

Abstract
We have previously reported that stabilization of the G-quadruplex structures in the HIV-1 long terminal repeat (LTR) promoter suppresses viral transcription. Here we sought to develop new G-quadruplex ligands to be exploited as antiviral compounds by enhancing binding toward the viral G-quadruplex structures. We synthesized naphthalene diimide derivatives with a lateral expansion of the aromatic core. The new compounds were able to bind/stabilize the G-quadruplex to a high extent, and some of them displayed clear-cut selectivity toward the viral G-quadruplexes with respect to the human telomeric G-quadruplexes. This feature translated into low nanomolar anti-HIV-1 activity toward two viral strains and encouraging selectivity indexes. The selectivity depended on specific recognition of LTR loop residues; the mechanism of action was ascribed to inhibition of LTR promoter activity in cells. This is the first example of G-quadruplex ligands that show increased selectivity toward the viral G-quadruplexes and display remarkable antiviral activity.
Introduction
G-quadruplexes (G4s) are nucleic acids secondary structures that may form in single-stranded G-rich sequences under physiological conditions.1−3 Four Gs bind via Hoogsteen-type hydrogen bonds base-pairing to yield G-quartets, which in turn stack on top of each other to form the G4. G4 structures may significantly vary, both in terms of strand stoichiometry (forming both inter- and intramolecular structures) and strand orientation/topology. The presence of K+ cations specifically supports G4 formation and stability.4−6 In the human genome and in prokaryotes, G4 DNA motifs have been found in telomeres, G-rich micro- and mini-satellites, near oncogene promoters, and within the rDNA.7−12 Human G4 DNA motifs have been reported to be associated with recombination prone regions13 and to show mutational patterns that preserve the potential to form G4 DNA structures.9 Misregulation of these G4 structures has been associated with relevant human neurological disorders.14−18 The identification of G4 binding proteins19 and their visualization in cells with antibody-based technology20,21 have also provided convincing evidence of the existence of cellular G4s in vivo.
Recently, the presence of G4s in viruses and their involvement in virus key steps has also been provided.22 G4s have been implicated in pathogenic mechanisms of the Epstein–Barr virus23,24 and the SARS coronavirus.25 We and other groups have identified functionally significant G4s in the Nef coding region26 and the unique long terminal repeat (LTR) promoter27−29 of the human immunodeficiency virus (HIV), the etiologic agent of the acquired immune deficiency syndrome (AIDS). These studies have shown that G4 folding at the LTR promoter decreased viral transcription with an effect that was augmented by a G4 ligand.27,30
The involvement of G4 structures in several human diseases propelled the development of G4 ligands. Several aromatic cores with protonable side chains bind the G4 conformation, such as porphyrins,31,32 acridines, such as 1 (BRACO-19, Scheme 1),33,34 fluoroquinolones35 anthraquinones, phenanthrolines, quinacridines, carbazoles, bis-indole carboxamides, triazoles, and benzimidazoles;36 natural compound derivatives, such as berberine,37 telomestatin,38 quindolin,39 several tetra-substituted perlyene tetracarboxylic40 and water-soluble naphthalene diimide (NDIs).41−46 So far, the vast majority of molecules has been tested against cellular G4s implicated in tumor pathogenesis: some compounds showed interesting antiproliferative properties;36 in particular, quarfloxin proceeded into phase II clinical trials, but its limited bioavailability prevented further progress.35 Few molecules have been tested also as antiviral agents:23,27,30,47 despite the low selectivity indexes, these studies have shown the possibility to develop antiviral molecules with a G-quadruplex-mediated mechanism of action.
Scheme 1. Structure of BRACO-19 and Core-Extended NDIs (c-exNDIs) Reported in the Literature,44,48−51 and the New Synthesized Water-Soluble c-exNDIs (lower right panel).

The lateral expansion of the NDI core provided by our investigation may offer an additional opportunity to enhance both G4 binding potency and selectivity, discriminating G4 structures with different accessibility to the edge quartets. Examples that comprise the annulation of five- and six-membered heterocyclic rings either on one side of the NDI48,49 or on both sides have been reported (Scheme 1).50,51 Surprisingly, only one class of these core-extended NDIs (c-exNDIs) has been engineered water-soluble to be investigated as G4 ligand,44 the others have found applications in the field of material chemistry and as pH sensors.49−51
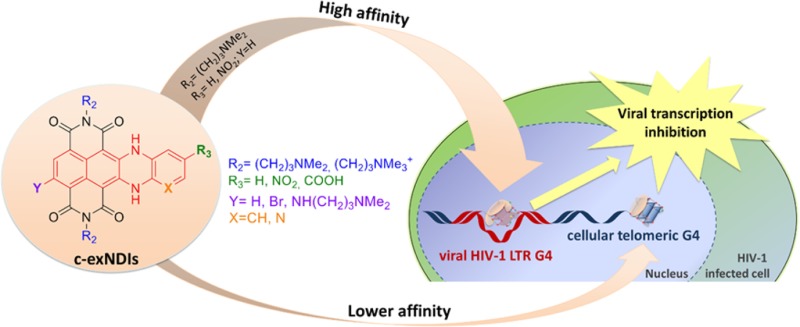
Because of the possibility to block HIV-1 transcription by stabilizing the G4 structures present in the unique viral LTR promoter, we here synthesized and tested a series of novel c-exNDIs for both their binding to the LTR G4s and anti-HIV-1 activity. In these new structures, the NDI core has been merged to the dihydrobenzophenazine polynuclear heterocycle (highlighted in red in Scheme 1). We proved that these derivatives display binding and stabilization selectivity toward the viral G4s as compared to a telomeric G4 and that this translates into a therapeutic window where the compounds are active against the virus but not toxic for the cell.
Results
Design and Synthesis of c-exNDIs
We have designed and synthesized a small library of new and water-soluble c-exNDIs (2–11, Scheme 2), where the NDI core has been merged to the dihydrobenzophenazine polynuclear heterocycle (highlighted in red in Scheme 1). The choice has been made based on the pKa values of the N,N-dimethylpropan-amine side chains: the pKa values are above 7 (pKa1 = 8.1, pKa2 = 8.6),52 suggesting that the side chains are mainly protonated under physiological conditions. This aspect ensures good solubilizing properties, reducing self-aggregations. We discharged the shorter N,N-dimethylthyl-amine side chains, as their pKa values are much lower (pKa1 = 5.6, pKa2 = 7.4). The c-exNDIs are structurally characterized by a larger flat core than the NDIs. This feature should improve π-stacking interaction implementing the binding to the G4. Indeed, DFT calculation performed at B3LYP/6-31+G(d,p) level of theory in gas phase indicated that the dimension of the c-exNDI core supported a more extensive overlap to the quartet of the G4 structure (Supporting Information, Figure S1). Enhancement of the ligand steric hindrance accessing the outside G4 quartets might offer an opportunity to implement selectivity as well. Furthermore, the proposed structural extension on the NDI scaffold should merge the useful optoelectronic properties (absorption, emission, and singlet oxygen sensitizer) of functionalized NDIs to those of substituted dihydrobenzophenazines. Compounds with both H and Br substituents at the Y position were considered to evaluate the effect of a bulky substituent (Br) at this position. To synthesize the c-exNDIs, we developed the synthetic protocol highlighted in Scheme 2. According to published methods, the imidation reaction of the readily available 2,6-dibromo-1,4,5,8-naphthalenetetracarboxylic dianhydride yielded the 2,6-dibromo-substituted NDI, under acidic conditions, in almost quantitative yield. Subsequently, a two-step protocol starting from a nucleophilic aromatic substitution (SNAr) in the presence of an excess (2.5 equiv) of ortho-diaminophenyl derivatives in DMF as solvent at 70 °C for 16 h afforded a 60:40 mixture of monobrominated and debrominated products in an overall quantitative yield. The ring closing reaction following the SNAr in one pot (step a, in Scheme 2) occurred through a Tchitchibabin mechanism, with the elimination of hydride favored by the formation of a six-membered ring. The second SNAr on the remaining aromatic bromide was carried out by dissolving 3 in neat N1,N1-dimethylpropane-1,3-diamine. To improve the reaction yield, we optimized a microwave-assisted protocol (150 °C, 200 psi, 250 bar, 200 W, 3 min, sealed reaction vessels) to give product 9, which crystallized from the reaction mixture. We achieved the synthesis of the permanently charged quaternary ammonium salts 10 and 11 with a classical exhaustive methylation, in the presence of MeI at rt for 16 h. All the synthesized ligands (2–11) were purified by preparative HPLC (CH3CN:H2O and 0.1% CF3COOH as eluent). Further anion exchange yielded 2–9 as hydrochloride salts. On the contrary, 10 and 11 were investigated as iodide quaternary ammonium salts.
Scheme 2. Optimized Synthetic Protocol for the Preparation of Water-Soluble 2–11.

(a) DMF, 70°C, 16 h, under N2; (b) N,N-dimethylpropylamine, MW, 150°C 3 min, closed vessel; (c) MeI, CHCl3, rt, 16 h.
c-exNDIs Greatly Stabilize G4 with a Preference for the LTR Conformations vs the Telomeric Sequence
To detect the ability of the new c-exNDIs to bind and stabilize the G4 conformations of the HIV-1 LTR promoter, two G4-forming LTR sequences were employed: LTR-III and LTR-IV, both composed of four GGG tracts (Figure 1A). When embedded within the full-length G-rich sequence of the LTR promoter, LTR-III is the most stable G4, while LTR-IV folds when induced by G4 ligands.27 A FRET melting assay was performed to assess the degree of stabilization of the LTR-III and LTR-IV G4 by c-exNDIs. The minimal intramolecular G4-forming human telomeric (hTel) sequence (Figure 1A) was employed to evaluate the activity of the c-exNDIs on one of the most highly represented and accessible cellular G4: in fact, the TTAGGG repeat is present as 2000–3000 double-stranded (ds) and 50–200 single-stranded sequences at the human telomeres.53 A double-stranded (ds) DNA was also added to address specificity toward the G4 conformation (Supporting Information, Table S1). While this kind of assay is typically performed using molar excess of the tested compounds, we here initially employed equimolar amounts of the target DNA sequence and compounds (i.e., 0.25 μM) to better differentiate the stabilization efficiency among the different c-exNDIs. The melting temperatures of the LTR-III, LTR-IV, hTel, and dsDNA G4 conformations were sufficiently similar (66.9, 61.9, 66.9, and 66.5 °C, respectively) to allow a meaningful comparison. In the presence of the c-exNDIs, a marked increase in the stabilization of the G4-forming oligonucleotides was observed (Figure 1B and Supporting Information, Table S2). Among the tested c-exNDIs, 2, 3, 4, 10, and 11 were the most efficient ligands, with a stabilization on LTR-IV G4 of 10–16 °C at equimolar amounts. At 4-fold c-exNDI:DNA molar ratio, the stabilization increased to above 25 °C, which is the maximum increase measurable in these settings (data not shown). The other c-exNDIs showed intermediate (4 °C ≤ ΔTm < 7 °C, 5, 6, and 7), or low (ΔTm < 4 °C, 8 and 9) level of stabilization on LTR-IV G4. Compounds 2 and 10 stabilized LTR-III G4 above 10 °C. Among the G4 structures, hTel G4 was the least efficiently stabilized, whereas stabilization on dsDNA was negligible. Only compounds 10 and 11 displayed a significant stabilization on dsDNA, indicating that the presence of two permanent charges (the quaternary ammonium moieties) favors an unspecific binding to the negatively charged nucleic acid. In general, the H series of all derivatives (Y = H, 2, 4, 6, 10; Scheme 2) was more active and more selective for the viral G4s compared to the Br substituted ones (Y = Br, 3, 5, 7, 8; Scheme 2). Compounds 2, 3, and 10 exhibited a difference in stabilization of LTR-IV G4s vs hTel G4 equal or above 6 °C, which roughly corresponds to 40% of the overall stabilization efficiency (Figure 1B and Supporting Information, Table S2).
Figure 1.

G4 stabilization by the c-exNDIs. (A) G4-forming sequences used in this study. (B) FRET analysis. ΔTm values of HIV-1 LTR G4-folded sequences (LTR-III and LTR-IV), the G4-folded telomeric sequence (hTel), and a control double-stranded sequence (dsDNA) obtained upon addition of equimolar amounts of 2–11 (0.25 μM). (C) Taq polymerase stop assay. Taq polymerization was performed in the presence/absence of K+ and 2, as indicated, on the hTel, LTR-III, and LTR-IV templates, which were previously folded into G4. A control template unable to fold into G4 was also used (non-G4 cnt). Stop regions are highlighted by vertical bars. (D) Quantification of the intensity of the stop bands obtained in the Taq polymerase stop assay. Stop band intensity of the treated samples were normalized vs the untreated sample.
Because oligonucleotides used for FRET analysis display fluorophore-modified ends that may influence the measured stabilization, additional techniques using label-free oligonucleotides were employed. Compound 2, a representative of the best ligands, was assayed by circular dichroism (CD) on LTR-IV, LTR-III, and hTel G4s. At 4:1 c-exNDI:DNA ratio, a stabilization higher than 27 °C was obtained on both LTR-III and LTR-IV (Supporting Information, Figures S2A and S2B), while hTel G4s was stabilized by only 19.9 °C (Supporting Information, Figure S2C), confirming a preference for the viral G4s. A Taq polymerase stop assay was performed on the LTR-III, LTR-IV, and hTel templates. A control template unable to fold into G4 was used to exclude unspecific inhibition of the polymerase enzyme by the c-exNDI. Compound 2 induced an intense stop site at the most 3′ G tract in each G4-forming template (Figure 1C), indicating effective stabilization of the G4 conformation by the compound. This effect was significantly more relevant on the viral sequences vs the telomeric template (Figure 1D), confirming again a preferential stabilization of the viral G4s.
The above techniques evaluated stabilization of the G4 conformation as an indicator of the efficiency of the interaction of the ligands with the tested G4 structures. Binding affinity was next directly assessed by two different methods.
Competition electrospray ionization mass spectrometry (ESI-MS) experiments54−56 allowed establishment of the relative affinity of the c-exNDIs toward the G4 sequences. We first confirmed by CD analysis that in the buffer conditions used for the ESI/MS experiments the topology of the G4-folded oligonucleotides did not significantly modify (Supporting Information, Figure S3) Equimolar amounts of LTR-III/hTel, LTR-IV/hTel, and LTR-III/LTR-IV G4s were incubated with 2-fold molar excess of compounds 2 or 4. Because ESI/MS transfers intact biomolecules complexes in the gas phase and signal intensity is proportional to the abundance of each species in solution, the ratio between m/z signals of each bound species and the corresponding bound and free G4 (Figure 2A) was calculated (Table 1). While LTR-III and LTR-IV were detected mostly as all-bound species, hTel was mostly unbound (Figure 2A, and Supporting Information, Figures S4A and S4B). This analysis clearly shows that our c-exNDIs preferentially bind the two viral sequences over the telomeric sequence. To corroborate the selectivity toward the viral sequences, two additional cellular G4 folding sequences were evaluated: c-kit257 and c-myc.32 The LTR G4s were preferred over c-kit2 G4 by both compounds; in the case of c-myc G4, compound 4 preferentially bound to the LTR G4s, whereas compound 2 showed more intense binding to c-myc (Table 1). Therefore, we confirmed a general, even though not absolute, selectivity toward the LTR G4s. It is also possible that the much higher stability of the c-myc G4 (Tm > 95 °C) vs the stability of the other tested G4s (LTR-III 66.9 °C, LTR-IV 61.9 °C, c-kit2 72.1 °C) played a role in this result. When the two LTR G4s were competed with each other, the LTR-III sequence was preferred. At 2-fold excess of compound, LTR-III bound up to two molecules of compounds, whereas only one molecule was obtained for both LTR-IV and hTel (Figure 2, and Supporting Information, Figure S5). At saturating concentrations (10-fold molar excess), three molecules were bound to LTR-III G4, whereas up to two molecules could bind LTR-IV and hTel G4s (Supporting Information, Figure S5).
Figure 2.

Affinity and binding site analysis. (A) ESI/MS competition assay. Representative spectrum. Equimolar amounts of LTR-III and hTel were incubated with 2-fold excess of 2 and analyzed by ESI/MS. The identity of m/z signals is shown above each signal. RI stands for relative intensity. (B) SPR analysis of 2 binding toward the LTRs and hTel G4s. Representative sensorgram of 2 binding to LTR-III G4. Biosensor data were collected at 25 °C; the compound was injected at increasing concentrations (26, 40, 59, 89, 133, 200 nM). Sensorgrams are shown as gray lines. Dotted black lines represent local 1:1 binding fits obtained for a calculated Rmax. (C,D) CL-mediated footprinting. LTR-IV (C) and LTR-III (D) oligonucleotides were G4 folded, incubated with increasing amounts of 2 (lanes 5–13), alkylated with CL (CL lanes), and subjected to hot piperidine treatment (PIP lanes) to induce cleavage of the phosphodiester bond at the alkylated bases. (C) Lanes 1–4 are control lanes not treated with the c-exNDI. The symbols * and ○ indicate base protection and exposure, respectively. M is a marker lane obtained by the Maxam and Gilbert sequencing of the oligonucleotides. The oligonucleotide sequence is shown on the left. The vertical dotted bar indicates the region of the LTR-III shared with LTR-IV. (E) Schematics of the LTR-IV and LTR-III G4 conformations. The represented LTR-IV G4 topology has been solved by NMR (PDB code 2N4Y); the shown LTR-III G4 conformation derives from previous low resolution structural data.27
Table 1. Relative Binding Affinity of 2 and 4 for the LTR-III, LTR-IV, hTel, c-kit2 (kit), and c-myc (myc) G4-Folded Oligonucleotides.
| binding affinity68 |
||||||
|---|---|---|---|---|---|---|
|
c-exNDI2 |
c-exNDI4 |
|||||
| competing G4s | LTR-III | LTR-IV | cell G4 (hTel, kit, or myc) | LTR-III | LTR-IV | cell G4 (hTel, kit, or myc) |
| LTR-III/hTel | 80 | 10 | 82 | 10 | ||
| LTR-IV/hTel | 52 | 25 | 75 | 18 | ||
| LTR-III/LTR-IV | 71 | 25 | 80 | 68 | ||
| LTR-III/kit | 33 | 12 | 54 | 18 | ||
| LTR-IV/kit | 25 | 9 | 55 | 23 | ||
| LTR-III/myc | 39 | 45 | 51 | 37 | ||
| LTR-IV/myc | 25 | 44 | 71 | 45 | ||
Next, absolute affinity of 2 was measured by surface plasmon resonance (SPR) toward the three G4-folded sequences. This analysis confirmed that LTR-III G4 displays the highest binding affinity (KD 3.2 nM), followed by LTR-IV G4 (KD 6.1 nM); hTel G4 exhibited the lowest affinity (KD 13.1 nM) (Figure 2B, and Supporting Information, Figure S6). The binding affinity of the Br derivatives with each of the substituents in position R against LTR-IV was next assessed. Compounds 3 and 5 had KD values similar to that of 2 (KD 6.6 and 5.9 nM, respectively), 10 displayed an increased binding affinity (KD 1.7 nM), whereas 7 and 11 showed the lowest affinity (KD 21.6 and 12.9 nM, respectively). These data are in line with the stabilization properties observed for these derivatives by FRET and CD analysis and confirm the improved and reduced recognition properties of the c-exNDIs exhibiting permanent dicationic (10) and neutral or monocationic charge states (7), respectively, under physiological conditions. They also confirm that a bulky substituent in position Y of the NDI core (11, Scheme 2) highly reduces binding, whereas a NO2 group in position R maintains excellent binding properties (5, Scheme 2).
We reasoned that the observed biased affinity for the LTR sequences could derive by specific binding of the c-exNDIs to structural/sequence features shared by the LTR G4s. To this end, we evaluated the protection exerted by compound 2 over residues of the LTR G4s against clerocidin (CL). This molecule has been shown to alkylate DNA single-stranded G, C, and A bases58,59 and to be able to predict G4 structural folding.60 Labeled oligonucleotides were folded, treated with increasing amounts of 2, exposed to CL, and further treated with piperidine to highlight CL-alkylated sites. In these settings, we expected to visualize only loop bases bound by the c-exNDI because base-paired Gs involved in G-quartet formation would not be available to CL alkylation, independently of the presence of the compound. The NMR structure of LTR-IV has been solved (PDB code 2N4Y), and therefore we were able to assess that nucleotides affected by the presence of the c-exNDI were in two loops of the structure: in particular, in the ACTG loop the A base was protected, whereas C and G were exposed; in the 1-base-loop, the C base was exposed (Figure 2C and 2E). The structure of LTR-III has not been solved yet, however, previous data indicated that this G4 can fold into different conformations, the main one of which is likely the one depicted in Figure 2E, as detected by low-resolution structural data.27 In this case, we observed protection of the C base in the ACTG loop (Figure 2D), which is shared with the LTR-IV sequence (see Figure 1A); three additional residues (C, C, and G) were protected in the 11-base-loop (Figure 2D and 2E). Protection and exposure of residues indicated specific interaction of 2 at the affected LTR sites. Interestingly, unique LTR loops and in particular the ACTG loop shared by both LTR-III and LTR-IV, and absent in the telomeric sequence, were involved, indicating that these are likely the moieties that induce selectivity. We cannot exclude that other regions are bound by the c-exNDI, but probably the interaction at other sites is less stable or more dynamic so that a clear protection/exposure could not be observed.
The Extended Core NDIs Display Anti-HIV-1 Activity at Nanomolar Concentrations
We have previously shown that the HIV-1 viral promoter folds into dynamic G4s and that stabilization of these G4 conformations by the G4 ligand 1 represses viral transcription and virus production in infected cells with IC50 (the concentration required to inhibit 50% of virus production) in the low μM range.27,30 The average selectivity index (SI) for 1 was 12. Given the c-exNDI binding/stabilization selectivity observed in vitro for the viral G4s vs the cellular G4, we set out to investigate the anti-HIV-1 activity of the newly synthesized c-exNDI derivatives. Compound entry was preliminarily measured by phase contrast and fluorescence microscopy, taking advantage of the blue-colored c-exNDI dyes (λabs = 555 and 600 nm, for the unsubstituted 2) and their red fluorescence emission (λem = 610 and 655 nm, for 2). After 30 min of incubation, compounds concentrated into the cell (Supporting Information, Figure S7). We next treated infected cells with increasing amounts of compounds and measured virus production; concurrently, the cytotoxic concentration (CC50) of the c-exNDIs in the same conditions but on uninfected cells was measured by MTT assay. All compounds were tested against the HIV-1 NL4-3 strain, which is a X4 strain. Results are reported in Table 2. Compounds 2, 3, 4, and 5 displayed inhibitory concentration (IC50) values below 25 nM. Compound 2 displayed a promising SI of 40, the best among the tested c-exNDIs, whereas SI values of compound 3, 4, and 5 were around 20, again superior to 1. Derivatives 6 and 7 exhibited low activity and low cytotoxicity, while 9 showed the worst combination with low activity, high cytotoxicity, and SI of 2.2. We also evaluated the range of concentration (therapeutic window or TW), at which we obtained antiviral activity paralleled by less than 80% cytotoxicity. As shown in Figure 3A, 2 displayed the widest TW (3.9–250 nM); 4 and 5 also showed interesting TW values; TW concentrations for 6 and 7, which are likely to be mainly monoanionic under physiological conditions as both the (CH2)3NHMe2+ moieties exhibit a pKa >8.1,52 were in the μM range; no TW was observed for 8 and 9 (Table 1). Finally, 10 and 11, containing two quaternary ammonium moieties, did not display any antiviral or cytotoxic activity likely due to their inability to enter the cells, as already shown for NDIs with permanent positive charges.61 Compound 2 and 3 were also tested on the HIV-1 BaL strain, a R5 strain with a different tropism. In this case, IC50 values below 40 nM were obtained, with SI of 14 for the H derivative (Table 1). Note that the H series of all derivatives was more active than the Br substituted one. Moreover, all compounds displayed CC50 values higher than 2 μM against a human fibroblast cell line.
Table 2. Antiviral Effects of the c-exNDI Derivatives and the Control 1 against Two HIV-1 Strains in TZM-bl Cellsa.
| IC50 (nM) |
SI |
TW (nM) |
|||||
|---|---|---|---|---|---|---|---|
| c-exNDI | HIV-1 strain NL4-3 | HIV-1 strain BaL | CC50 (nM) | HIV-1 strain NL4-3 | HIV-1 strain BaL | HIV-1 strain NL4-3 | HIV-1 strain BaL |
| 2 | 13.2 ± 0.1 | 36.2 ± 4.3 | 532.0 ± 11.7 | 40 | 14 | 3.9–250.0 | 15.6–250.0 |
| 3 | 15.7 ± 2.1 | 32.5 ± 7.1 | 343.2 ± 32.2 | 22 | 11 | 7.8–125.0 | 15.6–125.0 |
| 4 | 11.0 ± 0.3 | nd | 188.0 ± 4.08 | 17 | nd | 3.9–125.0 | nd |
| 5 | 23.0 ± 1.3 | nd | 439.0 ± 10.7 | 19 | nd | 15.6–250.0 | nd |
| 6 | 975.3 ± 16.2 | nd | > 5000 | >5 | nd | 800–5000 | nd |
| 7 | 862.6 ± 31.5 | nd | > 5000 | >6 | nd | 800–5000 | nd |
| 8 | 63.5 ± 4.8 | nd | 458.1 ± 54.0 | 7.2 | nd | no TW | nd |
| 9 | 814.3 ± 36.4 | nd | 1759.9 ± 62.5 | 2.2 | nd | no TW | nd |
| 10 | >5000 | nd | >5000 | nd | no TW | nd | |
| 11 | > 000 | nd | >5000 | nd | no TW | nd | |
| 1 | 3400 ± 140b | nd | >12000b | >3.5 | nd | 3000–12000 | nd |
NL4–3 and BaL are X4 and R5, respectively, HIV-1 strains. IC50 is the compound concentration required to inhibit 50% of HIV-1 production; CC50 is the compound concentration at which 50% of cell toxicity is observed; SI is the selectivity index; TW is the therapeutic window concentration. nd stands for “not determined”.
indicates values previously reported.27
Figure 3.

Antiviral activity of c-exNDIs and mechanism of action. (A) TZM-bl cells were infected with HIV-1 NL4-3 strain and treated with increasing concentrations (3.9–1000 nM) of the c-exNDIs. Uninfected cells were treated in the same conditions. Antiviral activity, expressed as relative luciferase units (RLU), was measured on infected cells, while cell viability was in parallel obtained with a MTT assay on the uninfected cells. The vertical dotted lines define the therapeutic window (TW) where the compound is active against the virus and not toxic against the cells. This figure is representative of the antiviral/cytotoxicity/TW experiments performed with the whole series of c-exNDI compounds. (B) Entry assay. TZM-bl cells were treated with the compound 2 (50–100 nM), AZT (0.5 μg/mL), or the entry inhibitor DS (100 μg/mL) at 0, 1, or 2 h pi. Virus production was measured at 31 h pi as RLU. The control depicts cells infected with the virus lacking compound treatment. (C) Effect of 2 on the LTR promoter activity compared to cytotoxicity. HeLa cells with a stably transfected LTR-EGFP reporter plasmid were treated with increasing concentrations of 2 (62.5–250 nM). EGFP mean was measured by FACS analysis. Cytotoxicity was estimate in parallel by gating on forward scatter (FSC) vs side scatter (SSC) using untreated cells as reference. (D) Specificity of the 2-induced inhibition of LTR promoter activity on the G4 LTR conformation. 293T cells were transiently transfected with plasmids with the wt or M4 + 5 mutant LTR promoter sequence upstream a luciferase reporter gene. Luciferase signals were measured as a function of increasing 2 amounts and normalized to total protein content.
The NDIs Derivatives Inhibit HIV-1 LTR Promoter Activity
It has been reported that compounds with multiple positive charges can block viral entry by interfering with the CXCR4 coreceptor.62 To rule out this unspecific mechanism of action of the c-exNDI, we treated TZM-bl cells at 0, 1, and 2 h post infection (pi) with compound 2 and tested virus production at 31 h pi. No significant difference in the activity of the compound was observed at the tested times (Figure 3B). Zidovudine (AZT), a reference drug that targets the reverse transcriptase step,63 displayed a behavior similar to that of the c-exNDI, maintaining activity from 0 to 2 h pi. In contrast, dextran sulfate (DS), a reference entry inhibitor,64 exhibited antiviral effect only when added at 0 h pi. (Figure 3B). These data indicate that the initial steps are not targeted by 2, which is thus active at/after 2 h pi, i.e., after the entry process has occurred.
We next tested the ability of 2 to reduce the activity of the HIV-1 LTR promoter in an EGFP reporter assay in HeLa-tat-III cells, which is a cell line stably transfected with a plasmid embedding the EGFP coding region under the control of the HIV-1 LTR promoter. Cells were treated with increasing concentrations of the c-exNDI, and the intensity of EGFP fluorescence was assessed by FACS analysis. A steady decrease in the mean levels of EGFP was observed at noncytotoxic concentrations of 2 (up to 60% of inhibition) (Figure 3C). To check if the activity of the c-exNDI at the LTR promoter was specific for the G4 conformation, two point mutations that abolish G4 folding were introduced in the LTR sequence (M4 + 5 LTR).27 The wt LTR and M4 + 5 LTR luciferase reporter plasmids were transiently transfected into 293T cells either alone or in the presence of increasing amounts of compound 2, and the luciferase signal was measured. Again, an important inhibition of LTR activity was observed on the wt LTR (up to 65% inhibition), whereas no effect was detected on the non-G4-forming mutant LTR sequence (Figure 3D). These data indicate a G4-mediated inhibition of the viral promoter by the c-exNDI.
Discussion and Conclusions
We have here shown the synthesis, G4 binding properties, and antiviral activity of a new series of NDI derivatives with an extended aromatic core. First, most of the new compounds displayed improved stabilization on G4 structures, with substantial increments in Tm above values previously reported for NDI derivatives.41−46 This feature is an indication that the expansion of the aromatic surface allows a more effective recognition of the G4 structure. This may be due to a more extensive overlap to the quartet of the G4 structure, as inferred by the dimension of the c-exNDI core, evaluated by DFT calculation (Supporting Information, Figure S1). Second, these compounds exhibited a very promising antiviral activity. Compared to 1,30 the best antiviral 2–5 showed improved IC50 values (in the low nM vs low μM range). This feature may derive from the selectivity observed in vitro for the LTR G4s over the telomeric G4 structures. The viral and telomeric G4s are likely the two most abundant DNA G4 species in the cell during infection, therefore a preferential effect on the viral G4s would result in higher antiviral activity and lower cytotoxicity, as observed. The c-exNDIs displayed both selective binding affinity and stabilization for the viral G4s. Between the two LTR G4s, the LTR-III sequence was most efficiently bound by 2, as measured by ESI/MS and SPR. Even though LTR-III (38 nts) and LTR-IV (29 nts) display an identical number of G-quartets, LTR-III offers longer loops that may accommodate additional molecules and therefore may display increased binding affinity. Indeed, at saturating concentrations, binding of up to three c-exNDI molecules was observed by ESI/MS with LTR-III compared to two molecules for LTR-IV. In addition, two loops in both LTR-III and LTR-IV G4s were shown to be involved in c-exNDI interaction by footprinting analyses. We suggest that the compounds interact with the edge G-quartets through the core NDI planar aromatic surface, while the extended moiety and side chains interact with the side loops, affording selectivity toward the LTR G4s. To our knowledge, this is the first example of this type of selectivity based on chemical recognition of specific residues; in fact, previously reported selectivity rather relied on the recognition of a G4 conformation.65
Analysis of the binding activity of the series of c-exNDIs led us to the following structure–activity relationship (SAR) conclusions: (i) the nonextended side of the c-exNDI core [position Y (Scheme 1)], lacking substituents as the introduction of an aminoalkyl side chain, highly hinders stabilization of the G4 structures (i.e., 9); (ii) Br is allowed as Y substituent, but it lowers binding and selectivity toward the viral sequences. The Br group may both hinder specific interactions (hence lower selectivity) and partially distort the aromatic core from planarity (hence lower G4 binding); (iii) the pyridine ring in place of the benzene in the extended core is strongly detrimental (compound 8). The fair antiviral activity is paralleled by moderate cytotoxicity, allowing no TW, possibly due to off-target activity; (iv) the NO2 moiety is allowed (compounds 4 and 5) as compound 4 is the most powerful and selective ligand toward LTR-III; (v) the negatively charged carboxylate group decrease the electrostatic component of the binding to the DNA G4s (compounds 6 and 7); (vi) on the contrary, the permanently positively charged ammonium groups extensively increase binding to the target G4s by adding electrostatic interaction (compounds 10 and 11). However, compounds with permanent charges are not suitable for cell entry and hence cannot be exploited for antiviral purposes. A scheme of the chemical properties that lead to high-affinity recognition and selectivity of the c-exNDI binders is reported in the graphical abstract.
In conclusion, we have reported a new class of G4 ligands able to discriminate G4 structures based on the chemical recognition of structural features that are unique to the selected G4 sequences. On one hand this approach opens the possibility to develop anti-HIV compounds with a new mechanism of action that may complement current clinical AIDS therapies; on the other hand, our work clearly highlights the possibility to selectively recognize G4 structures with small molecules, prompting the search and development of G4 ligands specific for G4s implicated in several important human diseases.
Experimental Section
Materials and General Procedures
Reagents, oligonucleotides, solvents, and chemicals were purchased from Alfa Aesar or Sigma-Aldrich and were used as supplied without further purification. TLC analysis was carried out on silica gel (Merck 60F-254) with visualization at 254 and 366 nm. HPLC analysis and purifications were performed using two different HPLC: Waters system combining a Delta 600 PUMP, a 2489 UV/vis detector and Fraction Collector III (for preparative and analytical), and an Agilent system series 1260 (for analytical). The analytical column was XSelect CSH Phenyl-Hexyl (150 mm × 4.6 mm) (Waters). The preparative column was XSelect CSH Prep phenyl–hexyl 5 μm (150 mm × 30 mm) (Waters). Flows were 1 or 1.4 mL/min for analytical and 27 mL/min for preparative. For the analytical analysis were used the following method A: (aqueous solvent: 0.1% trifluoroacetic acid in water; organic solvent, acetonitrile); method A = 1 mL/min, isocratic flow over 2 min 95% of aqueous solvent; gradient, 95% aqueous, gradually to 40% aqueous over 12 min and at the end an isocratic flow over 4 min. For the preparative purification was used the following method B: (aqueous solvent: 0.1% trifluoroacetic acid in water; organic solvent, acetonitrile); Method B = 27 mL/min, isocratic flow over 4 min 95% of aqueous solvent; gradient, 95% aqueous, gradually to 70% aqueous over 16 min and at the end an isocratic flow over 4 min). 1H , 13C NMR spectra were recorded on a Bruker ADVANCE 300 MHz spectrometer. The potentiometric titrations were made with a Radiometer TitraLab 90 titration system. UV/vis spectra were run on a Varian Cary 100 SCAN spectrophotometer with quartz cuvettes of the appropriate path length (0.1–1 cm) at 25.0 ± 0.1 °C. Emission spectra were recorded on a PerkinElmer LS 50B instrument.
Chemistry. General Methods
Synthesis of Intermediates and Final Ligands
Br2NDI has been synthesized according to the published procedure.52 The new ligands (2–11) were analyzed by HPLC (see Supporting Information, HPLC Purity Data), confirming ≥97.6% purity.
Nucleophilic Aromatic Substitution/Tchichibabin Reaction: Synthesis of 2–5, 8
The starting NDI (0.5 mmol) was dissolved into 20 mL of DMF in a round-bottom flask together with 1.0 mmol of o-phenyl-diamine derivatives, the mixture was stirred at 45 °C for 16 h under argon. The resulting dark-violet solution was basified with sodium bicarbonate aqueous solution and extracted with three portion of CHCl3 (50 mL). The organic layers were collected, dried on NaSO4, and evaporated under vacuo. The crude product was analyzed and purified by HPLC chromatography (CH3CN:H2O 0.1%TFA) according to analytical method A and preparative method B. HCl 1 M solution was added to each chromatographic portion. Solvent evaporation under vacuum afforded the product as hydrochloride salt.
2·2HCl
Yield = 25.2%; dark-violet solid; mp dec >200 °C. 1H NMR (300 MHz, CD3OD): 7.74 (s, 2H), 7.11 (dd, J = 3.3, 5.7 Hz, 2H), 6.89 (dd, J = 3.3, 5.7 Hz, 2H), 4.06 (m, 4H); 3.26 (m, 4H), 2.94 (s, 12H), 2.14 (m, 4H). 13C NMR (75 MHz, CD3OD): 166.4, 164.3, 143.2, 128.7, 128.0, 127.0, 125.9, 123.0, 118.2, 96.0, 57.0, 56.0, 43.9, 38.5, 38.2, 24.9, 24.2. Anal. Calcd for C30H34Cl2N6O4: C, 58.73; H, 5.59; Cl, 11.56; N, 13.70; O, 10.43. Found: C, 58.87; H, 5.51; N, 13.52.
3·2HCl
Yield = 19.4%; dark-violet solid; mp dec >200 °C. 1H NMR (300 MHz, CD3OD): 8.1 (s, 1H), 7.15 (dd, J = 3.4, 6.0 Hz, 2H), 7.03 (dd, J = 3.4, 6.0 Hz, 2H), 4.17 (m, 4H), 3.25 (m, 4H), 2.94 (s, 12H), 2.16 (m, 4H). 13C NMR (75 MHz, CD3OD): 166.5, 166.1, 163.9, 163.6, 152.4, 144.3, 132.5, 130.3, 129.0, 128.9, 128.3, 128.1, 126.6, 123.1, 121.1, 120.4, 118.4, 118.2, 97.0, 79.8, 57.1, 43.9, 38.7, 38.5, 24.9, 24.7. Anal. Calcd for C30H33BrCl2N6O4: C, 52.04; H, 4.80; Br, 11.54; Cl, 10.24; N, 12.14; O, 9.24. Found: C, 52.17; H, 4.81; N, 12.12.
4·2HCl
Yield = 11.2%; dark-violet solid; mp dec >200 °C. 1H NMR (300 MHz, CD3OD): 8.04 (s, 2H), 7.88 (d, J = 6.6 Hz, 1H), 7.84 (1H, s), 7.07 (d, J = 6.5 Hz, 1H), 4.2 (bs, 4H), 3.27 (m, 4H), 2.94 (s, 12H), 2.17 (m, 4H). 13C NMR (75 MHz, CD3OD):166.9, 166.7, 164.5, 134.6, 132.1, 129.6, 127.3, 127.2, 126.9, 126.6, 124.3, 124.1, 122.8, 117.8, 112.9, 99.6, 99.4, 57.0, 43.8, 38.4, 24.8. Anal. Calcd for C30H33Cl2N7O6: C, 54.72; H, 5.05; Cl, 10.77; N, 14.89; O, 14.58. Found: C, 54.80; H, 5.01; N, 14.92.
5·2HCl
Yield = 18.9%; dark-violet solid; mp dec >200 °C. 1H NMR (300 MHz, CD3OD): 8.00 (s, 1H), 7.87 (d, J = 6.6 Hz, 1H), 7.79 (1H, s), 7.0 (d, J = 6.5 Hz, 1H), 3.98 (bs, 4H), 3.14 (m, 4H), 2.83 (s, 12H), 2.01 (m, 4H). 13C NMR (75 MHz, CD3OD):162.0, 161.0, 144.6, 141.4, 140.7, 132.1, 127.1, 124.1, 122.9, 122.1, 121.2, 120.7, 118.9, 111.9, 55.1, 48.7, 46.5, 42.6, 37.5, 37.2, 22.5, 22.4. Anal. Calcd for C30H32BrCl2N7O6: C, 48.86; H, 4.37; Br, 10.84; Cl, 9.61; N, 13.30; O, 13.02. Found: C, 48.79; H, 4.31; N, 13.22.
8·2HCl
Yield = 18%; dark-violet solid; mp dec >200 °C. 1H NMR (400 MHz, CD3OD): 7.85 (s, 1H), 7.76 (s, 1H), 7.20 (m, 1H), 6.95 (bs, 1H), 3.91 (m, 4H), 2.70 (m, 4H), 2.52 (m, 12H), 1.93 (m, 4H). 13C NMR (100 MHz, CD3OD):165.8, 162.5, 161.5, 146.5, 143.8, 141.9, 133.2, 128.7, 125.5, 125.1, 123.4, 123.1, 121.9, 120.5, 97.8, 57.9, 45.2, 45.1, 39.9, 39.7, 26.0, 25.9. Anal. Calcd for C29H32BrCl2N7O4: C, 50.23; H, 4.65; Br, 11.52; Cl, 10.22; N, 14.14; O, 9.23. Found: C, 50.17; H, 4.71; N, 14.08.
Synthesis of 6 and 7
NDI 1 (0.5 mmol) was dissolved into 40 mL of CH3CN in a round-bottom flask together with 1.0 mmol of 3,4-diaminobenzoic acid, the mixture was stirred at 85 °C for 72 h under argon. The resulting dark-violet solution was cooled at r.t. to induce precipitation of the product. The crude powder was filtered and purified by HPLC chromatography, (CH3CN:H2O 0.1%TFA) according to analytical method A and preparative method B, affording the title NDIs 6 (27%) and 7 (30.5%).
6·2HCl
Yield = 27%; dark-violet solid; mp dec >200 °C. 1H NMR (400 MHz, CD3OD): 7.86 (s, 2H), 7.62 (d, J = 8.0 Hz, 1H), 7.39 (s, 1H), 6.89 (d, J = 8.0 Hz, 1H), 4.16 (m, 4H), 3.26 (m, 4H), 2.95 (s, 12H), 2.16 (m, 4H). 13C NMR (100 MHz, CD3OD):168.2, 166.6, 166.5, 164.3, 164.2, 143.1, 142.8, 132.6, 129.8, 129.2, 128.6, 127.0, 126.7, 126.6, 126.4, 123.6, 123.5, 119.0, 117.7, 98.3, 97.7, 57.0, 43.8, 40.7, 38.4; 24.8. Anal. Calcd for C31H34Cl2N6O6: C, 56.63; H, 5.21; Cl, 10.78; N, 12.78; O, 14.60. Found: C, 56.77; H, 5.31; N, 12.72.
7·2HCl
Yield = 30.5%; dark-violet solid; mp dec >200 °C. 1H NMR (300 MHz, CD3OD): 8.37 (s, 2H), 7.85 (d, J = 8.0 Hz, 1H), 7.73 (s, 1H), 7.21 (d, J = 8.0 Hz, 1H), 4.35 (m, 4H), 3.41 (m, 4H), 3.05 (s, 12H), 2.29 (m, 4H). 13C NMR (75 MHz, CD3OD): 168.3, 166.2, 163.5, 161.8, 161.7, 160.8, 152.2, 133.2, 132.6, 130.1, 129.4, 126.5, 123.4, 123.3, 121.6, 119.12, 118.21, 117.9, 98.3, 97.6, 57.04, 43.9, 38.3, 38.5, 24.8, 24.7. Anal. Calcd for C31H33BrCl2N6O6: C, 50.56; H, 4.52; N, 11.41; O, 13.03. Found: C, 50.34; H, 4.61; N, 11.29
Microwave Assisted (MW) Nucleophilic Aromatic Substitution: Synthesis of 9
Compound 3 (0.5 mmol) was dissolved into 4 mL of N,N-dimethyl-propan-1,3-diamine. The mixture was stirred and heated in a microwave reactor, according to a closed vessel protocol, at 150 °C, 250 psi, 200 W, for 3 min. The resulting green solution was cooled at rt to induce precipitation of the product. The resulting deep-green powder was filtered and washed by water afforded 1 (Yield 92%). Further HPLC preparative purification (CH3CN:H2O and 0.1% CF3COOH as eluent), and final trifluoroacetate-chloride anion exchange, by addition of 1 mL HCl 1M, yielded 9 as trihydrochloride (9·3HCl); deep-green solid; mp dec >200 °C. 1H NMR (300 MHz, CD3OD): δ = 7.10 (m, 1H), 6.96 (m, 2H), 6.69 (m, 2H), 4.11 (m, 4H), 3.42 (m, 2H), 3.26 (m, 6H), 2.99 (s, 6H), 2.93 (s, 12H), 2.18 (bs, 2H), 2.11 (bs, 4H). 13C NMR (75 MHz, CD3OD): δ = 166.7, 166.4, 164.5, 144.2, 139.7, 129.8, 129.0, 128.9, 127.5, 126.6, 125.0, 117.6, 117.1, 115.8, 108.2, 104.5, 98.1, 97.7, 63.4, 57.0, 54.0, 53.8, 43.8, 41.1, 38.5, 38.1, 25.7, 24.9, 24.8, 24.1. Anal. Calcd for C35H47Cl3N8O4: C, 56.04; H, 6.32; Cl, 14.18; N, 14.94; O, 8.53. Found: C, 56.17; H, 6.39; N, 14.90.
Exhaustive Methylation: Synthesis of 10 and 11
The c-exNDIs (2 or 3) purified as hydrochlorides were dissolved in a NaHCO3 solution and extracted 3 times with CH2Cl2. The recovered organic layers have been dried on Na2SO4 and the solvent evaporated under reduced pressure. The collected amine (2.5 mmol) was suspended in 50 mL of CHCl3 and 1.2 g (8.5 mmol) of CH3I were added. This suspension was stirred under nitrogen atmosphere, at room temperature for 12 h. After this period the resulting dark-violet powder was filtered and dried, afforded the product as pure di-iodide salt.
10
Yield = 99%; dark-violet crystals; mp dec >200 °C. 1H NMR (400 MHz, CD3OD): δ = 7.94 (s, 2H), 7.12 (dd, J = 3.3, 6.0 Hz, 2H), 6.97 (dd, J = 3.3, 6.0 Hz, 2H), 4.14 (m, 4H), 3.49 (m, 4H), 3.15 (m, 18H), 2.21 (m, 4H). 13C NMR (100 MHz, D2O): δ = 166.9, 165.8, 143.8, 129.1, 128.1, 127.6, 126.3, 123.5, 118.3, 97.5, 66.1, 54.1, 38.6, 23.6. Anal. Calcd for C32H38I2N6O4: C, 46.62; H, 4.65; I, 30.78; N, 10.19; O, 7.76. Found: C, 46.53; H, 4.59; N, 10.22.
11
Yield = 99%; dark-violet crystals; mp dec >200 °C. 1H NMR (400 MHz, CD3OD): δ = 8.04 (s, 1H), 7.15 (bs, 2H), 7.00 (bs, 2H), 4.12 (m, 4H), 3.51 (m, 4H), 3.15 (s, 18H), 2.20 (bs, 2H). 13C NMR (100 MHz, CD3OD): δ = 166.3, 165.9, 163.2, 162.2, 145.1, 144.1, 143.0, 132.5, 130.1, 128.8, 128.7, 128.3, 128.2, 126.4, 123.0, 121.1, 120.2, 118.3, 118.2, 96.9, 65.8, 53.9, 38.8, 38.6, 23.3, 23.2. Anal. Calcd for C32H37BrI2N6O4: C, 42.55; H, 4.13; Br, 8.84; I, 28.09; N, 9.30; O, 7.08. Found: C, 42.60; H, 4.18; N, 9.39.
Spectroscopic Techniques (FRET and CD)
All oligonucleotides used in this study were from Sigma-Aldrich (Milan, Italy). For fluorescence melting curves, equimolar amounts of the compounds were added to each FAM (6-carboxyfluorescein) 5′-end- and TAMRA (6-carboxy-tetramethylrhodamine) 3′-end-labeled oligonucleotide (0.25 uM) folded in the lithium cacodylate buffer supplemented with potassium (100 mM). After stabilization at 4 °C, samples were processed in a LightCycler II (Roche, Milan, Italy) or LightCycler 480 (Roche, Milan, Italy), and the oligonucleotide melting was monitored by observing 6-carboxyfluorescein (6-FAM) emission in the temperature range of 30– 95 °C with 1 °C/min gradient. Melting profiles were normalized as previously described.66Tm was defined as the temperature corresponding to the 0.5 fraction of the normalized fluorescence.
For CD analysis, oligonucleotides were diluted to a final concentration of 4 μM in lithium cacodylate buffer (10 mM, pH 7.4) and KCl 100 mM. After annealing step (95 °C for 5 min), DNA samples were gradually cooled to room temperature and compounds added from stock at final concentration of 16 μM. CD spectra were recorded on a Chirascan-Plus (Applied Photophysisics, Leatherhead, UK) equipped with a Peltier temperature controller using a quartz cell of 5 mm optical path length and an instrument scanning speed of 50 nm/min over a wavelength range of 230–320 nm. The reported spectrum of each sample represents the average of 2 scans is baseline-corrected for signal contributions due to the buffer. Observed ellipticities were converted to mean residue ellipticity (θ) = deg × cm2 × dmol–1 (mol ellip). For the determination of Tm, spectra were recorded over a temperature range of 20–95 °C, with temperature increase of 5 °C/min. Tm values were calculated according to the van’t Hoff equation, applied for a two-state transition from a folded to unfolded state, assuming that the heat capacity of the folded and unfolded states are equal.67
Taq Polymerase Stop Assay
Taq polymerase stop assay was carried out as previously described.27,30 Briefly, the 5′-end labeled primer was annealed to its template (Supporting Information, Table S1) in lithium cacodylate buffer in the presence or absence of KCl 100 mM and by heating at 95 °C for 5 min and gradually cooling to room temperature. Where specified, samples were incubated with 2 (3.1–25.0 nM). Primer extension was conducted with 2 U of AmpliTaq Gold DNA polymerase (Applied Biosystem, Carlsbad, California, USA) at 47 °C for 30 min. Reactions were stopped by ethanol precipitation, primer extension products were separated on a 15% denaturing gel, and finally visualized by phosphorimaging (Typhoon FLA 9000).
Mass Spectrometric (MS) Competition Assay
Oligonucleotides were heat-denatured and folded in 10 mM KCl, 150 mM trimethylammonium acetate (TMAA), pH 6.8, overnight at room temperature. The oligonucleotides where diluted to final concentration of 10 μM and incubated with the tested compound at ratio DNA:compound 1:1 overnight at 4 °C.
Samples were analyzed by direct infusion electrospray ionization (ESI) on a Xevo G2-XS QTof mass spectrometer (Waters, Manchester, UK). The injection was automatically performed by an Agilent 1290 Infinity HPLC (Agilent Technologies, Santa Clara, CA, US) equipped with an auto sampler; the carrying buffer was TMAA 100 mM. The absence of potassium in the carrying buffer allows the dilution of the salts before MS analysis decreasing the signal suppression. Up to 5 μL samples were typically injected for each analysis. The electrospray capillary was at 1.8 kV, the source and desolvation temperatures were 45 and 65 °C, respectively, and the sampling cone was at 65 V. All these parameters ensured minimal fragmentation of the DNAs complexes. The instrument was calibrated using a 2 mg/mL solution of sodium iodide in 50% 2-propanol. Additionally, the use of the LockSpray during the analysis provided a typical <2 ppm mass accuracy. The internal standard LockSpray consisted in a solution of leu-enkephalin 1 μg/mL in acetonitrile/water (50:50, v/v) containing 0.1% formic acid. Binding affinities were calculated for each experiment using the reconstructed-ion chromatogram area for each species calculated by MassLynx V4.1: this analysis was made possible by the experimental design that used an HPLC system to inject the samples in the mass spectrometer. The binding affinity was calculated with the following formula: [BA = (ΣG4b/(ΣG4f + ΣG4b)) × 100], where BA is the binding affinity, G4b is chromatogram area of bound G4 DNA, and G4f is the chromatogram area of free G4 DNA.68 G4b comprises DNA with one or two bound ligands, where present. Signals with charge states 6–, 5–, and 4– were used for the free and bound hTel DNA and c-myc, charge states 6– and 5– for LTR-III, charge states 5– and 4– for c-kit2, and charge state 4– for LTR-IV. Free DNA mono and dipotassium adducts were included in the calculation. For stoichiometric calculation, oligonucleotides were folded in 10 mM KCl, 150 mM TMAA pH 6.8 overnight at rt. The oligonucleotides, diluted to final concentration of 10 μM, were incubated with the tested compound (ratio 1:5, 1:10, 1:20, and 1:50) at 4 °C and analyzed by ESI-MS.
SPR Analysis
SPR was performed on the Biacore T100 platform (GE Healthcare). 5′-Biotinylated LTR-III, LTR-IV, and hTel oligonucleotides were heated at 95 °C for 5 min and cooled down at room temperature to allow G4 folding. Immobilization was performed in HEPES-KCl running buffer (0.01 M HEPES pH 7.4, 0.2 M KCl, 3 mM EDTA) on a steptavidin coated surface (SA sensor chip, Biacore). Oligonucleotides were diluted in HEPES-KCl running buffer to a concentration of 30 nM and injected to reach the response of around 500 RU. Flow cell 1 was left empty to allow reference subtraction. C-exNDIs binding analysis was performed at a flow rate of 30 μL/min, with contact time of 280 s and dissociation time of 360 s in HEPES-KCl buffer. Sensorgrams were obtained in the concentration range of 25–200 nM. After each compound injection the chip surface was regenerated with glycine 10 mM pH 2.0 solution (GE Healthcare). All sensorgrams were corrected by buffer injection response. Data were fitted to a 1:1 binding model with Rmax initial parameter set to theoretical calculated Rmax using BIAevaluation software (GE Healthcare). All experiments were performed independently at least twice, and in each instance χ2 values, which indicate the goodness of fitting, were below 0.2.
CL-Mediated Footprinting
All oligonucleotides were gel-purified before use and prepared in desalted/lyophilized form. Oligonucleotides were 5′-end-labeled with [γ- 32P]ATP by T4 polynucleotide kinase and purified by MicroSpin G-25 columns (GE Healthcare, Europe). They were next resuspended in lithium cacodylate 10 mM, pH 7.4, and KCl 100 mM, heat-denatured, and folded. Reactions of the labeled G-quadruplex folded oligonucleotides (4 pmol/sample) with increasing amounts of compound 2 (4–100 nM) were performed at 20 °C for 24 h in resuspension buffer. Samples were then reacted with CL (100 μM) at 37 °C for 24 h. Reactions were stopped by ethanol precipitation, resuspended, and either kept on ice or treated at 90 °C for 30 min with 1 M piperidine to complete strand scission according to the Maxam and Gilbert protocol. Samples were then lyophilized, resuspended in formamide gel loading buffer, and heated at 95 °C for 3 min. Reaction products were analyzed on 20% denaturing polyacrylamide gels and visualized by Typhoon FLA 9000 phosphorimaging analysis (GE Healthcare).
Reporter Assays
The HIV-1 promoter activity in live cells in the presence of 2 was monitored in HeLa-Tat-III/LTR/d1EGFP cells (obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from Dr. Satoh). This cell line derives from HeLa-tat-III and is transfected with d1EGFP under the control of HIV-1 LTR promoter. Then 8 ×104 of HeLa-Tat-III/LTR/d1EGFP cells were seeded in a 12-well plate in 1 mL of DMEM/10% FBS medium supplemented with Geneticin selective antibiotic (1 mg/mL, Life Technologies, Monza, Italy) and incubated overnight. Cells were next treated with increasing concentrations of 2 (0–62.5–125–250 nM) and incubated at 37 °C. After 48 h, cells were washed with PBS 1× and resuspended in 500 μL of PBS 1×. To evaluate the mean of EGFP fluorescence, a total of 10000 events were acquired for each sample with an LRS 2 instrument using FACS DIVA Software (BD Bioscience) and analyzed with Flow Jo (Tree Star). Cell debris and dead cells were excluded from the analysis by gating on forward scatter (FSC) versus side scatter (SSC) and were quantified to estimate the cytotoxicity.
For the luciferase assay, HIV-1 LTR region (wt or M4 + 5) was inserted into the promoterless luciferase reporter vector pGL4.10-Luc2 (Promega Italia, Italy), as previously reported.27 For assessment of luciferase activity, human embryonic kidney 293T (HEK293T) cells were seeded in a 24-well plate (9 × 104 cells/well). Then 24 h later, cells were transfected with 100 ng/well of pGL4.10-LTRwt or of pGL4.10-LTR-M4 + 5 using TransIT-293 transfection reagent (Mirus Bio LLC, Madison, WI, USA), according to the manufacturer’s protocol. After 1 h, cells were treated with 2 for 24 h at various concentrations. Luciferase activity was measured using the britelite plus Reporter Gene Assay System (PerkinElmer Inc., Milan, Italy) at a Victor X2 multilabel plate reader (PerkinElmer Inc., Milan, Italy), according to the manufacturer’s instructions. Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.2, 150 mM NaCl, 1% Igepal, 0.1% SDS) and protein concentration was determined by BCA assay (Thermo Scientific Pierce, Monza, Italy). Luciferase signals were subsequently normalized to total protein content, according to the manufacturer’s protocol (http://ita.promega.com/~/pdf/resources/pubhub/cellnotes/normalizing-genetic-reporter-assays/).
Antiviral Assay in HIV-1 Infected TZM-bl Cells
HIV-1 infectivity was measured using the TZM-bl reporter cell line (obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from Dr. J.C. Kappes, Dr. X. Wu, and Tranzyme Inc.). TZM-bl is a HeLa cell line stably expressing large amounts of CD4 and CCR5 and containing integrated copies of the luciferase and β-galactosidase genes under control of the HIV-1 promoter. TZM-bl were grown in DMEM supplemented with 10% FBS. Cells (1 ×104 cells/well) were seeded in 96-well plates and grown overnight to permit adherence prior to treatment and viral infection. Cells were next infected with HIV-1 NL4-3 strain or BaL strain at a MOI of 0.5, treated with serial dilutions of tested compounds, and incubated at 37 °C. After 48 h, cells were washed with PBS 1× and HIV-1 production was assessed following the LTR-luciferase signal using the britelite plus Reporter Gene Assay System (PerkinElmer, Waltman, MA, USA) according to the manufacturer’s protocol.
Cytotoxicity Assay
Cytotoxicity of tested compounds was tested in parallel to antiviral and reporter assays by MTT. The 50% cytotoxic concentration (CC50) was defined as the concentration of test compound that was able to reduce the absorbance of the mock-infected cells by 50%. The 50% inhibitory concentration (IC50) was defined as the concentration of test compound that inhibit 50% of LTR-Luciferase signal. The selectivity index (SI) is the relative effectiveness of the tested compound in inhibiting viral replication compared to inducing cell death (CC50 value/IC50 value). The therapeutic window (TW) is the concentration range at which the compound shows antiviral activity paralleled by less than 80% cytotoxicity.
HIV-1 Entry Assay
TZM-bl cells (1 × 104 cells/well) were seeded in 96-well plates and grown overnight to permit adherence prior to treatment and viral infection. Cells were next infected with HIV-1 NL4-3 (MOI 0.5) and incubated at 37 °C. Compound 2 (50–100 nM) and the reference compounds DS (100 μg/mL, Sigma-Aldrich) and AZT (0.5 μg/mL, Sigma-Aldrich) were added at different hours (0, 1, 2 h) pi. After 31 h, cells were washed with PBS 1× and HIV-1 production was assessed following the LTR-luciferase signal using the britelite plus Reporter Gene Assay System (PerkinElmer, Waltman, MA, USA) according to the manufacturer’s protocol.
Computational Method
DFT calculations were performed with the Gaussian 09, revision B.01 software package.69 Gas-phase geometry optimizations were carried out using the B3LYP functional,70 with 6-31+G(d,p) basis set for all the atoms.
Acknowledgments
This work was supported by the Bill and Melinda Gates Foundation (GCE grant numbers OPP1035881, OPP1097238) to S.N.R., the European Research Council (ERC Consolidator grant 615879) to S.N.R. and M.F., and the Italian Ministry of University and Research (FIRB-Ideas RBID082ATK) to S.N.R. and M.F. We thank Prof. Manlio Palumbo, University of Padua, for helpful comments on the manuscript.
Glossary
Abbreviations Used
- G4
G-quadruplex
- NDI
naphthalene diimide
- c-exNDI
core-extended NDI
- LTR
long terminal repeat
- ds
double-stranded
- Tm
melting temperature
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b01283.
Author Contributions
§ R.P., F.D., and E.B. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Lipps H. J.; Rhodes D. G-quadruplex structures: in vivo evidence and function. Trends Cell Biol. 2009, 19, 414–422 10.1016/j.tcb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Sen D.; Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Brosh R. M. Jr. G-quadruplex nucleic acids and human disease. FEBS J. 2010, 277, 3470–3488 10.1111/j.1742-4658.2010.07760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D.; Gilbert W. A sodium-potassium switch in the formation of four-stranded G4-DNA. Nature 1990, 344, 410–414 10.1038/344410a0. [DOI] [PubMed] [Google Scholar]
- Taylor J. P. Neurodegenerative diseases: G-quadruplex poses quadruple threat. Nature 2014, 507, 175–177 10.1038/nature13067. [DOI] [PubMed] [Google Scholar]
- Campbell N. H.; Neidle S. G-quadruplexes and metal ions. Met. Ions Life Sci. 2012, 10, 119–134 10.1007/978-94-007-2172-2_4. [DOI] [PubMed] [Google Scholar]
- Eddy J.; Maizels N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006, 34, 3887–3896 10.1093/nar/gkl529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert J. L.; Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakken S.; Rognes T.; Hovig E. The disruptive positions in human G-quadruplex motifs are less polymorphic and more conserved than their neutral counterparts. Nucleic Acids Res. 2009, 37, 5749–5756 10.1093/nar/gkp590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogoi S.; Xodo L. E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006, 34, 2536–2549 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy J.; Maizels N. Conserved elements with potential to form polymorphic G-quadruplex structures in the first intron of human genes. Nucleic Acids Res. 2008, 36, 1321–1333 10.1093/nar/gkm1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A.; Yadav V. K.; Basundra R.; Kumar A.; Chowdhury S. Evidence of genome-wide G4 DNA-mediated gene expression in human cancer cells. Nucleic Acids Res. 2009, 37, 4194–4204 10.1093/nar/gkn1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani P.; Yadav V. K.; Das S. K.; Chowdhury S. Genome-wide analyses of recombination prone regions predict role of DNA structural motif in recombination. PLoS One 2009, 4, e4399. 10.1371/journal.pone.0004399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler A. R.; Donnelly C. J.; Periz G.; Simko E. A.; Shaw P. G.; Kim M. S.; Maragakis N. J.; Troncoso J. C.; Pandey A.; Sattler R.; Rothstein J. D.; Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P.; O’Day E.; Emara M. M.; Wagner G.; Lieberman J.; Anderson P. G-quadruplex structures contribute to the neuroprotective effects of angiogenin-induced tRNA fragments. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18201–18206 10.1073/pnas.1407361111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sket P.; Pohleven J.; Kovanda A.; Stalekar M.; Zupunski V.; Zalar M.; Plavec J.; Rogelj B. Characterization of DNA G-quadruplex species forming from C9ORF72 G4C2-expanded repeats associated with amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Neurobiol. Aging 2015, 36, 1091–1096 10.1016/j.neurobiolaging.2014.09.012. [DOI] [PubMed] [Google Scholar]
- Fisette J. F.; Montagna D. R.; Mihailescu M. R.; Wolfe M. S. A G-rich element forms a G-quadruplex and regulates BACE1 mRNA alternative splicing. J. Neurochem. 2012, 121, 763–773 10.1111/j.1471-4159.2012.07680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M.; Loeb L. A. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 4950–4954 10.1073/pnas.91.11.4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sissi C.; Gatto B.; Palumbo M. The evolving world of protein-G-quadruplex recognition: a medicinal chemist’s perspective. Biochimie 2011, 93, 1219–1230 10.1016/j.biochi.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G.; Tannahill D.; McCafferty J.; Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson A.; Wu Y.; Huang Y. C.; Chavez E. A.; Platt J.; Johnson F. B.; Brosh R. M. Jr.; Sen D.; Lansdorp P. M. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 2014, 42, 860–869 10.1093/nar/gkt957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metifiot M.; Amrane S.; Litvak S.; Andreola M. L. G-quadruplexes in viruses: function and potential therapeutic applications. Nucleic Acids Res. 2014, 42, 12352–12366 10.1093/nar/gku999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat P.; Zhong J.; Lekieffre L.; Cowieson N. P.; Clancy J. L.; Preiss T.; Balasubramanian S.; Khanna R.; Tellam J. G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol. 2014, 10, 358–364 10.1038/nchembio.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norseen J.; Johnson F. B.; Lieberman P. M. Role for G-quadruplex RNA binding by Epstein-Barr virus nuclear antigen 1 in DNA replication and metaphase chromosome attachment. J. Virol. 2009, 83, 10336–10346 10.1128/JVI.00747-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J.; Vonrhein C.; Smart O. S.; Bricogne G.; Bollati M.; Kusov Y.; Hansen G.; Mesters J. R.; Schmidt C. L.; Hilgenfeld R. The SARS-unique domain (SUD) of SARS coronavirus contains two macrodomains that bind G-quadruplexes. PLoS Pathog. 2009, 5, e1000428. 10.1371/journal.ppat.1000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone R.; Nadai M.; Poe J. A.; Frasson I.; Palumbo M.; Palu G.; Smithgall T. E.; Richter S. N. Formation of a unique cluster of G-quadruplex structures in the HIV-1 Nef coding region: implications for antiviral activity. PLoS One 2013, 8, e73121. 10.1371/journal.pone.0073121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone R.; Nadai M.; Frasson I.; Poe J. A.; Butovskaya E.; Smithgall T. E.; Palumbo M.; Palu G.; Richter S. N. A dynamic G-quadruplex region regulates the HIV-1 long terminal repeat promoter. J. Med. Chem. 2013, 56, 6521–6530 10.1021/jm400914r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrane S.; Kerkour A.; Bedrat A.; Vialet B.; Andreola M. L.; Mergny J. L. Topology of a DNA G-quadruplex structure formed in the HIV-1 promoter: a potential target for anti-HIV drug development. J. Am. Chem. Soc. 2014, 136, 5249–5252 10.1021/ja501500c. [DOI] [PubMed] [Google Scholar]
- Piekna-Przybylska D.; Sullivan M. A.; Sharma G.; Bambara R. A. U3 region in the HIV-1 genome adopts a G-quadruplex structure in its RNA and DNA sequence. Biochemistry 2014, 53, 2581–2593 10.1021/bi4016692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone R.; Butovskaya E.; Daelemans D.; Palu G.; Pannecouque C.; Richter S. N. Anti-HIV-1 activity of the G-quadruplex ligand BRACO-19. J. Antimicrob. Chemother. 2014, 69, 3248–3258 10.1093/jac/dku280. [DOI] [PubMed] [Google Scholar]
- Cogoi S.; Paramasivam M.; Spolaore B.; Xodo L. E. Structural polymorphism within a regulatory element of the human KRAS promoter: formation of G4-DNA recognized by nuclear proteins. Nucleic Acids Res. 2008, 36, 3765–3780 10.1093/nar/gkn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui-Jain A.; Grand C. L.; Bearss D. J.; Hurley L. H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 11593–11598 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell N. H.; Parkinson G. N.; Reszka A. P.; Neidle S. Structural basis of DNA quadruplex recognition by an acridine drug. J. Am. Chem. Soc. 2008, 130, 6722–6724 10.1021/ja8016973. [DOI] [PubMed] [Google Scholar]
- Leonetti C.; Amodei S.; D’Angelo C.; Rizzo A.; Benassi B.; Antonelli A.; Elli R.; Stevens M. F.; D’Incalci M.; Zupi G.; Biroccio A. Biological activity of the G-quadruplex ligand RHPS4 (3,11-difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium methosulfate) is associated with telomere capping alteration. Mol. Pharmacol. 2004, 66, 1138–1146 10.1124/mol.104.001537. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S.; Hurley L. H.; Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy?. Nat. Rev. Drug Discovery 2011, 10, 261–275 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A.; Bhattacharya S. DNA binders in clinical trials and chemotherapy. Bioorg. Med. Chem. 2014, 22, 4506–4521 10.1016/j.bmc.2014.05.030. [DOI] [PubMed] [Google Scholar]
- Franceschin M.; Rossetti L.; D’Ambrosio A.; Schirripa S.; Bianco A.; Ortaggi G.; Savino M.; Schultes C.; Neidle S. Natural and synthetic G-quadruplex interactive berberine derivatives. Bioorg. Med. Chem. Lett. 2006, 16, 1707–1711 10.1016/j.bmcl.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Kim M. Y.; Vankayalapati H.; Shin-Ya K.; Wierzba K.; Hurley L. H. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular g-quadruplex. J. Am. Chem. Soc. 2002, 124, 2098–2099 10.1021/ja017308q. [DOI] [PubMed] [Google Scholar]
- Dai J.; Carver M.; Hurley L. H.; Yang D. Solution structure of a 2:1 quindoline-c-MYC G-quadruplex: insights into G-quadruplex-interactive small molecule drug design. J. Am. Chem. Soc. 2011, 133, 17673–17680 10.1021/ja205646q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoroff O. Y.; Salazar M.; Han H.; Chemeris V. V.; Kerwin S. M.; Hurley L. H. NMR-Based model of a telomerase-inhibiting compound bound to G-quadruplex DNA. Biochemistry 1998, 37, 12367–12374 10.1021/bi981330n. [DOI] [PubMed] [Google Scholar]
- Di Antonio M.; Doria F.; Richter S. N.; Bertipaglia C.; Mella M.; Sissi C.; Palumbo M.; Freccero M. Quinone methides tethered to naphthalene diimides as selective G-quadruplex alkylating agents. J. Am. Chem. Soc. 2009, 131, 13132–13141 10.1021/ja904876q. [DOI] [PubMed] [Google Scholar]
- Collie G. W.; Promontorio R.; Hampel S. M.; Micco M.; Neidle S.; Parkinson G. N. Structural basis for telomeric G-quadruplex targeting by naphthalene diimide ligands. J. Am. Chem. Soc. 2012, 134, 2723–2731 10.1021/ja2102423. [DOI] [PubMed] [Google Scholar]
- Micco M.; Collie G. W.; Dale A. G.; Ohnmacht S. A.; Pazitna I.; Gunaratnam M.; Reszka A. P.; Neidle S. Structure-based design and evaluation of naphthalene diimide G-quadruplex ligands as telomere targeting agents in pancreatic cancer cells. J. Med. Chem. 2013, 56, 2959–2974 10.1021/jm301899y. [DOI] [PubMed] [Google Scholar]
- Doria F.; Nadai M.; Sattin G.; Pasotti L.; Richter S. N.; Freccero M. Water soluble extended naphthalene diimides as pH fluorescent sensors and G-quadruplex ligands. Org. Biomol. Chem. 2012, 10, 3830–3840 10.1039/c2ob07006e. [DOI] [PubMed] [Google Scholar]
- Doria F.; Oppi A.; Manoli F.; Botti S.; Kandoth N.; Grande V.; Manet I.; Freccero M. A naphthalene diimide dyad for fluorescence switch-on detection of G-quadruplexes. Chem. Commun. (Cambridge, U. K.) 2015, 51, 9105–9108 10.1039/C5CC01536G. [DOI] [PubMed] [Google Scholar]
- Nadai M.; Doria F.; Germani L.; Richter S. N.; Freccero M. A photoreactive G-quadruplex ligand triggered by green light. Chem. - Eur. J. 2015, 21, 2330–2334 10.1002/chem.201405215. [DOI] [PubMed] [Google Scholar]
- Artusi S.; Nadai M.; Perrone R.; Biasolo M. A.; Palu G.; Flamand L.; Calistri A.; Richter S. N. The Herpes Simplex Virus-1 genome contains multiple clusters of repeated G-quadruplex: Implications for the antiviral activity of a G-quadruplex ligand. Antiviral Res. 2015, 118, 123–131 10.1016/j.antiviral.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria F.; di Antonio M.; Benotti M.; Verga D.; Freccero M. Substituted heterocyclic naphthalene diimides with unexpected acidity. Synthesis, properties, and reactivity. J. Org. Chem. 2009, 74, 8616–8625 10.1021/jo9017342. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Li Y.; Zhao Y.; Zhang J.; Yang W.; Li Y. An unusual addition reaction for constructing a novel pH-controlled fluorescence switch. Org. Lett. 2011, 13, 292–295 10.1021/ol102742f. [DOI] [PubMed] [Google Scholar]
- Suraru S. L.; Zschieschang U.; Klauk H.; Wurthner F. A core-extended naphthalene diimide as a p-channel semiconductor. Chem. Commun. (Cambridge, U. K.) 2011, 47, 11504–11506 10.1039/c1cc15144d. [DOI] [PubMed] [Google Scholar]
- Langhals H.; Kinzel S. Laterally extended naphthalene tetracarboxylic bisimides. J. Org. Chem. 2010, 75, 7781–7784 10.1021/jo101520a. [DOI] [PubMed] [Google Scholar]
- Doria F.; Manet I.; Grande V.; Monti S.; Freccero M. Water-soluble naphthalene diimides as singlet oxygen sensitizers. J. Org. Chem. 2013, 78, 8065–8073 10.1021/jo401347z. [DOI] [PubMed] [Google Scholar]
- Palm W.; de Lange T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- Rosu F.; Gabelica V.; Houssier C.; De Pauw E. Determination of affinity, stoichiometry and sequence selectivity of minor groove binder complexes with double-stranded oligodeoxynucleotides by electrospray ionization mass spectrometry. Nucleic Acids Res. 2002, 30, e82. 10.1093/nar/gnf081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G.; Zhang Q.; Zhou J.; Li H. Mass spectrometry of G-quadruplex DNA: formation, recognition, property, conversion, and conformation. Mass Spectrom. Rev. 2011, 30, 1121–1142 10.1002/mas.20315. [DOI] [PubMed] [Google Scholar]
- Turner K. B.; Hagan N. A.; Fabris D. Inhibitory effects of archetypical nucleic acid ligands on the interactions of HIV-1 nucleocapsid protein with elements of Psi-RNA. Nucleic Acids Res. 2006, 34, 1305–1316 10.1093/nar/gkl004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando H.; Reszka A. P.; Huppert J.; Ladame S.; Rankin S.; Venkitaraman A. R.; Neidle S.; Balasubramanian S. A conserved quadruplex motif located in a transcription activation site of the human c-kit oncogene. Biochemistry 2006, 45, 7854–7860 10.1021/bi0601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadai M.; Palu G.; Palumbo M.; Richter S. N. Differential targeting of unpaired bases within duplex DNA by the natural compound clerocidin: a valuable tool to dissect DNA secondary structure. PLoS One 2012, 7, e52994. 10.1371/journal.pone.0052994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter S. N.; Menegazzo I.; Nadai M.; Moro S.; Palumbo M. Reactivity of clerocidin towards adenine: implications for base-modulated DNA damage. Org. Biomol. Chem. 2009, 7, 976–985 10.1039/b819049f. [DOI] [PubMed] [Google Scholar]
- Nadai M.; Sattin G.; Palu G.; Palumbo M.; Richter S. N. Clerocidin-mediated DNA footprinting discriminates among different G-quadruplex conformations and detects tetraplex folding in a duplex environment. Biochim. Biophys. Acta, Gen. Subj. 2013, 1830, 4660–4668 10.1016/j.bbagen.2013.05.039. [DOI] [PubMed] [Google Scholar]
- Doria F.; Nadai M.; Folini M.; Di Antonio M.; Germani L.; Percivalle C.; Sissi C.; Zaffaroni N.; Alcaro S.; Artese A.; Richter S. N.; Freccero M. Hybrid ligand-alkylating agents targeting telomeric G-quadruplex structures. Org. Biomol. Chem. 2012, 10, 2798–2806 10.1039/c2ob06816h. [DOI] [PubMed] [Google Scholar]
- Wilkinson R. A.; Pincus S. H.; Shepard J. B.; Walton S. K.; Bergin E. P.; Labib M.; Teintze M. Novel compounds containing multiple guanide groups that bind the HIV coreceptor CXCR4. Antimicrob. Agents Chemother. 2011, 55, 255–263 10.1128/AAC.00709-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman P. A.; Fyfe J. A.; St. Clair M. H.; Weinhold K.; Rideout J. L.; Freeman G. A.; Lehrman S. N.; Bolognesi D. P.; Broder S.; Mitsuya H. Phosphorylation of 3′-azido-3′-deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 8333–8337 10.1073/pnas.83.21.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuya H.; Looney D. J.; Kuno S.; Ueno R.; Wong-Staal F.; Broder S. Dextran sulfate suppression of viruses in the HIV family: inhibition of virion binding to CD4+ cells. Science 1988, 240, 646–649 10.1126/science.2452480. [DOI] [PubMed] [Google Scholar]
- Czerwinska I.; Juskowiak B. Photoisomerizable arylstilbazolium ligands recognize parallel and antiparallel structures of G-quadruplexes. Int. J. Biol. Macromol. 2012, 51, 576–582 10.1016/j.ijbiomac.2012.06.027. [DOI] [PubMed] [Google Scholar]
- Rachwal P. A.; Fox K. R. Quadruplex melting. Methods 2007, 43, 291–301 10.1016/j.ymeth.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Greenfield N. J. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat. Protoc. 2007, 1, 2527–2535 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W.; Zhou J.; Yuan G. Electrospray ionization mass spectrometry probing of binding affinity of berbamine, a flexible cyclic alkaloid from traditional Chinese medicine, with G-quadruplex DNA. Rapid Commun. Mass Spectrom. 2014, 28, 143–147 10.1002/rcm.6763. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr., Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision B.01; Gaussian, Inc.: Wallingford CT, 2010.
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652 10.1063/1.464913. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.