

Abstract
Statins, which specifically inhibit HMG Co-A reductase, the rate-limiting step of cholesterol biosynthesis, are widely prescribed to reduce serum cholesterol and cardiac risk, but many other effects are seen. We now show an effect of these drugs to induce profound changes in the step-wise synthesis of glycosphingolipids (GSLs) in the Golgi. Glucosylceramide (GlcCer) was increased several-fold in all cell lines tested, demonstrating a widespread effect. Additionally, de novo or elevated lactotriaosylceramide (Lc3Cer; GlcNAcβ1-3Galβ1-4GlcCer) synthesis was observed in 70%. Western blot showed that GlcCer synthase (GCS) was elevated by statins, and GCS and Lc3Cer synthase (Lc3S) activities were increased; however, transcript was elevated for Lc3S only. Supplementation with the isoprenoid precursor, geranylgeranyl pyrophosphate (GGPP), a downstream product of HMG Co-A reductase, reversed statin-induced glycosyltransferase and GSL elevation. The Rab geranylgeranyl transferase inhibitor 3-PEHPC, but not specific inhibitors of farnesyl transferase, or geranylgeranyl transferase I, was sufficient to replicate statin-induced GlcCer and Lc3Cer synthesis, supporting a Rab prenylation-dependent mechanism. While total cholesterol was unaffected, the trans-Golgi network (TGN) cholesterol pool was dissipated and medial Golgi GCS partially relocated by statins. GSL-dependent vesicular retrograde transport of Verotoxin and cholera toxin to the Golgi/endoplasmic reticulum were blocked after statin or 3-PEHPC treatment, suggesting aberrant, prenylation-dependent vesicular traffic as a basis of glycosyltransferase increase and GSL remodeling. These in vitro studies indicate a previously unreported link between Rab prenylation and regulation of GCS activity and GlcCer metabolism.
Keywords: glucosylceramide, Golgi, lactotriaosylceramide, Verotoxin-cholera toxin retrograde transport, UGCG
Introduction
Biosynthesis of glycosphingolipids (GSLs) is a complex process that spans the secretory pathway, beginning with endoplasmic reticulum (ER) synthesis of ceramide (Gault et al. 2010). The activity and location of the glycosyltransferases, which catalyze subsequent step-wise sugar additions is highly organized within the Golgi membranes. The pathway has several branch points; ceramide may be directed toward sphingomyelin (SM) or glucosylceramide (GlcCer) synthesis, and lactosylceramide is the precursor of globo-, lacto-, neolacto- and ganglio- and acidic GSL synthesis. These synthetic decisions are poorly understood and not based merely on enzyme availability (Uliana et al. 2006; Tuuf and Mattjus 2014).
Control of GSL and cholesterol biosynthesis are closely linked (Glaros et al. 2005; Bijl et al. 2009; Davidson et al. 2009; Ishitsuka et al. 2009; Bietrix et al. 2010), and GSLs and cholesterol are the defining components of membrane lipid rafts (Quinn 2010). The interaction between cholesterol and membrane GSLs results in a change in GSL carbohydrate from a membrane perpendicular to parallel conformation which can result in restricted trans ligand access (Mahfoud et al. 2010; Lingwood et al. 2011). The cholesterol gradient within the Golgi (Orci et al. 1981) could therefore affect precursor GSL carbohydrate conformation to regulate GSL biosynthetic pathways.
Inhibitors of HMG Co-A reductase, the rate-limiting step of cholesterol biosynthesis, are known as statins; these drugs are extremely effective to lower serum cholesterol levels and widely prescribed to counter risk of cardiac events (Correale et al. 2012; Paliani and Ricci 2012). Although specificity of clinical statins for HMG Co-A reductase is high, other lipid biosynthetic pathways utilize the enzyme product, mevalonate, to generate protein-modifying geranylgeranyl and farnesyl isoprenoids, dolichol and co-enzyme Q (Beltowski et al. 2009). Statins have other bioactivities (Liu et al. 2009). These “pleiotropic” effects of are increasingly attributed to reduction of the non-sterol bioactive lipids, with inhibition of prenylation a key contributor (Cordle et al. 2005).
In using statins to probe the possible role of cholesterol masking in GSL biosynthesis, we have identified an overlooked pleiotropic effect of statins to induce profound, isoprenylation-dependent changes in cellular GSL biosynthesis. Statin-induced GSL remodeling was reversed by addition of the isoprenoid precursor, geranylgeranyl pyrophosphate, which is depleted by statin treatment. A specific inhibitor of Rab geranylgeranyl transferase 3-PEHPC, recapitulated the statin-induced elevation of GlcCer and Lc3Cer.
Our current studies demonstrate a previously unknown link between Rab prenylation and regulation of GSL glycosyltransferase/GSL activity and trafficking in the Golgi.
Results
Effect of HMG-CoA inhibitors on steady-state GSLs levels
The efficacy of statins to modulate the GSL content of cultured cells was first compared using a pill used clinically (Crestor™; rosuvastatin), and a lipophilic statin, lovastatin. Both had the same dramatic effect on the GSL content of A431G cells (Figure 1A), most notably to increase a ceramide monohexoside (CMH) which was identified as GlcCer by co-migration with standard on borate-impregnated thin layer chromtography (TLC) plates (Kean 1966; results not shown). An increase in a ceramide trihexoside was also noted. In A431S cells (unlike A431G, this strain of A431 cells expresses Gb3), this trihexoside migrated slightly below Gb3 and a substantial elevation of lactosylceramide was also evident (Figure 1B). U18666A, a downstream inhibitor of cholesterol synthesis (Sexton et al. 1983), and intracellular cholesterol traffic (Tepper et al. 2000), had a distinct effect on the GSL profile; GlcCer was increased, but significantly less than by statin treatment, LacCer was increased, and Gb3 was reduced (Figure 1B). The statin-induced trihexoside species was identified as lactotriaosyl ceramide (Lc3Cer; GlcNAcβ1-3Galβ1-4GlcCer) by reaction with anti-Lc3Cer but not anti-Gg3 (Figure 1C), and matrix-assisted laser desorption/ionization mass spectrometry (MALDI MS) (Figure 1D). Detection of Gb3 using Verotoxin B-subunit revealed a statin-induced decrease in Gb3 in A431S cells (Figure 1C).
Fig. 1.
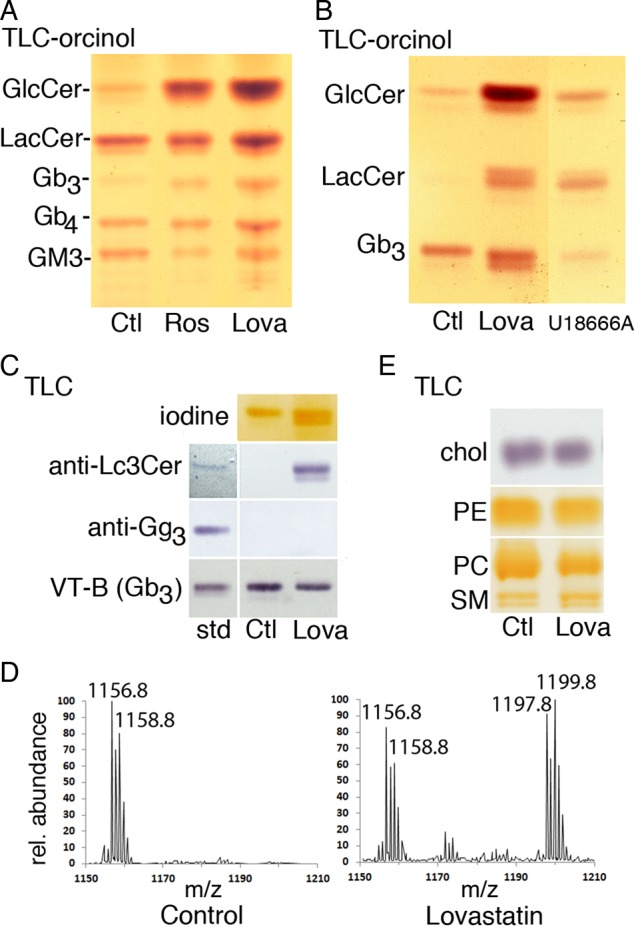
Statins increase GlcCer and Lc3Cer in A431 cells. (A) Total GSL extracts from 106 A431G cells cultured with Crestor™ pill extract (theoretical rosuvastatin concentration 20 µM) or 5 µM lovastatin for 72 h were separated by TLC in solvent C and detected by orcinol spray for carbohydrate. Migration of standards is indicated; GlcCer, lactosylceramide, Gb3 and Gb4 (globo-series GSLs, globotriaosyl or tetraosylceramide, respectively), GM3 (monosialoganglioside). Statins substantially increase GlcCer and a ceramide trihexoside. (B) Neutral GSLs from A431S cells grown ± 10 µM lovastatin or U18666A for 48 h were separated in solvent B and detected with orcinol. GSL changes induced by U18666A are distinct from statin effects. (C) The TLC trihexoside region is shown; A431S cells were treated with lovastatin or vehicle control as in (B), then TLC plates were probed with anti-Lc3Cer, anti-Gg3, or VT1 B-subunit for Gb3. Antibody–ligand binding to standard GSLs is shown left. (D) MALDI MS of neutral GSLs of A431S cells treated as in (B). The ceramide trihexoside (Gb3) C24:1/24:0 fatty acid region is shown, and sodiated peaks for C24:0 (m/z 1158.8) and C24:1 (1156.8) are labeled. On lovastatin treatment, the sodiated C24:0/C24:1 peaks of Lc3Cer are apparent (m/z 1199.8/1197.9, respectively). (E) TLC analysis of cholesterol and phospholipid fractions of A431S cells treated as in (B). Lipids were identified by co-migration with standards. Cholesterol from 2 × 105 cells was run in solvent D and detected with FeCl3 spray; PE, PC, and SM from 5 × 105 cells run in solvent B were detected by iodine vapor. This figure is available in black and white in print and in colour at Glycobiology online.
Qualitative TLC analysis showed no gross changes in SM, major phospholipids or free cholesterol (Figure 1E). Mammalian cells derive the majority of cholesterol from low density lipoprotein (LDL) uptake and our statin treatments were performed in serum-containing media. It is known that low ER cholesterol, caused by inhibition of cholesterol synthesis, triggers the sterol sensing machinery to increase LDL receptor expression and maintain the cellular cholesterol level (Cole et al. 2005).
Elevation of GlcCer by statins is a general effect
To establish the scope of statin-GSL regulation, a panel of human cell lines, representing a variety of tissue origins and GSL profiles, was treated with lovastatin and analyzed by TLC (Figure 2A). In each case, lovastatin elevated the steady-state expression of GlcCer, the simplest GSL and precursor to most complex neutral and acidic GSLs. GlcCer is the major CMH in these cell lines, but PC-3 cells showed a complex CMH pattern (Figure 2A) so that the extracts were run on borate-impregnated TLC plates to separate GlcCer and GalCer (Supplementary data, Figure S1). GlcCer was increased by statin treatment while GalCer (20% of total CMH) was not (Supplementary data, Figure S1). Many cell lines showed an increase in LacCer, perhaps reflecting increased substrate availability. Downstream GSLs showed some cell type-dependent changes but were not globally increased or decreased, suggesting that additional factors contribute to the statin-induced changes.
Fig. 2.

Lovastatin elevates GlcCer in all cell lines tested. (A, upper panel) Total saponified lipids, from cells treated 48 h ± 5–10 µM lovastatin, were separated by TLC in solvent B. GSLs are detected by orcinol spray, migration of standards is shown on left. GlcCer is increased in all cell lines tested. (A, lower panel) TLC immunoblot; replicate plate probed with anti-Lc3Cer shows increased/induced Lc3Cer in ACHN, DU145, HeLa, U87MG, HEK, and PC-3 cells. (B) Long-term, low-dose lovastatin titration in PC-3 cells shows both GlcCer and Lc3Cer (detected with orcinol and anti-Lc3Cer, respectively) are elevated at 500 nM lovastatin dose. (C) Time course treatment with 10 µM lovastatin. GSL changes are apparent at 24 h in PC-3 cells and 48 h in A431S cells. GSLs from 106 cells separated by TLC in solvent (C). (D) Identification of slow-migrating lactosamine-containing species. Total neutral GSL fraction from A431G cells treated 48 h ± 20 µM rosuvastatin was separated by TLC in solvent C and probed with anti-Lc3Cer (MAC-1; terminal GlcNAcβ1-3Gal), anti-nLc4 (FE-A5; terminal Galβ1-4GlcNAcβ1-3) or anti-SSEA1 (Lewis × determinant; Galβ1-4(Fucα1-3)GlcNAcβ1-3). (E) Identification of GM3 in A431G cells. The acidic fraction of cells from panel (D) was detected with orcinol or probed with mAb anti-GM3 (clone DH2). Three major acidic GSLs were detected including GM3, which was increased in statin-treated cells. This figure is available in black and white in print and in colour at Glycobiology online.
Since Lc3Cer was particularly upregulated in both A431 cell strains (Figure 1A–C), the cell panel was further examined by TLC-overlay immuno-staining (Figure 2A, lower panel). The prostate carcinoma lines DU145 and PC-3 had a detectable basal level of Lc3Cer, which was substantially elevated by lovastatin. Lc3Cer was detected after lovastatin treatment of ACHN, HeLa, U87-MG and HEK cells, but not Daudi, A549 or MCF-7 cells (Figure 2A, lower panel). In general, induction/increase of Lc3Cer was observed in concert with LacCer increase. A prolonged (7 days) low-dose titration study showed 500 nM lovastatin was sufficient to increase both GlcCer and Lc3Cer in PC-3 cells (Figure 2B), suggesting a cumulative effect. At high dose, the treatment time required to elevate GlcCer and Lc3Cer varied by cell type, being evident at 24 h in PC-3 cells or 48 h in A431S (Figure 2C). We noticed on immunoblots that a polar, anti-Lc3Cer reactive GSL was concurrently expressed in statin-treated A431 cells. We suspected that this species may be nLc5, which is also a product of Lc3Cer synthase, has the same terminal GlcNAcβ(1–3) Gal as Lc3Cer and is recognized by anti-Lc3Cer MAC-1 (Togayachi et al. 2001; Nozaki et al. 2010). To confirm this, and identify additional Lc3Cer-related slow-migrating complex GSLs, a total neutral GSL fraction was prepared from A431G cells and probed by TLC immunoblot with anti-Lc3Cer (MAC-1), anti-nLc4 (FE-A5) or anti-SSEA1 (MC-480) (Figure 2D). Several species induced by rosuvastatin were observed. A 4-sugar neutral GSL abundantly expressed in A431 cells was identified as nLc4 by reaction with the type II lactosamine-specific mAb FE-A5 (Fenderson et al. 1983) (Figure 2D). Despite the large increase in precursor Lc3Cer, nLc4 was only slightly elevated by statin treatment. Additional FE-A5 reactive, complex neutral GSLs were observed which varied in their responsiveness to statin treatment. The increased band immediately below nLc4 has migration consistent with Galβ(1-4)GlcNAc-terminating nLc6, but these species were not further characterized (Figure 2B). Likewise, multiple anti-SSEA1-reactive complex GSLs were observed, some substantially increased by statin treatment (Figure 2B). The acidic GSL fraction of the same cells was also isolated and analyzed by TLC (Figure 2E). GM3, identified by reaction with mAb DH2 (Dohi et al. 1988), is the major ganglioside of A431G cells and was increased approximately 2-fold by statin treatment (Figure 2E).
GlcCer and Lc3Cer synthases are increased by statin treatment
GlcCer is synthesized in vivo by the action of a single enzyme, GlcCer synthase (GCS, GlcT-1) (Marks et al. 2001) while Lc3Cer synthase (Lc3CerS, B3GnT5) catalyzes addition of GlcNAc to LacCer (and nLc4) (Togayachi et al. 2001), the rate-limiting step in all lacto and neo-lacto GSL synthesis (Nakamura et al. 1992). Expression of GSL synthases was assessed using qualitative RT-PCR (Supplementary data, Figure S2). We found the transcript for Lc3CerS was increased in lovastatin-treated PC-3 cells while transcripts for GCS, the two LacCer synthases, and the GlcCer transport protein, Fapp2 were not (Supplementary data, Figure S2). These results suggest that statins affect the turnover of select GSL glycosyltransferases. ACHN GlcCer was increased by three different statins (Figure 3A), and we detected a corresponding increase in GCS total protein by western blot (Figure 3A) and enzyme activity in cell lysates (Figure 3A). Increases in GCS protein and activity were also seen in A431S and PC-3 cells (not shown), suggesting that this may be a common mechanism for GlcCer elevation. Increased Lc3S activity was also detected in treated cells, at acute or low, prolonged statin doses (Figure 3B). Western blot analysis of Lc3Cer expression was hampered by the inadequate specificity of commercial antibodies. Exogenous cholesterol was available during statin treatment so we explored the possibility that inhibition of isoprenylation, rather than cholesterol synthesis, leads to increased GlcCer/Lc3Cer. Supplementation with GGPP (10 µM GGPP), the isoprenoid precursor depleted by HMG Co-A reductase inhibition, countered the statin-induced increase in GCS enzyme activity (Figure 3C) and GCS protein in ACHN cell lysates (Figure 3D), and reversed the lovastatin-induced GlcCer and Lc3Cer elevation in A431S cells (Figure 3E).
Fig. 3.

Statin-induced increase in GlcCer results from prenylation-dependent GCS elevation. (A) ACHN cells were cultured with 10 µM lovastatin, 20 µM rosuvastatin or 10 µM atorovastatin for 48 h. GlcCer was detected by TLC/orcinol (upper panel), GCS by western blot (middle panel; 20 µg protein lysate) and GCS enzyme activity was monitored by conversion of NBD-Cer to NBD-GlcCer (bottom panel; 3 h reaction, GlcCer:Cer band intensity ratios are shown). (B) Lc3Cer synthase activity, monitored by conversion of NBD-LacCer to NBD-Lc3Cer, increased in lysates of PC-3 cells treated with low dose (0.5 or 1.0 µM) lovastatin for 4 days, or acute treatment (10 µM, 48 h). (C) ACHN cells were treated with 20 µM rosuvastatin ± 10 µM GGPP for 48 h and analyzed for GCS enzyme activity. Bar graph shows average GlcCer:Cer ratio±range of duplicate samples. (D) Western blot of ACHN cell lysates, treatment as in (C). (E) TLC analysis of GSLs from A431S cells treated with vehicle control or 10 µM lovastatin ± 10 µM GGPP for 48 h (orcinol detection). This figure is available in black and white in print and in colour at Glycobiology online.
Examination of sphingolipid metabolites by quantitative mass spectroscopy
GCS can deplete ceramide induced by cytotoxic drugs and “ceramide glycosylation” has been proposed as a mechanism of drug resistance in cancer cells (Liu et al. 2008). Transcriptional up-regulation of GCS in response to elevated ceramide has been shown (Uchida et al. 2004). Since inhibition of prenylation by statins is a known inducer of apoptosis (Perez-Sala and Mollinedo 1994), a potential effect on ceramide levels was explored. The pathways linking Cer, GlcCer and SM are summarized in Figure 4A.
Fig. 4.

Effect of statin treatment on sphingolipid metabolite levels. (A) Summary of sphingolipid metabolism pathways. CerS, ceramide synthases 1–5, which vary in fatty acid chain length preference; DES, dhCer desaturase; SMS or SMase, sphingomyelin synthase or sphingomyelinase, respectively; Cerase, ceramidase; GCC; glucocerobrosidase (cytosolic and lysosomal forms). Extracts of ACHN cells treated with 20 µM rosuvastatin for 0, 24 or 48 h were analyzed by quantitative LC–MS-MS for; (B) precursor Sa and dhCer species and (C) total Cer, GlcCer and SM. In (D) the individual fatty acid species for Cer, GlcCer and SM are shown. Quantitative measurement of some species was not available at the time of analysis; all species analyzed are shown. Bar graphs represent average ng/106 cells ± range of duplicate samples divided by two. This figure is available in black and white in print and in colour at Glycobiology online.
Quantitative sphingolipid LC–MS-MS analysis of sphingolipids from control, or 20 µM rosuvastatin-treated ACHN cells was performed. Levels of the Cer precursors, sphinganine (Sa) and dihydroceramides (dhCer) are a measure of de novo sphingolipid synthesis (Figure 4A), while sphingosine (So), formed by cleavage of Cer, is a salvage product. Sa, was reduced at 24 h, and increased 1.6-fold after 48 h of statin treatment. However, elevation of dhCer at 48 h was only seen for a single species, the anti-apoptotic C16:0 dhCer (Siddique et al. 2012) (Figure 4B). Total Cer and SM were unchanged, while GlcCer increased 2-fold after 24 h and 6-fold after 48 h (Figure 4B). The increase was similar for the GlcCer fatty acid isoforms analyzed (Figure 4D). GalCer was identified but represented <1% of the total CMH fraction (not shown). Ceramide is a branch point in sphingolipid formation (Figure 4A). Once formed in the ER, Cer is transported to the Golgi by CERT for SM synthesis (Perry and Ridgway 2005) and possibly by vesicular trafficking for GlcCer synthesis (Giussani et al. 2008). Levels of SM species remained constant, despite the abundant GlcCer (Figure 4C and D). There was a 40% decrease in So (Figure 4D), which may indicate the accumulated GlcCer, which appears largely unavailable for downstream GSL synthesis, is also less available for degradation. A 30% increase in C16:0 Cer was matched by a 35% decrease in C24:0 Cer (Figure 4D). Thus, the statin-induced increases in GCS and GlcCer in ACHN cells do not appear to be driven by elevated ceramide (Figure 4C and D).
Rab prenylation inhibition recapitulates statin effects on GSL metabolism
GGPP supplied during statin treatment reversed the rise in GlcCer, GCS protein and activity, and Lc3Cer (Figure 3C–E) so the effect of specific prenylation inhibitors, acting downstream of statins, was monitored. Prenylation is carried out by geranylgeranyl transferase I and II (GGTase I and II, respectively), which utilize GGPP, or farnesyl transferase (FTase), which utilizes farnesyl pyrophosphate (FPP), the precursor of GGPP. Statin treatment depletes the geranyl-PP precursor to both (Figure 5A). We analyzed the neutral GSLs of treated ACHN cells by TLC and found that only the GGTase II inhibitor, 3-PEHPC (Coxon et al. 2005), increased GlcCer (and LacCer), similar to statin treatment (Figure 5B, upper “TLC” panel), whereas GGTase I or FTase inhibition, with GGTI-2133 or FTI-277, respectively, did not. The same was found for elevation of GCS, analyzed by western blot of treated cell lysates (Figure 5B, lower “WB” panel). The activity and specificity of each inhibitor was verified by monitoring the prenylation status of known substrates of each prenyl transferase by western blot (Kinsella and Maltese 1991); Rab6 for GGTase II (3-PEHPC), Rap1A for GGTase I (GGTI-2133) and HDJ2 for FTase (FTI-277) (Davis et al. 1998) (Figure 5B, lower “WB” panel). We noted that unprenylated HDJ2 was detected in FTI-277, but not rosuvastatin, treated cells. Interestingly, GCS detected by western blot was increased to a similar degree by rosuvastatin or 3-PEHPC treatment, however the glycosphingolipid product, GlcCer, was elevated to a lesser degree by 3-PEHPC (Figure 5B). Triple inhibition of GGTase I, II and FTase did not further increase GlcCer in these cells (Figure 5B, upper “TLC” panel), suggesting that reduced prenylation of GGTase I and FTase substrates do not contribute to the statin-induced GlcCer increase. A similar experiment was conducted in A431S cells in which Lc3Cer is readily monitored by TLC. Lovastatin-induced a dose-dependent increase in GlcCer, LacCer and Lc3Cer that was replicated by 3 mM 3-PEHPC (Figure 5C). The activity of Lc3CerS in cell lysates was similarly elevated (Figure 5D). GGTase II, the target of 3-PEHPC, is also known as Rab geranylgeranyl transferase. The enzyme catalyzes transfer of (in most cases) two prenyl groups to the C-terminus of Rab proteins, which facilitates their membrane association and activity. Thus, these results suggest the statin effects on GSL expression and synthase activity result from reduced Rab protein prenylation.
Fig. 5.

Rab prenylation inhibitor replicates statin effects on GSLs. (A) Scheme for effect of statins or specific inhibitors of farnesyl transferase, geranylgeranyl transferase −1 or geranylgeranyl transferase II on protein prenylation. (B) Effects of specific prenylation inhibitors on GSL and GCS expression were determined. The Rab geranylgeranyl transferase inhibitor, 3-PEHPC, was effective to increase GlcCer (TLC, upper panel) and GCS (western blot, lower panel). ACHN cell treatment was 48 h with 3 mM 3-PEHPC, 15 µM FT1-277, 10 µM GGTI-2133 or 20 µM rosuvastatin. Inhibition of prenylation by 3-PEHPC and FTI-277 are indicated by appearance of the slower migrating, unprenylated forms of Rab6 (25 kD) and the co-chaperone HDJ2 (40 kD), respectively. Inhibition of GGTase I is confirmed using an antibody specific for unprenylated Rap1A (21 kD). Blots were probed with anti-GAPDH to verify equal sample loading and transfer. (C) Neutral GSLs of A431S cells treated with the inhibitor panel as in (A). A dose–response between 0 and 10 µm lovastatin was seen for GlcCer and Lc3Cer elevation and 1 mM 3-PEHPC caused similar increases within this range. (D) Lc3CerS enzyme activity in lysates of A431S cells treated as in (B). Band intensity (arbitrary units) of NBD-Lc3Cer was quantified using Image J; error bars represent the range of duplicate samples divided by two. This figure is available in black and white in print and in colour at Glycobiology online.
Statins alter intracellular cholesterol and GCS distribution
Rab GTPases regulate intracellular vesicular trafficking events spanning the endocytic and exocytic pathways, including intra-Golgi cysternal transport (Zerial and McBride 2001; Cottam and Ungar 2012; Pfeffer 2012). To establish whether perturbed vesicular trafficking alters distribution of GCS, the subcellular location of endogenous GCS was first characterized. ACHN cells were fixed, permeablized with filipin and labeled with anti-GCS 1.2 antiserum specific for the exposed, cytosolic C-terminal region (Marks et al. 1999). Permeablization with filipin allowed us to visualize the distribution of cholesterol that was found concentrated in the plasma membrane and trans-Golgi network (TGN), as reported (Mukherjee et al. 1998). GCS was detected mainly in punctate perinuclear structures that were identified as Golgi (Figure 6A). Anti-GCS stained vesicular assemblies associated with the GM130-labeled cis-Golgi but appeared more closely coincident with the Golgi SNARE GS15, reported to be found mainly in the medial Golgi (Xu et al. 2002) (Figure 6A). GCS labeling was mainly excluded from the filipin-stained TGN (Figure 6A, inset). Filipin effectively permeablized the plasma membrane to allow detection of cytosol-exposed epitopes but did not permeablize the TGN to allow wheat germ agglutinin (WGA) staining (Figure 6A). Following rosuvastatin or 3-PEHPC treatment, GCS became more widely distributed (Figure 6B) and TGN cholesterol was lost. GM130 (Figure 6B) or Giantin (not shown) labeling revealed more dispersed structures reminiscent of Golgi mini-stacks. WGA labeling of detergent-permeablized cells (Tartakoff and Vassalli 1983) showed that 80% of WGA TGN was retained after 48 h of statin treatment, suggesting that reduced TGN cholesterol was not due to dispersal of this organelle (Supplementary data, Figure S3). Cells treated with the GCS inhibitor P4 did not show appreciable changes in Golgi GCS (Figure 6B). After Brefeldin A treatment both GCS and GM130-labeled structures were dispersed to small, largely non-coincident vesicles (Figure 6B). These results suggest that GCS is mainly in the medial Golgi cisternae associated with the high cholesterol containing TGN. Statin or Rab inhibitor treatment reduced TGN cholesterol and GCS and Golgi markers become more disperse (Figure 6B).
Fig. 6.

Golgi GCS is relocalized after statin or 3-PEHPC treatment. (A) After permeablization with 50 µg/mL filipin cells were double-labeled with rabbit anti-GCS 1.2 antiserum and mAb GM130 (cis-Golgi) or mAb GS15 (medial Golgi). GCS was localized in punctate Golgi structures partially overlapping with GS15, and proximal but largely distinct from GM130 and filipin-labeled TGN (pseudo-colored red in the bottom panel for clarity). Scale bar = 6 µm. (B) ACHN cells were treated 48 h with vehicle, 10 µM rosuvastatin, 1 mM 3-PEHPC, 2 µM P4 or 30 min with 10 µM BFA, prior to fixation and labeling. After statin or 3-PEHPC treatment, GCS became more widely distributed, the TGN cholesterol pool (but not PM cholesterol) was absent, anti-GM130-labeled dispersed Golgi “mini-stacks”. Scale bar = 10 µm.
In addition, 3-PEHPC mimicked rosuvastatin to disperse the elevated TGN cholesterol pool and prevent the Golgi/ER targeting of internalized Verotoxin or Cholera toxin B-subunits (Figure 7A). In control cells, VT-B and CT-B were rapidly internalized to the Golgi. Though some double-stained cells were evident, most cells bound either VT-B or CT-B, consistent with previously reported cell-cycle-dependent receptor Gb3 and GM1 expression (Majoul et al. 2002). In double-labeled ACHN cells, VT-B and CT-B are co-localized. In statin or 3-PEHPC-treated cells however, the distinct TGN cholesterol pool is diminished and VT-B/CT-B are detected more diffusely throughout the cell (Figure 7A). These changes were not observed in cells treated with FTI-277 or GGTI-2133. Multiple Rab GTPases have been implicated in this GSL-mediated transport route (White et al. 1999; Sandvig et al. 2004; Fuchs et al. 2007).
Fig. 7.

Rab prenylation inhibitor mimics statins to alter intracellular traffic. (A) ACHN cells were pre-treated 48 h with vehicle, rosuvastatin, 1 or 3 mM 3-PEHPC, 15 µM FTI-277 or 10 µM GGT1-2133, then Alexa488-VT1 B subunit and Cy3-CT B subunit were internalized together at 37°C for 1 h. TGN cholesterol and the Golgi targeting of VT-B and CT-B are apparent in control, GGTI-2133 and FTI-277 treated cells. In statin or 3-PEHPC-treated cells, intracellular cholesterol is dispersed and internalized VT-B and CT-B became more diffuse within the cell and retention at the plasma membrane was increased. Scale bar = 21 µm. (B) Solid lines/square symbols, VT1; dashed lines/triangles, VT2; solid symbols, 1 pg/mL VT; open symbols, 100 pg/mL VT.
Statins doses which impair prenylation block Verotoxin cytotoxicity
Since GSL-mediated VT-B trafficking to the Golgi was disrupted, the cytotoxicities of VT1 and VT2 holotoxins were compared after 24 h statin pre-treatment. These E. coli-derived toxins are the primary cause of hemolytic uremic syndrome (Page and Liles 2013), and while closely related, have subtle differences in their internalization/retrograde transport pathways (Tam et al. 2008). Golgi membrane association of endogenous Rab6 (an indicator of prenylation status), but not WGA TGN staining, was substantially reduced in statin-treated cells (Supplementary data, Figure S3). Rosuvastatin, at 5 µM, was able to confer significant and similar cell protection against both toxins (Figure 7B), but 20 µM treatment was more effective against VT1, suggesting that VT2 may have access to an additional non-Golgi/ER-dependent pathway, as previously suggested (Saito et al. 2012).
Discussion
Glycosphingolipids, particularly within lipid microdomains (Furukawa et al. 2012), play important roles in cell signaling. Studies using GCS inhibitors have shown that abberent GSL synthesis plays a key role in many human diseases and their animal models (Bietrix et al. 2010; Karman et al. 2010; Yew et al. 2010). Several specific GSLs serve as disease cofactors (Tagami et al. 2002; Vieira et al. 2008; Kawashima et al. 2009; Mazzulli et al. 2011). Understanding the regulation of precursor and complex GSL synthesis is therefore of wide potential therapeutic utility.
In this study, we looked at the effect of statins on GSL synthesis in cultured cells. Membrane cholesterol can alter GSLs carbohydrate conformation to restrict ligand binding (Mahfoud et al. 2010; Yahi et al. 2010; Lingwood et al. 2011; Novak et al. 2013), and we speculated that this interaction might also influence the binding of substrate GSLs with glycosyltransferases or binding proteins to affect cellular GSL metabolism. We observed that lovastatin caused a marked elevation in GlcCer, the simplest GSL, in all cell lines tested. Another unusually elevated GSL (70% of cell lines tested) was identified as Lc3Cer. In A431 cells selected for more detailed GSL study, we demonstrated that statins increased GM3 ganglioside and detected increased species corresponding to nLc5, nLc6 and several SSEA-1-related complex GSL species. In the cell lines assayed, the activities of both GCS and Lc3Cer synthase were found to be increased by statins, and in the case of GCS, for which an antibody was available, increased protein was detected by western blot.
Unexpectedly, these GSL and glycosyltransferase changes were independent of cholesterol synthesis inhibition. Instead, the GSL effects were shown to result from reduced isoprenoid biosynthesis from mevalonate, the product of HMG-CoA reductase; supplementation with the isoprenoid precursor GGPP, which acts downstream of cholesterol biosynthesis in the mevalonate pathway, reversed the statin-induced elevation of GlcCer and Lc3Cer.
To identify the target of GGPP rescue, and rule out a possible non-prenylation-related function of GGPP, cells were treated with specific inhibitors of the three protein prenyltransferases. GGPP is a substrate for GGTase I and GGTase II (Rab geranylgeranyltransferase), while FTase farnesylates proteins use FPP, the precursor of GGPP. (Lobell et al. 2001). GGTase I and FTase are more closely related and show some substrate and inhibitor cross-reactivity, particularly at high doses of the latter, while Rab GGTase is mechanistically unique. Using a standard western blotting technique, we found the inhibitors were specific for their targets at the doses used. Only Rab GGTase inhibition with 3-PEHPC (Coxon et al. 2005) reproduced the effect of statins to increase GSLs, and elevate synthase activity. Taken together, the results showing reversal of GSL and glycosyltransferase increase by GGPP, and replication of statin effects by 3-PEHPC alone, strongly suggest the effects of statins on GSL metabolism result from inhibition of Rab GTPase prenylation.
Rab GTPases function as molecular switches controlling the direction of endocytic and exocytic vesicular budding, trafficking and fusion (Hutagalung and Novick 2011). Studies using C-terminal cysteine prenylation site mutants (Gomes et al. 2003) or drugs blocking prenylation (Coxon et al. 2005) have shown that unprenylated Rab proteins do not associate with membranes and are inactive. Since several of the 60+ Rab GTPases identified in the human genome (Stenmark and Olkkonen 2001) function in the Golgi, the site of GSL glycosyltransferase activity, and can be inactivated by statins (Kinsella and Maltese 1992), we investigated whether statins or Rab GGTase inhibition affected the subcellular location of GCS within the TGN/Golgi.
GCS is unusual among Golgi-resident glycosyltransferases in having a C-terminal cytosol-facing active site (Futerman and Pagano 1991; Jeckel et al. 1992), and (predicted) multiple membrane interacting C-terminal domains (Marks et al. 2001). This unique topology underlies the complex fate of the GCS product, GlcCer, which may be membrane translocated to the Golgi lumen for downstream GSL synthesis, or moved by protein or vesicular-mediated traffic to other cellular locations for translocation (Halter et al. 2007; D'Angelo et al. 2013). By immunofluorescence, we detected endogenous GCS in medial-cis-Golgi structures adjacent to the cholesterol-enriched filipin-labeled TGN (Mukherjee et al. 1998). This is consistent with reports of GCS detected biochemically, and by immuno-EM in both early and distal Golgi fractions (Jeckel et al. 1992; Halter et al. 2007).
After 48 h statin or 3-PEHPC treatment, GCS was more widely dispersed in the cell and GM130, a Rab-interacting Golgi matrix protein and regulator of glycosylation (Nakamura 2010), was redistributed to a lesser extent. By EM, statins were shown to swell Golgi cisternae, but their stacking was preserved (Ivessa et al. 1997). Several Rab GTPases are now known to be important regulators of Golgi organization and integrity, functioning in the continual anterograde and retrograde membrane recycling of this organelle (White et al. 1999; Liu and Storrie 2012). Our immunofluorescence results suggest that the Golgi disruption caused by statin or 3-PEPHC is due to inhibition of Rab function. Specific inhibition of several Golgi Rabs (e.g. Rab1a, Rab1, 2, 8) leads to Golgi fragmentation (Wilson et al. 1994; Rendon et al. 2013). Defective ER/Golgi or Golgi/lysosome vesicular transport could compromise the turnover of GCS/Lc3S enzymes (or interacting partners) to increase their expression and activity. Post-translational increase in GCS activity via direct interaction with the ER reticulon, RTN1C, involved in ER/Golgi vesicle traffic, has been described (Di Sano et al. 2003).
The TGN cholesterol pool was dispersed by (statins and) Rab GGTase inhibition. Cholesterol accumulation itself can reduce Rab GTPase dissociation from target membranes (Takahashi and Kobayashi 2009) to change membrane traffic. Whether cholesterol TGN accumulation is a consequence of Rab-dependent traffic, lost following prenylation blockade or whether cholesterol redistribution allows a previously inhibited Rab GTPase membrane dissociation, provides the basis for the statin-dependent change in GSL synthesis observed, requires future definition of the Rab GTPase(s) involved.
Intracellular immunohistochemistry of endogenous GCS has not been previously reported. In our study, endogenous GCS was readily detected in ACHN Golgi membranes following plasma membrane permeabilization with filipin, or saponin (not shown), both cholesterol binding agents. We found detergent (Triton X-100) permeabilization was incompatible with specific GCS immunolocalization using a C-terminus-specific antibody. From the observed statin-induced redistribution of intracellular cholesterol and GCS, it is tempting to speculate that the GCS C-terminal domain could interact with cholesterol (Fantini and Barrantes 2013) to affect Golgi retention.
The accumulation of GlcCer after statin treatment was common to all cell lines investigated, and to our knowledge, has not been reported previously. The glycosyltransferases GCS and Lc3CerS are both rate-limiting enzymes (Radin 1994; Merrill 2011), so the accumulation of their products suggests that normal downstream anabolism is blocked by Rab inhibition. In the case of GlcCer, previous studies have shown that a major pool of synthesized GlcCer is largely unavailable for conversion to downstream GSLs (Warnock et al. 1994; Maxzud and Maccioni 2000). Furthermore, GCS inhibitors, such as P4 (Lee et al. 1999), lead to a rapid reduction in GlcCer (our unpublished results), which suggests that normally GlcCer is rapidly metabolized in the cell. GCS protein (and activity) is increased in statin or 3-PEHPC-treated cells, which may account for much of the GlcCer elevation seen. However, the GCS is also redistributed from a partially disrupted Golgi. The striking accumulation of GlcCer could therefore arise if the GlcCer membrane translocase or cytosolic glucocerebrosidase (Boot et al. 2007; Dekker et al. 2011) were deficient in these novel GCS locations, or prenylation-dependent vesicular traffic may be required to shuttle GlcCer to sites of further anabolism. Complex GSL synthesis was shown to depend on protein-mediated transfer of GlcCer by Fapp2, rather than vesicular traffic (D'Angelo et al. 2007). Our finding that, in general, GSLs downstream of GlcCer were not reduced by statins or 3-PEHPC is consistent with such a model. However, GM3 ganglioside synthesis, apparently independent of Fapp2 (D'Angelo et al. 2013), was also increased. GSL elevation itself could also contribute to decreased degradation; GlcCer has been implicated in the endolysosomal traffic of proteins (Sillence et al. 2002) and increased GlcCer can raise the lysosomal pH (Sillence 2013).
Disregulation of GCS by statins gives new insight into the physiological control of this enzyme. The 3-PEHPC Rab geranylgeranyl transferase inhibition studies show that one or more Rab prenylation-dependent processes regulate intracellular GSL trafficking, trafficking/turnover of GCS, and its product, GlcCer. Statin/3-PEHPC induced redistribution of GM130-labeled Golgi structures implies changes in cysternal fusion, consistent with the redistribution of GCS, and Golgi organization as a regulator of glycosylation (Nakamura 2010). WGA TGN staining was largely statin-resistant, but the punctate Rab6 Golgi staining was reduced after 24 h and lost after 48 h statin treatment, consistent with the redistribution of GCS from the medial Golgi.
Lc3Cer is the precursor for all lacto and neo-lacto GSLs and is typically rapidly converted to these more complex GSLs. Transcriptional regulation of Lc3Cer synthase is evident during embryonic development in mice (Henion et al. 2001) and during myeloid differentiation in humans (Wang et al. 2012). Interestingly, a human mutation in Sec23b, involved in COPII mediated ER-Golgi vesicular traffic, is the cause of congenital diserythropoietic anemia type II (CDA II). The disease affects glycosylation of erythrocyte proteins and lipids and a phenotype feature is 10-fold elevated Lc3Cer (Bouhours et al. 1985; Bianchi et al. 2009).
Since statins are prescribed to millions worldwide, it is encumbant to consider the potential clinical relevance of these findings. Epidemiologically, statin treatment may have a beneficial effect on cancer prognosis (Nielsen et al. 2012). We speculate that this could, in part, be due to lowering of cholesterol levels and exposure of cholesterol-masked tumor-associated GSL antigens for immune recognition (Novak et al. 2013). Additionally, Lc3Cer and Lc3S are markedly increased in colon cancer (Holmes et al. 1987) and acute myeloid leukemia (Wang et al. 2012). By increasing Lc3Cer synthesis, statins may promote an antineoplastic immune response; Lc3Cer is the precursor to SSEA1, a tumor stem cell marker (Son et al. 2009) (SSEA-1 related GSLs are increased by statins) and the Lewis blood group antigens, also associated with cancer (Le Pendu et al. 2001). By inhibiting prenylation, statins directly induce acute myeloid leukemia apoptosis in vitro (Wong et al. 2001), which may have therapeutic value in vivo (Minden et al. 2001). Investigations of the antineoplastic potential of high dose lovastatin in vivo have achieved safe serum values in the range of 0.3–12 µM (Thibault et al. 1996; Holstein et al. 2006), within the dose range used in the current work. Serum statin concentrations within this range, or specific Rab GTPase inhibitors (Agola et al. 2011), could prove protective against the pathology of Verotoxin or cholera toxin which require Rab-dependent GSL-mediated retrograde traffic to the Golgi/ER to effect toxicity.
Statins increase the risk of type 2 diabetes (Carter et al. 2013) and GSLs, particularly ganglioside GM3 (Tagami et al. 2002), play a role in this disease. GlcCer is the precursor of GM3, which increases when GlcCer accumulates in Gaucher disease (Ghauharali-van der Vlugt et al. 2008) to increase insulin resistance (Langeveld et al. 2008). The statin-increased GM3 we found, if manifest in vivo, could contribute to the increased diabetic risk.
Low serum GlcCer levels are a potential risk factor for venous thrombosis (Deguchi et al. 2001). GlcCer can directly bind to activated protein C and enhance its anticoagulant activity (Yegneswaran et al. 2003). There is mixed evidence that statins reduce the incidence of venous thromboembolic events (Rahimi et al. 2012), but studies have convincingly shown reduced platelet activity in statin-treated subjects (Miller et al. 2013). It is intriguing to consider that statins could elevate serum GlcCer levels and reduce risk of venous thrombosis in a subset of at-risk individuals.
We found the statin-induced GlcCer elevation is isoprenoid, rather than cholesterol dependent. Cholesterol distribution may be prenylation/Rab dependent since cholesterol can inhibit Rab dissociation (Takahashi and Kobayashi 2009). In the current study, most experiments were performed at 10–20 µM statin, where global inhibition of Rab prenylation is expected (Ostrowski et al. 2007). Future work will aim to establish the Golgi target(s) of prenylation blockade that increase/relocate GCS and Lc3S activity and increase their products.
Materials and methods
Materials
Lovastatin (lactone form), rosuvastatin and atorovastatin were from Toronto Research Chemicals. In some experiments, lovastatin was activated by base hydrolysis of the lactone ring as described (Morimoto et al. 2006). U18666A, GGPP, FTI-277, GGTI-2133, cholesterol, UDP-sugars (galactose, glucose and N-acetylglucosamine) were from Sigma-Aldrich. 3-PEHPC (formerly referred to as NE10790) was from Aroz Technologies. Lyso-LacCer and soy GlcCer were purchased from Avanti Polar Lipids. Lyso-GlcCer was prepared in our laboratory. d-sphingosine was from Matreya. Fluorescent C6-NBD derivatives were prepared in our laboratory by reaction of lyso-SLs with succinimidyl 6-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)hexanoate (NBD-X-SE, NHS ester; Anaspec). NBD derivatives were purified by silica gel chromatography and stored under N2 in CHCl3–MeOH, 2:1, at −20°C. Machery Nagel SilG plastic-backed TLC plates were from Caledon Labs. GSL standards from the following sources were purified in our lab by silica gel chromatography; Lc3Cer from bovine erythrocytes, Gg3 from guinea pig erythrocytes, and Gb3 and Gb4 from human kidney.
Antibodies and ligands
Mouse anti-Lc3Cer (clone MAC-1) was a generous gift of Dr Yasunori Kushi, Nihon University, Tokyo (Nozaki et al. 2010). Mouse anti-GM3, clone DH2 was generously provided by Sen-itiroh Halkomori (Pacific Northwest Research Laboratory, Seattle, WA) (Dohi et al. 1988). Mouse anti-Gg3 hybridoma clone 2D4 was from ATCC. Mouse mAb FE-A5 (Galβ1-4GlcNAc; (Fenderson et al. 1983) and MC-480 (SSEA1; Solter and Knowles 1978) were from the Developmental Studies Hybridoma Bank at the University of Iowa. Rabbit antisera 1.2 (immunofluorescence dilution, 1/600) and 6.2 (western blot dilution, 1/1000) against peptide fragments of human GCS have been previously characterized (Marks et al. 1999). Rabbit anti-Rab6 (C-19) and goat antisera specific for unprenylated Rap1A (C-17) were from Santa Cruz, rabbit anti-HDJ2/Hsp40 was from Enzo, mouse anti-GM130 (clone 35/GM130) and mouse anti-GS15 (clone 19/GS15) were from BD Biosciences and anti-GAPDH (clone 6C5) was from Abcam. Purified Verotoxin-1 B-subunit (VT1-B) VT1, VT2 and anti-VT1B were prepared in our lab (Boyd et al. 1991; Nutikka et al. 2003). Cholera toxin B-subunit was from List Labs. Cy3 and Alexa488 labeling kits were from Amersham and Molecular Probes, respectively. Alexa 594 WGA, Alexa488 and Alexa 594 goat anti-rabbit and mouse IgG were from Molecular Probes. horseraddish peroxidase (HRP) goat anti-mouse and rabbit IgG were from Bio-Rad, HRP donkey anti-goat IgG was from Santa Cruz and HRP goat anti-mouse IgM from Jackson labs.
Cell culture and drug treatments
The following human cell lines and culture conditions were used in this study; ACHN (epithelial carcinoma from renal tubular adenocarcinoma pleural metastasis) grown in Ehrlich's minimal essential medium, A431 (epidermoid squamous skin carcinoma), MCF-7 (hormone-responsive breast adenocarcinoma from pleural metastasis), DU145 (androgen-independent prostate carcinoma from brain metastasis) and HeLa (cervical epithelial carcinoma) cells in DMEM, PC-3 (prostate small cell adenocarcinoma from bone metastasis), Daudi (B-cell Burkitt's lymphoma) and Jurkat E6 (T-cell lymphoblastoid leukemia) cells in RPMI, A549 (alveolar basal epithelial adenocarcinoma) cells in F12K, U-87MG (epithelial grade IV glioblastoma-astrocytoma) cells in αMEM. Two strains of A431 cells, we designated G (ATCC) or S (kindly provided by Dr Kai Simons, Max Planck Institute, Dresden, Germany), were used. Unlike A431G, A431S cells have high basal Gb3 synthase expression. All drug and control treatments were performed in media containing 10% fetal bovine serum unless otherwise indicated. For each experiment, 3-PEHPC solid was freshly dissolved in culture medium and adjusted to neutral pH by addition of 2 N NaOH. Other drugs were diluted from stock solutions in dimethyl sulfoxide (DMSO). Statin stock solutions (10–20 mM in DMSO) were stored at −20°C for up to 1 month. A 10 mg Crestor™ tablet was dissolved overnight in phosphate buffered saline (PBS), vortexed and centrifuged. The resulting rosuvastatin concentration in the extract was assumed to be 0.5 mM. Matching vehicle control treatments were performed as appropriate.
Lipid extraction and TLC analysis
Cellular lipids were extracted as described (Kamani et al. 2011). Briefly, lipids were extracted with CHCl3–MeOH 2:1 and where indicated, phospholipids degraded by saponification prior to desalting with C18 cartridges (Waters). Lipid classes were isolated from non-saponified extracts using silica gel chromatography. For separation of neutral and acidic GSL, non-saponified total extracts were passed through DEAE sephadex. The neutral GSL “flow-though” was saponified and desalted using C18 cartridges. Bound gangliosides were eluted with 0.2 M sodium acetate and desalted. For TLC analysis of GSLs, extracts were separated on silica TLC plates in solvent system A (CHCl3:MeOH; 98:2), dried and re-chromatographed in solvent system B (CHCl3:MeOH:H2O; 65:25:4) or C (CHCl3:MeOH:H2O; 60:35:8) as indicated. For cholesterol and ceramide analysis, lipids were chromatographed in solvent system D (CHCl3:MeOH:H2O; 190:9:1). Cholesterol was detected with ferric chloride (Lowry 1968), Cer and SM by iodine vapor and GSLs by orcinol spray. GSLs were detected by specific antibodies or ligands after TLC separation as described (Kamani et al. 2011). Binding was detected using either the colourimetric HRP substrate 4-chloro-1-naphthol or enhanced chemiluminescent (ECL) substrate and imaging as described below for western blotting.
Western blotting
Proteins in post-nuclear cell lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (10–12% gels), then transferred to nitrocellulose. Blots were probed with primary antibodies, HRP species-specific secondary antibodies then developed with SuperSignal West Fempto ECL reagent (Thermo Scientific) and bands recorded using a LiCor Odyssey Fc imager (Mandel Scientific). Prestained molecular weight protein markers (Fermentas) were imaged on the 700 nm channel.
Glycosyltransferase enzyme assays
Detergent-free post-nuclear cell homogenates prepared in 10 mM Tris–HCl, pH 7.2, 10 mM NaCl were used as enzyme source for GCS. Detergent lysates in 2% Triton X-100, 25 mM 4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid (HEPES), pH 7.0 were used for Lc3S. Protein content was determined by the BCA protein assay (Thermo Scientific, Rockford, IL). All protein preparations contained an ethylenediaminetetraacetic acid (EDTA)-free, protease inhibitor cocktail containing 1 mM 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride, 0.5 µM aprotinin, 15 µM bestatin, 10 µM E64, 0.1 mM leupeptin and 1 µM pepstatin A (BioShop Canada Inc.). GCS activity was measured as described (Ichikawa et al. 1996). Briefly, 100 µL reactions contained 25 mM Tris–HCl, pH 7, 1 mM EDTA, 50 µg cell protein lysate, 500 µM UDP-Glc, 1 mM conduritol epoxide to inhibit glucocerobrosidase activity and 10 µL of liposomes containing 0.5 µg NBD-Cer and 5 µg PC. Reactions were stopped after 1–4 h by the addition of 600 µL CHCl3:CH3OH (2:1, v/v) and 20 µL water, vortexed vigorously, then centrifuged. The lower phase was dried under N2 and analyzed by TLC in solvent system B. Lc3Cer synthase (B3GnT5) reactions used NBD-LacCer dissolved in 2% Triton X-100 as substrate and contained 100 µg protein, 5 mM MnCl2, 50 mM HEPES, pH 6.8, 500 µM UDP-GlcNAc. Incubation was 16 h at 37°C. Bands were separated in TLC solvent system A. Fluorescent bands were visualized using a Typhoon FLA 9500 image analyzer (GE Healthcare Bioscience), and intensities were quantified using Image J software.
Fluorescence microscopy
Cells grown on glass coverslips were treated as indicated then fixed with 3% paraformaldehyde/PBS. The standard permeablization protocol used 0.2% Triton X-100, 1% bovine serum albumen/PBS for 12 min at ambient temperature. This method was ineffective for GCS detection with either antiserum 6.2 or 1.2 however, and an alternate method in which fixed cells were permeabilized with the fluorescent sterol-binding polyene macrolide filipin as described (Neufeld et al. 1999), was employed. Filipin appeared to poorly permeabilize organelles, as little labeling of intra-organelle targets (WGA and protein disulfide isomerase) was seen after filipin permeablization, however cytosol-exposed epitopes were readily detected (GCS 1.2, Rab6, GM130, GS15, Giantin). Another cholesterol binding agent, saponin (0.05%), was also effective to selectively permeablize the plasma membrane for GCS detection. For immunofluorescence the following reagents were used; anti-GCS 1.2 serum, or control rabbit serum (1/600), rabbit anti-Giantin (1/250; cis/medial Golgi) (Linstedt and Hauri 1993), mouse anti-GM130 (1/200; cis-Golgi; Nakamura et al. 1995), rabbit anti-Rab6 (1/200, Golgi marker), mouse anti-GS15 (1/100), Alexa594-WGA 2 µg/mL (Tartakoff and Vassalli 1983) (5 µg/mL; Invitrogen) (PM/TGN marker), antibodies were detected with Alexa488 or Alexa594-conjugated goat anti-rabbit or mouse antibodies. Images were obtained using an Olympus IX81 inverted fluorescence microscope (60× oil immersion, NA 1.35) equipped with a Hamamatsu C9100-13 back-thinned EM-CCD camera and Yokogawa CSU X1 spinning disk confocal scan head. Image acquisition, deconvolution and cropping were done using Volocity software. Composite images were assembled using Adobe Photoshop.
Quantitative MS analysis
A GlcCer internal standard (IS; C17:0/18:2 sphingosine) was synthesized from soy lyso-GlcCer as described (Kamani et al. 2011). Cells were treated with statin or vehicle control in 10 cm dishes and harvested by trypsinization at the indicated times. Fifty nanograms of IS were added to a portion of cell homogenate prior to lipid extraction. To separate GlcCer and GalCer, LC–MS-MS was performed using a Phenomenex Kinetex HILIC, 2.6 µm, 100 A, 50 × 4.6 mm column with mobile phase: A = water; B = 2.5/97.5 water/80:20 acetonitrile:tetrahydrofuran, both A and B containing 2.5 mM ammonium formate, pH 3.2. MS–MS detector was a Sciex 5500 with ESI ionization in positive mode. MS–MS acquisition used the following ions for Glc/GalCer (M + H; secondary diagnostic ion for endogenous Glc/GalCer is from the 18:1 long chain base): (IS) 17:0/18:2 = 712.6–>262.4, 12:0 = 644.7–>264.3, 16:0 = 700.6–>264.3, 18:0 = 728.6–>264.3, 18:1 = 726.6–>264.3, 20:0 = 756.7–>264.3, 22:0 = 784.7–>264.3, 24:0 = 812.7–>264.3, 24:1 = 810.7–>264.3.
Verotoxin cytoxicity assay
ACHN cells grown in 96 well plates were pre-incubated for 24 h with 0–20 µM rosuvastatin prior to addition of Verotoxin 1 or 2 (1 pg/mL or 100 pg/mL). After 72 h of VT treatment, live adherent cells were fixed, stained with Crystal Violet and the absorbance read at 570 nm. Percent cell survival was calculated using the absorbance of wells treated with VT only (0% protection) or rosuvastatin only (100% protection). Rosuvastatin treatment was maintained for the duration of the experiment (96 h).
Supplementary data
Supplementary data for this article are available online at http://glycob.oxfordjournals.org/.
Conflict of interest statement
None declared.
Funding
This work was supported by a Canadian Institutes of Health Research team grant in Lysosomal Storage Disease (C.A.L.) and by an NSERC award on GSL metabolism (C.A.L).
Abbreviations
CMH, ceramide monohexoside; dhCer, dihydroceramides; DMSO, dimethyl sulfoxide; EDTA, ethylenediaminetetraacetic acid; ER, endoplasmic reticulum; FTase, farnesyl transferase; GCS, GlcCer synthase; GGPP, geranylgeranyl pyrophosphate; GGPP, geranylgeranyl pyrophosphate; GGTase I, geranylgeranyl transferase I; GGTase II, geranylgeranyl transferase II; GlcCer, glucosylceramide; GSLs, glycosphingolipids; HEPES, 4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid; HRP, horseraddish peroxidase; Lc3, lactotriaosyl ceramide; Lc3S, Lc3Cer synthase; LDL, low density lipoprotein; MALDI MS, matrix-assisted laser desorption/ionization mass spectrometry; PBS, phosphate buffered saline; Sa, sphinganine; SM, sphingomyelin; So, sphingosine; TGN, trans-Golgi network; TLC, thin layer chromtography; WGA, wheat germ agglutinin.
Supplementary Material
Acknowledgements
We thank Michael Leadley and Denis Reynaud for LC–MS-MS analysis at AFBM of the Centre for the Study of Complex Childhood Diseases at the Hospital for Sick Children, Toronto. CSCCD is supported by the Canadian Foundation for Innovation.
References
- Agola JO, Jim PA, Ward HH, Basuray S, Wandinger-Ness A. 2011. Rab GTPases as regulators of endocytosis, targets of disease and therapeutic opportunities. Clin Genet. 80(4):305–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltowski J, Wojcicka G, Jamroz-Wisniewska A. 2009. Adverse effects of statins—Mechanisms and consequences. Current Drug Safety. 4(3):209–228. [DOI] [PubMed] [Google Scholar]
- Bianchi P, Fermo E, Vercellati C, Boschetti C, Barcellini W, Iurlo A, Marcello AP, Righetti PG, Zanella A. 2009. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Human Mutation. 30(9):1292–1298. [DOI] [PubMed] [Google Scholar]
- Bietrix F, Lombardo E, van Roomen CP, Ottenhoff R, Vos M, Rensen PC, Verhoeven AJ, Aerts JM, Groen AK. 2010. Inhibition of glycosphingolipid synthesis induces a profound reduction of plasma cholesterol and inhibits atherosclerosis development in APOE*3 Leiden and low-density lipoprotein receptor–/– mice. Arterioscler Thromb Vasc Biol. 30(5):931–937. [DOI] [PubMed] [Google Scholar]
- Bijl N, van Roomen CP, Triantis V, Sokolovic M, Ottenhoff R, Scheij S, van Eijk M, Boot RG, Aerts JM, Groen AK. 2009. Reduction of glycosphingolipid biosynthesis stimulates biliary lipid secretion in mice. Hepatology. 49(2):637–645. [DOI] [PubMed] [Google Scholar]
- Boot RG, Verhoek M, Donker-Koopman W, Strijland A, van Marle J, Overkleeft HS, Wennekes T, Aerts JM. 2007. Identification of the non-lysosomal glucosylceramidase as beta-glucosidase 2. J Biol Chem. 282(2):1305–1312. [DOI] [PubMed] [Google Scholar]
- Bouhours JF, Bouhours D, Delaunay J. 1985. Abnormal fatty acid composition of erythrocyte glycosphingolipids in congenital dyserythropoietic anemia type II. J Lipid Res. 26(4):435–441. [PubMed] [Google Scholar]
- Boyd B, Richardson S, Gariepy J. 1991. Serological responses to the B subunit of Shiga-like toxin 1 and its peptide fragments indicate that the B subunit is a vaccine candidate to counter the action of the toxin. Infect Immun. 59(3):750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AA, Gomes T, Camacho X, Juurlink DN, Shah BR, Mamdani MM. 2013. Risk of incident diabetes among patients treated with statins: Population based study. BMJ. 346:f2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SL, Grudzien A, Manhart IO, Kelly BL, Oakley H, Vassar R. 2005. Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanism. J Biol Chem. 280(19):18755–18770. [DOI] [PubMed] [Google Scholar]
- Cordle A, Koenigsknecht-Talboo J, Wilkinson B, Limpert A, Landreth G. 2005. Mechanisms of statin-mediated inhibition of small G-protein function. J Biol Chem. 280(40):34202–34209. [DOI] [PubMed] [Google Scholar]
- Correale M, Abruzzese S, Greco CA, Concilio M, Di Biase M, Brunetti ND. 2012. Statins in heart failure. Curr Vasc Pharmacol. [PubMed] [Google Scholar]
- Cottam NP, Ungar D. 2012. Retrograde vesicle transport in the Golgi. Protoplasma. 249(4):943–955. [DOI] [PubMed] [Google Scholar]
- Coxon FP, Ebetino FH, Mules EH, Seabra MC, McKenna CE, Rogers MJ. 2005. Phosphonocarboxylate inhibitors of Rab geranylgeranyl transferase disrupt the prenylation and membrane localization of Rab proteins in osteoclasts in vitro and in vivo. Bone. 37(3):349–358. [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Polishchuk E, Di Tullio G, Santoro M, Di Campli A, Godi A, West G, Bielawski J, Chuang CC, van der Spoel AC et al. 2007. Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature. 449(7158):62–67. [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Uemura T, Chuang CC, Polishchuk E, Santoro M, Ohvo-Rekila H, Sato T, Di Tullio G, Varriale A, D'Auria S et al. 2013. Vesicular and non-vesicular transport feed distinct glycosylation pathways in the Golgi. Nature. 501(7465):116–120. [DOI] [PubMed] [Google Scholar]
- Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, Ory DS, Vanier MT, Walkley SU. 2009. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One. 4(9):e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AR, Alevy YG, Chellaiah A, Quinn MT, Mohanakumar T. 1998. Characterization of HDJ-2, a human 40 kD heat shock protein. Int J Biochem Cell Biol. 30(11):1203–1221. [DOI] [PubMed] [Google Scholar]
- Deguchi H, Fernandez JA, Pabinger I, Heit JA, Griffin JH. 2001. Plasma glucosylceramide deficiency as potential risk factor for venous thrombosis and modulator of anticoagulant protein C pathway. Blood. 97(7):1907–1914. [DOI] [PubMed] [Google Scholar]
- Dekker N, Voorn-Brouwer T, Verhoek M, Wennekes T, Narayan RS, Speijer D, Hollak CE, Overkleeft HS, Boot RG, Aerts JM. 2011. The cytosolic beta-glucosidase GBA3 does not influence type 1 Gaucher disease manifestation. Blood Cells Mol Dis. 46(1):19–26. [DOI] [PubMed] [Google Scholar]
- Di Sano F, Fazi B, Citro G, Lovat PE, Cesareni G, Piacentini M. 2003. Glucosylceramide synthase and its functional interaction with RTN-1C regulate chemotherapeutic-induced apoptosis in neuroepithelioma cells. Cancer Res. 63(14):3860–3865. [PubMed] [Google Scholar]
- Dohi T, Nores G, Hakomori S. 1988. An IgG3 monoclonal antibody established after immunization with GM3 lactone: Immunochemical specificity and inhibition of melanoma cell growth in vitro and in vivo. Cancer Res. 48(20):5680–5685. [PubMed] [Google Scholar]
- Fantini J, Barrantes FJ. 2013. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front Physiol. 4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenderson BA, Hahnel AC, Eddy EM. 1983. Immunohistochemical localization of two monoclonal antibody-defined carbohydrate antigens during early murine embryogenesis. Dev Biol. 100(2):318–327. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Haas AK, Spooner RA, Yoshimura S, Lord JM, Barr FA. 2007. Specific Rab GTPase-activating proteins define the Shiga toxin and epidermal growth factor uptake pathways. J Cell Biol. 177(6):1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Ohkawa Y, Yamauchi Y, Hamamura K, Ohmi Y. 2012. Fine tuning of cell signals by glycosylation. J Biochem. 151(6):573–578. [DOI] [PubMed] [Google Scholar]
- Futerman AH, Pagano RE. 1991. Determination of the intracellular sites and topology of glucosylceramide synthesis in rat liver. Biochem J. 280(Pt 2):295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault CR, Obeid LM, Hannun YA. 2010. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv Exp Med Biol. 688:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghauharali-van der Vlugt K, Langeveld M, Poppema A, Kuiper S, Hollak CE, Aerts JM, Groener JE. 2008. Prominent increase in plasma ganglioside GM3 is associated with clinical manifestations of type I Gaucher disease. Clin Chim Acta. 389(1–2):109–113. [DOI] [PubMed] [Google Scholar]
- Giussani P, Colleoni T, Brioschi L, Bassi R, Hanada K, Tettamanti G, Riboni L, Viani P. 2008. Ceramide traffic in C6 glioma cells: Evidence for CERT-dependent and independent transport from ER to the Golgi apparatus. Biochim Biophys Acta. 1781(1–2):40–51. [DOI] [PubMed] [Google Scholar]
- Glaros EN, Kim WS, Quinn CM, Wong J, Gelissen I, Jessup W, Garner B. 2005. Glycosphingolipid accumulation inhibits cholesterol efflux via the ABCA1/apolipoprotein A-I pathway: 1-Phenyl-2-decanoylamino-3-morpholino-1-propanol is a novel cholesterol efflux accelerator. J Biol Chem. 280(26):24515–24523. [DOI] [PubMed] [Google Scholar]
- Gomes AQ, Ali BR, Ramalho JS, Godfrey RF, Barral DC, Hume AN, Seabra MC. 2003. Membrane targeting of Rab GTPases is influenced by the prenylation motif. Mol Biol Cell. 14(5):1882–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halter D, Neumann S, van Dijk SM, Wolthoorn J, de Maziere AM, Vieira OV, Mattjus P, Klumperman J, van Meer G, Sprong H. 2007. Pre- and post-Golgi translocation of glucosylceramide in glycosphingolipid synthesis. J Cell Biol. 179(1):101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henion TR, Zhou D, Wolfer DP, Jungalwala FB, Hennet T. 2001. Cloning of a mouse beta 1,3-N-acetylglucosaminyltransferase GlcNAc(beta 1,3)Gal(beta 1,4)Glc-ceramide synthase gene encoding the key regulator of lacto-series glycolipid biosynthesis. J Biol Chem. 276(32):30261–30269. [DOI] [PubMed] [Google Scholar]
- Holmes EH, Hakomori S, Ostrander GK. 1987. Synthesis of type 1 and 2 lacto series glycolipid antigens in human colonic adenocarcinoma and derived cell lines is due to activation of a normally unexpressed beta 1–3N-acetylglucosaminyltransferase. J Biol Chem. 262(32):15649–15658. [PubMed] [Google Scholar]
- Holstein SA, Knapp HR, Clamon GH, Murry DJ, Hohl RJ. 2006. Pharmacodynamic effects of high dose lovastatin in subjects with advanced malignancies. Cancer Chemother Pharmacol. 57(2):155–164. [DOI] [PubMed] [Google Scholar]
- Hutagalung AH, Novick PJ. 2011. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 91(1):119–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S, Sakiyama H, Suzuki G, Hidari KI, Hirabayashi Y. 1996. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc Natl Acad Sci USA. 93(10):4638–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishitsuka R, Hirabayashi Y, Kobayashi T. 2009. Glycosphingolipid deficiency increases the sterol regulatory element-mediated gene transcription. Biochem Biophys Res Commun. 378(2):240–243. [DOI] [PubMed] [Google Scholar]
- Ivessa NE, Gravotta D, De Lemos-Chiarandini C, Kreibich G. 1997. Functional protein prenylation is required for the brefeldin A-dependent retrograde transport from the Golgi apparatus to the endoplasmic reticulum. J Biol Chem. 272(33):20828–20834. [DOI] [PubMed] [Google Scholar]
- Jeckel D, Karrenbauer A, Burger KN, van Meer G, Wieland F. 1992. Glucosylceramide is synthesized at the cytosolic surface of various Golgi subfractions. J Cell Biol. 117(2):259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamani M, Mylvaganum M, Tian R, Binnington B, Lingwood C. 2011. Adamantyl glycosphingolipids provide a new approach to the selective regulation of cellular glycosphingolipid metabolism. J Biol Chem. 286:21413–21426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karman J, Tedstone JL, Gumlaw NK, Zhu Y, Yew N, Siegel C, Guo S, Siwkowski A, Ruzek M, Jiang C et al. 2010. Reducing glycosphingolipid biosynthesis in airway cells partially ameliorates disease manifestations in a mouse model of asthma. Int Immunol. 22(7):593–603. [DOI] [PubMed] [Google Scholar]
- Kawashima N, Yoon SJ, Itoh K, Nakayama K. 2009. Tyrosine kinase activity of epidermal growth factor receptor is regulated by GM3 binding through carbohydrate to carbohydrate interactions. J Biol Chem. 284(10):6147–6155. [DOI] [PubMed] [Google Scholar]
- Kean EL. 1966. Separation of gluco- and galactocerebrosides by means of borate thin-layer chromatography. J Lipid Res. 7(3):449–452. [PubMed] [Google Scholar]
- Kinsella BT, Maltese WA. 1991. rab GTP-binding proteins implicated in vesicular transport are isoprenylated in vitro at cysteines within a novel carboxyl-terminal motif. J Biol Chem. 266(13):8540–8544. [PubMed] [Google Scholar]
- Kinsella BT, Maltese WA. 1992. rab GTP-binding proteins with three different carboxyl-terminal cysteine motifs are modified in vivo by 20-carbon isoprenoids. J Biol Chem. 267(6):3940–3945. [PubMed] [Google Scholar]
- Langeveld M, Ghauharali KJ, Sauerwein HP, Ackermans MT, Groener JE, Hollak CE, Aerts JM, Serlie MJ. 2008. Type I Gaucher disease, a glycosphingolipid storage disorder, is associated with insulin resistance. J Clin Endocrinol Metab. 93(3):845–851. [DOI] [PubMed] [Google Scholar]
- Le Pendu J, Marionneau S, Cailleau-Thomas A, Rocher J, Le Moullac-Vaidye B, Clement M. 2001. ABH and Lewis histo-blood group antigens in cancer. APMIS. 109(1):9–31. [DOI] [PubMed] [Google Scholar]
- Lee L, Abe A, Shayman JA. 1999. Improved inhibitors of glucosylceramide synthase. J Biol Chem. 274(21):14662–14669. [DOI] [PubMed] [Google Scholar]
- Lingwood D, Binnington B, Róg T, Vattulainen I, Grzybek M, Coskun U, Lingwood C, Simons K. 2011. Cholesterol modulates glycolipid conformation and receptor activity. Nat Chem Biol. 7:260–262. [DOI] [PubMed] [Google Scholar]
- Linstedt AD, Hauri HP. 1993. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol Biol Cell. 4(7):679–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PY, Liu YW, Lin LJ, Chen JH, Liao JK. 2009. Evidence for statin pleiotropy in humans: Differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation. 119(1):131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Storrie B. 2012. Are Rab proteins the link between Golgi organization and membrane trafficking? Cell Mol Life Sci. 69(24):4093–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YY, Yu JY, Yin D, Patwardhan GA, Gupta V, Hirabayashi Y, Holleran WM, Giuliano AE, Jazwinski SM, Gouaze-Andersson V et al. 2008. A role for ceramide in driving cancer cell resistance to doxorubicin. FASEB J. 22(7):2541–2551. [DOI] [PubMed] [Google Scholar]
- Lobell RB, Omer CA, Abrams MT, Bhimnathwala HG, Brucker MJ, Buser CA, Davide JP, deSolms SJ, Dinsmore CJ, Ellis-Hutchings MS et al. 2001. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 61(24):8758–8768. [PubMed] [Google Scholar]
- Lowry RR. 1968. Ferric chloride spray detector for cholesterol and cholesteryl esters on thin-layer chromatograms. J Lipid Res. 9(3):397. [PubMed] [Google Scholar]
- Mahfoud R, Manis A, Binnington B, Ackerley C, Lingwood CA. 2010. A major fraction of glycosphingolipids in model and cellular cholesterol containing membranes are undetectable by their binding proteins. J Biol Chem. 285(46):36049–36059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoul I, Schmidt T, Pomasanova M, Boutkevich E, Kozlov Y, Soling HD. 2002. Differential expression of receptors for Shiga and Cholera toxin is regulated by the cell cycle. J Cell Sci. 115(Pt 4):817–826. [DOI] [PubMed] [Google Scholar]
- Marks DL, Dominguez M, Wu K, Pagano RE. 2001. Identification of active site residues in glucosylceramide synthase. A nucleotide-binding catalytic motif conserved with processive beta-glycosyltransferases. J Biol Chem. 276(28):26492–26498. [DOI] [PubMed] [Google Scholar]
- Marks DL, Wu K, Paul P, Kamisaka Y, Watanabe R, Pagano RE. 1999. Oligomerization and topology of the Golgi membrane protein glucosylceramide synthase. J Biol Chem. 274(1):451–456. [DOI] [PubMed] [Google Scholar]
- Maxzud MK, Maccioni HJ. 2000. Glucosylceramide synthesized in vitro from endogenous ceramide is uncoupled from synthesis of lactosylceramide in Golgi membranes from chicken embryo neural retina cells. Neurochem Res. 25(1):145–152. [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. 2011. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 146(1):37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill AH., Jr 2011. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. 111(10):6387–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, DiNicolantonio JJ, Can M, Grice R, Damoulakis A, Serebruany VL. 2013. The effects of ezetimibe/simvastatin versus simvastatin monotherapy on platelet and inflammatory biomarkers in patients with metabolic syndrome. Cardiology. 125(2):74–77. [DOI] [PubMed] [Google Scholar]
- Minden MD, Dimitroulakos J, Nohynek D, Penn LZ. 2001. Lovastatin induced control of blast cell growth in an elderly patient with acute myeloblastic leukemia. Leuk Lymphoma. 40(5–6):659–662. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Janssen WJ, Fessler MB, McPhillips KA, Borges VM, Bowler RP, Xiao YQ, Kench JA, Henson PM, Vandivier RW. 2006. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J Immunol. 176(12):7657–7665. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Zha X, Tabas I, Maxfield FR. 1998. Cholesterol distribution in living cells: Fluorescence imaging using dehydroergosterol as a fluorescent cholesterol analog. Biophys J. 75(4):1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N. 2010. Emerging new roles of GM130, a cis-Golgi matrix protein, in higher order cell functions. J Pharmacol Sci. 112(3):255–264. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis T, G. W. 1995. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 131:1715–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Tsunoda A, Sakoe K, Gu J, Nishikawa A, Taniguchi N, Saito M. 1992. Total metabolic flow of glycosphingolipid biosynthesis is regulated by UDP-GlcNAc:lactosylceramide beta 1–>3N-acetylglucosaminyltransferase and CMP-NeuAc:lactosylceramide alpha 2–>3 sialyltransferase in human hematopoietic cell line HL-60 during differentiation. J Biol Chem. 267(33):23507–23514. [PubMed] [Google Scholar]
- Neufeld EB, Wastney M, Patel S, Suresh S, Cooney AM, Dwyer NK, Roff CF, Ohno K, Morris JA, Carstea ED et al. 1999. The Niemann-Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J Biol Chem. 274(14):9627–9635. [DOI] [PubMed] [Google Scholar]
- Nielsen SF, Nordestgaard BG, Bojesen SE. 2012. Statin use and reduced cancer-related mortality. N Engl J Med. 367(19):1792–1802. [DOI] [PubMed] [Google Scholar]
- Novak A, Binnington B, Ngan B, Chadwick K, Fleshner N, Lingwood CA. 2013. Cholesterol masking membrane glycosphingolipid tumor-associated antigens reduces their immunodetection in human cancer biopsies. Glycobiology. 23(11):1230–1239. [DOI] [PubMed] [Google Scholar]
- Nozaki H, Yanagida M, Koide K, Shiotani K, Kinoshita M, Kobayashi Y, Watarai S, Nakamura K, Suzuki A, Ariga T et al. 2010. Production and characterization of monoclonal antibodies specific to lactotriaosylceramide. Glycobiology. 20(12):1631–1642. [DOI] [PubMed] [Google Scholar]
- Nutikka A, Binnington-Boyd B, Lingwood CA. 2003. Methods for the purification of Shiga toxin 1. In: Philpot D, Ebel F, editors. Methods Mol Med. 73 E. coli Shiga Toxin Methods and Protocols. Totowa, NY, Humana Press; p. 187–195. [DOI] [PubMed] [Google Scholar]
- Orci L, Montesano R, Meda P, Malaisse-Lagae F, Brown D, Perrelet A, Vassalli P. 1981. Heterogeneous distribution of filipin–cholesterol complexes across the cisternae of the Golgi apparatus. Proc Natl Acad Sci USA. 78(1):293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski SM, Wilkinson BL, Golde TE, Landreth G. 2007. Statins reduce amyloid-beta production through inhibition of protein isoprenylation. J Biol Chem. 282(37):26832–26844. [DOI] [PubMed] [Google Scholar]
- Page AV, Liles WC. 2013. Enterohemorrhagic Escherichia coli infections and the hemolytic-uremic syndrome. Med Clin N Am. 97(4):681–695, xi. [DOI] [PubMed] [Google Scholar]
- Paliani U, Ricci S. 2012. The role of statins in stroke. Intern Emerg Med. 7(4):305–311. [DOI] [PubMed] [Google Scholar]
- Perez-Sala D, Mollinedo F. 1994. Inhibition of isoprenoid biosynthesis induces apoptosis in human promyelocytic HL-60 cells. Biochem Biophys Res Commun. 199(3):1209–1215. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Ridgway ND. 2005. Molecular mechanisms and regulation of ceramide transport. Biochim Biophys Acta. 1734(3):220–234. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR. 2012. Rab GTPase localization and Rab cascades in Golgi transport. Biochem Soc Trans. 40(6):1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn PJ. 2010. A lipid matrix model of membrane raft structure. Prog Lipid Res. 49(4):390–406. [DOI] [PubMed] [Google Scholar]
- Radin NS. 1994. Rationales for cancer chemotherapy with PDMP, a specific inhibitor of glucosylceramide synthase. Mol Chem Neuropathol. 21(2–3):111–127. [DOI] [PubMed] [Google Scholar]
- Rahimi K, Bhala N, Kamphuisen P, Emberson J, Biere-Rafi S, Krane V, Robertson M, Wikstrand J, McMurray J. 2012. Effect of statins on venous thromboembolic events: A meta-analysis of published and unpublished evidence from randomised controlled trials. PLoS Med. 9(9):e1001310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendon WO, Martinez-Alonso E, Tomas M, Martinez-Martinez N, Martinez-Menarguez JA. 2013. Golgi fragmentation is Rab and SNARE dependent in cellular models of Parkinson's disease. Histochem Cell Biol. 139(5):671–684. [DOI] [PubMed] [Google Scholar]
- Saito M, Mylvaganum M, Tam P, Binnington B, Lingwood C. 2012. Structure-dependent pseudo-receptor intracellular traffic of adamantyl globotriaosyl ceramide mimics. J Biol Chem. 287(20):16073–16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig K, Spilsberg B, Lauvrak SU, Torgersen ML, Iversen TG, van Deurs B. 2004. Pathways followed by protein toxins into cells. Int J Med Microbiol. 293(7–8):483–490. [DOI] [PubMed] [Google Scholar]
- Sexton RC, Panini SR, Azran F, Rudney H. 1983. Effects of 3 beta-[2-(diethylamino)ethoxy]androst-5-en-17-one on the synthesis of cholesterol and ubiquinone in rat intestinal epithelial cell cultures. Biochemistry. 22(25):5687–5692. [DOI] [PubMed] [Google Scholar]
- Siddique MM, Bikman BT, Wang L, Ying L, Reinhardt E, Shui G, Wenk MR, Summers SA. 2012. Ablation of dihydroceramide desaturase confers resistance to etoposide-induced apoptosis in vitro. PLoS One. 7(9):e44042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillence DJ. 2013. Glucosylceramide modulates endolysosomal pH in Gaucher disease. Mol Genet Metab. 109:194–200. [DOI] [PubMed] [Google Scholar]
- Sillence DJ, Puri V, Marks DL, Butters TD, Dwek RA, Pagano RE, Platt FM. 2002. Glucosylceramide modulates membrane traffic along the endocytic pathway. J Lipid Res. 43(11):1837–1845. [DOI] [PubMed] [Google Scholar]
- Solter D, Knowles BB. 1978. Monoclonal antibody defining a stage-specific mouse embryonic antigen (SSEA-1). Proc Natl Acad Sci USA. 75(11):5565–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son MJ, Woolard K, Nam DH, Lee J, Fine HA. 2009. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 4(5):440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Olkkonen VM. 2001. The Rab GTPase family. Genome Biology. 2(5):Reviews 3007.1- reviews3007.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami S, Inokuchi Ji J, Kabayama K, Yoshimura H, Kitamura F, Uemura S, Ogawa C, Ishii A, Saito M, Ohtsuka Y et al. 2002. Ganglioside GM3 participates in the pathological conditions of insulin resistance. J Biol Chem. 277(5):3085–3092. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Kobayashi T. 2009. Cholesterol regulation of rab-mediated sphingolipid endocytosis. Glycoconj J. 26(6):705–710. [DOI] [PubMed] [Google Scholar]
- Tam P, Mahfoud R, Nutikka A, Khine A, Binnington B, Paroutis P, Lingwood C. 2008. Differential intracellular trafficking and binding of verotoxin 1 and verotoxin 2 to globotriaosylceramide-containing lipid assemblies. J Cell Physiol. 216:750–763. [DOI] [PubMed] [Google Scholar]
- Tartakoff AM, Vassalli P. 1983. Lectin-binding sites as markers of Golgi subcompartments: Proximal-to-distal maturation of oligosaccharides. J Cell Biol. 97(4):1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper AD, Diks SH, van Blitterswijk WJ, Borst J. 2000. Glucosylceramide synthase does not attenuate the ceramide pool accumulating during apoptosis induced by CD95 or anti-cancer regimens. J Biol Chem. 275(44):34810–34817. [DOI] [PubMed] [Google Scholar]
- Thibault A, Samid D, Tompkins AC, Figg WD, Cooper MR, Hohl RJ, Trepel J, Liang B, Patronas N, Venzon DJ et al. 1996. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res. 2(3):483–491. [PubMed] [Google Scholar]
- Togayachi A, Akashima T, Ookubo R, Kudo T, Nishihara S, Iwasaki H, Natsume A, Mio H, Inokuchi J, Irimura T et al. 2001. Molecular cloning and characterization of UDP-GlcNAc:lactosylceramide beta 1,3-N-acetylglucosaminyltransferase (beta 3Gn-T5), an essential enzyme for the expression of HNK-1 and Lewis X epitopes on glycolipids. J Biol Chem. 276(25):22032–22040. [DOI] [PubMed] [Google Scholar]
- Tuuf J, Mattjus P. 2014. Membranes and mammalian glycolipid transferring proteins. Chem Phys Lipids. 178:27–37. [DOI] [PubMed] [Google Scholar]
- Uchida Y, Itoh M, Taguchi Y, Yamaoka S, Umehara H, Ichikawa S, Hirabayashi Y, Holleran WM, Okazaki T. 2004. Ceramide reduction and transcriptional up-regulation of glucosylceramide synthase through doxorubicin-activated Sp1 in drug-resistant HL-60/ADR cells. Cancer Res. 64(17):6271–6279. [DOI] [PubMed] [Google Scholar]
- Uliana AS, Crespo PM, Martina JA, Daniotti JL, Maccioni HJ. 2006. Modulation of GalT1 and SialT1 sub-Golgi localization by SialT2 expression reveals an organellar level of glycolipid synthesis control. J Biol Chem. 281(43):32852–32860. [DOI] [PubMed] [Google Scholar]
- Vieira KP, de Almeida e Silva Lima Zollner AR, Malaguti C, Vilella CA, de Lima Zollner R. 2008. Ganglioside GM1 effects on the expression of nerve growth factor (NGF), Trk-A receptor, proinflammatory cytokines and on autoimmune diabetes onset in non-obese diabetic (NOD) mice. Cytokine. 42(1):92–104. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wen L, Ma X, Chen Z, Yu Y, Zhu J, Wang Y, Liu Z, Liu H, Wu D et al. 2012. High expression of lactotriaosylceramide, a differentiation-associated glycosphingolipid, in the bone marrow of acute myeloid leukemia patients. Glycobiology. 22(7):930–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock D, Lutz M, Blackburn W, Young W, Baenziger J. 1994. Transport of newly synthesized glucosyl ceramide to the plasma membrane by a non-Golgi pathway. Proc Natl Acad Sci USA. 91:2708–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J, Johannes L, Mallard F, Girod A, Grill S, Reinsch S, Kelles P, Echard A, Goud B, Stelzer EHK. 1999. Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J Cell Biol. 147:743–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BS, Nuoffer C, Meinkoth JL, McCaffery M, Feramisco JR, Balch WE, Farquhar MG. 1994. A Rab1 mutant affecting guanine nucleotide exchange promotes disassembly of the Golgi apparatus. J Cell Biol. 125(3):557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WW, Tan MM, Xia Z, Dimitroulakos J, Minden MD, Penn LZ. 2001. Cerivastatin triggers tumor-specific apoptosis with higher efficacy than lovastatin. Clin Cancer Res. 7(7):2067–2075. [PubMed] [Google Scholar]
- Xu Y, Martin S, James DE, Hong W. 2002. GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Mol Biol Cell. 13(10):3493–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahi N, Aulas A, Fantini J. 2010. How cholesterol constrains glycolipid conformation for optimal recognition of Alzheimer's beta amyloid peptide (Abeta1–40). PLoS One. 5(2):e9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yegneswaran S, Deguchi H, Griffin JH. 2003. Glucosylceramide, a neutral glycosphingolipid anticoagulant cofactor, enhances the interaction of human- and bovine-activated protein C with negatively charged phospholipid vesicles. J Biol Chem. 278(17):14614–14621. [DOI] [PubMed] [Google Scholar]
- Yew NS, Zhao H, Hong EG, Wu IH, Przybylska M, Siegel C, Shayman JA, Arbeeny CM, Kim JK, Jiang C et al. 2010. Increased hepatic insulin action in diet-induced obese mice following inhibition of glucosylceramide synthase. PLoS One. 5(6):e11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M, McBride H. 2001. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2:107–117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.