

Abstract
Congenital adrenal hyperplasia (CAH) is a group of hereditary diseases, which are autosomal recessive. CAH occurs due to defect in one of the cortisol coding genes and often clinically presents itself with signs of androgen overproduction. In this article, we report a case of CAH and Schmid metaphyseal dysplasia. Our literature review indicated that this report is the first attempt on CYP11B1 and Schmid dysplasia in a child. The specific diagnosis of 11-β-hydroxylase deficiency can be determined using high basal levels of deoxycorticosterone and/or 11-deoxycortisol serums.
Keywords: Adrenal hyperplasia, Congenital, Osteochondrodysplasias, Humans, Male
What’s Known
11-β-hydroxylase deficiency is a rare disorder and must be considered in patients with precocious puberty and secondly presented with hypertension.
What’s New
This is the first report about the occurrence of Schmid-type metaphyseal chondrodysplasia and congenital adrenal hyperplasia.
Introduction
Congenital maternal hyperplasia (CAH) is a group of autosomal recessive disease in which dysfunction of one of the five cortisol coding genes occurs and causes enzymatic defects in cortisol synthesis cycle from cholesterol.1 Each enzymatic defect leads to imbalanced ratio hormone precursor and final hormone. The most common form of the disease is 21-hydroxilase deficiency, which accounts for 90-95% of the CAH cases. In the 1950’s, it was found that there were a small group of patients that developed hypertension and responded to glucocorticoid. These patients suffered from a distinct metabolic disorder, the 11-ß-hydroxilase deficiency. Clinical manifestations of 11-ß-hydroxilase deficiency include, hypertension (roughly occurs in 75% of patients and generally diagnosed during childhood) and other signs related to overproduction of mineralocorticoids such as hypokalemia or muscle weakness, salt loss, and virilization. Signs of androgen excessive secretion include early closure of the epiphysis (short stature). The hypothalamic-pituitary-gonadal axis might cause amenorrhea or spermatogenesis disorders or hirsutism and acne.2
Schmid type is an autosomal dominant skeletal dysplasia in which the commonest form is metaphyseal chondrodysplasia. Its incidence is 3 to 6 cases per one million. The clinical pattern varies widely.3 The Schmid metaphyseal chondrodysplasia is characterized by short stature, but bone maturation process is normal. However, bowed legs, coxa vara, and specific metaphyseal changes are seen on radiographs.4
In this article, we report a case of a 4-year-old boy with CAH and Schmid metaphyseal chondrodysplasia. Our literature survey confirmed that it is the first reported case of coexistence of these two rare diseases.
Case Report
A 4-year-old boy with increased amount of pubic hair was referred to the Children’s Endocrinology Clinic. He was the first child of a family with normal growth and development until one year after birth. His parents were not related and there was no positive familial history.
An increase in pubic and axillary hair developed after he reached the age of one and was concurrent with penis enlargement. He also had a history of high blood pressure. The number and quality of other body hair were also transformed. The patient was brought to the Children’s Endocrinology Clinic after a long delay. Four months later, he was treated for CAH with 5 mg hydrocortisone, three times a day.
Another problem with this child was gait disorder and bowed knee (figure 1). This condition worsened from the age of three. There was a similar history with his cousin, considering that the cousin was affected by skeletal dysplasia.
Figure 1.
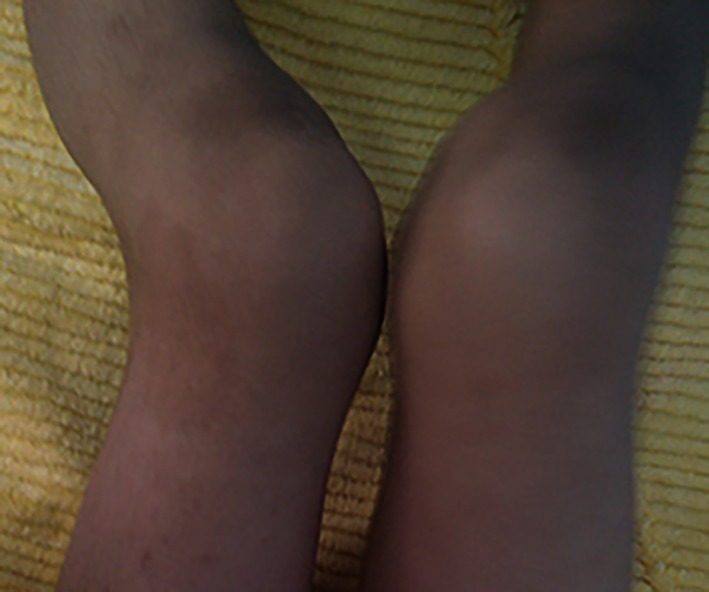
Bowed knee in the patient with congenital adrenal hyperplasia.
On clinical examination, his growth curve (height and weight) was normal. Pubic and axillary hairs were in stage 2-3 of puberty with macrophallus. There was no evidence of acne in the skin. The result of patient’s laboratory tests is shown in table 1. DOC level was 543 (ng/ml) and ACTH, DHEA, 17-OHD, testosterone and androstenedione levels were higher than the normal limits. VBG test result was PH=7.43, PCO2=42, and HCO3=28.3. Biochemistry tests such as BUN, Cr, Na, P, K, and ALKP were normal.
Table 1.
Laboratory test results
| Test | Result | Normal limits |
|---|---|---|
| ACTH | 237 | 7.20-63.30 pg/ml |
| DHEA | 243 | 3.4-124 mcg/dl |
| 17-OHD | 17 | 0.07-1.7 ng/ml |
| Androstenedione | 9.9 | 0.08-2.5 ng/ml |
| Aldosterone | 23.7 | Supine: 20-180 pg/ml Upright: 30-400 pg/ml |
| PRA | 0.1 | 0.2-1.9 pg/ml |
| Testosterone | 182 | 2.5-6.1 ng/dl |
Radiological examination (plain plantar view and knee radiography) estimated the bone age to be about 10 years and 3 months. Metaphyseal irregularity was suggestive of Schmid metaphyseal chondrodysplasia (figure 2).
Figure 2.
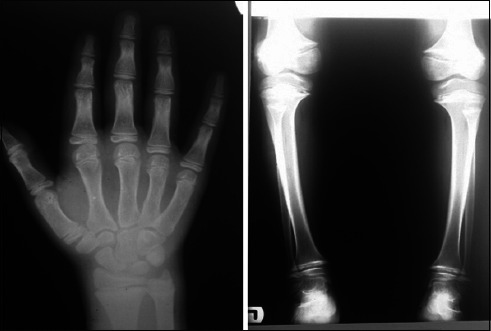
Radiographic evaluation indicated metaphyseal irregularity and bowed knee.
Finally, the patient was treated for precocious puberty due to 11-ß-hydroxilase deficiency and Schmid metaphyseal chondrodysplasia. After 5 months, testosterone level decreased to lower than 10 ng/dl and DHEA reached 27 mcg/dl.
Discussion
Schmid type metaphyseal chondrodysplasia is an autosomal dominant disorder and can be caused by various mutations in the COL10A1 gene.5 Its diagnosis is hard due to the rarity of the disease and its similarity to rickets, particularly vitamin D resistant type.6 However, rickets and Schmid can be differentiated with normal bone density and irregular dense zone in Schmid chondrodysplasia.7 In our case, Schmid metaphyseal chondrodysplasia was considered by clinical manifestation and was confirmed with radiology and laboratory tests. Bowed knee was the dominant characteristic sign and bone related biochemical tests such as CA, P and ALKP were normal. The most important point in Schmid disease is that over-treating by vitamin D, which can lead to toxicity, must be avoided. In some cases, immobilization had a dramatic effect on healing of growth plate. This condition may explain why deformities are more sever in weight bearing limbs.8
Congenital adrenal hyperplasias are a group of metabolic disorder diseases that lead to enzymatic defects in the biosynthesis of cortisol from cholesterol. CAH is an inherited disease with an autosomal recessive manner. Clinical features of 11-β-hydroxylase deficiency in childhood might be premature pubarche and accelerated bone age, all of which occurred in our patient.
The exact diagnosis of 11-β-hydroxylase deficiency can be performed by the high basal levels of deoxycorticosterone and or 11-deoxycortisol serums or tetra-hydrometabolites, which might be found during a 24-hour urine test. This disorder should be considered in patients with elevated serum levels of ACTH, about three times higher than the 95th percentile predicted for patient’s age.9
11-β-hydroxylase deficiency or CYP11B1 is the second cause of CAH. Its prevalence is 1 in 100,000 population and can be presented during childhood or adolescence.10 Its gene is located on 8q21-q22.3 Because of familial marriage in Iran, CAH incidence is high. According to a study by Qaemi et al,10 its incidence in Mashhad is 10.3%, in Shiraz 13% and in Tehran 19%. Rohani11 showed that precocious puberty could be a manifestation of non-classical form of CAH and its early diagnosis and treatment is important. In some cases, the first and mortal feature is adrenal crisis.2,11 Ambiguous genitalia, sodium, and potassium homeostasis disorders are signs of CAH. Long-term complications of adrenal hyperplasia are short stature, infertility, gender identity disorders, and death. Female neonates with ambiguous genitalia and male with precocious puberty are in the highest risk category to develop HTN because of the high secretion of deoxycorticosterone (DOC). In our patient, the first sign was puberty deviations. Clinical suspicion is very important in diagnosis of CAH. In our patient, mild HTN was present. Although CAH can be associated with other genetic disorders, but this is the first report on the association between CAH and Schmid metaphyseal chondrodysplasia.
Conclusion
11-β-hydroxylase deficiency is a rare disorder and must be considered in patients with precocious puberty and secondly presented with hypertension.
Conflict of interest: None declared.
References
- 1.Harde V, Müller M, Sippell WG, Schwarz T, Fölster-Holst R. Acne infantum as presenting symptom of congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency. J Dtsch Dermatol Ges. 2006;4:654–7. doi: 10.1111/j.1610-0387.2006.06016.x. [DOI] [PubMed] [Google Scholar]
- 2.Isiavwe AR, Ekpebegh CO, Fasanmade OA, Ohwovoriole AE. Steroid responsive hypertension secondary to 11-beta hydroxylase deficiency--a case report. West Afr J Med. 2008;27:182–5. [PubMed] [Google Scholar]
- 3.Savarirayan R, Cormier-Daire V, Lachman RS, Rimoin DL. Schmid type metaphyseal chondrodysplasia: a spondylometaphyseal dysplasia identical to the “Japanese” type. Pediatr Radiol. 2000;30:460–3. doi: 10.1007/s002470000181. [DOI] [PubMed] [Google Scholar]
- 4.Dahl M, Birkebaek N. Metaphyseal chondrodysplasia as differential diagnosis to rickets. Ugeskrift for laeger. 1996;158:1683–4. [PubMed] [Google Scholar]
- 5.Mäkitie O, Susic M, Cole WG. Early-onset metaphyseal chondrodysplasia type Schmid associated with a COL10A1 frame-shift mutation and impaired trimerization of wild-type α1 (X) protein chains. Journal of Orthopaedic Research. 2010;28:1497–501. doi: 10.1002/jor.21161. [DOI] [PubMed] [Google Scholar]
- 6.Woelfle JV, Brenner R, Zabel B, Reichel H, Nelitz M. Schmid-type metaphyseal chondrodysplasia as the result of a collagen type X defect due to a novel COL10A1 nonsense mutation. Journal of Orthopaedic Science. 2011;16:245–9. doi: 10.1007/s00776-011-0021-y. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Y, Li L, Zhou L, Mei H, Jin K, Liu K, et al. A novel mutation leading to elongation of the deduced α1 (X) chain results in Metaphyseal Chondrodysplasia type Schmid. Clinica Chimica Acta. 2011;412:1266–9. doi: 10.1016/j.cca.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 8.New MI. An update of congenital adrenal hyperplasia. Ann N Y Acad Sci. 2004;1038:14–43. doi: 10.1196/annals.1315.009. [DOI] [PubMed] [Google Scholar]
- 9.Sathya A, Ganesan R, Kumar A. Congenital adrenal hyperplasia masquerading as periodic paralysis in an adolescent girl. Singapore Medical Journal. 2012;53:E148–E9. [PubMed] [Google Scholar]
- 10.Qaemi N, Vakili R. Congenital adrenal hyperplasia. Medical Journal Of Mashhad University of Medical Sciences. 2004;47:407–12. Persian. [Google Scholar]
- 11.Rohani F. A case report on nonclassical 3B#-hydroxysteroid dehydrogenase deficiency. Iranian Journal of Endocrinology and Metabolism. 2002;4:59–62. [Google Scholar]