslinking.
Solution Disruption Experiments Reveal TNH Subcomplexes αβγ and αβ
Solution disruption studies have successfully predicted the subcomplexes in several protein complex systems. Hernández et al.,40 for example, were able to derive a three-dimensional interaction map of yeast exosome, which contains 10 different subunits, by generating subcomplexes from a highly pure native protein complex via methanol and DMSO solution disruption. As an alternative to adding organic solvent to perturb the native complexes, ionic strength can also be manipulated to generate protein subcomplexes. Zhou et al.41 observed a series of subcomplexes from elF3, a complex containing 13 subunits, with increasing ammonium acetate (AmAc) concentration from 0.25 to 0.5 M. It was suggested that electrostatic interactions play an important role in sustaining the whole complex, and thus it can be disrupted by high ionic strength.
Our previous energy resolved-SID on a QTOF platform showed that the TNH hexamer is a dimer composed of two αβγ trimers, with subunits α and β interacting strongly.39 Solution disruption was used here as an alternative tool to generate subcomplexes for comparison with our previous SID results (Scheme 1A). Increasing ionic strength for the TNH protein complex by raising the AmAc concentration up to 1 M did not induce the dissociation of TNH hexamer (spectra shown in Supplementary Figure S-1). This suggests that the strongest interactions between subunits are most likely to be hydrophobic interactions, which would not be disrupted with higher ionic strength solutions, but which might be disrupted at higher percentages of organic solvents.42 For this reason, and also because TNH is maximally active (∼120 s–1) in up to 30% methanol, and maintains significant activity (∼70 s–1) at 60% methanol (Supplementary Figure S-2 and supplementary methods), methanol was used for the solution disruption experiment. As shown in Figure 1A, increasing the percentage of methanol in the sample solution leads to the dissociation of TNH hexamer (dark striped area decreases). The subcomplexes generated from solution disruption studies can provide insight into the arrangement of subunits in the intact complex. For TNH, the αβγ trimer and αβ dimer (black solid and purple crosshatch areas, respectively) are observed as the methanol content increases. In addition, the single subunits of α, β, and γ can also be observed with increasing methanol percentage (red, blue, and green areas, respectively). The presence of the αβγ trimer and αβ dimer subcomplexes from methanol disruption agrees very well with our previous SID-MS results from a QTOF platform; thus a partial interaction map can be generated from solution disruption (Figure 1B). The hexamer is clearly composed of two αβγ trimers. Within the trimer, the α–β interaction is the strongest, enabling these subcomplexes to be preserved. However, the solution disruption experiments reveal neither how γ interacts with α and β within the trimer, nor how the two trimers interact.
Figure 1.
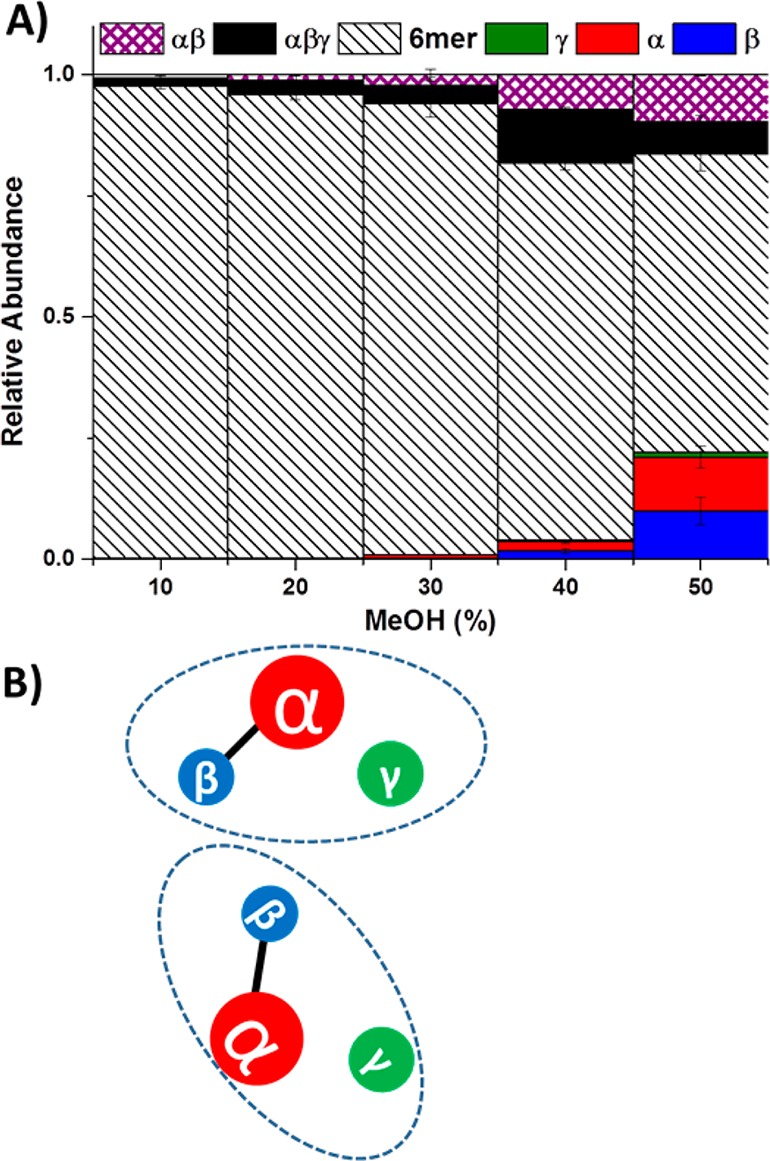
(A) Methanol-induced solution disruption of TNH hexamer. The detection of the TNH complex and its subcomplexes are by nanoelectrospray MS. Observation of αβ subcomplex indicates that α and β are in contact and their interaction is strong so that the dimer can be preserved upon methanol disruption. Observation of αβγ subcomplexes indicates that γ can interact with αβ, but does not indicate whether it is interacting with α or β or both. (B) A cartoon showing the connectivity of subunits in TNH by methanol disruption results. The hexamer is composed of two αβγ trimers and within the trimer, the α–β interaction is the strongest.
SID/IM and SID/IM/SID Reveal Additional Information on Subunit Arrangement
SID is a gas phase dissociation method that involves collision of an analyte with a surface. The dissociation has been shown for small molecules to occur over a very fast time scale (picoseconds),18 and it has the advantage that, because of the more massive target, greater energy is transferred in a single collision with the target than in collision induced dissociation (CID) with a smaller gaseous target. The SID energy is not deposited stepwise in multiple very small increments as it is in CID. Furthermore, Zhou et al.43 have shown that SID of reduced charge state complexes better preserves subunit contacts of products than dissociation of the higher charge states observed by directly spraying from AmAc. SID can access more dissociation pathways and provide more information about the contacts within a protein complex,13−18 making identification of weaker and stronger interfaces possible as collision energy is raised.44 The CID product profile of the TNH hexamer for a range of precursor charge states (+14 to +19) is dominated by monomer ejection. However, surface induced dissociation of +14 TNH hexamer (charge-reduced precursor) generates several subcomplexes that provide connectivity information (Scheme 1B). The major products observed in low energy SID coupled with ion mobility (SID/IM) are αβγ trimer (Figure 2A, shown with a square root intensity scale). At higher SID energies, αβ dimer (32%, if we assign αβγ trimer as 100%) and αβγ2 tetramer (60%) become more abundant (Figure 2B). The simplest interpretation of this observation is that the two γ subunits are in contact and may provide a significant interface between the two trimers. The interaction between α and β is the strongest within the αβγ trimer. With the separation provided by IM and a square root intensity scale, some less abundant subcomplexes can also be observed clearly in higher energy SID/IM (Figure 2B), namely, the γ2 dimer (5%), βγ dimer (1%), αγ dimer (10%), αγ2 trimer (8%), αβ2 trimer (2%), α2β2 tetramer (2%), and αβ2γ tetramer (2%). These products appear only at the higher SID energies, suggesting that their dissociation from the complex may involve breaking a greater number of interfaces or stronger interfaces. The presence of αβ, αγ, and βγ suggests that the αβγ trimer may adopt a trigonal arrangement, with any two of the three subunits in contact. The presence of γ2 dimer and αγ2 trimer suggests that two γ subunits are in contact. The γ2 dimer and α2β2 tetramer may come from dissociation of the precursor with multiple interfaces broken at the same time. The αγ dimer and αγ2 trimer may be generated from secondary fragmentation from αβγ trimer and αβγ2 tetramer, respectively. Alternatively, αγ dimer, αγ2 trimer might be generated directly from the hexamer, yielding complementary αβ2γ tetramer (2%) and αβ2 trimer (2%). These complementary oligomers might then undergo secondary dissociation to αβγ trimer and αβ dimer. The observation of αβ2 trimer, αβ2γ tetramer, α2β2 tetramer, and αβ2γ2 pentamer makes it highly likely that the two β subunits are in weak contact. No α2 dimer is detected, and all the subcomplexes containing two α, which are α2β2 tetramer (2%), α2βγ2, and α2β2γ pentamer (3% each), can be explained by the contact of two β and/or two γ. In addition, no α2 dimer has been observed when the recombinant α, which is catalytically active, is expressed alone.45 Thus, the SID and expression data both strongly suggest that the two α subunits are not in contact in the TNH hexamer.
Figure 2.
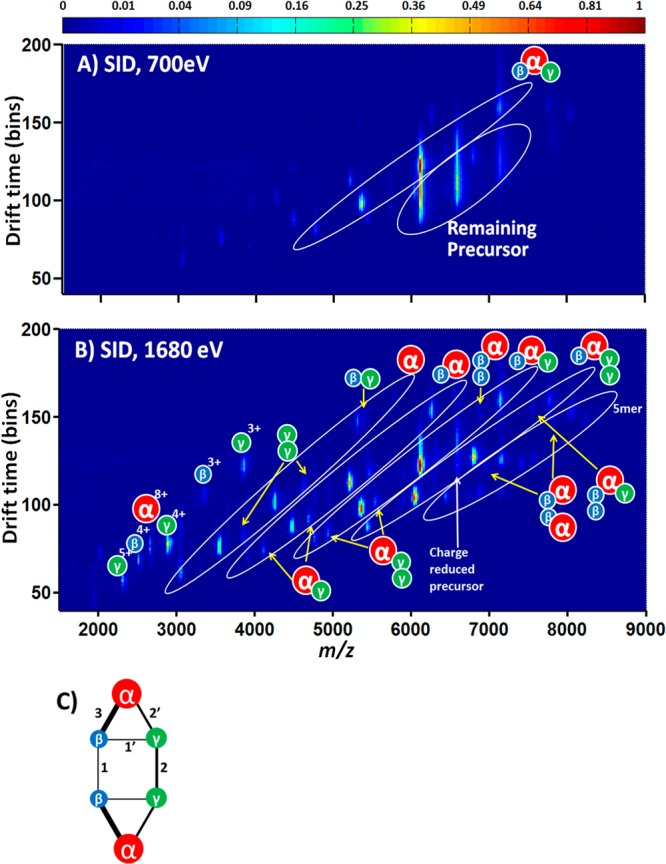
(A) SID/IM/MS plot (700 eV) and (B) SID/IM/MS plot (1680 eV) for +14 TNH hexamer. The intensity of spots shows the normalized abundance of the ion species (square root scale). (C) A cartoon showing the connectivity of subunits in TNH based on the SID/IM/MS results. In (A), the most abundant species are αβγ trimer (average charge ∼7) and remaining precursors with charge reduction. In (B), the most abundant species in oval areas are α monomer (33%, average charge ∼5), αβ dimer (32%, average charge ∼6), αβγ trimer (100%, average charge ∼7), αβγ2 tetramer (60%, average charge ∼8), and αβ2γ2 pentamer (11%, average charge ∼9), respectively. There are also species with lower abundance in oval regions which correspond to γ2 dimer (5%), βγ dimer (1%), αγ dimer (10%), αγ2 trimer (8%), αβ2 trimer (2%), α2β2 (2%), and αβ2γ (2%) tetramer. In (C), the lines represent contacts between pairs of subunits, and the thicknesses denote the strength of noncovalent interactions: 1 ≈ 1′ < 2 ≈ 2′ < 3.
The information obtained on the contact between subunits from SID experiments enables the composition of this complex to be visualized, as shown in the cartoon representation given in Figure 2C. The stronger noncovalent interaction of subunits, based on intensities of subcomplexes, is represented by thicker lines connecting spheres. The strongest interaction is α–β, followed by α–γ and γ–γ, while the weakest interactions are between β–β and β–γ with strength of interaction inferred from ion intensities in the SID/IM data of Figure 2B.
The αβγ trimer is the most abundant product from the hexamer upon SID at the lowest energy onset. In order to understand the relative interface strengths within this trimer, a pseudo MS3 experiment can be performed with two SID devices (SID/IM/SID).46 The first SID occurs after the selection of the precursor (+17 hexamer). Following separation in the IM cell, the ions are subjected to a second SID event. Manual extraction of a specific drift time of a trimer from the 2D mobiligram can identify dissociation products of this trimer following increasing secondary SID energies. The energy resolved SID results of either the +8 or +9 αβγ (Supplementary Figure S-3) highlight that the trimer abundance decreases with increasing SID acceleration voltage, as expected. The most abundant products are γ and its complementary dimer αβ across the different SID acceleration voltages. This directly shows that within the αβγ trimer, the interaction between α and β is much stronger than α–γ or β–γ, as the earlier SID/IM experiments inferred. This result is also consistent with direct SID of the trimer formed by solution disruption. SID of that trimer also yields αβ dimer and complementary γ.
Predicting the TNH Structure via Collisional Cross-Section Constraints on Coarse-Grained Models
Good agreement has been reported between experimental collisional cross sections (CCSs) of subcomplexes generated from solution perturbation and theoretical CCSs calculated from structural information in PDB files.32 It has been shown that the CCS of a set of protein complexes represented by overlapping spheres (representing subunits) is in very close agreement to the CCS calculated from high-resolution atomic structural information.32 Pukala et al.31 applied CCS constraints to predict the topology of two 3-unit subcomplexes, f:h:m and e:l:k from human eukaryotic initiation factor 3, the former of which adopts a trigonal geometry, while the latter is linearly arranged. Bernstein et al.47 utilized CCSs of oligomers of amyloid-β proteins to determine the qualitative structure of each of the aggregates, and their results showed that two isoforms of amyloid-β proteins aggregate differently.
The previous successes in correlating CCS methods in predicting complex structures clearly demonstrate that this approach can be very useful in structural characterization of unknown systems, and hence the approach is applied here to the hexameric TNH complex (Scheme 1C). The CCSs of the α and β subunits were measured from the IM/MS spectra obtained from the 50% methanol perturbed solution, which disrupts the complex without significant unfolding of the subunits (Supplementary Figure S-4). The γ subunit, however, is not observed reproducibly under these conditions. By contrast, the γ subunit is observed readily in high energy SID. Thus, the CCS of the γ subunit, along with CCSs of multiple other product species could be obtained from SID/IM/MS. The experimental CCSs of the three subunits measured from methanol disruption and separately by SID are shown in Supplementary Table S-2. There is no significant difference between the CCSs measured by methanol disruption and SID fragmentation for the subunits α and β; thus we make the assumption that it is valid to use the CCS measured in SID for the γ subunit. The CCS values of individual subunits were used to simulate the radius of the corresponding sphere model as described in the Methods section. The radii of the spheres representing different subunits are shown in Supplementary Table S-2. The CCSs of the αβ dimer and the αβγ trimer were used as constraints to limit the possible arrangement of the subunit spheres within the subcomplexes. The experimental CCSs used as constraints of the two subcomplexes αβ and αβγ were measured from 50% methanol perturbation and the full IM/MS scan. There is no significant difference in CCSs measured for αβ dimer by methanol perturbation full MS and SID tandem MS (Supplementary Table S-2). However, the CCS measured for αβγ by SID tandem MS is slightly smaller (5.5%) than the CCS measured by methanol perturbation (Supplementary Table S-2). This may be due to the subcomplexes collapsing slightly following SID of hexamer, which has been reported previously by Zhou et al., for other systems and in this case the cross section from the MeOH disruption was used in the modeling.43 The simulation of αβ dimer was based on optimizing the distance, d, between the two centers to achieve the experimental CCS. When d equals 0.66(rα + rβ), or the αβ linear overlap is 34%, the simulated model reaches the same CCS as the experimental value. After determination of the coarse-grained αβ dimer model, the location of γ was explored to achieve a CCS that matches the experimental trimer CCS. As Figure 3A illustrates, in the plane defined by the centers of α, β, and γ with the coordinates α and β fixed, the location of γ can be represented by a center (xγ, yγ) value and a known radius. At each (xγ, yγ), a CCS of the trimer was calculated and its difference from the experimental methanol disruption CCS value is shown as the color of the (xγ, yγ) spot, with the more intense green demonstrating a closer representation of the experimental CCS. It was previously reported that the average linear overlap derived from a series of coarse-grained models generated with known crystallographic positions with atoms was 29 ± 10%.32 Thus, together with the restriction that linear overlap must be over 20% and below 40%, the center of γ was determined to be at the position indicated by the red arrow in Figure 3A (Xγ, Yγ) = (18, 21). The model results show that αβγ adopts a trigonal topology, rather than a linear arrangement, in good agreement with SID/IM predictions.
Figure 3.
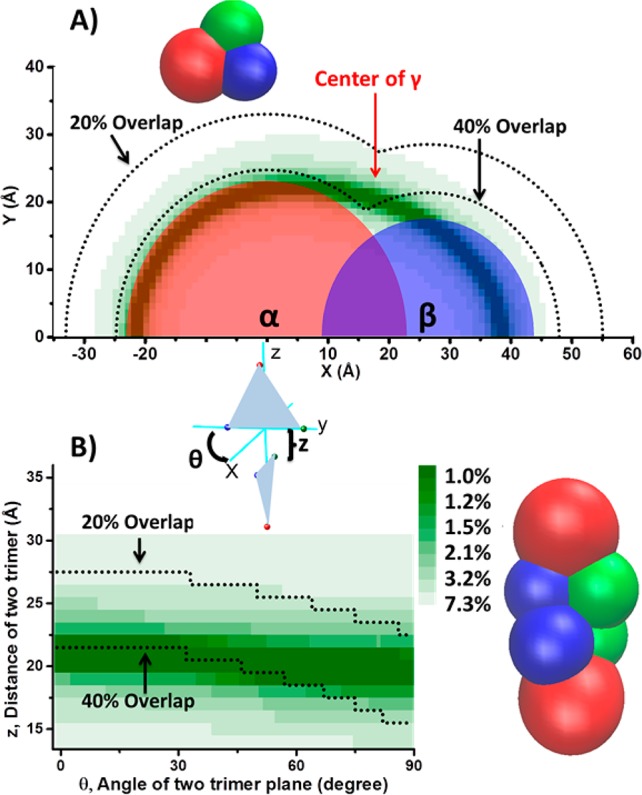
(A) Possible location of the center of a coarse-grained sphere γ subunit in the plane defined by the centers of coarse-grained spheres α (0, 0), β (0, 26.5) and γ (Xγ, Yγ). (Xγ, Yγ) is varied to obtain relative difference of modeled CCS from the experimentally measured αβγ CCS, as shown by the intensity of the green. More intense green shows closer approximation to the measured CCS as shown by the color bar. The white area represents a CCS error over 7.3%. The dashed curve shows the 40% and 20% linear overlap limits of the γ center from α and β. Considering both the CCS constraints and overlap constraints, the most likely location of the γ center is highlighted by the red arrow. (B) A possible arrangement of the hexamer by docking the two trimers represented by sphere models. The two variables are the angle (θ) of the two trimer plane and the distance (z) between the two trimers.
After assembly of the αβγ trimer, the next step is to associate the two trimers into a hexamer and use the hexamer CCS to constrain the possible structure. SID/IM experiments showed that the two γ subunits are in close contact and also that two β subunits are in contact. Most protein complexes adopt symmetry to some extent,23 so here we initially assume that the sphere-modeled hexamer will follow C2 symmetry. If the first trimer is fixed in the yz plane and the center of βγ is the origin, the second trimer triangle can be initiated as a mirror image of the first triangle along the xy plane. Then the second trimer needs to be rotated along the z axis (θ degree) and translated along the z axis (z angstrom) to accommodate/reduce overlap of spherical units. A 2D color map shows a combination of θ and z and their corresponding absolute relative CCS error compared with the measurement (Figure 3B). The experimental CCS can be achieved, and a resulting possible arrangement of TNH hexamer represented by stacked spheres is also shown in Figure 3B.
Phenylglyoxal Surface Mapping of TNH to Identify Surface-Exposed Arg Residues
To obtain additional constraints to help in constructing an atomic model of the TNH structure, we carried out surface mapping experiments (Scheme 1D). Surface mapping involves covalently labeling solvent-exposed amino acids with specific functional groups. Thus, structural information on the protein in native solution can be preserved postdigestion, in the form of modified peptides. Lysine is a target heavily used for covalent labeling in protein chemistry due to its reactive free amine group in the side chain.33 There are, however, only four lysines in the whole TNH complex, which makes lysine a less favorable target in our analysis. In TNH, arginine accounts for 95% percent of the basic residues (K and R), and hence a reagent that targets arginine is more appropriate for our analysis. Phenylglyoxal (PGO) is reported to react with guanidinium groups.48Supplementary Scheme S-1A shows the experimental procedure for surface mapping experiments, and the reaction of PGO and an arginine is shown in supplementary Scheme S-1B. To demonstrate its sensitivity and selectivity toward exposed arginine versus buried arginine, glutamate dehydrogenase (GDH) hexamer, which has a known X-ray crystal structure (PDB: 3MVO), was reacted with PGO as a control. GDH is a homohexamer with D3 symmetry. Among the 30 arginines in each chain, 20 of them have a relative solvent accessible surface area compared to Gly-Arg-Gly (%SASA) of over 30% as calculated by GETAREA49 software, which is a way of assessing if a residue is exposed.36Supplementary Table S-3 shows calculated %SASA and the experimentally determined labeling results at five concentrations of PGO, ranging from 0.5 to 7 mM. Bottom-up proteomics indicates a high level of labeling of the 17 covered Arg residues that have over 30% SASA at all concentrations of PGO employed, as shown in Supplementary Figure S-5. By contrast, buried or solvent inaccessible Arg residues are only labeled at higher concentrations of PGO or are not labeled at all. For example R396, which is buried in the structure of the GDH complex (<30% SASA), did not get labeled at any of the levels of PGO concentrations studied. R86 (<30% SASA), however, is labeled at high concentrations of PGO. The threshold for reaction between PGO and solvent inaccessible Arg residues in GDH appeared to be ∼2 mM.
Treatment of TNH with PGO leads to modification of a number of Arg residues in TNH. Sixteen out of 41 Arg residues are modified regardless of the concentration of PGO employed (Supplementary Table S-4). Use of covalent labeling (surface mapping) approaches requires consideration of whether the introduction of modifications can alter the structure of the analytes. Thus, the structural integrity of the TNH complex after labeling was checked by native MS to see whether the complex has dissociated upon labeling. As shown in Supplementary Figure S-6, as the concentration of labeling reagent PGO increases, the hexamer abundance decreases slightly and αβγ trimer and αβ dimer increase slightly. Therefore, it is possible to have buried Arg residues labeled at higher concentrations of PGO, disrupting the overall structure. The labeled residues at low PGO concentration were used to provide constraints for high resolution models.
High-Resolution Structural Models for TNH Hexamer
The data on TNH and its subcomplexes obtained from the SID experiments described above and the coarse-grained modeling can be combined with constraints from the PGO labeling experiments to infer high-resolution structural information on TNH by computer modeling (Scheme 1E). To identify a good starting model for the computations, the amino acid sequences of TNH subunits were examined by Protinfo PPC50 (protinfo.compbio.washington.edu/ppc/), which exports starting models for protein complexes based on sequence similarity searches and comparison to known high resolution structures. The TNH hexamer studied here has some homology to other nitrile hydratases that have been studied previously. However, unlike others which are tetramers, TNH consists of three subunits that are together homologous to the αβ dimeric subunits of the prototypical tetrameric nitrile hydratase. ProtinfoPPC did not return any hits when the sequences of the α, β, γ subunits were all input together. However, when we used the sequences of the αβ or αγ subunits, several hits were obtained. When the sequences of α and β subunits were submitted, five homologues with known PDB structures were identified (1ugq, 3hht, 1ugs, 2d0q, and 1ahj). The submission of α and γ sequences returned six structural homologues (3hht, 2cz6, 1ahj, 1ugp, 1ugq and 1ugs). As expected, the N- and C-terminal halves of the β subunits of known tetramer α2β2 nitrile hydratases are homologous to the β and γ subunits of hexamer α2β2γ2 TNH, respectively.38
The four αβγ trimer models that were common to ProtinfoPPC searches conducted with both αβ and αγ are 1ugq, 3hht, 1ahj and 1ugs (shown above in boldface); the nature of these homologues and TNH sequence alignment to these homologues are provided in Supplementary Table S-5. Each of these models was subjected to a 1000-step energy minimization and 10 ns equilibration in a water box or vacuum by NAMD software51 to reduce any steric clashes and also test model stability (detailed description in Methods). The calculated root-mean-square deviation (RMSD) of backbone atoms (relative to the initial trimer PDB) over the simulation time frame for each candidate is shown in Supplementary Figure S-7. Models based on 3hht and 1ahj maintain lower RMSD throughout the course of the simulation as compared to 1ugq and 1ugs, suggesting that they are more stable. We repeated the simulation in water boxes three additional times to confirm that 3hht and 1ahj maintain lower RMSD (Supplementary Figure S-8). Furthermore, the CCSs of candidates in dynamic simulations were calculated by the scaled projection approximation (PA) method30 and compared with the experimental αβγ trimer CCS from solution disruption. The relative CCS deviations of atomic coordinates of every 2 ns simulation in either a periodic water box or vacuum are plotted in Supplementary Figure S-9. For all the candidate structures, the CCS in water equilibrium is greater than the measured αβγ trimer CCS, and the CCS in vacuum equilibrium is smaller than the measured αβγ trimer CCS (by methanol disruption), but within 2% of the trimer CCS produced by SID. The candidate structure based on 3hht has the closest CCS to the αβγ trimer CCS formed by methanol disruption and is 7% higher in water equilibrium and 3% lower in vacuum equilibrium than the measured value.
Covalent labeling results from surface mapping experiments are further used to eliminate models that possess buried Arg residues at positions that we know likely are not buried because they are modified even at the lowest concentration of PGO (Supplementary Table S-4). Because the PGO labeling experiments are performed in solution phase, the NAMD simulation employing a periodic water box were used for comparison with the experimental results. The model based on 3hht is most consistent with the experimental labeling results (Figure 4). For each arginine, the greatest %SASA among 2, 4, 6, 8, 10, 12, and 14 ns simulation frames of the model based on 3hht is shown in Supplementary Table S-4. Arg residues that have %SASA over 30% are considered to be exposed. The table indicates that none of the buried arginines in the model based on 3hht are labeled with PGO at low concentration. There are some arginines that are predicted to be exposed by modeling and simulation of the trimer. They are not labeled in the hexamer by PGO at any concentration level, and are α134, β2, and γ26 (Supplementary Table S-4, Figure 4). We cannot exclude the possibility that the local environment around these residues, though accessible to solvent, occludes the larger, hydrophobic PGO label. We are, however, not trying to predict labeling patterns in the trimer but rather in the hexamer, which is what was labeled experimentally; Arg residues that are predicted to be exposed on the surface of trimer but not labeled experimentally are likely involved in trimer–trimer interactions. The locations β2 and γ26 are highly likely to be involved in the trimer–trimer interface based on the SID/IM experiments combined with coarse-grained modeling (Scheme 1C, Figure 2C, and Figure 3B).
Figure 4.
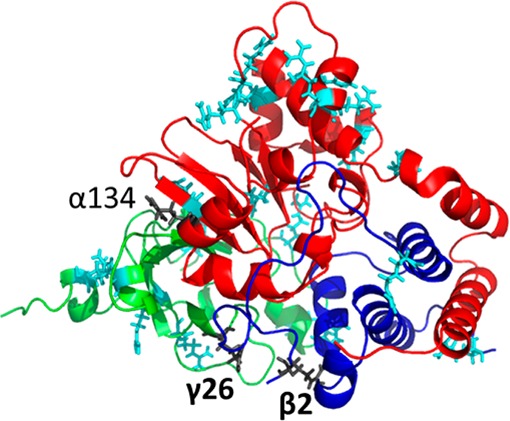
Modeled TNH αβγ trimer structure based on homologue PDB 3hht and equilibrated in a periodic water box for 10 ns. Subunits α, β, and γ are in red, blue, and green, respectively. The exposed arginines are in stick representation. Labeled arginines are in cyan color. Arginine β2 and γ26 in dark gray are not labeled and thus postulated to be involved in trimer–trimer interactions to form hexamer.
On the basis of the above results, two identical modeled TNH αβγ trimer structures (Figure 4) based on homologue PDB 3hht and equilibrated in a periodic water box for 10 ns were subjected to docking using the program ZDOCK52 (http://zdock.umassmed.edu/) with β2 and γ26 Arg residues selected as contacting residues. Ten hexamer models were returned and further subjected to a 1000-step energy minimization, followed by 10 ns water equilibrium. Hexamers 1, 2, and 3 are the most stable structures as evidenced by their low RMSD (<3.5 Å, Supplementary Figure S-10) throughout the course of the simulation. The calculated CCSs of the energy-minimized (0 ns) hexamer structures 1, 2, and 3 are within 5% error compared with the experimental hexamer CCS (Supplementary Figure S-11). Furthermore, although the %SASA of β2 of the hexamer is ∼50%, it is smaller than the %SASA of the corresponding trimer β2 (∼65%). The %SASA of γ26 Arg is below 30% among all the three structures, indicating that it is buried upon hexamer formation. Therefore, the surface mapping experiments and homology modeling, together with SID/IM constraints, predict a tentative model αβγ trimer (Figure 4) and three possible hexameric structures (Figure 5). Given that the TNH sample is hard to crystallize, it is highly possible that the hexamer is flexible, having several different trimer–trimer interconverting conformations. The MS/computational modeling approach used here thus may be providing insights into the dynamic nature of TNH.
Figure 5.

Possible TNH hexamer structures (hexamer 1 in green, 2 in magenta, and 3 in blue) with docking of αβγ trimer structure based on homologue PDB 3hht and equilibrated in periodic water box for 10 ns. The difference between the three structures is the rotation angle between the top and bottom trimers. The left shows alignment by the top trimers and the right shows alignment by the bottom trimers.
To further cross-validate the interfacial strengths within the proposed hexamer structures and the predicted interfacial strengths from the SID/IM experiments, the interfacial areas of the three modeled hexamer structures after energy minimization and 10 ns vacuum equilibrium were calculated via PDBePISA53 (www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver) and are shown in Supplementary Figure S-12. If we assume the interfacial strength is proportional to the interfacial area, the interfacial strengths of the three proposed hexamer structures trend with the SID/IM prediction (the strongest interaction is α–β, followed by α–γ and γ–γ, and the weakest interaction is β–γ and β–β). As a final validation of the proposed structures, we also performed chemical cross-linking experiments (cross-links indicated in Scheme 1F, details in methods) with the amine-to-amine cross-linkers, BS2G and BS3 with two different lengths (7.7 and 11.4 A). Cross-linked peptides (at α N-terminus to α N-terminus, α N-terminus to β N-terminus) consistent with two of the three structures proposed from homology modeling were detected (Supplementary Figure S-13), validating these structures. It is noteworthy that the homologue 3hht, which was used as the starting point for the homology modeling, could not produce the cross-links shown here, which are unique to the computed structures and consistent with the experimental results.
Conclusions
Multiple MS based approaches have been applied to refine the structure of hexameric TNH. The results from both the previously reported SID experiments performed on a QTOF platform and the current solution-phase organic solvent disruption native MS experiments suggest that TNH is a dimer composed of αβγ trimers, and that α–β interact strongly. Other subcomplexes in addition to αβγ and α–β provide contact information for TNH and were generated in surface induced dissociation experiments coupled with ion mobility MS (SID/IM), which enables the construction of a complete connectivity map with relative interfacial strengths. The results reveal that γ–γ subunits are the primary contact between the two trimers, and weak interactions between the two β subunits may also exist. None of our data suggest that the two α subunits interact within the hexamer. The trimer–trimer contact information is critical for building either the coarse-grained or the atomic level TNH hexamer models. Collisional cross sections measured from IM experiments of both solution and SID disrupted complexes were used as constraints for postulating the arrangement of the subunits represented by coarse-grained spheres. Homology modeling was utilized to propose a possible atomic structure of the TNH αβγ trimer with necessary constraints from covalent labeling, SID, and solution disruption. Trimers were combined via docking software to generate three possible atomic structures of the TNH hexamer. Chemical cross-linking provided experimental results consistent with two of the proposed hexamer models (cross-linking the amino termini of the two α subunits in one model and cross-linking the α and β amino termini in a second proposed model). The fact that two of the similar proposed model structures fit the cross-linking data may suggest some motion of the complex. In addition to providing structural information on TNH that can be used for future protein engineering, the combined results from different MS approaches and computational modeling provide a robust framework for analysis of other protein complexes that cannot be characterized by X-ray crystallography, NMR, or cryo-EM.
Methods
Complex Analysis by Native Mass Spectrometry
TNH from Streptomyces rimosus was prepared by the Bandarian lab at the University of Arizona and was expressed and purified as written in Supplemental Methods, Supporting Information. To perform solution disruption and SID/IM experiments, the sample was first buffer exchanged into 100 mM ammonium acetate (AmAc, Sigma-Adrich, St. Louis, MO) via Micro Bio-Spin 6̅ Columns (Bio-Rad, Hercules, CA). The final concentration of the protein complex was 16 μM. Solution disruption was performed by adding 10%, 20%, 30%, 40%, and 50% methanol (Fisher Scientific, Pittsburgh, PA) to buffer exchanged protein but keeping the ionic strength (100 mM AmAc) and protein complex concentration (8 μM) constant. Charge reduction was performed by adding triethylammonium acetate (TEAA, Sigma-Adrich) to sample solution to a final concentration of 30 mM with AmAc at 100 mM.
Samples were nanoelectrosprayed into a SYNAPT G2-S mass spectrometer (Waters Corporation, Manchester, UK) with an SID device incorporated after the trap cell and before the ion mobility cell (SID/IM experiments).15,43 The trap traveling wave ion guide (TWIG) was previously truncated to accommodate the SID cell. Under transfer mode, the voltages applied to the 10 lenses of the SID cell were adjusted to transmit without surface collisions. In SID mode, the ions were steered toward the surface by the voltages on the lenses relative to upstream voltages. All of the electrostatic lenses are tuned to achieve good precursor transmission and product collection. For voltages applied to lenses at specific conditions from 30 to 180 V SID acceleration voltages, see Supplementary Table S-1. Collision energy is charge times acceleration voltage. Glass capillaries for nanoelectrospray ionization were pulled on a P-97 micropipette puller (Sutter Instruments, Hercules, CA) and filled with sample solution. A 0.8–1.1 kV ionization voltage was applied to a platinum wire inserted into the back of the capillary. Typical instrument settings for SID/IM experiments were sampling cone 20 V, source offset 10 V, source temperature 25 °C, trap gas flow 2 mL/min, helium cell gas flow 180 mL/min, IMS gas flow 60 mL/min; trap DC entrance 0 V, trap DC bias 83 to 233 V (corresponding to SID acceleration voltages of 30 V-180 V), trap DC −2 V, trap DC exit 0 V, trap wave velocity 160 m/s, trap wave height 4 V; IMS DC entrance 10 V, helium cell DC 25 V, helium exit −5 V, IMS DC bias 5 V, IMS DC exit 0 V, IMS wave velocity 300 m/s, IMS wave height 20 V; transfer DC entrance 2 V, transfer DC exit 15 V, transfer wave velocity 50 m/s, transfer wave height 4 V.
Collisional Cross-Section Measurement and Spherical Subunit Modeling
The CCS was measured following a published protocol.28 The calibrants used for α, β, γ monomer and αβ dimer CCS measurement were +3, + 4 melittin from honey bee venom, + 6, + 7 cytochrome c from equine heart, + 8, + 9 β-lactoglobulin monomer and +11 to +13 β-lactoglobulin dimer from bovine milk. The calibrants used for αβγ trimer and (αβγ)2 hexamer were +14 to +16 transthyretin from human plasma, + 15 to +18 avidin from egg white, + 20 to +23 concanavalin A, + 23 to +26 alcohol dehydrogenase. All the protein standards were purchased from Sigma-Aldrich (St. Louis, MO) and prepared at 10 μM in 100 mM AmAc.
The CCSs of each subunit were used to generate the spherical models. The approach used was described by Hall et al.,32 the radius of a spherical model r = (CCS/π)0.5 – rHe, in which rHe = 1.4 Å. The projection approximation (PA) in the Mobcal program54,55 and a scaled PA method56 were used to perform theoretical CCS calculations. The mobcal.f (downloaded from http://www.indiana.edu/~nano/software.html) was modified to calculate the CCS of a complex of spherical models as explained in a published protocol.28 The spherical model was generating by adding a subunit as an atom in the file but specifying the mass and radius derived from IM/MS analysis. (The specific lines added are shown in Supplemental Methods, Supporting Information.) The coordinates of the centers of the spheres (in the .mfj file) determine how the spheres are stacked together. An example of an .mfj file is shown in Supplemental Methods, Supporting Information.
Bottom-up Approach for Surface Mapping Modification Localization
A flowchart showing the experimental procedure for surface mapping experiments is shown in Supplementary Scheme S-1A, and the reaction used to label amino acids side chain is shown in Supplementary Scheme S-1B. The surface mapping of arginines in TNH or control protein glutamate dehydrogenase (GDH, Sigma-Adrich, St. Louis, MO) was performed at room temperature for 14 h. 1 mM, 2 mM, 4 mM, and 7 mM of phenylglyoxal (PGO, Sigma-Adrich, St. Louis, MO) were chosen to react with 8 μM of TNH in 25 mM HEPES (pH 7.5), yielding 125:1, 250:1, 500:1, and 875:1 molar ratios of phenylglyxal to TNH. The ratios of PGO to arginine targets in TNH were 1.5:1, 3:1, 6:1, and 11:1, respectively. To keep approximately the same PGO to arginine ratios for GDH, the concentration used was 4 μM GDH. The reaction was stopped by depleting PGO through buffer exchanging to 100 mM AmAc via Micro Bio-Spin 6̅ Columns. A part of the sample was subjected to native MS analysis to check hexamer integrity, and the rest was subjected to bottom-up experiments to localize the modified arginine.
The bottom-up approach to identify arginine modifications was performed by in-solution pepsin digestion of PGO labeled samples with LC-MS/MS analysis on a linear ion trap mass spectrometer (Velos Pro, ThermoFisher Scientific Inc., Waltham, MA) coupled to a UPLC (nanoACQUITY, Waters Corporation, Manchester, UK). Pepsin digestion was performed first by adjusting the pH of the labeled sample solution with 1 M HCl (Sigma-Adrich, St. Louis, MO) to below 4, followed by adding pepsin (Promega, Madison, WI) at a 1:10 enzyme/protein ratio. The digestion was performed at 37 °C, on a 150 rpm thermomixer for 7 h. The reaction was stopped by heating on a 95 °C block for 5 min. Prior to LC-MS/MS injection, the sample was centrifuged at 10000g for 10 min. The amount of injection onto the UPLC was 0.4 μg.
LC-MS/MS data analysis was performed by SEQUEST HT in Proteome Discoverer software (ThermoFisher Scientific Inc., Waltham, MA). Searching parameters were precursor mass ranging from 400 to 5000 Da, peptide length ranging from 4 to 144, precursor mass tolerance 1.5 Da, and fragment mass tolerance 0.8 Da. The database searched included the sequences of α, β, γ of TNH and pig pepsin. No enzymes were specified for in silico digestion. Dynamic modifications were methionine oxidiation (M + 15.995) and phenylglyoxal reacted arginine (R + 116.026 or 134.0368). Peculator was used to validate the identities of peptides with a 1% false discovery rate (FDR).
Molecular Modeling
The structural model candidates for the TNH αβγ trimer are generated based on four templates (PDB ID: 3hht, 1ugq, 1ugs, and 1ahj) with dimer models predicted by protinfo ppc server.50 A 10 ns all-atom molecular dynamics simulation with periodic water box was conducted using NAMD 2.951 with the CHARMM force field.57,58 The periodic water box was set up as a layer of 10 Å water from the atom with the largest coordinate in xyz dimensions. The charges were neutralized by 0.15 M NaCl. Long-range forces in the periodic system were calculated using the particle mesh Ewald (PME)59 with 1.0 grid spacing. Linear bonds involving hydrogens are treated as rigid bonds to allow the simulation to be performed at 2 fs steps. Different trimer candidates were first subjected to 1000 steps of energy minimization, followed by equilibrium simulation in constant temperature, 310 K, and constant pressure, 1 atm, using the Langevin piston method.60
Also, a 1000-step energy minimization and 10 ns all-atom molecular dynamics simulation in a vacuum were conducted using NAMD 2.951 with the CHARMM force field.57,58 In this case, none of periodic boundary conditions, PME, and constant pressure was used.
Chemical Cross-Linking Experiments
The amine-to-amine cross-linking was performed by adding 2 mM BS2G or BS3 cross-linker (Thermo Fisher Scientific, Waltham, MA) to 10 μL 200 μM TNH hexamer in 25 mM HEPES solution and reacted at room temperature for 2 h. The reaction was quenched by adding 50 mM Tris·HCl (Sigma-Aldrich, St. Louis, MO). The control was performed by replacing cross-linker solution by water. One sixth volume of the control and cross-linked samples were mixed with 4× Laemmli protein sample buffer followed by heating at 95 °C for 5 min. The samples were then loaded on a 4–20% precast protein gel (Mini-PROTEAN TGX, Bio-Rad, Hercules, CA) together with 10 μL of Precision Plus Protein standard (Bio-Rad) in the adjacent lane.
Two bands that appeared in the cross-linked sample, but were absent in the control sample have masses corresponding to 43 kDa (Band 1) and 30 kDa (Band 2). They were excised and subjected to an in-gel trypsin digestion. One third of the digest was subjected to an LC-MS/MS analysis on an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) coupled to a nanoACQUITY UPLC (Waters). Cross-linked peptides were searched in Thermo Discoverer (V1.4) by adding masses of N-terminal tryptic peptides or K-containing tryptic peptides, together with cross-linker added mass (C5H4O2 96.0211 Da for BS2G and C8H10O2, 138.0681 Da for BS3) as dynamic modifications.
Acknowledgments
Research reported in this paper was supported by the National Institutes of Health under Award Numbers R01GM113658 to V.H.W. and R01GM72623 to V.B. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Also, this material is based upon work supported by the National Science Foundation under Grant No. 0923551 (V.H.W.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. We thank the Ohio Supercomputer Center for computational resources. Dr. Marcos Sotomayor is also thanked for discussions on coarse-grained sphere modeling and atomic level modeling. Sophie R. Harvey and Royston S. Quintyn are thanked for setting up instrumentation for performing SID/IM/SID experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.5b00251.
Supporting Information includes supplementary figures, tables and methods (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Aebersold R.; Mann M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.; Tomas H.; Havlis J.; Olsen J. V.; Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protocols 2007, 1, 2856–2860. 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Domon B.; Aebersold R. Mass Spectrometry and Protein Analysis. Science 2006, 312, 212–217. 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Vahidi S.; Sowole M. A. Mass Spectrometry Methods for Studying Structure and Dynamics of Biological Macromolecules. Anal. Chem. 2014, 86, 213–232. 10.1021/ac4039306. [DOI] [PubMed] [Google Scholar]
- Heck A. J. R.; van den Heuvel R. H. H. Investigation of intact protein complexes by mass spectrometry. Mass Spectrom. Rev. 2004, 23, 368–389. 10.1002/mas.10081. [DOI] [PubMed] [Google Scholar]
- Ruotolo B. T.; Giles K.; Campuzano I.; Sandercock A. M.; Bateman R. H.; Robinson C. V. Evidence for Macromolecular Protein Rings in the Absence of Bulk Water. Science 2005, 310, 1658–1661. 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- Sharon M.; Robinson C. V. The Role of Mass Spectrometry in Structure Elucidation of Dynamic Protein Complexes. Annu. Rev. Biochem. 2007, 76, 167–193. 10.1146/annurev.biochem.76.061005.090816. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Robinson C. V. When proteomics meets structural biology. Trends Biochem. Sci. 2010, 35, 522–529. 10.1016/j.tibs.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Marcoux J.; Robinson C. V. Twenty years of gas phase structural biology. Structure 2013, 21, 1541–1550. 10.1016/j.str.2013.08.002. [DOI] [PubMed] [Google Scholar]
- Shapovalov M. V.; Wang Q.; Xu Q.; Andrake M.; Dunbrack R. L. BioAssemblymodeler (BAM): user-friendly homology modeling of protein homo- and hetero-oligomers. PLoS One 2014, 9, e98309. 10.1371/journal.pone.0098309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis A.; Stengel F.; Hall Z.; Hernandez H.; Leitner A.; Walzthoeni T.; Robinson C. V.; Aebersold R. A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat. Methods 2014, 11, 403–406. 10.1038/nmeth.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall Z.; Hernández H.; Marsh; Joseph A.; Teichmann; Sarah A.; Robinson C. V. The role of salt bridges, charge density, and subunit flexibility in determining disassembly routes of protein complexes. Structure 2013, 21, 1325–1337. 10.1016/j.str.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Wysocki V. H. Surface Induced Dissociation: Dissecting Noncovalent Protein Complexes in the Gas phase. Acc. Chem. Res. 2014, 47, 1010–1018. 10.1021/ar400223t. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Huang C.; Wysocki V. H. Surface-induced dissociation of ion mobility-separated noncovalent complexes in a quadrupole/time-of-flight mass spectrometer. Anal. Chem. 2012, 84, 6016–6023. 10.1021/ac300810u. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Dagan S.; Wysocki V. H. Protein Subunits Released by Surface Collisions of Noncovalent Complexes: Nativelike Compact Structures Revealed by Ion Mobility Mass Spectrometry. Angew. Chem., Int. Ed. 2012, 51, 4336–4339. 10.1002/anie.201108700. [DOI] [PubMed] [Google Scholar]
- Dodds E. D.; Blackwell A. E.; Jones C. M.; Holso K. L.; O’Brien D. J.; Cordes M. H. J.; Wysocki V. H. Determinants of gas-phase disassembly behavior in homodimeric protein complexes with related yet divergent structures. Anal. Chem. 2011, 83, 3881–3889. 10.1021/ac2003906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. M.; Beardsley R. L.; Galhena A. S.; Dagan S.; Cheng G.; Wysocki V. H. Symmetrical gas-phase dissociation of noncovalent protein complexes via surface collisions. J. Am. Chem. Soc. 2006, 128, 15044–15045. 10.1021/ja064586m. [DOI] [PubMed] [Google Scholar]
- Beardsley R. L.; Jones C. M.; Galhena A. S.; Wysocki V. H. Noncovalent protein tetramers and pentamers with ″n″ charges yield monomers with n/4 and n/5 charges. Anal. Chem. 2009, 81, 1347–1356. 10.1021/ac801883k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X.; Zhou M.; Wysocki V. Surface induced dissociation yields quaternary substructure of refractory noncovalent phosphorylase B and glutamate dehydrogenase complexes. J. Am. Soc. Mass Spectrom. 2014, 25, 368–379. 10.1007/s13361-013-0790-y. [DOI] [PubMed] [Google Scholar]
- May J. C.; McLean J. A. Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Anal. Chem. 2015, 87, 1422–1436. 10.1021/ac504720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Jones C. M.; Wysocki V. H. Dissecting the large noncovalent protein complex GroEL with surface-induced dissociation and ion mobility–mass spectrometry. Anal. Chem. 2013, 85, 8262–8267. 10.1021/ac401497c. [DOI] [PubMed] [Google Scholar]
- Hernandez H.; Robinson C. V. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat. Protoc. 2007, 2, 715–726. 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- Levy E. D.; Erba E. B.; Robinson C. V.; Teichmann S. A. Assembly reflects evolution of protein complexes. Nature 2008, 453, 1262–1265. 10.1038/nature06942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J. A.; Hernández H.; Hall Z.; Ahnert; Sebastian E.; Perica T.; Robinson; Carol V.; Teichmann S. A. Protein Complexes Are under Evolutionary Selection to Assemble via Ordered Pathways. Cell 2013, 153, 461–470. 10.1016/j.cell.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine S. J.; Kulchania M.; Barnes C. A. S.; Clemmer D. E. Multidimensional separations of complex peptide mixtures: a combined high-performance liquid chromatography/ion mobility/time-of-flight mass spectrometry approach. Int. J. Mass Spectrom. 2001, 212, 97–109. 10.1016/S1387-3806(01)00511-5. [DOI] [Google Scholar]
- Wu C.; Siems W. F.; Asbury G. R.; Hill H. H. Electrospray Ionization High-Resolution Ion Mobility Spectrometry–Mass Spectrometry. Anal. Chem. 1998, 70, 4929–4938. 10.1021/ac980414z. [DOI] [PubMed] [Google Scholar]
- Wyttenbach T.; Kemper P. R.; Bowers M. T. Design of a new electrospray ion mobility mass spectrometer. Int. J. Mass Spectrom. 2001, 212, 13–23. 10.1016/S1387-3806(01)00517-6. [DOI] [Google Scholar]
- Ruotolo B. T.; Benesch J. L.; Sandercock A. M.; Hyung S. J.; Robinson C. V. Ion mobility-mass spectrometry analysis of large protein complexes. Nat. Protoc. 2008, 3, 1139–1152. 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- Bush M. F.; Hall Z.; Giles K.; Hoyes J.; Robinson C. V.; Ruotolo B. T. Collision Cross Sections of Proteins and Their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology. Anal. Chem. 2010, 82, 9557–9565. 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- Benesch J. L. P.; Ruotolo B. T. Mass spectrometry: come of age for structural and dynamical biology. Curr. Opin. Struct. Biol. 2011, 21, 641–649. 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukala T. L.; Ruotolo B. T.; Zhou M.; Politis A.; Stefanescu R.; Leary J. A.; Robinson C. V. Subunit architecture of multiprotein assemblies determined using restraints from gas-phase measurements. Structure 2009, 17, 1235–1243. 10.1016/j.str.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Hall Z.; Politis A.; Robinson C. V. Structural Modeling of Heteromeric Protein Complexes from Disassembly Pathways and Ion Mobility-Mass Spectrometry. Structure 2012, 20, 1596–1609. 10.1016/j.str.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Mendoza V. L.; Vachet R. W. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom. Rev. 2009, 28, 785–815. 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamoto K.; Chance M. R. Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 251–276. 10.1146/annurev.biophys.35.040405.102050. [DOI] [PubMed] [Google Scholar]
- Hambly D.; Gross M. Laser flash photochemical oxidation to locate heme binding and conformational changes in myoglobin. Int. J. Mass Spectrom. 2007, 259, 124–129. 10.1016/j.ijms.2006.08.018. [DOI] [Google Scholar]
- Mendoza V. L.; Vachet R. W. Protein Surface Mapping Using Diethylpyrocarbonate with Mass Spectrometric Detection. Anal. Chem. 2008, 80, 2895–2904. 10.1021/ac701999b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar H.; Clarke K. G.; Preez R. d.; Cowan D. A.; Burton S. G. Developments in nitrile and amide biotransformation processes. Trends Biotechnol. 2010, 28, 561–569. 10.1016/j.tibtech.2010.08.004. [DOI] [PubMed] [Google Scholar]
- McCarty R. M.; Bandarian V. Deciphering Deazapurine Biosynthesis: Pathway for Pyrrolopyrimidine Nucleosides Toyocamycin and Sangivamycin. Chem. Biol. 2008, 15, 790–798. 10.1016/j.chembiol.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell A. E.; Dodds E. D.; Bandarian V.; Wysocki V. H. Revealing the quaternary structure of a heterogeneous noncovalent protein complex through surface-induced dissociation. Anal. Chem. 2011, 83, 2862–2865. 10.1021/ac200452b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández H.; Dziembowski A.; Taverner T.; Séraphin B.; Robinson C. V. Subunit architecture of multimeric complexes isolated directly from cells. EMBO Rep. 2006, 7, 605–610. 10.1038/sj.embor.7400702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Sandercock A. M.; Fraser C. S.; Ridlova G.; Stephens E.; Schenauer M. R.; Yokoi-Fong T.; Barsky D.; Leary J. A.; Hershey J. W.; Doudna J. A.; Robinson C. V. Mass spectrometry reveals modularity and a complete subunit interaction map of the eukaryotic translation factor eIF3. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 18139–18144. 10.1073/pnas.0801313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Hamdan S. M.; Dixon N. E.; Sheil M. M.; Beck J. L. Application of electrospray ionization mass spectrometry to study the hydrophobic interaction between the ε and θ subunits of DNA polymerase III. Protein Sci. 2004, 13, 2878–2887. 10.1110/ps.04889604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Dagan S.; Wysocki V. H. Impact of charge state on gas-phase behaviors of noncovalent protein complexes in collision induced dissociation and surface induced dissociation. Analyst 2013, 138, 1353–1362. 10.1039/c2an36525a. [DOI] [PubMed] [Google Scholar]
- Quintyn R. S.; Yan J.; Wysocki V. H. Surface-Induced Dissociation of Homotetramers with D2 Symmetry Yields their Assembly Pathways and Characterizes the Effect of Ligand Binding. Chem. Biol. 2015, 22, 583–592. 10.1016/j.chembiol.2015.03.019. [DOI] [PubMed] [Google Scholar]
- Nelp M. T.; Astashkin A. V.; Breci L. A.; McCarty R. M.; Bandarian V. The Alpha Subunit of Nitrile Hydratase Is Sufficient for Catalytic Activity and Post-Translational Modification. Biochemistry 2014, 53, 3990–3994. 10.1021/bi500260j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintyn R. S.; Harvey S. R.; Wysocki V. H. Illustration of SID-IM-SID (surface-induced dissociation-ion mobility-SID) mass spectrometry: homo and hetero model protein complexes. Analyst 2015, 140, 7012–7019. 10.1039/C5AN01095K. [DOI] [PubMed] [Google Scholar]
- Bernstein S. L.; Dupuis N. F.; Lazo N. D.; Wyttenbach T.; Condron M. M.; Bitan G.; Teplow D. B.; Shea J.-E.; Ruotolo B. T.; Robinson C. V.; Bowers M. T. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 2009, 1, 326–331. 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krell T.; Chackrewarthy S.; Pitt A. R.; Elwell A.; Coggins J. R. Chemical modification monitored by electrospray mass spectrometry: a rapid and simple method for identifying and studying functional residues in enzymes. J. Pept. Res. 1998, 51, 201–209. 10.1111/j.1399-3011.1998.tb01217.x. [DOI] [PubMed] [Google Scholar]
- Fraczkiewicz R.; Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 1998, 19, 319–333. . [DOI] [Google Scholar]
- Kittichotirat W.; Guerquin M.; Bumgarner R. E.; Samudrala R. Protinfo PPC: A web server for atomic level prediction of protein complexes. Nucleic Acids Res. 2009, 37, W519–W525. 10.1093/nar/gkp306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce B. G.; Wiehe K.; Hwang H.; Kim B.-H.; Vreven T.; Weng Z. ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Mesleh M. F.; Hunter J. M.; Shvartsburg A. A.; Schatz G. C.; Jarrold M. F. Structural Information from Ion Mobility Measurements: Effects of the Long-Range Potential. J. Phys. Chem. 1996, 100, 16082–16086. 10.1021/jp961623v. [DOI] [Google Scholar]
- Shvartsburg A. A.; Jarrold M. F. An exact hard-spheres scattering model for the mobilities of polyatomic ions. Chem. Phys. Lett. 1996, 261, 86–91. 10.1016/0009-2614(96)00941-4. [DOI] [Google Scholar]
- Hall Z.; Politis A.; Bush M. F.; Smith L. J.; Robinson C. V. Charge-State Dependent Compaction and Dissociation of Protein Complexes: Insights from Ion Mobility and Molecular Dynamics. J. Am. Chem. Soc. 2012, 134, 3429–3438. 10.1021/ja2096859. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiórkiewicz-Kuczera J.; Yin D.; Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Mackerell A. D.; Feig M.; Brooks C. L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. 10.1063/1.470648. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.