

Abstract
Current treatments that use hematopoietic progenitor cell (HPC) transplantation in acute myeloid leukemia (AML) patients substantially reduce the risk of relapse, but are limited by the availability of immune compatible healthy HPCs. Although cellular reprogramming has the potential to provide a novel autologous source of HPCs for transplantation, the applicability of this technology toward the derivation of healthy autologous hematopoietic cells devoid of patient-specific leukemic aberrations from AML patients must first be evaluated. Here, we report the generation of human AML patient-specific hematopoietic progenitors that are capable of normal in vitro differentiation to myeloid lineages and are devoid of leukemia-associated aberration found in matched patient bone marrow. Skin fibroblasts were obtained from AML patients whose leukemic cells possessed a distinct, leukemia-associated aberration, and used to create AML patient-specific induced pluripotent stem cells (iPSCs). Through hematopoietic differentiation of AML patient iPSCs, coupled with cytogenetic interrogation, we reveal that AML patient-specific HPCs possess normal progenitor capacity and are devoid of leukemia-associated mutations. Importantly, in rare patient skin samples that give rise to mosaic fibroblast cultures that continue to carry leukemia-associated mutations; healthy hematopoietic progenitors can also be generated via reprogramming selection. Our findings provide the proof of principle that cellular reprogramming can be applied on a personalized basis to generate healthy HPCs from AML patients, and should further motivate advances toward creating transplantable hematopoietic stem cells for autologous AML therapy. Stem Cells 2013;33:1839–1849
Keywords: Acute myeloid leukemia, Chromosome aberrations, Human induced pluripotent stem cells, Hematopoietic progenitor cells, Reprogramming
Introduction
Acute myeloid leukemia (AML) is characterized by the rapid growth of nonfunctional immature myeloid cells (AML blasts) in the bone marrow (BM) and peripheral blood (PB) of patients, leading to anemia, bleeding, increased risk of infection, and ultimately death 1,2. Accumulated clinical data have identified recurrent leukemia-associated genomic aberrations in 50%–60% of AML patients 3–5, and these mutations are used as informative diagnostic and prognostic markers that are useful in managing patient therapy. Current treatments achieve high rates of remission, but subsequent relapse contributes to a reduction to 20%–30% of patients who attain disease-free survival 6,7.
Although hematopoietic progenitor cell (HPC) transplantation during consolidation therapy significantly reduces relapse 8, safe autologous sources of HPCs required for normal hematopoietic recovery are limited, and include concerns of reinfusion of leukemic cells with genomic abnormalities. Unfortunately, current graft purging methods 9 do not alleviate the risk of leukemic cell reinfusion and relapse in autologous BM transplantation settings 10–12. Alternatively, use of allogeneic blood sources to avoid leukemic abnormalities (BM, mobilized PB, and cord blood) 13 for transplantation in AML patients is restricted by the availability of matched donors, and the long-term complications associated with an inability to separate graft-versus-host disease from the beneficial graft-versus-leukemia effect 6,14,15. Furthermore, alternative efforts over the past decades to increase the low numbers of HPCs that can be obtained for the management of a single patient 16 by ex vivo expansion have had variable success 13,17, where recent clinical trials question the benefits of expanded HPCs 17. As such, the generation of novel autologous sources of HPCs to circumvent limited availability and complications associated with current transplant sources could benefit patient survival, and thus deserves deeper investigation.
The ability to generate induced pluripotent stem cell (iPSCs) that share phenotypic, molecular, and functional hallmarks with human embryonic stem cells 18–22 provides an opportunity to develop renewable sources of immune-compatible cells. In the context of AML, generation of AML patient-specific HPCs that are devoid of the leukemic aberration(s) that affect the patient’s hematopoietic tissue would provide a transformative approach in establishing a healthy autologous blood source for transplantation during AML therapy. Although robust long-term engraftment of PSC-derived HPCs in murine xenografts has not been fully demonstrated 23,24, incremental advances have been made 25–27. However, multiple studies have delineated protocols to differentiate human PSCs to HPCs that possess in vitro multipotent functionality 28–31. Independent of advancements required for the generation of transplantable long-term HPCs from hPSCs, the potential of using reprogramming to generate healthy blood cells from an AML patient has yet to be explored and it remains unclear whether generation of AML patient HPCs is even possible. To this end, we obtained dermal fibroblasts from human AML patients whose leukemic cells possessed known leukemia-associated genomic aberration, and used reprogramming technology to generate HPCs. By probing for the absence of this aberration, in conjunction with immunophenotypical, functional, and morphological in vitro assessments as compared to the patients’ AML blasts, we provide evidence that derivation of healthy autologous sources of blood using cellular reprogramming is possible.
Materials and Methods
Human Patient Samples
Individual disease cases were assessed to determine patient eligibility based on the following criteria: (a) disease was clinically classified as AML; (b) AML blasts possessed a recurrent leukemia-associated genomic aberration; (c) AML blasts were obtained by BM aspiration; and (d) patient consented to provide one dermal fibroblast skin biopsy. Informed consent was obtained from all sample donors in accordance with Research Ethics Board-approved protocols at McMaster University. BM aspirates were obtained from consenting leukemic patients at the Juravinski Cancer Center (Hamilton, Canada) as available, and from healthy patients (Lonza, Basel, Switzerland, http://www.lonza.com). Primary BM mononuclear cells were prepared using density gradient centrifugation (20 minutes, 1,500 rpm) in Ficoll-Paque Premium (GE Healthcare Life Sciences, Piscataway, NJ, http://www.gelifesciences.com), and ammonium chloride treatment (Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com) for 5 minutes at 4°C. Samples were assessed by flow cytometry for cell surface hematopoietic markers. Dermal skin biopsies (5 mm × 5 mm) were obtained from the forearm of consenting patients at the Juravinski Cancer Center. Primary human fibroblast cultures were established as described 32, and assessed by flow cytometry.
Human Cell Culture
Human dermal adult forearm fibroblasts were cultured in Fib media (Dulbecco’s modified Eagle’s medium [DMEM] with 10% vol/vol fetal bovine serum [Neonatal Bovine Serum, HyClone, Logan, UT, http://www.hyclone.com], 1% vol/vol nonessential amino acid [Gibco, Grand Island, NY, http://www.invitrogen.com], and 1 mM l-glutamine [Gibco]). Patient-specific iPSCs were derived and cultured on irradiated mouse embryonic fibroblasts (iMEFs) in F-12 iPSC media (DMEM/F-12 [Gibco] with 20% knockout serum replacement [Gibco], 100 µM β-mercaptoethanol, 100 µM nonessential amino acid [Gibco], 1 mM l-glutamine [Gibco]) supplemented with 10 ng/ml basic fibroblast growth factor; F-12 iPSC media were not supplemented with antibiotics. iPSC-derived embryoid bodies (EBs) were cultured in hematopoietic differentiation media (KO-DMEM [Gibco] with 20% knockout serum replacement [Gibco], 100 µM β-mercaptoethanol, 100 µM nonessential amino acid [Gibco], 1 mM l-glutamine [Gibco]) supplemented with 50 ng/ml granulocyte colony-stimulating factor (Amgen, Inc., Thousand Oaks, CA, http://www.amgen.com), 300 ng/ml stem cell factor (Amgen, Inc.), 10 ng/ml interleukin-3 (IL-3; R&D Systems, Minneapolis, MN, http://www.rndsystems.com), 10 ng/ml interleukin-6 (IL-6; R&D Systems), 25 ng/ml bone morphogenetic protein 4 (BMP4; R&D Systems), and 300 ng/ml Flt-3 ligand (Flt-3L; R&D Systems).
Patient-Specific iPSC Generation
Plasmids pSIN4-EF2-O2S and pSIN-EF2-K2M developed by James A. Thomson (University of Madison-Wisconsin) were obtained from Addgene (Cambridge, MA, http://www.addgene.org). Virus containing plasmid was produced from HEK 293FT Cells with second generation pMD2.G and psPAX2 packaging plasmids. Viral supernatants were harvested 72 hours after transfection and concentrated by ultracentrifugation. Human adult dermal fibroblasts (105) were incubated with concentrated lentiviral vectors in Fib media supplemented with 8 µg/ml polybrene (Sigma-Aldrich) for 48 hours, then washed, and fed fresh Fib media. Ninety-six hours after initial lentiviral transduction, Fibs were dissociated and seeded on 150,000 iMEFs and maintained in F-12 iPSC media conditions. iPSC colonies emerged between days 16 and 25 post-transduction, and were individually isolated, expanded on iMEFs, and verified for TRA-1–60 expression through live staining using TRA-1–60 DyLight 488 (Stemgent, Cambridge, MA, http://www.stemgent.com). For immunocytochemistry, iPSCs were fixed in 4% paraformaldehyde, permeabilized using BD permeabilization buffer (if required), stained with TRA-1–60, OCT4, SOX2, or NANOG antibodies (BD Biosciences, San Jose, CA, http://www.bdbiosciences.com), and counterstained with 4′,6-diamidino-2-phenylindole (DAPI).
Teratoma Assay
The developmental potential of human AML Fib iPSCs in vivo was assessed by teratoma assay. Briefly, confluent undifferentiated iPSC cultures were treated with 200 U/ml collagenase IV (Invitrogen, Carlsbad, CA, http://www.invitrogen.com) for 2 minutes at 37°C, scraped into clumps using a 5 ml pipette, collected and centrifuged at 1,000 rpm for 10 seconds, resuspended in 30 µl of media, and injected into the testicle of NOD/SCID mice. One well of a six-well plate (equivalent to 700,000–900,000 cells, as determined by cell count) was injected per mouse. Teratomas were harvested after 8–10 weeks, sectioned, and stained by hematoxylin and eosin. Images were acquired using ScanScope CS digital slide scanner with Aperio ImageScope software (Leica Biosystems, Nussloch, Germany, http://www.leicabiosystems.com).
Hematopoietic Differentiation of iPSCs
EBs were generated by suspension culture as previously described 33. Briefly, confluent undifferentiated iPSC cultures were treated with 200 U/ml collagenase IV (Invitrogen) for 2 minutes at 37°C, scraped into clumps using a 5 ml pipette, and transferred to 6- or 12-well ultralow attachment plates (Corning Inc., Corning, NY, http://www.corning.com) to form EBs. EBs were cultured for 15 days in hematopoietic differentiation media with medium changes every 4–5 days, and dissociated into single-cell suspensions by 0.4 U/ml collagenase B (Roche Life Science, Indianapolis, IN, http://www.lifescience.roche.com) treatment for 2 hours at 37°C. Total single-cell suspensions were collected for flow cytometric analysis or colony-forming unit (CFU) plating.
Clonogenic CFU Assay
Clonogenic colony-forming capacities of healthy BM and mobilized PB (10,000–30,000 cells), AML BM mononuclear cells (10,000–30,000 cells), and total dissociated EB cell suspensions (20,000–30,000 cells) plated in Methocult H4434 medium (Stem Cell Technologies, Vancouver, Canada, http://www.stemcell.com) were monitored between days 7 and 16, and colonies were quantified based on morphology between 14 and 16 days. Individual colonies were isolated and assessed for single-cell morphology, and full wells were collected for fluorescence in situ hybridization (FISH) analysis. Depending on number of colonies generated in CFU assay, single-cell morphologies of at least three colonies were analyzed to confirm colony quantification criteria and evaluate the maturity of colonies. Briefly, colonies were isolated and resuspended in 100 µl PBS and spun onto microscope slides using the Shandon Cytospin 3 (Block Scientific, Inc., Bellport, NY, http://www.blockscientific.com). Morphological features were visualized by Giemsa-Wright staining performed using Shandon Kwik-Diff Stain Kit (Thermo Scientific, Waltham, MA, http://www.thermoscientific.com). Images were acquired using ScanScope CS digital slide scanner with Aperio ImageScope software (Leica Biosystems).
Fluorescence In Situ Hybridization
t(9;11)(p22;q23), del(5)(q13q33), +4, del(16)(q22), and +8 leukemic aberrations were investigated using commercially available, validated FISH probes (Abbott Molecular, Abbott Park, IL, http://www.abbott.com). Cells incubated in 0.075 M KCl (37°C, 15 minutes) were fixed in Carnoy’s Solution. Slide preparations and probes were denatured (73°C, 5 minutes), followed by overnight hybridization in humid 37°C [Locus-specific identifier (LSI) probes] or 42°C [Chromosome enumeration probes (CEP)] incubators. Posthybridization washes were performed in 0.4× SSC/0.3% Nonidet P40, pH 7.0 (73°C, 2 minutes), followed by 2× SSC/0.1% Nonidet P40, pH 7.0 (RT, 1 minute), and mounted with DAPI II counterstain (Abbott Molecular). Visualization and analysis was performed using a fluorescence microscope equipped with appropriate filters using MetaMorph software (Molecular Devices, Sunnyvale, CA, http://www.moleculardevices.com). To confirm absence of aberration, ≥500 nuclei were analyzed. False positive events, detected below threshold of aberration detection established in normal samples, are not depicted in scoring plots.
Flow Cytometry
Single-cell suspensions were stained using combinations of the following antibodies: CD13-FITC, CD33-PE, CD34-PE, CD45-APC (Miltenyi Biotech, Bergisch Gladbach, Germany, http://www.miltenyibiotec.com) for hematopoietic phenotyping; SSEA3-PE and TRA-1–60-Alexa Fluor 647 (BD Pharmingen) for live extracellular pluripotent phenotyping; and OCT4-Alexa Fluor 488, SOX2-Alexa Fluor 647, and NANOG-PE (BD Biosciences) for intracellular pluripotent phenotyping of cells fixed and permeabilized using the BD Cytofix/Cytoperm kit. Flow cytometry was performed using the LSRII Flow Cytometer with FACSDiva software (Becton-Dickinson, Franklin Lakes, NJ, http://www.bd.com) and analyzed by FlowJo software (Tree Star, Inc., Ashland, OR, http://www.treestar.com).
Statistical Analysis
Data are presented as mean ± SEM. Prism software (version 5.0a; GraphPad, La Jolla, CA, http://www.graphpad.com) was used for all statistical analyses, and the criterion for statistical significance was p < .05.
Results
The Majority of AML Patient-Derived Fibroblasts Do Not Share Leukemia-Associated Aberration(s) Detected in Patient BM
Although a number of known leukemia-associated genomic abnormalities are not shared in nonhematopoietic BM cells derived from leukemic patients 34, this has not been established in dermal skin-derived fibroblasts in culture. To examine this further, dermal skin biopsies were obtained from four human leukemic patients diagnosed with AML carrying the t(9;11)(p22;q23) 35, del(5)(q13q33) 36, trisomy 4 (+4) 37 and del(16)(q22) 38, or trisomy 8 (+8) 39 leukemia-associated aberration(s), respectively (Table1 and Supporting Information Fig. S1). These detectable genetic markers enabled us to investigate whether nonhematopoietic dermal skin cells possessed leukemia-associated aberration. Accordingly, AML patient skin fibroblast cultures (AML Fibs) were established from patient skin biopsies 32, with the lack of CD45+ cells indicating the absence of leukemic skin infiltrates (Supporting Information Fig. S2A–S2C) 40. AML Fibs possessed bipolar, elongated morphologies similar to healthy patient-derived Fibs (Supporting Information Fig. S2B, S2D, S2E). Next, we used diagnostic fluorescence in situ hybridization (FISH) probes (Fig. 1A) to assess early passage AML Fibs in comparison to BM mononuclear cells isolated from each patient (AML BM, Supporting Information Fig. S2F, S2G). Analysis of 500 cell nuclei per AML Fib culture revealed that AML Fibs derived from patient #1 to 3 were completely devoid of the aberration(s) that was readily detected in matched AML BM (Fig. 1B–1D). In contrast, the leukemia-associated aberration detected in patient #4 AML BM (+8) was shared in 8.4% of their AML Fibs (Fig. 1E), indicating that a genetically mosaic AML Fib culture had been established from the skin biopsy. Taken together, these data indicate that although the majority of human AML Fibs are devoid of AML-specific aberration(s), it is possible for them to share aberrations found in the AML BM. Our findings here using human AML Fibs provide further evidence that leukemia-associated aberration can be harbored in nonhematopoietic cells 34.
Table 1.
Clinical disease classification of enrolled patients and leukemia-associated aberration(s) detected in their AML blast cells
| Disease classification | Aberration | |
|---|---|---|
| Patient #1 | AML M5, Monocytic | t(9;11)(p22;q23) |
| Patient #2 | AML, NOS | del(5)(q13q33) |
| Patient #3 | AML M4, Myelomonocytic | +4 and del(16)(q22)a |
| Patient #4 | MDS → AML | +8 |
Two distinct aberrations detected; not observed in the same nuclei.
Abbreviations: AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; MDS → AML, MDS progressed to AML; NOS, not otherwise specified.
Figure 1.

The majority of acute myeloid leukemia (AML) Fibs are devoid of leukemia-associated aberration. (A): Schematics illustrating patient-specific leukemic aberration(s) identified in AML blast nuclei. Fluorescence in situ hybridization (FISH) probe hybridization regions are indicated (green/red) on affected chromosomes. (B–E): FISH performed in AML patient-derived (i) Fibs and (ii) bone marrow (BM) mononuclear cells (scale bars represent 100 µm); n = 1 per AML patient. Aberrations were detected in each patient AML BM, and a population of patient #4 AML Fibs. Red arrows denote probe separation associated with translocation in patient #1 AML BM. Adjacent plots depict the frequency of detection of patient-specific, leukemia-associated aberration; blue circles represent number of nuclei analyzed. Blue circles with either one red dot or three green dots represent del(16)(q22) and +4 events in patient #3 AML BM, respectively; aberrations were never detected in the same nuclei. 500 nuclei were analyzed to exclude 1% genetic mosaicism in AML patient #1–3 Fibs with 99% confidence 41.
AML Patient-Specific iPSCs Exhibit Functional Pluripotency and Lack Leukemia-Associated Aberration
It has been previously demonstrated that healthy patient skin-derived Fibs can be reprogrammed to the pluripotent state 18,19,22, but this remained to be demonstrated in human AML patients. Toward establishing and characterizing patient-specific iPSC platforms for derivation of hematopoietic cells, we generated iPSCs from AML patient #1 to 4 Fib cultures using well-established reprogramming methods 18,42. AML Fib iPSC cultures consisted of flat colonies of densely packed single cells with large nuclei and scant cytoplasm (Fig. 2A), and were indistinguishable from healthy Fib iPSCs (Supporting Information Fig. S3A). To assess whether AML Fib iPSCs possessed biomolecular hallmarks of pluripotency similar to healthy Fib iPSCs, we performed immunocytochemistry and flow cytometric analyses. Like healthy Fib iPSCs 18, proteins that regulate the core intracellular pluripotency network OCT4, SOX2, and NANOG 43, and extracellular pluripotency markers SSEA3 and TRA-1–60 44 were expressed and localized to AML Fib iPSC colonies (Fig. 2B and Supporting Information Fig. S3B, S3C). Next, we subjected AML Fib iPSCs to in vivo teratoma formation assays to assess their functional pluripotency capacity. Following intratesticular injection, AML Fib iPSCs demonstrated in vivo pluripotent potential 45 by generating teratomas that possessed early tissue derivatives of the three embryonic germ layers (ectoderm, endoderm, and mesoderm), as evaluated by morphological assessment of hematoxylin and eosin stained tumor sections (Fig. 2C). Together, these results indicate that AML Fibs can be reprogrammed to functional iPSCs that are morphologically, molecularly, and functionally indistinguishable from healthy Fib iPSCs based on standard criteria of human pluripotency 18,19,22.
Figure 2.

Characterization of acute myeloid leukemia (AML) patient-specific Fib iPSCs. (A): Representative images of iPSC colonies generated from AML patient Fibs. Highlighted areas are displayed at higher magnification in the right, adjacent images. Scale bars represent 100 µm. (B): Representative immunofluorescence staining of pluripotency markers OCT4, SOX2, NANOG, and TRA-1–60 expressed in AML Fib iPSCs. All pluripotent markers were assessed in 12 total iPSC lines (six iPSC lines from each of patient #1 and #2), and TRA-1–60 expression was confirmed in at least three iPSC lines derived from each of AML patient #3 and #4. Scale bar represents 100 µm. (C): Representative teratoma-forming capacity of AML Fib iPSCs. Two independent iPSC lines (one from each of patient #1 and #2) were subjected to teratoma assay, each in triplicate. AML Fib iPSC-derived teratoma 10 weeks post-IT injection (top left). Hematoxylin and eosin stained sections of teratoma sections displaying early tissue derivatives of ectoderm (skin cells), endoderm (gut-like goblet cells), and mesoderm (cartilage). Arrows indicate denoted cell types. (D): Fluorescence in situ hybridization performed in patient-specific AML Fib iPSCs (n = 1 iPSC line per AML patient). Aberration identified in matched patient AML bone marrow was not detected. Adjacent plots depict the number of nuclei (blue circle) scored; 500 nuclei were analyzed to exclude 1% genetic mosaicism with 99% confidence 41. Abbreviation: iPSCs, induced pluripotent stem cells.
To probe AML Fib iPSCs for patient-specific, leukemia-associated aberration(s), we performed FISH and analyzed 500 nuclei per iPSC line. All patient-specific AML Fib iPSCs were devoid of the abnormality (Fig. 2D) that was detected in matched AML BM (Fig. 1B–1E). Interestingly, the +8 aberration harbored in a subpopulation of AML patient #4 Fibs (Fig. 1E) was not detected in the 500 iPSC nuclei analyzed by FISH (Fig. 2D), suggesting that the aberration was lost during reprogramming 46 or that the reprogramming process favors iPSC generation from genetically normal cells. Together, these data indicate that functional iPSCs devoid of the patient-specific leukemia-associated aberration(s) can be generated from AML Fibs. Moreover, these results demonstrate that the presence of genetic mosaicism in a starting cell population does not affect derivation of genomically normal iPSCs, and is consistent with previous results where reprogramming selects for normal cells 47.
AML Fib iPSC-Derived Hematopoietic Progenitors Are Devoid of Leukemia-Associated Aberration and Exhibit Normal Differentiation Capacity
Since AML Fib iPSCs did not possess leukemia-associated aberration, they represented potential cellular platforms from which to derive healthy hematopoietic cells. Toward establishing whether AML patient-specific iPSCs possessed the capacity to give rise to normal HPCs, characterized by CD34+CD45+ coexpression and functional in vitro colony-forming unit capacity, we subjected AML Fib iPSCs to an EB based in vitro hematopoietic differentiation assay 31. Three-dimensional EBs derived from AML Fib iPSC aggregates gave rise to cells coexpressing CD34+CD45+ (Fig. 3A, 3B and Supporting Information Fig. S4A), similar to healthy Fib iPSC-derived EBs 42. These results suggest that AML Fib iPSCs possess normal differentiation capacity toward the hematopoietic lineage and, based on CD34+CD45+ coexpression, are able to generate putative HPCs. Next, we performed FISH in EB-derived cells to probe for AML patient-specific aberration. Positive events were not detected in 500 cell nuclei analyzed per EB-cell-derived population (Fig. 3C), which contrasted that of the patients’ AML BM (Fig. 1B–1E) and indicated that AML patient-specific putative HPCs did not harbor leukemia-associated aberration. To further evaluate whether generation of putative HPCs devoid of leukemia-associated aberration was possible from multiple iPSC lines derived from a single patient, we performed flow cytometric and FISH analyses on EBs derived from additional patient #4 AML Fib iPSC lines. The use of these iPSC lines, derived from the genetically mosaic AML Fib culture (Fig. 1E), also provided further biological replicates from which to assess if chromosomal abnormalities are lost during the reprogramming process. Consistent with our initial findings, EBs derived from patient #4 AML Fib iPSC lines gave rise to putative HPCs expressing CD34+CD45+ (Supporting Information Fig. S4B, S4C), indicating that hematopoietic differentiation potential was not limited to a single iPSC line. Furthermore, the AML BM-specific +8 aberration was not detected (Supporting Information Fig. S4D), providing further evidence that genetically normal cells could be derived from a genetically mosaic AML Fib culture. Taken together these data demonstrate that putative HPCs devoid of leukemia-associated aberration can be generated from AML patient-specific iPSCs.
Figure 3.
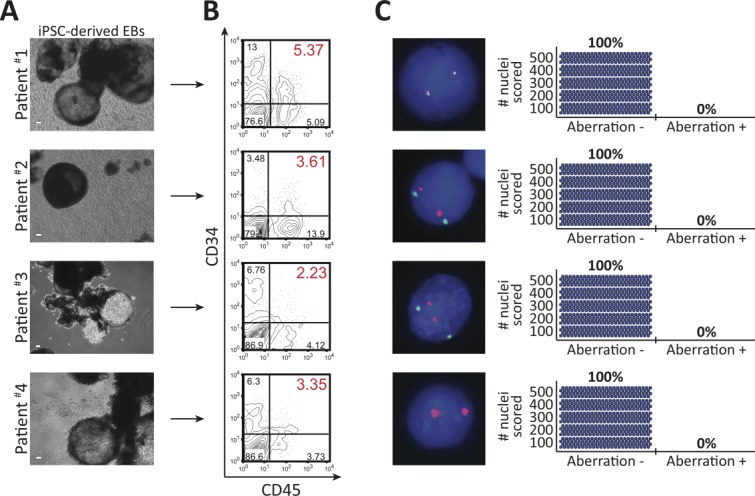
Acute myeloid leukemia (AML) patient-specific putative hematopoietic progenitor cells are devoid of leukemia-associated aberration. (A): Representative embryoid bodies (EBs) derived from AML patient Fib iPSCs, in hematopoietic differentiation conditions. Scale bar represents 100 µm. (B): Representative plots of flow cytometric analyses used to detect the generation and presence of CD34+CD45+ putative hematopoietic progenitors. Flow cytometric analysis for CD34+CD45+ expression was performed on a minimum of three independent hematopoietic differentiation experiments for each indicated iPSC line. Percentages represent frequency of total live cells with indicated cell surface phenotype. (C): Fluorescence in situ hybridization performed in patient-specific, EB-derived cells from one iPSC line per AML patient. Aberration identified in matched patient AML bone marrow was not detected. Adjacent plots depict the number of nuclei (blue circle) scored; 500 nuclei were analyzed to exclude 1% genetic mosaicism with 99% confidence 41. Abbreviation: iPSCs, induced pluripotent stem cells.
We next assessed and compared the functional capacity of putative HPCs to that of matched AML BM using the in vitro colony-forming unit (CFU) assay (Fig. 4A) to evaluate whether they had normal or leukemic features. First, we characterized the CFU capacity of AML BM to establish baseline criteria for identification of leukemic cells. Despite the inherent diversity and heterogeneity of AML samples 3, patient #1–4 AML BM possessed at least one of the following dysfunctional features suggestive of a leukemic phenotype: impaired CFU capacity characterized by an inability to generate the granulocytic lineage and the presence of persisting single cells 48, presence of cells with immature blast morphology as assessed by clinical standard Giemsa-Wright staining, and/or presence of leukemia-associated aberration as detected by FISH (Table2, Supporting Information Fig. S5B, S5D–S5F). Importantly, a small population of AML blast progenitors (patient #4, 1.7%, Fig. 1E) could be detected by CFU and FISH assays (Supporting Information Fig. S5F), illustrating the sensitivity of leukemic cell detection in the CFU assay. Together these results established criteria for detecting leukemic cells in the CFU assay. On this basis, AML patient-specific putative HPCs were subjected to CFU assay and evaluated for normal versus leukemic capacity. Consistent with healthy Fib iPSC-derived HPCs, all AML patient-specific HPCs exhibited functional capacity to generate multiple myeloid lineages as evidenced by formation of erythroid, granulocytic, and monocytic colonies (Fig. 4B, 4C and Supporting Information Fig. S6A–S6C). Although frequency of colony types showed variability between patient-specific HPCs, this result is similar to that observed in healthy BM and mobilized PB samples (Supporting Information Fig. S5A). Next, we assessed individual colonies for normal hematopoietic maturation according to clinically established morphological criteria 49. Accordingly, Giemsa-Wright staining performed on individual colonies revealed that AML patient-specific HPCs had the capacity to differentiate to mature cells (Fig. 4D and Supporting Information Fig. S6C), while immature blasts were not detected. Together, these results directly contrasted the dysfunctional differentiation capacity of AML BM and were consistent with results obtained using healthy BM and mobilized PB (Table2 and Supporting Information Fig. S5), suggesting that AML patient-specific HPCs possessed normal functional capacity. Next, we performed FISH on total mature hematopoietic colonies to probe for leukemia-associated aberration. Scoring of 500 nuclei per AML patient-specific HPC line revealed that mature hematopoietic colonies derived from HPCs did not possess leukemia-associated aberration(s) that was detected in AML BM CFU (Fig. 4E, Table2 and Supporting Information Figs. S5F, S6D). Based on the demonstrated high sensitivity of FISH to detect a small percentage of AML progenitors harboring leukemic aberration (Supporting Information Fig. S5F), coupled with the rigor of our scoring analyses that exceeded clinical requirements and excluded a 1% chance of genetic mosaicism with a 99% confidence level 41, these results provide substantial evidence that functional, AML patient-specific HPCs are completely devoid of leukemia-associated aberration carried in the patients’ own blood cells. Taken together, our data establish that AML Fib iPSC-derived, functional HPCs (CD34+CD45+ coexpression and the capacity to generate mature cells of multiple myeloid lineages) are devoid of leukemia-associated aberration; directly contrasting features of patients’ original leukemic cells.
Figure 4.

Acute myeloid leukemia (AML) patient-specific hematopoietic progenitor cells (HPCs) are capable of normal in vitro differentiation to mature blood cells and are devoid of leukemia-associated aberration. (A): Experimental strategy used to characterize patient-specific, putative HPCs in vitro. Methodologies used to assess normal hematopoietic functional capacity are indicated in red. (B): Putative HPC functionality assessed by multilineage differentiation capacity in in vitro colony-forming unit assay. Bars represent mean frequencies of mature hematopoietic colonies generated + SEM (n = 3 independent experiments per patient-specific HPC line). AML patient Fib iPSC-derived HPCs generate all mature lineages, consistent with healthy patient Fib iPSC-derived HPCs. (C): Representative mature hematopoietic colonies derived from patient-specific HPCs. Scale bars represent 100 µm. (D): Representative single-cell morphologies following Giemsa-Wright staining performed on individual hematopoietic colonies (n ≥ 3 colonies analyzed per patient-specific HPC line). Scale bars represent 10 µm. (E): Fluorescence in situ hybridization performed in total mature hematopoietic colonies derived from patient-specific HPCs. Aberration identified in matched patient AML bone marrow was not detected. Adjacent plots depict the number of nuclei (blue circle) scored; 500 nuclei were analyzed to exclude 1% genetic mosaicism with 99% confidence 41.
Table 2.
Characterizations of colony-forming unit (CFU) assays performed using indicated source of blood cells
| Healthy patient | Patient #1 | Patient #2 | Patient #3 | Patient #4 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BM | HPC | BM | HPC | BM | HPC | BM | HPC | BM | HPC | |
| Impaired differentiation capacity? | No | No | Yes | No | No | No | Yes | No | No | No |
| Immature single-cell morphology detected? | No | left/T | a | No | Yes | No | No | No | No | No |
| Leukemia-associated aberration detected? | N/A | N/A | a | No | Yes | No | Yes | No | Yes | No |
CFU capacity insufficient for further analysis.
Abbreviations: BM, bone marrow; HPC, induced pluripotent stem cell-derived hematopoietic progenitor cells; N/A, not applicable; N/T, not tested.
Discussion
This study reveals that cellular reprogramming allows for generation of human AML patient-specific hematopoietic progenitors that are devoid of leukemia-associated aberration and are capable of normal in vitro clonogenic differentiation, in direct contrast to matched patient leukemic cells. Given that current sources of healthy blood used for hematopoietic recovery during AML therapy are limited 6,10–12,14,15, we provide initial proof of principle toward generation of novel iPSC-derived, autologous blood sources devoid of leukemia-associated aberration that should enable more AML patients to receive safe transplantations during consolidation therapy and thereby increase the rate of disease-free survival (Fig. 5).
Figure 5.

Utilization of AML-specific genetic markers to interrogate cell populations throughout reprogramming toward generation of healthy blood cells for transplantation. (A): Experimental strategy developed here to generate and characterize AML patient-specific HPCs that are capable of normal in vitro differentiation to the myeloid lineage and are devoid of leukemia-associated aberration found in matched patient bone marrow. As represented by dashed arrow, technological advances in cellular reprogramming may provide novel autologous blood sources for transplantation that circumvent limitations associated with current transplantation options used during AML therapy (denoted in red font). FISH results associated with presence (+) versus absence (−) of leukemia-associated aberration are indicated above cell populations. Abbreviations: AML, acute myeloid leukemia; GVHD, graft-versus host disease; HSC, hematopoietic stem cell.
Based on previous work in the human system, the limited capacity of hPSC-derived HPCs to have transplantable hematopoietic stem cell (HSC) properties may be attributed to an inability to activate 24 or downregulate 23 regulatory somatic HSC molecular programs during differentiation 50. As such, the development of novel differentiation strategies that better specify the hematopoietic lineage from PSCs has been the focus of recent studies aimed at the generation of clinically transplantable HSCs. For instance, temporal inhibition of the early hematopoietic-regulating Hedgehog pathway during in vitro differentiation initiates adult hematopoietic gene expression programs 51. Similarly, in vivo differentiation conditions better mimic BM physiology and enable the generation of hematopoietic cells with multilineage reconstitution capacity in vivo, perhaps by providing cell extrinsic signals that regulate HSC molecular programs 25,26. Finally, forced exogenous expression of HSC-regulating transcription factors endows PSC-derived CD34+CD45+ hematopoietic cells with in vivo myeloid lineage reconstitution capacity 27. Together these recent efforts illustrate incremental advances toward the generation of PSC-derived bona fide HSCs 52. Our current findings suggest that reprogramming approaches could be used to generate healthy, transplantable sources of AML patient-specific HPCs that are capable of restoring normal, short-term myelopoiesis in AML patients to combat anemia, bleeding, and infection due to disease 1,2 and/or treatment-related myeloablation 53. Moreover, this study establishes an approach that motivates further effort for the derivation of clinically transplantable HSCs from iPSCs toward circumventing limitations of current hematopoietic sources and enabling long-term hematopoietic recovery in AML patients following therapy.
Given the diversity of AML-associated germline 54,55 and in utero/adult-acquired somatic mutations 3–5,56, and the rare presence of these mutations in nonhematopoietic tissues as demonstrated here and previously by Menendez et al. 34, it is possible that cases whereby AML Fibs share or independently acquire leukemia-associated aberration will be encountered when our strategy is applied to larger AML patient populations. Recent results indicate that large-scale chromosomal aberrations carried by Fib cultures derived from Miller Dieker Syndrome patients are lost and replaced by wild-type duplication during the reprogramming process 46. Potentially attributed to a similar loss of chromosomal aberration phenomenon during reprogramming, our current findings demonstrate that normal iPSCs can be generated from genetically mosaic AML Fib cultures. This result may also be due to the genetically normal AML Fib subpopulation preferentially reprogramming to the iPSC state. Independent of which hypothesis is true, our findings suggest that the generation of normal blood progenitors may still be feasible in cases whereby AML Fibs possess large-scale aberrations or heterogeneously harbor leukemia-associated aberration. Furthermore, cellular reprogramming may be a feasible technique toward purging chromosomal abnormalities and generating healthy cell types from cancer or disease patients that carry germline mutations, including AML patients.
It has been postulated that preleukemic mutations may predispose cells to genomic instability and increase their susceptibility to acquiring disease-specific secondary mutations 57,58 due to enhanced cell survival properties 59. Recent utilization of next-generation sequencing technologies has enabled identification of preleukemic mutations in HSCs 60,61, and forms the basis for future delineation of early genetic events that contribute to leukemogenesis. Pending further identification and annotation of these events, deep sequencing should provide further insight into the genomic integrity of patient-specific iPSCs and HPCs. By tracking the absence of leukemia-associated aberration in iPSCs and hematopoietic progenitors/mature cell derivatives, we demonstrate that the AML-specific-aberration did not arise at any stage of hematopoietic specification or maturation. This suggests that in addition to being devoid of the AML-specific aberration, AML Fib iPSCs may also be free of preleukemic mutations that predispose them to genetic instability upon in vitro hematopoietic differentiation; although, this does not preclude the possibility of long-term genetic instability. We envision that advances in genetic screening and PSC differentiation technologies that enable next-generation genetic characterization of AML patient-specific HPCs following long-term in vivo engraftment in preclinical mouse xenograft models will facilitate further efforts aimed at investigating the long-term safety and genetic stability of these cells prior to clinical application.
Given the potential of PSCs to generate multiple human tissues, cellular reprogramming may also allow for the generation of healthy cell types from other cancer or disease patients requiring transplantation. To date, we are unaware of studies that have used aberrations specific to patients’ cancer or disease as a marker to interrogate cell populations throughout the reprogramming process toward deriving and characterizing healthy cell types for transplantation purposes, as we have demonstrated here in the context of AML. As such, our study provides the proof of principle to formulate strategies toward developing healthy autologous cellular sources for AML patients, and also for other leukemia or cancer patients whereby distinct aberrations are harbored in the cancerous tissue.
Summary
Generation of AML patient-specific Fib iPSCs establishes a cellular platform from which to derive healthy HPCs that are devoid of leukemia-associated aberration detected in the patients’ BM. These autologous HPCs also possess normal in vitro differentiation capacity to multiple myeloid lineages as compared to the patients’ dysfunctional AML blasts. Our work provides proof of principle that derivation of healthy autologous sources of blood using cellular reprogramming is possible, and should enable more AML patients to receive safe transplantations during therapy toward increasing the rate of disease-free survival.
Acknowledgments
We thank Monica Graham, Jamie McNicol, Margaret Koletar, and Jenn Russell for technical support; Monika Lenkiewicz and Pamela O’Hoski for their roles as liaisons between the McMaster Stem Cell and Cancer Research Institute and the Juravinski Cancer Center; Drs. Catherine Ross and Michael R. Trus for providing clinical information; and Drs. Yannick D. Benoit and Luca Orlando, and Ryan R. Mitchell for their critical reading of the manuscript and other assistance and advice. K.R.S. is supported by NSERC (CREATE M3 Scholarship) and previously by an Ontario Graduate Scholarship, and M.B. by the Canada Research Chair Program, CRC Tier 1 in Human Stem Cell Biology, and Michael G. DeGroote School of Medicine.
Author Contributions
K.R.S.: conception and design, collection and assembly of data, data analysis and interpretation, and manuscript writing; J.-H.L.: conception and design, collection and assembly of data, and performing reprogramming; S.L., S.D., R.K., and A.F.-C.: collection of data; B.L. and R.F.: conception and provision of study material; A.D.C.: provision of study material; M.B.: conception and design, financial support, data analysis and interpretation, manuscript writing, and final approval of manuscript. K.R.S. and J.-H.L. contributed equally to this work.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article
Supporting Information Figures
Supporting Information Text
Supporting Information Figure S1
Supporting Information Figure S2
Supporting Information Figure S3
Supporting Information Figure S4
Supporting Information Figure S5
Supporting Information Figure S6
Supporting Information Figure Legends
References
- Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- Perl AE, Carroll M. Exploiting signal transduction pathways in acute myelogenous leukemia. Curr Treat Options Oncol. 2007;8:265–276. doi: 10.1007/s11864-007-0043-z. [DOI] [PubMed] [Google Scholar]
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood. 2009;114:937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: Analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998;92:2322–2333. [PubMed] [Google Scholar]
- Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–494. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106:1154–1163. doi: 10.1182/blood-2005-01-0178. [DOI] [PubMed] [Google Scholar]
- Shipley JL, Butera JN. Acute myelogenous leukemia. Exp Hematol. 2009;37:649–658. doi: 10.1016/j.exphem.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Burnett AK, Goldstone AH, Stevens RM, et al. Randomised comparison of addition of autologous bone-marrow transplantation to intensive chemotherapy for acute myeloid leukaemia in first remission: Results of MRC AML 10 trial. UK Medical Research Council Adult and Children’s Leukaemia Working Parties. Lancet. 1998;351:700–708. doi: 10.1016/s0140-6736(97)09214-3. [DOI] [PubMed] [Google Scholar]
- Alvarnas JC, Forman SJ, et al. Graft purging in autologous bone marrow transplantation: A promise not quite fulfilled. Oncology (Williston Park) 2004;18:867–876. discussion 876. [PubMed] [Google Scholar]
- Hagenbeek A, Martens AC. Reinfusion of leukemic cells with the autologous marrow graft: Preclinical studies on lodging and regrowth of leukemia. Leuk Res. 1985;9:1389–1395. doi: 10.1016/0145-2126(85)90127-4. [DOI] [PubMed] [Google Scholar]
- Linker CA. Autologous stem cell transplantation for acute myeloid leukemia. Bone Marrow Transplant. 2003;31:731–738. doi: 10.1038/sj.bmt.1704020. [DOI] [PubMed] [Google Scholar]
- Brenner MK, Rill DR, Moen RC, et al. Gene-marking to trace origin of relapse after autologous bone-marrow transplantation. Lancet. 1993;341:85–86. doi: 10.1016/0140-6736(93)92560-g. [DOI] [PubMed] [Google Scholar]
- Hodby K, Pamphilon D. Concise review: Expanding roles for hematopoietic cellular therapy and the blood transfusion services. Stem Cells. 2011;29:1322–1326. doi: 10.1002/stem.689. [DOI] [PubMed] [Google Scholar]
- Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- Li JM, Giver CR, Lu Y, et al. Separating graft-versus-leukemia from graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Immunotherapy. 2009;1:599–621. doi: 10.2217/imt.09.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop MR. Hematopoietic stem cell transplantation. Introduction. Cancer Treat Res. 2009;144:ix–x. [PubMed] [Google Scholar]
- Kelly SS, Parmar S, De Lima M, et al. Overcoming the barriers to umbilical cord blood transplantation. Cytotherapy. 2010;12:121–130. doi: 10.3109/14653240903440111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Aasen T, Raya A, Barrero MJ, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- Eminli S, Utikal J, Arnold K, et al. Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem Cells. 2008;26:2467–2474. doi: 10.1634/stemcells.2008-0317. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Risueno RM, Sachlos E, Lee JH, et al. Inability of human induced pluripotent stem cell-hematopoietic derivatives to downregulate microRNAs in vivo reveals a block in xenograft hematopoietic regeneration. Stem Cells. 2012;30:131–139. doi: 10.1002/stem.1684. [DOI] [PubMed] [Google Scholar]
- Wang L, Menendez P, Shojaei F, et al. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J Exp Med. 2005;201:1603–1614. doi: 10.1084/jem.20041888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Yamazaki S, Yamaguchi T, et al. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol Ther. 2013;21:1424–1431. doi: 10.1038/mt.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amabile G, Welner RS, Nombela-Arrieta C, et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood. 2013;121:1255–1264. doi: 10.1182/blood-2012-06-434407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S, Vo LT, Chou SS, et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell. 2013;13:459–470. doi: 10.1016/j.stem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KD, Vodyanik M, Slukvin II. Hematopoietic differentiation and production of mature myeloid cells from human pluripotent stem cells. Nat Protoc. 2011;6:296–313. doi: 10.1038/nprot.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KD, Yu J, Smuga-Otto K, et al. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells. 2009;27:559–567. doi: 10.1634/stemcells.2008-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerdan C, Rouleau A, Bhatia M. VEGF-A165 augments erythropoietic development from human embryonic stem cells. Blood. 2004;103:2504–2512. doi: 10.1182/blood-2003-07-2563. [DOI] [PubMed] [Google Scholar]
- Chadwick K, Wang L, Li L, et al. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood. 2003;102:906–915. doi: 10.1182/blood-2003-03-0832. [DOI] [PubMed] [Google Scholar]
- Villegas J, McPhaul M, et al. Establishment and culture of human skin fibroblasts. Curr Protoc Mol Biol. 2005;28 doi: 10.1002/0471142727.mb2803s71. Chapter. [DOI] [PubMed] [Google Scholar]
- Hong SH, Werbowetski-Ogilvie T, Ramos-Mejia V, et al. Multiparameter comparisons of embryoid body differentiation toward human stem cell applications. Stem Cell Res. 2010;5:120–130. doi: 10.1016/j.scr.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Menendez P, Catalina P, Rodriguez R, et al. Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J Exp Med. 2009;206:3131–3141. doi: 10.1084/jem.20091050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch C, Schnittger S, Klaus M, et al. AML with 11q23/MLL abnormalities as defined by the WHO classification: Incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood. 2003;102:2395–2402. doi: 10.1182/blood-2003-02-0434. [DOI] [PubMed] [Google Scholar]
- Giagounidis AA, Germing U, Aul C. Biological and prognostic significance of chromosome 5q deletions in myeloid malignancies. Clin Cancer Res. 2006;12:5–10. doi: 10.1158/1078-0432.CCR-05-1437. [DOI] [PubMed] [Google Scholar]
- Bains A, Lu G, Yao H, et al. Molecular and clinicopathologic characterization of AML with isolated trisomy 4. Am J Clin Pathol. 2012;137:387–394. doi: 10.1309/AJCP7ZC9YQERSKGX. [DOI] [PubMed] [Google Scholar]
- Marlton P, Keating M, Kantarjian H, et al. Cytogenetic and clinical correlates in AML patients with abnormalities of chromosome 16. Leukemia. 1995;9:965–971. [PubMed] [Google Scholar]
- Paulsson K, Johansson B. Trisomy 8 as the sole chromosomal aberration in acute myeloid leukemia and myelodysplastic syndromes. Pathol Biol (Paris) 2007;55:37–48. doi: 10.1016/j.patbio.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Ratnam KV, Su WP, Ziesmer SC, et al. Value of immunohistochemistry in the diagnosis of leukemia cutis: Study of 54 cases using paraffin-section markers. J Cutan Pathol. 1992;19:193–200. doi: 10.1111/j.1600-0560.1992.tb01658.x. [DOI] [PubMed] [Google Scholar]
- Hook EB. Exclusion of chromosomal mosaicism: Tables of 90%, 95% and 99% confidence limits and comments on use. Am J Hum Genet. 1977;29:94–97. [PMC free article] [PubMed] [Google Scholar]
- Hong SH, Lee JH, Lee JB, et al. ID1 and ID3 represent conserved negative regulators of human embryonic and induced pluripotent stem cell hematopoiesis. J Cell Sci. 2011;124:1445–1452. doi: 10.1242/jcs.077511. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adewumi O, Aflatoonian B, Ahrlund-Richter L, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol. 2007;25:803–816. doi: 10.1038/nbt1318. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Bershteyn M, Hayashi Y, Desachy G, et al. Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells. Nature. 2014;507:99–103. doi: 10.1038/nature12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein SM, Batada NN, Vuoristo S, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- Moore MA, Spitzer G, Williams N, et al. Agar culture studies in 127 cases of untreated acute leukemia: The prognostic value of reclassification of leukemia according to in vitro growth characteristics. Blood. 1974;44:1–18. [PubMed] [Google Scholar]
- Bain BJ, Clark DM, Wilkins BS. Bone Marrow Pathology. Oxford: Wiley; 2010. [Google Scholar]
- Schnerch A, Lee JB, Graham M, et al. Human embryonic stem cell-derived hematopoietic cells maintain core epigenetic machinery of the polycomb group/Trithorax Group complexes distinctly from functional adult hematopoietic stem cells. Stem Cells Dev. 2013;22:73–89. doi: 10.1089/scd.2012.0204. [DOI] [PubMed] [Google Scholar]
- McIntyre BA, Ramos-Mejia V, Rampalli S, et al. Gli3-mediated hedgehog inhibition in human pluripotent stem cells initiates and augments developmental programming of adult hematopoiesis. Blood. 2013;121:1543–1552. doi: 10.1182/blood-2012-09-457747. [DOI] [PubMed] [Google Scholar]
- Kushwah R, Guezguez B, Lee JB, et al. Pleiotropic roles of Notch signaling in normal, malignant, and developmental hematopoiesis in the human. EMBO Rep. 2014;15:1128–1138. doi: 10.15252/embr.201438842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Lazarus HM, Keating A. Myeloablative conditioning regimens for AML allografts: 30 years later. Bone Marrow Transplant. 2003;32:969–978. doi: 10.1038/sj.bmt.1704285. [DOI] [PubMed] [Google Scholar]
- Kazenwadel J, Secker GA, Liu YJ, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood. 2012;119:1283–1291. doi: 10.1182/blood-2011-08-374363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst T, Eyholzer M, Haefliger S, et al. Somatic CEBPA mutations are a frequent second event in families with germline CEBPA mutations and familial acute myeloid leukemia. J Clin Oncol. 2008;26:5088–5093. doi: 10.1200/JCO.2008.16.5563. [DOI] [PubMed] [Google Scholar]
- Greaves M. In utero origins of childhood leukaemia. Early Hum Dev. 2005;81:123–129. doi: 10.1016/j.earlhumdev.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Zuna J, Burjanivova T, Mejstrikova E, et al. Covert preleukemia driven by MLL gene fusion. Genes Chromosomes Cancer. 2009;48:98–107. doi: 10.1002/gcc.20622. [DOI] [PubMed] [Google Scholar]
- Brain JM, Goodyer N, Laneuville P. Measurement of genomic instability in preleukemic P190BCR/ABL transgenic mice using inter-simple sequence repeat polymerase chain reaction. Cancer Res. 2003;63:4895–4898. [PubMed] [Google Scholar]
- Hong D, Gupta R, Ancliff P, et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008;319:336–339. doi: 10.1126/science.1150648. [DOI] [PubMed] [Google Scholar]
- Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figures
Supporting Information Text
Supporting Information Figure S1
Supporting Information Figure S2
Supporting Information Figure S3
Supporting Information Figure S4
Supporting Information Figure S5
Supporting Information Figure S6
Supporting Information Figure Legends