

Abstract
Background
WRKY transcription factors (TFs) constitute one of the largest protein families in higher plants, and its members contain one or two conserved WRKY domains, about 60 amino acid residues with the WRKYGQK sequence followed by a C2H2 or C2HC zinc finger motif. WRKY proteins play significant roles in plant development, and in responses to biotic and abiotic stresses. Pear (Pyrus bretschneideri) is one of the most important fruit crops in the world and is frequently threatened by abiotic stress, such as drought, affecting growth, development and productivity. Although the pear genome sequence has been released, little is known about the WRKY TFs in pear, especially in respond to drought stress at the genome-wide level.
Results
We identified a total of 103 WRKY TFs in the pear genome. Based on the structural features of WRKY proteins and topology of the phylogenetic tree, the pear WRKY (PbWRKY) family was classified into seven groups (Groups 1, 2a–e, and 3). The microsyteny analysis indicated that 33 (32 %) PbWRKY genes were tandemly duplicated and 57 genes (55.3 %) were segmentally duplicated. RNA-seq experiment data and quantitative real-time reverse transcription PCR revealed that PbWRKY genes in different groups were induced by drought stress, and Group 2a and 3 were mainly involved in the biological pathways in response to drought stress. Furthermore, adaptive evolution analysis detected a significant positive selection for Pbr001425 in Group 3, and its expression pattern differed from that of other members in this group. The present study provides a solid foundation for further functional dissection and molecular evolution of WRKY TFs in pear, especially for improving the water-deficient resistance of pear through manipulation of the PbWRKYs.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2233-6) contains supplementary material, which is available to authorized users.
Keywords: Pear, WRKY transcription factor, Drought stress
Background
Transcriptional regulation of gene expression is one of the most important regulatory mechanisms and transcription factors (TFs) mediate transcriptional regulation in response to developmental and environmental changes in plants. WRKY family is one of the largest TF families in higher plants, but is absent from animals. Since the discovery of the WRKY domain with DNA-binding capability [1], members of the WRKY protein family have been found to have an ever increasing number of functions in essential physiological and developmental processes in plants [2]. WRKY proteins contain either one or two WRKY domains. The WRKY domain contains approximately 60 amino acids with the conserved amino acid sequence WRKYGQK at its N-terminus and a zinc finger motif, C2H2 (C–X4–5–C–X22–23–H–X–H) or C2HC (C–X7–C–X23–H–X–C), at the C-terminal region [3]. The WRKY family can be classified on the basis of both the number of WRKY domains and the features of their zinc-finger motif. WRKY proteins with two WRKY domains belong to Group 1, whereas proteins with one WRKY domain belong to Group 2 or 3. Generally, the WRKY domains of members of Groups 1 and 2 members have the same type of finger motif, C2H2. The single finger motif of Group 3 is distinct from that of members of Groups 1 and 2. Instead of a C2H2 pattern, their WRKY domains contain a C2HC motif. However, the WRKYGQK amino acid sequence of all members in three groups forms a β-strand that binds sequence-specifically to the DNA sequence motif (T)(T)TGAC(C/T), which is known as the W box [4].
Experimental evidences has shown that plant WRKY proteins are involved in responses to biotic and abiotic stresses, and in developmental processes [2]. WRKY proteins play an important role in plant defense against biotic stresses, such as bacterial, fungal, and viral pathogens [5, 6]. WRKY proteins are also involved in plant-specific processes, such as trichome development [7], embryogenesis [8], seed development [9], dormancy [10], and senescence [11]. They are also key components in some signal transduction processes mediated by plant hormones such as gibberellic acid [12], abscisic acid (ABA) [13], and salicylic acid [14]. It is also well documented that WRKY proteins are involved in responses to various abiotic stresses, such as salinity, drought, and cold [15, 16]. Accumulating evidences shows that WRKY genes play an important role in responses to drought stress. ABO3, a WRKY TF, mediates plant responses to ABA and drought tolerance in Arabidopsis [15]; 10 TaWRKY genes responsive to drought stress were identified in an RNA-seq experiment [17]; and 42 OsWRKY genes were inducible under drought treatment [18].
Pear (Pyrus bretschneideri) is one of the most important fruit crops in the world. In the field, pear frequently experiences abiotic stress, such as drought, which is a key factor affecting growth, development and productivity. Recently, the complete genome of pear was sequenced by the Centre of Pear Engineering Technology Research, Nanjing Agricultural University (http://peargenome.njau.edu.cn/) [19]. This completed genome provides an opportunity to better understand the evolution and function of the WRKY family at the whole-genome level. Many WRKY proteins have been reported to be involved in responses to drought stress and so our initial interest was in the drought-related WRKY genes in pear. In this study, we identified 103 pear WRKY (PbWRKY) genes from the pear genomic sequence and carried out phylogenetic analysis to determine the relationships among these pear genes. Analysis of protein motifs and intron/exon structures provided support for the classification of the WRKY family. Furthermore, we identified the duplication events that likely contributed to the expansion of the WRKY family. In addition, RNA-seq data showed the expression patterns of PbWRKYs in different water-deficient stress, and subsequent quantitative real-time PCR (qRT-PCR) analysis indicated that two groups (Groups 2a and 3) of this family responded to drought stress. Then, to examine the driving force for the evolution of function for genes in Groups 2a and 3, we further analyzed adaptive evolution at the amino acid level. Our systematic analysis provided a foundation for further functional dissection and molecular evolution of WRKY genes in pear, especially for improving the drought resistance of pear and through the manipulation of PbWRKYs.
Results
The PbWRKY protein family consists of at least 103 members
To obtain sequences of WRKY genes in the pear genome, we used a HMMER-BLASTP-InterProScan strategy to search for genes encoding proteins containing the Pfam PF03106 domain. In total, 103 PbWRKY genes were identified (Additional file 1). Of the 103 PbWRKY genes, 87 were mapped on all pear chromosomes except chromosome 14, and 16 PbWRKY genes were located on scaffold contigs (Fig. 1). Among the other 87 PbWRKY genes, there were 10 respectively situated on chromosomes 6, 12 and 15; nine on chromosome 9; seven on chromosome 13; six each on chromosomes 7 and 8; five respectively on chromosomes 3, 10 and 17; three respectively on chromosomes 1, 5 and 11; two on chromosomes 2; and only one gene respectively on chromosomes 4 and 16.
Fig. 1.
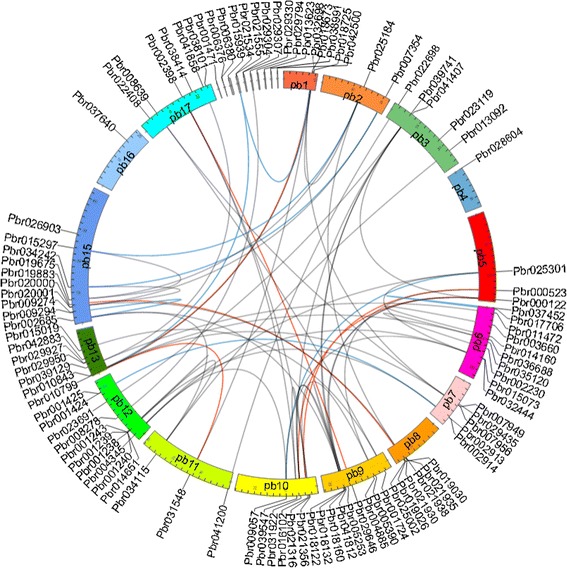
Localization and duplication of the WRKY genes in the pear genome. Circular visualization of the 103 WRKY genes was mapped on the different chromosomes in the pear genome using Circos software. Chromosome number is indicated on the chromosome. The microsynteny between each pair of WRKY genes were detected by using the MicroSyn software. The genes with synteny relationship are linked by lines. Red link: ≥30 anchors in a synteny block, blue link: 20–30 anchors, green link: 10–20 anchors, gray link: 5–10 anchors
Phylogenetic analysis of pear WRKY genes
In previous studies, WRKY TFs were classified into seven groups based on their number of WRKY domains and the pattern of their zinc finger motif [3]. Group 1 contains two WRKY domains (N-terminal and C-terminal), including a C2H2 motif, whereas Group 2a–e and 3 have only one domain. Group 3 has a distinct zinc finger motif, C2HC. To investigate the phylogenetic relationships of the WRKY genes in pear, we first constructed an un-rooted phylogenetic tree of 103 PbWRKY genes from the multiple sequence alignment of their WRKY domains. Three methods, Neighbor-Joining (NJ), Maximum Likelihood (ML), and Maximum Parsimony (MP) generated nearly identical topologies of phylogenetic trees (Additional files 2 and 3), although the support values at some inter nodes are different. Therefore, only the NJ tree was used for further analysis (Fig. 2). To better separate the groups and examine the evolutionary relationships of PbWRKY genes, we considered the tree topology , as well as the conserved sequence feature (i.e. number of WRKY domains and the pattern of zinc finger motif). The PbWRKY genes were first divided into three distinct groups: Groups 1, 2 and 3. This classification was consistent with results of previous studies. Group 1 (17 genes) contained two WRKY domains and was distinctly separated from other groups. However, Pbr029332 with only one WRKY domain was clustered with C-terminal WRKY domains of Group 1. Pbr029332 may have lost the N-terminal WRKY domain during evolution, or the N-terminal part of this gene was annotated incorrectly. Group 1 PbWRKY genes had a C2H2-type zinc-finger motif in the C-terminal WRKY domain. Group 2 contained 71 PbWRKYs, which possessed a single WRKY domain and a C2H2-type zinc-finger motif. Group 3 comprised 15 PbWRKY genes with a single WRKY domain. The C2HC zinc-finger structure in this group differed from those in other groups. Additionally, PbWRKY proteins in Group 2 had diverse sequences (Fig. 3). The structure and phylogenetic tree clearly indicated that Group 2 proteins could be divided into five distinct subgroups: a–e.
Fig. 2.
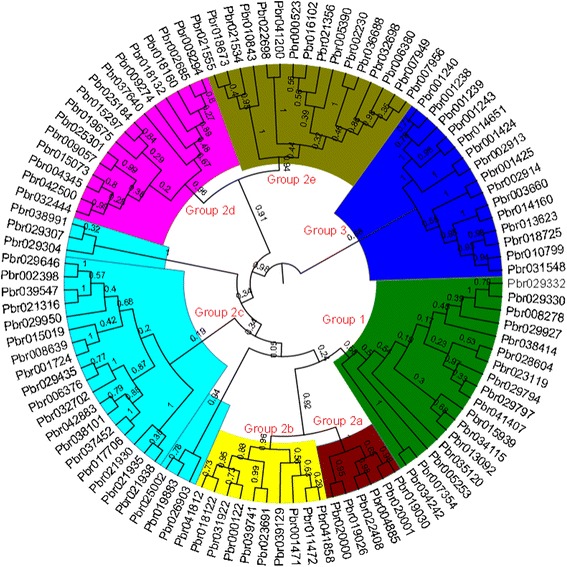
Phylogenetic trees of WRKY genes in pear. The un-rooted phylogenetic tree of WRKY domains was constructed with MEGA5.1 program with the NJ method. The numbers beside the branches represent bootstrap values based on 1000 replications. The name of groups (1, 2a-e, and 3) are shown at the inside of the circle. The groups of genes are shown in different colors
Fig. 3.

Schematic representations of the conserved motifs and exon–intron compositions. Names of genes are indicated on the left. a Conserved motifs in WRKY proteins. Different motifs are highlighted with different colored boxes with numbers 1 to 20. Lines represent protein regions without detected motif. b Exon–intron compositions in WRKY gene. Exons, represented by gray or red boxes, are drawn to scale. Dashed lines connecting two exons represent an intron. WRKY domain is marked in red
Conserved structural features of PbWRKY proteins
The most prominent feature of proteins in WRKY TFs is the WRKY domain, which preferentially binds to the promoter of their downstream target genes on a specific cis-element (e.g. W-box). We surveyed up to top 20 motifs in the 103 PbWRKY proteins using MEME (Fig. 3 and Additional file 4). Motifs 1, 2 and 6 were the conserved motifs at the N-terminus of the WRKY domain, while motifs 3–5 represented the zinc-finger motif at the C-terminus. The distribution of motifs outside the WRKY domain was highly conserved within groups. For example, motifs 9 and 12 only appeared in Groups 2a and 2b; and motifs 10 and 37 appeared exclusively in Group 2d.
Structure of PbWRKY genes
Since the intron/exon organizations and intron types and numbers are typical imprints of evolution within some gene families, we examined the PbWRKY gene structures to gain further insight into their evolutionary events. All WRKY genes in Groups 2 and 3 contained one intron in their WRKY domains except for two genes in Group 2e (Fig. 3). In addition, the exon/intron structures outside the WRKY domain were highly conserved within groups. Each group of PbWRKY genes mostly shared the same intron/exon structural pattern. One intron with phase 2 in the N-terminal existed in Groups 2d, 2e and 3; and there were three or four introns with phase 0 in Groups 2a and 2b.
Whole-genome duplication analysis of PbWRKY genes
It is thought that gene families evolved from a process of genome-wide duplication, segmental duplication and tandem duplication accompanied by post-duplication diversification [20–22]. Duplication events can result in a clustered occurrence of family members through tandem amplification, or a scattered occurrence through segmental duplication of chromosomal regions [20–22]. In this analysis, we focused on the tandem and segmental duplication modes of WRKY TFs in the whole pear genome. To identify the amplification patterns of the WRKY TFs, we first detected the existence of tandem duplications. We defined tandem duplication as one falling within 10 neighbors of another on genomic regions. Of the 103 PbWRKY genes, 33 (32 %) genes formed 15 tandemly duplicated clusters. The information concerning tandemly duplicated WRKY genes in pear is listed in Table 1, including four genes in Group 1, four in Group 2a, seven in Group 2c, six in Group 2d, two in Group 2b and seven in Group 3. No putative tandemly duplicated genes were found in Group 2d. To detect the segmental duplication events between two members in a gene family, the DNA sequences containing their neighboring genes were considered. Conserved, flanking collinear homologous gene pairs between the two genomic regions were searched for microsynteny to determine the segmental duplication events, controlled by a statistical distance function [23]. There were 61 pairs with collinear relationships detected and 57 genes (55.3 %) were involved in segment duplication (Table 2), suggesting that segmental duplication contributed to the expansion of the WRKY gene family in the pear genome. Taken together, tandem and segmental duplication events were involved in the expansion of the WRKY family in the pear genome.
Table 1.
Genes involved in tandem duplication
| Tandem duplicated genes | Group | Chromosome |
|---|---|---|
| Pbr029794, Pbr029797 | 1 | scaffold503.0 |
| Pbr029330, Pbr029332 | 1 | scaffold491.0 |
| Pbr019026, Pbr019030 | 2a | Chr8 |
| Pbr020001, Pbr020000 | 2a | Chr15 |
| Pbr029304, Pbr029307 | 2c | scaffold490.0 |
| Pbr021938, Pbr021935, Pbr021930 | 2c | Chr8 |
| Pbr006376, Pbr006380 | 2c | scaffold1326.0 |
| Pbr032698, Pbr032702 | 2c | scaffold591.0 |
| Pbr015073, Pbr032444 | 2d | Chr6 |
| Pbr018132, Pbr018122 | 2d | Chr9 |
| Pbr009294, Pbr009274 | 2d | Chr15 |
| Pbr007956, Pbr007949 | 2e | Chr7 |
| Pbr002913, Pbr002914 | 3 | Chr7 |
| Pbr001238, Pbr001243, Pbr001239, Pbr001240 | 3 | Chr12 |
| Pbr001424, Pbr001425 | 3 | Chr12 |
Table 2.
Synteny related to WRKY genes in pear
| Gene pairs | Anchors | E-value | Mean Ks | SD | Duplicated type |
|---|---|---|---|---|---|
| Pbr002398 vs. Pbr029646 | 33 | 8.58E-49 | 0.19 | 0.09 | segmental |
| Pbr000122 vs. Pbr031922 | 33 | 3.71E-49 | 0.18 | 0.08 | segmental |
| Pbr031548 vs. Pbr010799 | 33 | 9.59E-56 | 0.03 | 0.03 | segmental |
| Pbr000523 vs. Pbr016102 | 32 | 1.77E-48 | 0.19 | 0.05 | segmental |
| Pbr025002 vs. Pbr019883 | 32 | 2.91E-46 | 0.25 | 0.23 | segmental |
| Pbr018673 vs. Pbr010843 | 31 | 9.14E-46 | 0.26 | 0.35 | segmental |
| Pbr037452 vs. Pbr017706 | 29 | 6.97E-47 | 0.03 | 0.09 | segmental |
| Pbr007354 vs. Pbr034242 | 29 | 5.27E-48 | 0.21 | 0.1 | segmental |
| Pbr025301 vs. Pbr009057 | 28 | 5.33E-38 | 0.18 | 0.06 | segmental |
| Pbr018673 vs. Pbr021534 | 27 | 5.31E-41 | 0.05 | 0.05 | segmental |
| Pbr001424 vs. Pbr002913 | 26 | 1.11E-41 | 0.22 | 0.09 | segmental |
| Pbr002685 vs. Pbr009294 | 26 | 1.67E-45 | 0.01 | 0.01 | segmental |
| Pbr015297 vs. Pbr025184 | 24 | 8.01E-30 | 0.27 | 0.34 | segmental |
| Pbr010843 vs. Pbr021534 | 22 | 4.27E-33 | 0.19 | 0.05 | segmental |
| Pbr010799 vs. Pbr018725 | 19 | 9.26E-29 | 0.28 | 0.35 | segmental |
| Pbr039547 vs. Pbr021316 | 18 | 1.25E-33 | 0.03 | 0.12 | segmental |
| Pbr008639 vs. Pbr001724 | 15 | 6.05E-20 | 0.23 | 0.25 | segmental |
| Pbr013092 vs. Pbr034115 | 14 | 6.10E-16 | 0.55 | 0.92 | segmental |
| Pbr008278 vs. Pbr029330 | 14 | 2.54E-18 | 0.23 | 0.15 | segmental |
| Pbr031548 vs. Pbr018725 | 13 | 1.72E-22 | 0.2 | 0.07 | segmental |
| Pbr015073 vs. Pbr004345 | 12 | 4.56E-20 | 0.25 | 0.28 | segmental |
| Pbr011472 vs. Pbr039129 | 12 | 9.50E-12 | 1.38 | 0.42 | segmental |
| Pbr041812 vs. Pbr018132 | 12 | 5.92E-19 | 0.09 | 0.28 | segmental |
| Pbr019675 vs. Pbr025184 | 11 | 2.87E-10 | 1.27 | 0.48 | segmental |
| Pbr036688 vs. Pbr002230 | 11 | 3.00E-20 | 0.05 | 0.13 | segmental |
| Pbr019675 vs. Pbr015297 | 10 | 6.23E-10 | 1.36 | 0.75 | segmental |
| Pbr000122 vs. Pbr041812 | 10 | 4.80E-14 | 1.25 | 0.62 | segmental |
| Pbr039741 vs. Pbr031922 | 10 | 2.94E-15 | 1.49 | 0.95 | segmental |
| Pbr022698 vs. Pbr041200 | 10 | 1.01E-18 | 0.19 | 0.06 | segmental |
| Pbr000122 vs. Pbr039741 | 9 | 2.71E-14 | 1.57 | 0.9 | segmental |
| Pbr031922 vs. Pbr018132 | 9 | 2.06E-10 | 1.34 | 0.43 | segmental |
| Pbr019026 vs. Pbr020001 | 9 | 9.84E-16 | 0.35 | 0.52 | segmental |
| Pbr023691 vs. Pbr039741 | 8 | 4.43E-14 | 0.1 | 0.27 | segmental |
| Pbr000122 vs. Pbr018132 | 8 | 1.57E-09 | 1.57 | 0.43 | segmental |
| Pbr025301 vs. Pbr004345 | 8 | 1.33E-08 | 1.04 | 0.63 | segmental |
| Pbr041812 vs. Pbr031922 | 8 | 1.14E-11 | 1.36 | 0.35 | segmental |
| Pbr034242 vs. Pbr020001 | 8 | 3.02E-06 | 1.49 | 0.42 | segmental |
| Pbr002398 vs. Pbr039547 | 7 | 2.62E-06 | 1.57 | 0.46 | segmental |
| Pbr001238 vs. Pbr014651 | 7 | 2.56E-11 | 0.18 | 0.05 | segmental |
| Pbr039741 vs. Pbr041812 | 7 | 1.94E-10 | 1.27 | 0.25 | segmental |
| Pbr039741 vs. Pbr018132 | 7 | 1.79E-08 | 1.38 | 0.25 | segmental |
| Pbr041858 vs. Pbr039129 | 7 | 7.25E-09 | 1.33 | 0.78 | segmental |
| Pbr005390 vs. Pbr000523 | 6 | 5.22E-08 | 1.59 | 0.56 | segmental |
| Pbr004345 vs. Pbr009057 | 6 | 2.11E-07 | 1.11 | 0.47 | segmental |
| Pbr002685 vs. Pbr018132 | 6 | 5.22E-08 | 0.2 | 0.08 | segmental |
| Pbr018673 vs. Pbr007956 | 6 | 1.91E-06 | 1.15 | 0.15 | segmental |
| Pbr003660 vs. Pbr014160 | 6 | 2.09E-12 | 0.02 | 0.03 | segmental |
| Pbr007354 vs. Pbr020001 | 6 | 2.82E-05 | 1.57 | 0.44 | segmental |
| Pbr034115 vs. Pbr014651 | 6 | 5.51E-06 | 1.43 | 0.5 | segmental |
| Pbr002398 vs. Pbr021316 | 5 | 1.44E-05 | 1.66 | 0.5 | segmental |
| Pbr015073 vs. Pbr025301 | 5 | 3.91E-07 | 1.01 | 0.21 | segmental |
| Pbr015073 vs. Pbr009057 | 5 | 3.02E-05 | 1.18 | 0.38 | segmental |
| Pbr002913 vs. Pbr014651 | 5 | 5.94E-06 | 1.27 | 0.45 | segmental |
| Pbr036688 vs. Pbr010843 | 5 | 1.38E-04 | 1.51 | 0.26 | segmental |
| Pbr005390 vs. Pbr016102 | 5 | 1.64E-06 | 1.9 | 0.56 | segmental |
| Pbr002685 vs. Pbr034115 | 5 | 1.44E-03 | 1.36 | 0.71 | segmental |
| Pbr008639 vs. Pbr007956 | 5 | 4.32E-04 | 1.72 | 0.86 | segmental |
| Pbr001724 vs. Pbr015019 | 5 | 2.97E-05 | 1.51 | 0.42 | segmental |
| Pbr007956 vs. Pbr010843 | 5 | 1.13E-03 | 1.11 | 0.33 | segmental |
| Pbr025184 vs. Pbr019026 | 5 | 5.60E-03 | 1.59 | 0.49 | segmental |
| Pbr025184 vs. Pbr018725 | 5 | 3.98E-06 | 1.51 | 0.79 | segmental |
Expression of PbWRKY genes under drought stress
Some evidence has suggested that PbWRKY proteins are involved in signaling and responses to abiotic stimuli [15, 18], such as drought stress, but limited information is available on involvement of WRKY TFs in drought stress response of pear. In this study, RNA-seq data for short-term dehydration stress on pear seedling treatment were from parallel work (paper in preparation) aiming to study the response to water-deficiency stresses of pear. The heatmap was divided into six clusters (Fig. 4). Cluster 1 contained seven (6.8 %) detectable PbWRKY genes, which were significantly up-regulated by drought treatment at 3 and 6 h, except for Pbr037452. Cluster 2 contained 16 genes, which were also highly induced at 3 and 6 h after drought treatment, but their relative expression levels (compared with control) were lower than for genes in Cluster 1. In Clusters 3 (14 genes) and 4 (six genes), most genes were up-regulated after 3 and/or 6 h of drought treatment; however, some genes (Pbr041200, Pbr001471 and Pbr039741) were not induced by drought stress. Genes in Clusters 5 and 6 were down-regulated or not induced by drought treatment. Overall, 44 PbWRKY genes were up-regulated at least two-fold after drought treatment relative to controls and were within the range of 2–1024 fold; and 19 PbWRKY genes were down-regulated at least two-fold after drought treatment relative to controls within the range of 2–64 fold. We focused on the up-regulated genes of Clusters 1, 2 and 3 and the expression patterns in phylogenetic groups were surveyed.
Fig. 4.

Heat map of RNA-seq expression of PbWRKY gene in response to drought stress. Color scale of the dendrogram represents log2 ratio value of treated sample to control sample. D1, D3, D6 and D24, dehydrated for 1, 3 and 6 h in an ambient environment and recovered for 24 h in water, respectively. The colors of genes in Cluster 1–4 represent their groups in the phylogenetic tree in Fig. 2
We found that all six genes in Group 2a, 13 (86.7 %) in Group 3, four (40.0 %) in Group 2b, nine (37.5 %) in Group 2c, three (18.8 %) in Group 2e, three (17.6 %) in Group 1 and one gene (6.7 %) in Group 2d were up-regulated in response to drought stress. Genes in Cluster 1 were more significantly up-regulated than genes in the other clusters, and most genes in Cluster 1 belonged to Groups 2b and 3. This result suggests that PbWRKY genes in different groups were induced by water deficiency, and Groups 2a and 3 were mainly involved in the biological pathways responding to drought stress.
Orthologous genes are homologous genes that have diverged after a speciation event. Orthologous genes are generally assumed to retain equivalent functions in different organisms and to share other key properties. In this type of homologous gene, the ancestral gene and its function is maintained through a speciation event, although variations may arise within the gene after the point at which the species diverged [24]. In the published literature, 13 WRKY TFs have been shown to be involved in drought, salt and osmotic stresses [15, 25–34]. Nine of the 13 WRKY TFs located in eight orthologous groups were identified using InParanoid [35]. Six of the eight orthologous groups had 14 stress-responsive PbWRKY TFs (Additional file 5). These 14 stress-responsive PbWRKY TFs may retain equivalent functions to those in Arabidopsis. However, we found four WRKY TFs that were not located in orthologous groups, indicating they did not have equivalent PbWRKY TFs. Taken together, these results indicate that the functions of PbWRKY TFs were largely conserved.
In plants, transcriptional regulation is mediated by a large number (>1500) of TFs controlling the expression of tens or hundreds of target genes in various, sometimes intertwined, signal transduction cascades [36]. As TFs, WRKY TFs can bind to cis-elements to control the expression of tens or hundreds of target genes in plants. To understand the function of WRKY TFs at the system level, we investigated the gene co-expression clusters that had drought-responsive WRKY TFs. Nine co-expression networks were found to have drought-responsive WRKY TFs (Additional file 6). There were 1–22 drought-responsive WRKY TFs. Co-expression gene Cluster 3 had the greatest number of WRKY TFs (i.e. 22). Gene ontology (GO) analysis showed that genes in Cluster 3 were enriched in GO terms of regulation of macromolecule biosynthetic process, protein modification process, response to biotic stimulus, biological regulation, macromolecule modification, response to water stress, aromatic amino acid family biosynthetic process, chorismate metabolic process, phosphate metabolic process, phosphorus metabolic process, secretion, and secretion by cell. Furthermore, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis showed that genes located in co-expression gene Cluster 3 were enriched in plant hormone signal transduction and circadian entrainment. These two pathways are well-known to be related to drought stress [37, 38].
To validate the expression patterns of the 13 genes in Group 3 in the phylogenetic tree, we performed a qRT-PCR experiment on pear seedlings involving short-term drought stress. The result of qRT-PCR was highly consistent with RNA-seq data (Fig. 5), indicating that our RNA-seq data were reliable. Gene expression levels of all 13 genes increased to their highest level at either 3 or 6 h and then decreased by 24 h of recovery. Twelve genes exhibited the highest level at 6 h after drought stress treatment; and Pbr001425 showed the highest expression level at 3 h of drought treatment.
Fig. 5.
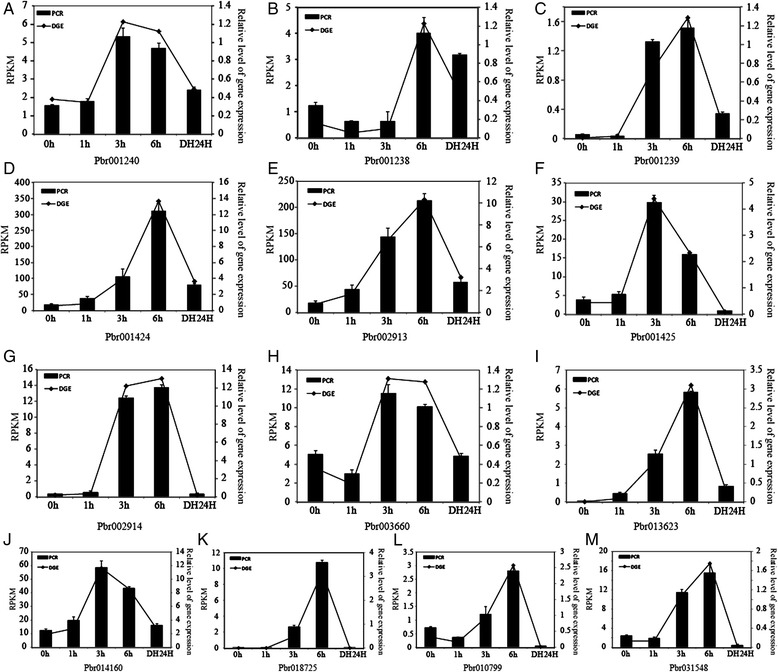
Quantitative RT-PCR analysis of PbWRKY gene expression in response to drought stress. D0, D1, D3, D6 and D24, dehydrated for 0, 1, 3 and 6 h in an ambient environment and recovered for 24 h in water, respectively
Detection of positive selection in Group 2A and subfamily Group 3 WRKY genes
Our initial interest in the WRKY proteins came from the proposed role of WRKY genes in response to drought stress. As indicated, Groups 2a and 3 were involved in the biological pathways in response to drought stress; therefore, we focused on these two groups. Positive selection is one of the major forces in the emergence of new motifs and functions in genes after duplication events. Selection pressure is measured by ω and the ratio of non-synonymous site (Ka) to synonymous substitution site (Ks), and if a proportion of sites in the sequence provides statistically significant support for ω > 1 along the lineages of interest, then episodic positive selection is inferred. Adaptive evolution frequently occurs in a few sites in a gene, and to a small number of lineages in a phylogenetic tree. In this study, branch-site random effects likelihood (REL) were implemented in Datamonkey [39] to test for positive selection of the WRKY genes within Groups 2a and 3. The results identified no WRKYs with positive selection in Group 2a. For branches in Group 3, we found one branch (Pbr001425) under episodic diversifying selection with corrected p-value < 0.0001. The ω value inferred from positively selected sites (ω+) along the branch of Pbr001425 was 1187.8, and the proportion of sites evolving at ω+ was 7.0 %. To validate the results from Datamonkey, we also performed analysis of positive selection using the branch-site model in PAML for Group 3. The branches being tested for positive selection are referred to as foreground branches, and all other branches on the phylogenetic tree are referred to as background branches. The branch of Pbr001425 was independently defined as a foreground branch. We used branch-site model A (model = 2, NSsites = 2, fix_omega = 0, omega = 1.5) as the alternative hypothesis. The model assumes four classes of sites: class 0 includes codons that are conserved throughout the tree, with 0 < ω0 < 1 estimated; class 1 includes codons that are evolving neutrally throughout the tree with ω1 < 1; and classes 2a and 2b include codons that are conserved or neutral on the background branches, but become under positive selection on the foreground branches with ω2 > 1. The null hypothesis is the branch-site model A, but with ω2 = 1 fixed. This null model allows sites to evolve under negative selection on the background lineages and to evolve neutrally on the foreground lineages. Significant positive selection was detected under the χ2 test (p < 0.01) (Table 3). The results from the two independent types of software indicated that Pbr001425 had undergone positive selection after gene duplication.
Table 3.
Parameters estimation and likelihood ratio tests for the branch-site models
| Hypothesis | lnL | Site class 0 | Site class 1 | Site class 2a | Site class 2b | 2ΔlnL |
|---|---|---|---|---|---|---|
| Alternative | −9212.750 | p0 = 0.36828 ω0(b)1 = 0.17271 ω0(f)2 = 0.17271 | p1 = 0.56715 | p2a = 0.02542 | p2b = 0.03915 | 35.237** |
| ω1(b) = 1 | ω2a(b) = 0.17271 | ω2b(b) = 1 | ||||
| ω1(f) = 1 | ω2a(f) = 999 | ω2b(f) = 999 | ||||
| Null | −9230.368 | p0 = 0.31965 | p1 = 0.45639 | p2a = 0.09225 | p2b = 0.13171 | |
| ω0(b=)0.17495 | ω1(b) = 1.00000 | ω2a(b) = 0.17495 | ω2b(b) = 1.00000 | |||
| ω0(f) = 0.17495 | ω1(f) = 1.00000 | ω2a(f) = 1.00000 | ω2b(f) = 1.00000 |
Note: **p < 0.01 (χ2 test); 1 Background ω; 2 Foreground ω
Discussion
In this study, a total of 103 PbWRKY genes were identified through genome-wide analysis. We adopted the classification scheme for the WRKY family of Eulgem et al. [3]. The PbWRKY genes were divided into three distinct clusters: Groups 1, 2 and 3. The Group 2 proteins were further divided into five distinct groups: a–e. However, the phylogenetic tree of PbWRKY genes clearly showed that Groups 2a and 2b, and Groups 2d and 2e seemed to form monophyletic clades, respectively. The motifs and exon/intron analysis indicated that Groups 2a, 2b, 2d and 2e were four distinct groups; whereas, Groups 2a and 2b, and Groups 2d and 2e had close phylogenetic relationships, respectively. Additionally, Group 2 was divided into five distinct groups (a–e) with good support values, except for Group 2c. However, the NJ, MP and ML trees consistently clustered Group 2c as a natural clade, supporting the classification of this group. Interestingly, for genes in Group 1, the C-terminal WRKY domain (CTWD) contained one intron, whereas the N-terminal WRKY domain (NTWD) had no introns. The pattern of intron number of the WRKY domain indicated that CTWDs in Group 1 could be ancestors of the WRKY genes in other groups, consistent with the phylogenetic analysis [40].
Gene duplication and divergence events have generally been viewed as a necessary source of evolutionary momentum [20, 21]. In our study, we found that a large fraction of WRKY TFs had arisen by either tandem or segmental duplication (Tables 1 and 2), consistent with results in grapevine [41]. The microsynteny analysis indicated that 33 (32 %) of PbWRKY genes were tandemly duplicated and 57 genes (55.3 %) were segmentally duplicated, implying low tandem and high segmental duplications in PbWRKY genes, consistent with results for both Arabidopsis and grapevine [41, 42]. Compared to tandem duplication, genes arising through segmental duplication may be detected more often in the genome due to sub-functionalization [43, 44]. Genes within a single genome can be classified as singletons, dispersed duplicates, proximal duplicates, tandem duplicates and segmental/WGD [45]. The expression of PbWRKY genes in response to drought stress was investigated using RNA-seq data and qRT-PCR. Overall, we found that 44 PbWRKY genes were up-regulated at least two-fold under drought treatment, and PbWRKY genes in different groups were induced by water deficit treatment, and Groups 2a and 3 were mainly involved in the biological pathways responding to drought stress. All members of Group 2a were up-regulated in response to drought stress. The adaptive evolution analysis showed that no WRKYs within Group 2a experienced positive selection, and so the drought stress-related function in Group 2a is highly conservative. In Group 3, 13 WRKY genes were induced under drought stress. Most PbWRKY genes were induced by drought stress with a peak of expression at 6 h. However, Pbr001425 was mainly up-regulated after 3 h of drought treatment and then expression level decreased after 6 h. Pbr001425 and Pbr001424 were tandemly duplicated genes; however, they had different expression patterns. Furthermore, significant positive selection was detected for Pbr001425. Therefore, we proposed that Pbr001425 underwent positive selection after gene duplication and obtained new functions during evolution.
As an abiotic stress, drought can cause loss of yield and quality of fruit trees [46–48]. In our study, we found 44 drought-responsive WRKY genes (Fig. 4). In Arabidopsis, four WRKY genes were reported to regulate drought response. AtWRKY57 can elevate ABA levels and so improve drought tolerance of Arabidopsis [25]. The grapevine VvWRKY11 is involved in the response to dehydration stress. Overexpression of VvWRKY11 in Arabidopsis led to more tolerance to water stress induced by mannitol than wild-type plants [49]. Similar to VvWRKY11, transgenic Arabidopsis lines overexpressing soybean GsWRKY20 also showed enhanced drought tolerance. Exposure to drought or salt stress triggers many common reactions in plants, such as cellular dehydration, which can lead to osmotic stress and the production of reactive oxygen species [50]. WRKY54 and WRKY70 regulate osmotic stress by working as negative regulators of stomata closure. The wrky54wrky70 double mutants exhibited clearly enhanced tolerance to osmotic stress [51]. We found that some PbWRKY TFs—orthologs of WRKY TFs that involved in drought, salt and osmotic stresses in Arabidopsis—were also responsive to drought stress in pear (Additional file 5), indicating that these PbWRKY TFs may have equivalent functions in pear compared to Arabidopsis. However, we also found that some of the orthologous PbWRKY TFs did not respond to drought stress and two of these Arabidopsis WRKY TFs did not have orthologous PbWRKY TFs in pear, indicating divergence of WRKY TFs between Arabidopsis and pear.
Methods
Gene identification
The complete genome, proteome sequences and GFF (General Feature Format) of Arabidopsis and pear downloaded from The Arabidopsis Information Resource (version 10; http://www.arabidopsis.org) and http://peargenome.njau.edu.cn, respectively. In proteome datasets, if two or more protein sequences at the same locus were identical where they overlapped, we selected the longest sequence. HMMER is used to search sequence databases for WRKY protein sequences. HMMER implements methods using probabilistic models called profile hidden Markov models (profile HMMs). A HMM profile for the WRKY domain (PF03106) was downloaded from the Pfam protein family database (http://pfam.sanger.ac.uk/). HMMER [52] was used to search a customized database containing the proteome with the threshold set of the Pfam GA gathering cutoff. The HMMER-selected proteins were used for a BLASTP query of the original protein database. Finally, the BLASTP hits were scanned for WRKY domains using InterProScan [53]. To confirm our data set of amino acid sequences as WRKYs, we manually examined the conserved amino acid sequence WRKYGQK at the N-terminal and the zinc-finger-like motif at the C-terminal region of the predicted WRKY domain. After removing truncated and pseudo genes, a total of 103 WRKY genes were assigned in pear. The CDS and protein sequences of these WRKY genes were stored in Additional files 7 and 8, respectively. These WRKY genes were named PbWRKY (Pyrus bretschneideri WRKY) genes and each given a number designation of 1–73 based on their E-value of InterProScan search in the order of increasing values. The nomenclature and corresponding information are listed in Additional file 1.
Three building and gene structure prediction
The starting point for our tree construction was the amino acid multiple sequence alignment created using MUSCLE [54] with the default parameters. The Jones, Taylor and Thorton (JTT) with an estimated γ-distribution parameter (G) was selected as the best-fitting amino acid substitution model with four categories using the Akaike information criterion implemented in Model Generator version 85 [55]. The ML analyses were performed using PHYML 3.0 [56], using the JTT + I + G model. Heterogeneity of amino acid substitution rates was corrected using a γ-distribution with five categories. Tree topology searching was optimized using the subtree pruning and regrafting option. The statistical support of the retrieved topology was assessed using a bootstrap analysis with 100 replicates. NJ and MP were implemented with MEGA 5.0 [57]. In NJ and MP, the ‘pairwise deletion’ setting was used. A bootstrap analysis with 1000 replicates was performed in each case. The conserved motifs in the proteins were detected by MEME (http://meme.nbcr.net/meme/cgibin/meme.cgi), with the following parameters: number of repetitions: any; maximum number of motifs: 20; and the optimum motif widths: 6–200 amino acid residues.
Chromosomal distribution and gene duplication
The genes were plotted separately onto the chromosomes according to gene location in the chromosome in the GFF file using a programmed Perl script. Genes within a maximum of 10 genes distance were considered to be tandem duplicates. The microsyntenies between each pair of members were detected using MicroSyn software [23]. The parameters were set as follows: window size of 50 genes, tandem gap value of 2, expected threshold value cut off of 0.01, and three homologous pairs to define a syntenic segment. Type of gene duplication was determined using the software MCScanX [58].
Adaptive evolution analysis
Episodic diversifying selection was performed on the Datamonkey web server (http://www.datamonkey.org/) [39], implementing a Branch-site REL approach [59]. Positive selection was validated using the CODEML program contained in the PAML 4 software package [60], using the branch-site model A.
Data analysis of Solexa/ Illumina sequencing
RNA-seq data of short-term dehydration stress on pear seedlings were obtained from parallel work (paper in preparation) aiming to study the response of pear to water-deficiency stress. Briefly, differentially expressed genes (DEGs) of Pyrus bretschneideri were determined using Solexa/Illumina sequencing. The total RNA was extracted from leaves sampled from the seedlings dehydrated for 0, 3, 6 and 24 h of recovery. The library products were ready for sequencing via Illumina HiSeqTM 2000 or other sequencers when necessary. The high-quality clean sequence reads were mapped onto the pear reference genome (http://peargenome.njau.edu.cn) to identify continuous gene regions using SOAP2 [61] and allowed no more than 2-nt mismatching. The unique mapped reads were used for further analysis. For gene expression level analysis, the number of unique-match reads was calculated and then normalized to RPKM (reads per kb per million reads). The gene expression levels were expressed as log(x/y), where x is the detection signal of the treatment sample and y is that of control or the mean of samples. Data were analyzed using Bioconductor.
Genes with similar expression patterns are usually functionally related. We performed a co-expression cluster analysis on the gene expression patterns using cluster software [62] (Additional file 9). In the gene expression profiling analysis, InterPro domains [63] were annotated using InterProScan Release 36.0 [64] and functional assignments were mapped onto Gene Ontology (GO) [65]. The GO classifications and GO enrichment were done using WEGO (Additional files 10 and 11) [66]. For the pathway enrichment analysis, genes in each cluster were mapped to terms in the Kyoto Encyclopedia of Genes and Genomes database (KEGG, release [67]) using BLASTX [68] at E values ≤ 1e-10 to identify significantly enriched KEGG terms. A Perl script was used to retrieve KO (KEGG Ontology) information from the BLAST search result so that pathway associations between unigenes and the database could be established (Additional file 12).
Heat maps were generated using the R package ‘gplots’ (http://www.bioconductor.org/). Clustering in the heat map was carried out using Hierarchical Clustering with the hclust function in R (http://www.r-project.org/).
Identification of orthologous genes between Arabidopsis and pear
To identify orthologous genes between Arabidopsis and pear, InParanoid was used with default settings [35]. During InParanoid analysis, an orthology group is initially composed of two so-called seed orthologs that are found by two-way best hits between two proteomes [35]. More sequences are added to the group if there are sequences in the two proteomes that are closer to the corresponding seed ortholog than to any sequence in the other proteome. These members of an orthology group are called inparalogs. In total, there were 118,087 orthology groups identified between Arabidopsis and pear, and included 22,580 pear and 15,988 Arabidopsis genes. The orthology groups with WRKY TFs involved in drought, salt and osmotic stresses were then extracted.
Gene expression analysis by qRT-PCR
RNA samples were used for cDNA synthesis using the ReverTra Ace-α First Strand cDNA Synthesis Kit (TOYOBO, TOYOBO Biotech Co. Ltd, Japan) following the manufacturer’s instructions. Primers were designed using Primer5 software based on the target genes (Additional file 13). The 10 μL qPCR solutions contained 5 μL of using SYBR® Green Premix kit (TaKaRa Biotechnology. Dalian, China), 0.25 μM forward and 0.25 μM reverse primer, and 50 ng cDNA templates. The quadruple qRT-PCR reactions were performed on an Lightcycle-480 (Roche) using the following cycling regime: 50 °C/2 min, 95 °C/10 min, followed by 40 cycles of 95 °C/15 s, and 58 °C/1 min. Relative expression levels of each gene were calculated using the 2-ΔΔCt algorithm by normalizing to expression of the pear tubulin gene (AB239681) [69], which was used as an internal control. Four technical replicates were used for each sample and the data are shown as means ± standard errors (SE) (n = 3). The source of variation resulted from technical errors, such as operational approach, equipment and reagent. The biological replicates were repeated three times for consistent results, the data were analyzed using analysis of variance (ANOVA) by SAS software (version 8.0, SAS Institute, NC, USA), and statistical differences were compared based on Fisher’s LSD test. The primer sequences used for qRT-PCR are listed in Additional file 13.
Conclusions
Genome-wide identification, evolutionary analysis, gene structure analysis and expression analysis of pear WRKY genes provide us a deep insight of this TF family and their potential roles in drought stress response. This will facilitate the further research on the biological functions of WRKY TFs in pear.
Acknowledgments
This work was supported by National Natural Science Foundation of China (31301758), the Research Fund for the Doctoral Program of Higher Education (130600661), the Jiangsu Provincial Natural Science Foundation (BK20130689, SBK2015042246), the National Postdoctoral Fund (2013 T60545, 2012 M521092, 2014 M551615), the Jiangsu Provincial Postdoctoral Fund (1201019B, 1401125C), and Science Foundation of Ministry of Education in China (14YJC630058). The Fundamental Research Funds for the Central Universities (KYTZ201401, SK2014007, KYTZ201401), the Ministry of education of Humanities and Social Science project (14YJC630058). We are also grateful to Dr. Bing Cai and Dr. Shaojun Xie for his assistance with data analysis.
Additional files
List of PbWRKY genes. (XLS 1392 kb)
Phylogenetic tree generated using the Maximum Likelihood (ML) method. (PDF 169 kb)
Phylogenetic tree generated using the Maximum Parsimony (MP) method. (PDF 186 kb)
Consensus sequences of motifs in PbWRKY TFs detected by MEME. (DOCX 16 kb)
List of othology groups with Arabidopsis WRKY TFs that involved in drought, salt or osmotic stress. (DOCX 26 kb)
List of drought responsive PbWRKY TFs that located in co-expression clusters. (XLS 31 kb)
CDS of PbWRKY genes. (GZ 13907 kb)
Protein sequences of PbWRKY TFs. (TXT 42 kb)
List of genes in co-expression gene clusters. (XLS 1128 kb)
GO enrichment analysis of co-expression gene clusters. (XLS 39 kb)
List of genes in enriched GO terms. (XLS 290 kb)
KEGG enrichment analysis of co-expression gene clusters. (XLS 50 kb)
Quantitative RT-PCR analysis of the PbWRKY gene expression in response to drought stress. (DOCX 17 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XH contributed to the experimental design and management, data analysis, and manuscript preparation. KL, XX contributed to proofreading and critical review of this manuscript. ZY and CJ contributed to genes expression analysis. XH and SZ designed and managed the experiments. All authors have read and approved the final manuscript.
Contributor Information
Xiaosan Huang, Email: huangxs@njau.edu.cn.
Kongqing Li, Email: likq@njau.edu.cn.
Xiaoyong Xu, Email: xyxu@yzu.edu.cn.
Zhenghong Yao, Email: 2014804187@njau.edu.cn.
Cong Jin, Email: 2012204008@njau.edu.cn.
Shaoling Zhang, Email: slzhang@njau.edu.cn.
References
- 1.Ishiguro S, Nakamura K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5’ upstream regions of genes coding for sporamin and beta-amylase from sweet potato. Mol Gen Genet. 1994;244(6):563–571. doi: 10.1007/BF00282746. [DOI] [PubMed] [Google Scholar]
- 2.Ulker B, Somssich IE. WRKY transcription factors: from DNA binding towards biological function. Curr Opin Plant Biol. 2004;7(5):491–498. doi: 10.1016/j.pbi.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Eulgem T, Rushton PJ, Robatzek S, Somssich IE. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000;5(5):199–206. doi: 10.1016/S1360-1385(00)01600-9. [DOI] [PubMed] [Google Scholar]
- 4.Yamasaki K, Kigawa T, Seki M, Shinozaki K, Yokoyama S. DNA-binding domains of plant-specific transcription factors: structure, function, and evolution. Trends Plant Sci. 2013;18(5):267–276. doi: 10.1016/j.tplants.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Eulgem T, Somssich IE. Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol. 2007;10(4):366–371. doi: 10.1016/j.pbi.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 6.Pandey SP, Somssich IE. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009;150(4):1648–1655. doi: 10.1104/pp.109.138990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson CS, Kolevski B, Smyth DR. TRANSPARENT TESTA GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant Cell. 2002;14(6):1359–1375. doi: 10.1105/tpc.001404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagacé M, Matton DP. Characterization of a WRKY transcription factor expressed in late torpedo-stage embryos of Solanum chacoense. Planta. 2004;219(1):185–189. doi: 10.1007/s00425-004-1253-2. [DOI] [PubMed] [Google Scholar]
- 9.Luo M, Dennis ES, Berger F, Peacock WJ, Chaudhury A. MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc Natl Acad Sci U S A. 2005;102(48):17531–17536. doi: 10.1073/pnas.0508418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pnueli L, Hallak-Herr E, Rozenberg M, Cohen M, Goloubinoff P, Kaplan A, et al. Molecular and biochemical mechanisms associated with dormancy and drought tolerance in the desert legume Retama raetam. Plant J. 2002;31(3):319–330. doi: 10.1046/j.1365-313X.2002.01364.x. [DOI] [PubMed] [Google Scholar]
- 11.Hinderhofer K, Zentgraf U. Identification of a transcription factor specifically expressed at the onset of leaf senescence. Planta. 2001;213(3):469–473. doi: 10.1007/s004250000512. [DOI] [PubMed] [Google Scholar]
- 12.Zhang ZL, Xie Z, Zou X, Casaretto J, Ho TH, Shen QJ. A rice WRKY gene encodes a transcriptional repressor of the gibberellin signaling pathway in aleurone cells. Plant Physiol. 2004;134(4):1500–1513. doi: 10.1104/pp.103.034967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rushton DL, Tripathi P, Rabara RC, Lin J, Ringler P, Boken AK, et al. WRKY transcription factors: key components in abscisic acid signalling. Plant Biotechnol J. 2012;10(1):2–11. doi: 10.1111/j.1467-7652.2011.00634.x. [DOI] [PubMed] [Google Scholar]
- 14.Du L, Chen Z. Identification of genes encoding receptor-like protein kinases as possible targets of pathogen- and salicylic acid-induced WRKY DNA-binding proteins in Arabidopsis. Plant J. 2000;24(6):837–847. doi: 10.1046/j.1365-313x.2000.00923.x. [DOI] [PubMed] [Google Scholar]
- 15.Ren XZ, Chen ZZ, Liu Y, Zhang HR, Zhang M, Liu QA, et al. ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant J. 2010;63(3):417–429. doi: 10.1111/j.1365-313X.2010.04248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou QY, Tian AG, Zou HF, Xie ZM, Lei G, Huang J, et al. Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol J. 2008;6(5):486–503. doi: 10.1111/j.1467-7652.2008.00336.x. [DOI] [PubMed] [Google Scholar]
- 17.Okay S, Derelli E, Unver T. Transcriptome-wide identification of bread wheat WRKY transcription factors in response to drought stress. Mol Genet Genomics. 2014;289(5):765–781. doi: 10.1007/s00438-014-0849-x. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Lai ZB, Shi JW, Xiao Y, Chen ZX, Xu XP. Roles of Arabidopsis WRKY18, WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 2010;10:281. doi: 10.1186/1471-2229-10-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J, Wang Z, Shi Z, Zhang S, Ming R, Zhu S, et al. The genome of the pear (Pyrus bretschneideri Rehd.) Genome Res. 2013;23(2):396–408. doi: 10.1101/gr.144311.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chothia C, Gough J, Vogel C, Teichmann SA. Evolution of the protein repertoire. Science. 2003;300(5626):1701–1703. doi: 10.1126/science.1085371. [DOI] [PubMed] [Google Scholar]
- 21.Ohno S, Wolf U, Atkin NB. Evolution from fish to mammals by gene duplication. Hereditas. 1968;59(1):169–187. doi: 10.1111/j.1601-5223.1968.tb02169.x. [DOI] [PubMed] [Google Scholar]
- 22.De Grassi A, Lanave C, Saccone C. Genome duplication and gene-family evolution: the case of three OXPHOS gene families. Gene. 2008;421(1–2):1–6. doi: 10.1016/j.gene.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Cai B, Yang X, Tuskan GA, Cheng Z-M. MicroSyn: a user friendly tool for detection of microsynteny in a gene family. BMC Bioinformatics. 2011;12:79. doi: 10.1186/1471-2105-12-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabaldon T, Koonin EV. Functional and evolutionary implications of gene orthology. Nat Rev Genet. 2013;14(5):360–366. doi: 10.1038/nrg3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang YJ, Liang G, Yu DQ. Activated expression of WRKY57 confers drought tolerance in Arabidopsis. Mol Plant. 2012;5(6):1375–1388. doi: 10.1093/mp/sss080. [DOI] [PubMed] [Google Scholar]
- 26.Sun YD, Yu DQ. Activated expression of AtWRKY53 negatively regulates drought tolerance by mediating stomatal movement. Plant Cell Rep. 2015;34(8):1295–1306. doi: 10.1007/s00299-015-1787-8. [DOI] [PubMed] [Google Scholar]
- 27.Vanderauwera S, Vandenbroucke K, Inze A, van de Cotte B, Muhlenbock P, De Rycke R, et al. AtWRKY15 perturbation abolishes the mitochondrial stress response that steers osmotic stress tolerance in Arabidopsis. Proc Natl Acad Sci U S A. 2012;109(49):20113–20118. doi: 10.1073/pnas.1217516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding ZJ, Yan JY, Xu XY, Yu DQ, Li GX, Zhang SQ, et al. Transcription factor WRKY46 regulates osmotic stress responses and stomatal movement independently in Arabidopsis. Plant J. 2014;79(1):13–27. doi: 10.1111/tpj.12538. [DOI] [PubMed] [Google Scholar]
- 29.Ding ZJ, Yan JY, Li CX, Li GX, Wu YR, Zheng SJ. Transcription factor WRKY46 modulates the development of Arabidopsis lateral roots in osmotic/salt stress conditions via regulation of ABA signaling and auxin homeostasis. Plant J. 2015;84(1):56–69. doi: 10.1111/tpj.12958. [DOI] [PubMed] [Google Scholar]
- 30.Babitha KC, Ramu SV, Pruthvi V, Mahesh P, Nataraja KN, Udayakumar M. Co-expression of AtbHLH17 and AtWRKY28 confers resistance to abiotic stress in Arabidopsis. Transgenic Res. 2013;22(2):327–341. doi: 10.1007/s11248-012-9645-8. [DOI] [PubMed] [Google Scholar]
- 31.Gong X, Zhang J, Hu J, Wang W, Wu H, Zhang Q, et al. FcWRKY70, a WRKY protein of Fortunella crassifolia, functions in drought tolerance and modulates putrescine synthesis by regulating arginine decarboxylase gene. Plant Cell Environ. 2015;38(11):2248–62. doi: 10.1111/pce.12539. [DOI] [PubMed] [Google Scholar]
- 32.Jiang Y, Deyholos MK. Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol Biol. 2009;69(1–2):91–105. doi: 10.1007/s11103-008-9408-3. [DOI] [PubMed] [Google Scholar]
- 33.Chen H, Lai Z, Shi J, Xiao Y, Chen Z, Xu X. Roles of Arabidopsis WRKY18, WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 2010;10:281. doi: 10.1186/1471-2229-10-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Aken O, Zhang B, Law S, Narsai R, Whelan J. AtWRKY40 and AtWRKY63 modulate the expression of stress-responsive nuclear genes encoding mitochondrial and chloroplast proteins. Plant Physiol. 2013;162(1):254–271. doi: 10.1104/pp.113.215996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ostlund G, Schmitt T, Forslund K, Kostler T, Messina DN, Roopra S, et al. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010;38(Database issue):D196–203. doi: 10.1093/nar/gkp931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wellmer F, Riechmann JL. Gene network analysis in plant development by genomic technologies. Int J Dev Biol. 2005;49(5–6):745–759. doi: 10.1387/ijdb.051991fw. [DOI] [PubMed] [Google Scholar]
- 37.Jensen AB, Busk PK, Figueras M, Alba MM, Peracchia G, Messeguer R, et al. Drought signal transduction in plants. Plant Growth Regul. 1996;20(2):105–110. doi: 10.1007/BF00024006. [DOI] [Google Scholar]
- 38.Legnaioli T, Cuevas J, Mas P. TOC1 functions as a molecular switch connecting the circadian clock with plant responses to drought. EMBO J. 2009;28(23):3745–3757. doi: 10.1038/emboj.2009.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26(19):2455–2457. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Wang L. The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol. 2005;5:1. doi: 10.1186/1471-2148-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang M, Vannozzi A, Wang G, Liang Y-H, Tornielli GB, Zenoni S, et al. Genome and transcriptome analysis of the grapevine (Vitis vinifera L.) WRKY gene family. Horticulture Research. 2014;1:14016. doi: 10.1038/hortres.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cannon SB, Mitra A, Baumgarten A, Young ND, May G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;4:10. doi: 10.1186/1471-2229-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lynch M, Conery JS. The evolutionary demography of duplicate genes. J Struct Funct Genomics. 2003;3(1–4):35–44. doi: 10.1023/A:1022696612931. [DOI] [PubMed] [Google Scholar]
- 44.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 45.Bowers JE, Chapman BA, Rong J, Paterson AH. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003;422(6930):433–438. doi: 10.1038/nature01521. [DOI] [PubMed] [Google Scholar]
- 46.Marsal J, Girona J. Effects of water stress cycles on turgor maintenance processes in pear leaves (Pyrus communis) Tree Physiol. 1997;17(5):327–333. doi: 10.1093/treephys/17.5.327. [DOI] [PubMed] [Google Scholar]
- 47.Naor A. Irrigation and crop load influence fruit size and water relations in field-grown ‘Spadona’ pear. J Am Soc Hortic Sci. 2001;126(2):252–255. [Google Scholar]
- 48.Lopez G, Larrigaudiere C, Girona J, Behboudian MH, Marsal J. Fruit thinning in ‘Conference’ pear grown under deficit irrigation: Implications for fruit quality at harvest and after cold storage. Sci Hortic-Amsterdam. 2011;129(1):64–70. doi: 10.1016/j.scienta.2011.03.007. [DOI] [Google Scholar]
- 49.Liu HY, Yang WL, Liu DC, Han YP, Zhang AM, Li SH. Ectopic expression of a grapevine transcription factor VvWRKY11 contributes to osmotic stress tolerance in Arabidopsis. Mol Biol Rep. 2011;38(1):417–427. doi: 10.1007/s11033-010-0124-0. [DOI] [PubMed] [Google Scholar]
- 50.Bartels D, Sunkar R. Drought and salt tolerance in plants. Crit Rev Plant Sci. 2005;24(1):23–58. doi: 10.1080/07352680590910410. [DOI] [Google Scholar]
- 51.Li J, Besseau S, Toronen P, Sipari N, Kollist H, Holm L, et al. Defense-related transcription factors WRKY70 and WRKY54 modulate osmotic stress tolerance by regulating stomatal aperture in Arabidopsis. New Phytol. 2013;200(2):455–472. doi: 10.1111/nph.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14(9):755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 53.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2012;40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McLnerney JO. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol. 2006;6:29. doi: 10.1186/1471-2148-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 57.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7) doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kosakovsky Pond SL, Murrell B, Fourment M, Frost SD, Delport W, Scheffler K. A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol. 2011;28(11):3033–3043. doi: 10.1093/molbev/msr125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 61.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 62.de Hoon MJL, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20(9):1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 63.Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Barrell D, Bateman A, et al. The interpro database, 2003 brings increased coverage and new features. Nucleic Acids Res. 2003;31(1):315–318. doi: 10.1093/nar/gkg046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17(9):847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 65.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ye J, Fang L, Zheng HK, Zhang Y, Chen J, Zhang ZJ, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]