

Abstract
The synthesis, structure and anion-recognition properties of a new strapped-porphyrin-containing [2]catenane anion host system are described. The assembly of the catenane is directed by discrete chloride anion templation acting in synergy with secondary aromatic donor–acceptor and coordinative pyridine–zinc interactions. The [2]catenane incorporates a three-dimensional, hydrogen-bond-donating anion-binding pocket; solid-state structural analysis of the catenane⋅chloride complex reveals that the chloride anion is encapsulated within the catenane’s interlocked binding cavity through six convergent CH⋅⋅⋅⋅Cl and NH⋅⋅⋅Cl hydrogen-bonding interactions and solution-phase 1H NMR titration experiments demonstrate that this complementary hydrogen-bonding arrangement facilitates the selective recognition of chloride over larger halide anions in DMSO solution.
Keywords: anion binding, catenanes, porphyrins, self-assembly, supramolecular chemistry
Introduction
[n]Catenanes, a class of mechanically bonded molecules comprising n interlocked ring components,1 have received ever-growing attention over recent decades on account of their interesting topologies and aesthetic appeal, in addition to their potential applications as molecular machines,2 imaging agents,3 host systems4 and functional nanomaterials.5 However, despite the widespread interest in catenane compounds, their synthesis remains challenging, usually relying on the use of interweaving templating interactions to organise the molecular precursor components in an orthogonal manner, before performing the final ring-closing step. Since Sauvage’s pioneering use of a CuI-directed orthogonal assembly strategy,6 much synthetic effort has been devoted to the development of new and efficient template-directed protocols for the preparation of catenanes. Although coordinate metal–ligand bonds remain the most widely exploited templating interactions,7 a variety of alternative noncovalent interactions, including π–π interactions,8 hydrogen bonding,b8c, 9 radical–radical interactions,10 halogen bonding,11 and solvatophobic effects,12 have been successfully applied to catenane synthesis.
Our group13 and others14 have demonstrated that anions can also be effectively employed as discrete interweaving templates during catenane synthesis, and that the preorganised three-dimensional binding pockets contained within the resultant interlocked architectures can subsequently be exploited for selective anion-recognition purposes.15 Incorporation of a suitable optical or redox-active reporter group can give rise to systems that produce a signalling response upon complexation of the target guest species. However, despite the promise of this approach, anion-templation strategies remain underdeveloped and examples of catenane-based host systems that are capable of optically or electrochemically sensing the presence of an anionic guest species are rare.16
Herein we describe the synthesis and solid-state structure of a new strapped-porphyrin-containing [2]catenane anion host system, which is assembled by using chloride anion templation in combination with aromatic donor–acceptor interactions and pyridine–zinc ligation.17 After removal of the templating anion, the catenane’s halide anion-recognition and sensing properties were probed by 1H NMR, UV/Vis and fluorescence titration experiments.
Results and Discussion
Design and synthetic strategy
We have previously employed a chloride-anion-templated, amide-condensation-based clipping strategy to assemble [2]catenane architectures in which bidentate hydrogen-bond-donor groups from each of the interlocked macrocyclic components converge towards a central, three-dimensional binding cavity, where the halide anion template is encapsulated. Upon removal of the template, the [2]catenanes were shown to recognise halide anions selectively over oxoanions in 1:1 CDCl3/CD3OD, which is primarily attributed to the optimal size- and shape-complementarity between the halide anions and the hosts’ interlocked binding domains.b13d In the current study we have modified our previous catenane design by incorporating a zinc(II) metalloporphyrin unit into one of the interlocked macrocyclic components, and a 3,5-pyridine bis(amide) motif into the second macrocycle component, in order to introduce an intercomponent pyridine–zinc interaction into the final interlocked structure (Figure 1).
Figure 1.
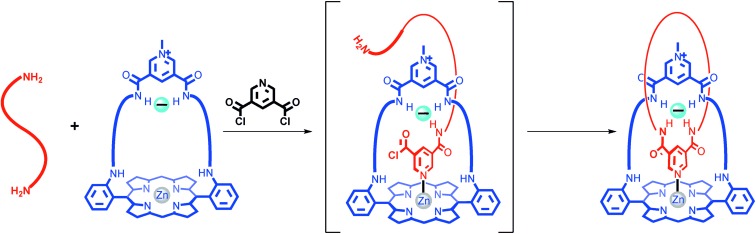
Cartoon representation of the intended synthetic route to the target strapped-porphyrin-containing [2]catenane, which incorporates an intercomponent pyridine–zinc coordinative bond.
We anticipated that this coordinative interaction could be exploited in several ways: as well as providing an auxiliary templating interaction during catenane assembly, the pyridine–zinc ligation process was predicted to enhance the anion recognition properties of the [2]catenane host system by increasing both the overall preorganisation of the host system and the acidity of the 3,5-pyridine bis(amide) hydrogen-bond-donor groups; it was also envisaged that the pyridine–zinc bond would create a direct through-bond communication pathway between the anion recognition site and the porphyrin chromophore, which could potentially enable the [2]catenane host system to optically sense the presence of an encapsulated guest anion.
Synthesis of pyridinium-strapped porphyrin macrocycle
The synthesis of the new pyridinium bis(amide)- strapped porphyrin macrocyclic fragment of the target [2]catenane was carried out as outlined in Schemes 1 and 2. Initially the bis(amino)porphyrin precursor Zn⋅3 a was prepared (Scheme 1). The trans-substituted dinitroporphyrin 218 was obtained from a BF3-catalysed [2+2] condensation reaction between meso-unsubstituted dipyrromethane 219 and 2-nitrobenzaldehyde, with subsequent p-chloranil oxidation.20 Compound 2 was obtained in 25 % yield, as an approximately equimolar mixture of the α,α- and α,β-atropisomeric forms 2 a and 2 b. Reduction of this isomeric mixture with SnCl2/HCl afforded the corresponding α,α- and α,β-bis(amino)porphyrin atropisomers 3 a and 3 b, which were separated by column chromatography.21 On the basis of molecular dipole considerations, and by analogy with related compounds in the literature,22 the more polar compound was assigned as the desired α,α-isomer 3 a, which was isolated in 37 % yield. The α,β-isomer 3 b was isolated in a comparable yield of 40 %.23 Metallation of the α,α-bis(amine) 3 a by treatment with excess Zn(OAc)2⋅2 H2O afforded the corresponding zinc(II) metalloporphyrin Zn⋅3 a in 91 % yield.
Scheme1.
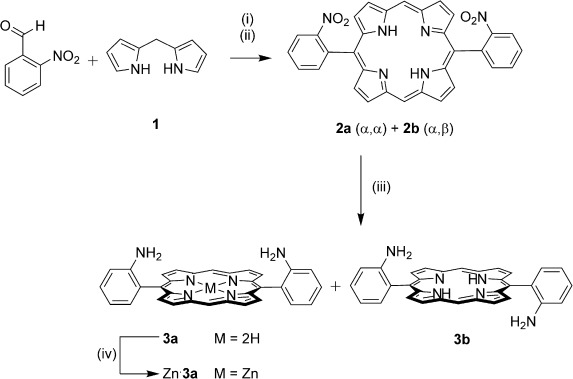
Synthesis of the bis(amino)porphyrin 3. Reagents and conditions: i) BF3⋅OEt2, EtOH, CH2Cl2, RT, 1 h; ii) p-chloranil, reflux, 1 h, 25 %; iii) SnCl2⋅2 H2O, 37% HCl(aq), RT, 16 h, 37 % (3 a) and 40 % (3 b); iv) Zn(OAc)2⋅2 H2O, CH2Cl2, MeOH, RT, 18 h, 91 %.
Scheme2.
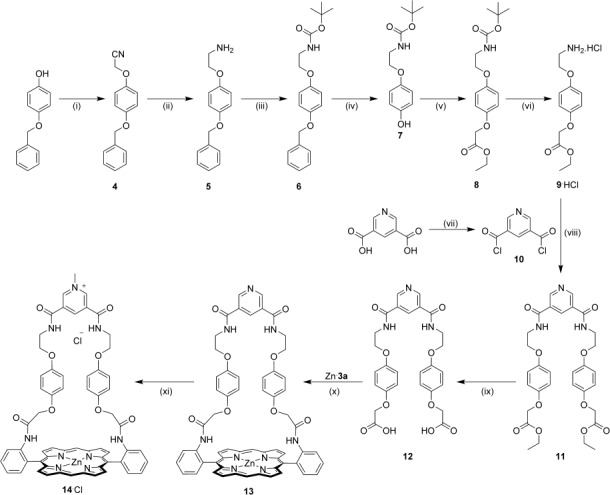
Synthesis of the pyridinium-strapped macrocycle 14⋅Cl. Reagents and conditions: i) Bromoacetonitrile, K2CO3, acetone, reflux, 18 h, 97 %; ii) LiAlH4, Et2O, reflux, 84 %; iii) Boc2O, Et3N, CH2Cl2, RT, 87 %; iv) H2, Pd/C, CHCl3, MeOH, RT, 95 %; v) ethylbromoacetate, NaH, THF, RT, 1 h, 50 °C, 12 h, 94 %; vi) HCl(g), Et2O, RT, 97 %; vii) (COCl)2, DMF (cat.), CH2Cl2, RT, quantitative yield; viii) Et3N, CH2Cl2, 0 °C to RT, 18 h, 73 %; ix) KOH, CH2Cl2, MeOH, H2O RT, 88 %; x) EDC⋅HCl, HOBt, DMAP, DMF, RT, 44 %; xi) MeI, NaHCO3, DMF, 80 °C, 2 h, then NH4Cl(aq), 99 %.
The assignment of the atropisomers was confirmed by solid-state structural characterisation of compounds Zn⋅3 a and 3 b (Figure 2). In both structures, the meso-aryl substituents are arranged almost orthogonally to the planes of the porphyrins (mean dihedral angle=86.7° and 67.8° for compounds Zn⋅3 a and 3 b respectively). For compound Zn⋅3 a, the desired cis arrangement of the amino groups is observed, whereas for compound 3 b the meso-substituents adopt a trans conformation, with the two amino groups diverging away from the porphyrin plane. For both compounds, the hybridization states of the aniline nitrogen atoms appear to be intermediate between sp3 and sp2, with all of the aniline nitrogen torsion angles falling within the range 150–165°.24 The structure of the metalloporphyrin Zn⋅3 a confirms that the zinc(II) cation adopts the expected five-coordinate square pyramidal coordination environment in the solid state, with an axially coordinated methanol molecule occupying the apical position of the square pyramid (mean Zn–N distance: 2.048(2) Å; Zn–O distance: 2.166(20) Å).25 The zinc(II) cation is located 0.174 Å above the mean plane of the porphyrin, which is significantly ruffled, in contrast to the highly planar free base porphyrin derivative 3 b.
Figure 2.
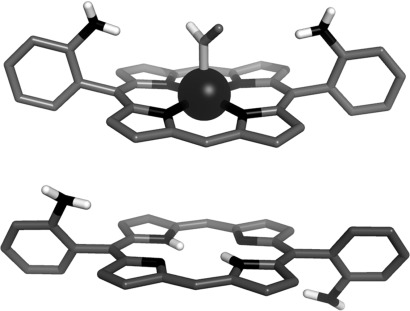
Solid-state structures of the α,α- and α,β-aminoporphyrin derivatives Zn(MeOH)⋅3 a (top) and 3 b (bottom). For clarity, nonpolar hydrogen atoms have been omitted and only one of the two fractional occupancies of the axially coordinated methanol solvate molecule is shown.
Having isolated the α,α-bis(amino)porphyrin precursor Zn⋅3 a, the strap component of the porphyrin-containing macrocycle was constructed in ten steps from commercially available 4-(benzyloxy)phenol. Reaction of this precursor with bromooacetonitrile, followed by cyano-group reduction, Boc-protection of the amino group and hydrogenative debenzylation afforded the phenol derivative 7,b13a which was condensed with ethyl bromoacetate to provide compound 8. Cleavage of the N-Boc protecting group by bubbling HCl(g) through a solution of compound 8 in Et2O yielded the corresponding amine as its hydrochloride salt, 9⋅HCl. This was condensed with 0.5 equivalents of the bis-acid chloride 10 to produce the bis-ester intermediate 11, which was subsequently converted into the bis-acid-functionalised strap precursor 12 in 88 % yield by base-mediated hydrolysis of the ester groups. An EDC-promoted coupling reaction [EDC=N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide, which was added to the reaction as a hydrochloride salt] between this bis-acid derivative and the α,α-bis(amino)porphyrin Zn⋅3 a in DMF afforded the pyridine-strapped macrocycle 13 in 44 % yield.26 Finally, alkylation of the pyridine group by treatment of macrocycle 13 with MeI in the presence of NaHCO327 in DMF, followed by repeated extraction with NH4Cl(aq), afforded the chloride salt of the target pyridinium-strapped porphyrin macrocycle, 14⋅Cl, in 99 % yield (Scheme 2).
Both of the new macrocycles 13 and 14⋅Cl were characterised by X-ray crystallography in the solid state. The crystal structure of the pyridine-strapped macrocycle 13 (Figure 3) reveals the existence of an intramolecular coordinative interaction between the pyridyl nitrogen atom and the zinc(II) cation (Zn–Npyr distance: 2.222(3) Å), which causes the strap to fold inwards, inducing a parallel stacking arrangement between the 1,4-hydroquinone and 3,5-pyridine bis(amide) moieties. A saddle distortion of the porphyrin unit is also apparent. For the N-methylpyridinium-strapped macrocycle 14⋅Cl, it is not possible for an analogous intramolecular pyridine–zinc interaction to occur, and the axial coordination site is instead occupied by a pyridine solvate molecule, which ligates to the outer face of the porphyrin unit (Zn–Npyr distance: 2.136(4) Å; Figure 4). The macrocycle’s 3,5-pyridinium bis(amide) group adopts a syn–syn conformation and the chloride counteranion is held within the resultant binding cleft by three short NH⋅⋅⋅Cl and CH⋅⋅⋅Cl contacts (N⋅⋅⋅Cl distances: 3.348(1) and 3.233(1) Å; C⋅⋅⋅Cl distance: 3.231(2) Å). Examination of the crystal packing reveals that the pyridinium macrocycle 14⋅Cl forms a head-to-tail dimer in the solid state. Each dimeric unit appears to be stabilised by two complementary intermolecular aromatic stacking interactions between the pyridinium and porphyrin groups, in addition to four NH⋅⋅⋅O amide–amide hydrogen bonds.
Figure 3.
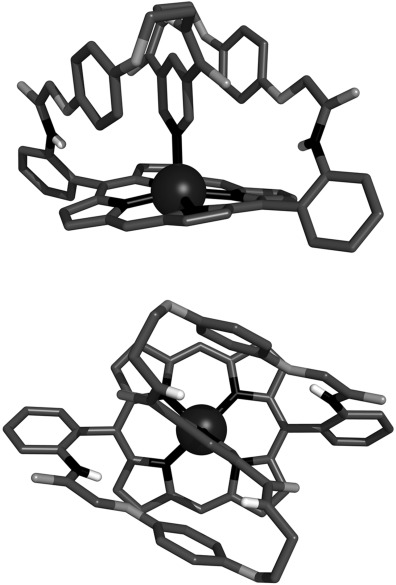
Orthogonal views of the solid-state structure of the pyridine- strapped porphyrin macrocycle 13. Solvent molecules and nonpolar hydrogen atoms have been omitted for clarity.
Figure 4.
Solid-state structure of the pyridine solvate of the pyridinium-strapped porphyrin macrocycle 14⋅Cl: content of the asymmetric unit (top) and view of the head-to-tail dimer which forms as a result of crystal packing (bottom). Nonpolar hydrogen atoms have been omitted for clarity. Hydrogen bonds are represented as dashed lines.
Synthesis of strapped-porphyrin-containing [2]catenane
Condensation of the bis(amine) derivative 1528 with 3,5-bis(chlorocarbonyl) pyridine 10 in the presence of the pyridinium-strapped porphyrin macrocycle 14⋅Cl in CH2Cl2 afforded the [2]catenane 16⋅Cl in 30 % yield after purification by preparative TLC and recrystallisation (Scheme 3). The chloride anion template was removed by stirring compound 16⋅Cl with AgNO3 in 95:5 DMSO/H2O, before precipitation of the hexafluorophosphate salt 16⋅PF6 in 73 % yield by addition of NH4PF6(aq).
Scheme3.
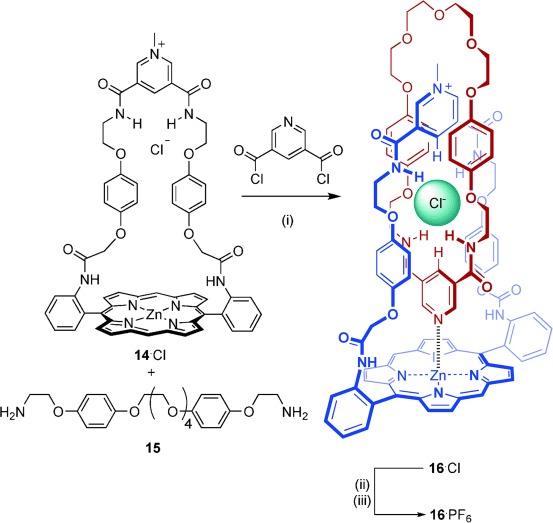
Synthesis of the [2]catenanes 16⋅Cl and 16⋅PF6. Reagents and conditions: i) Et3N, CH2Cl2, RT, 2 h, 30 %; ii) AgNO3, DMSO/H2O, RT, 45 min; iii) NH4PF6(aq), 73 %.
[2]Catenane characterisation
The [2]catenanes 16⋅Cl and 16⋅PF6 were fully characterised by electrospray mass spectrometry and 1H and 13C NMR spectroscopy techniques in solution, and by X-ray crystallography in the solid state. The 1H NMR spectrum of the [2]catenane 16⋅Cl in CD2Cl2 is compared with that of non-interlocked 3,5-pyridine bis(amide)-functionalised macrocycle 1729 in Figure 5.
Figure 5.
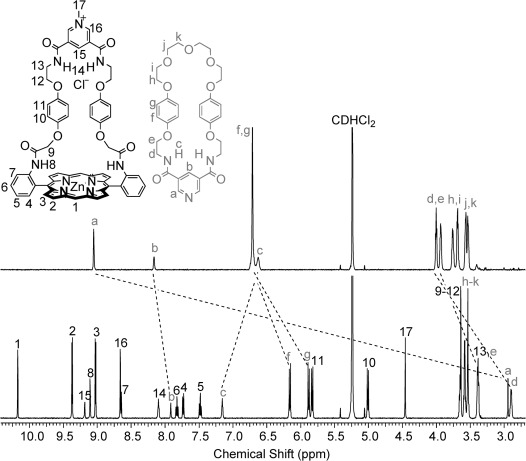
Partial 1H NMR spectra (500 MHz) of macrocycle 17 (top spectrum) and the [2]catenane 16⋅Cl (bottom spectrum) in CD2Cl2 at 293 K.
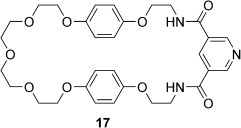 |
A dramatic >6 ppm upfield shift in the signal for the aromatic ortho pyridine proton a is observed upon incorporation of macrocycle 17 into the [2]catenane, with concomitant 0.3–1.2 ppm upfield shifts in the signals corresponding to the para pyridine proton b and aliphatic CH2 protons d and e. This indicates that the pyridyl group is located within the shielding region created by the porphyrin ring currents, and therefore strongly suggests that the [2]catenane is stabilised by an intercomponent pyridine–zinc interaction in solution. In contrast, the resonance for amide proton c shifts downfield, which is consistent with the pyridine bis(amide) group participating in NH⋅⋅⋅Cl hydrogen-bonding interactions with the chloride anion, since these interactions would be expected to polarise the amide NH bonds. The pronounced upfield shift and splitting of the resonances for hydroquinone protons f and g is diagnostic of secondary aromatic-donor–acceptor interactions between the electron-rich hydroquinone groups in the neutral macrocycle and the electron-deficient pyridinium component of the charged macrocycle.30 In addition, a number of through-space interactions between the two interlocked macrocyclic components of the catenane were observed by 2D 1H NMR ROESY spectroscopy in CD2Cl2 and [D6]DMSO, which provided further supportive evidence for the interlocked nature and proposed solution conformation of the [2]catenane (see the Supporting Information, Figures S20 and S24).
Single crystals of the [2]catenanes 16⋅Cl and 16⋅PF6 that were suitable for X-ray structural determination were grown by layered diffusion of hexane into a CH2Cl2/MeOH solution of the chloride salt 16⋅Cl, and by layered diffusion of diisopropyl ether into an acetone solution of the hexafluorophosphate salt 16⋅PF6. In both cases, the crystals were small and weakly diffracting, and X-ray diffraction data were collected by using synchrotron radiation.
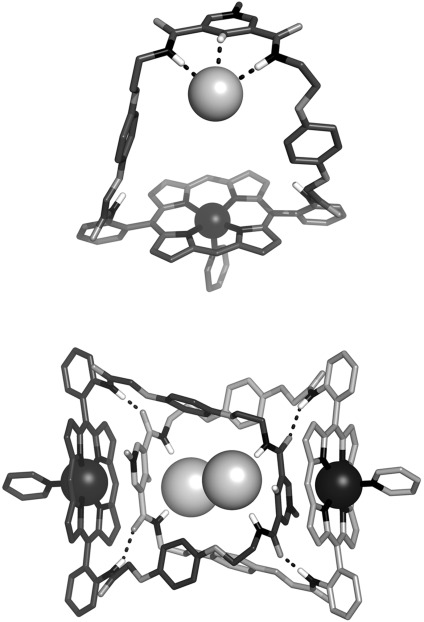
The crystal structure of the chloride-complexed catenane 16⋅Cl (Figure 6) confirms the existence of an intercomponent pyridine–zinc coordinative bond in the solid state (Zn–Npyr distance: 2.138(2) Å). The chloride anion is encapsulated within the pseudo-octahedral interlocked binding cavity defined by the orthogonally disposed 3,5-pyridinium bis(amide) and 3,5-pyridine bis(amide) moieties. The hydrogen atoms from each of the six aromatic CH and amide NH hydrogen-bond-donor groups are directed towards the encapsulated chloride anion, and six short X⋅⋅⋅Cl (X=C, N) contacts are observed, with X⋅⋅⋅Cl distances ranging from 3.285 to 3.353(2) Å and X–H⋅⋅⋅Cl angles ranging from 159 to 180°. A parallel donor–acceptor–donor aromatic stacking arrangement between the electron-rich 1,4- hydroquinone and electron-deficient pyridinium motifs is also observed, with centroid-to-centroid distances of 3.493 and 3.822 Å.
Figure 6.
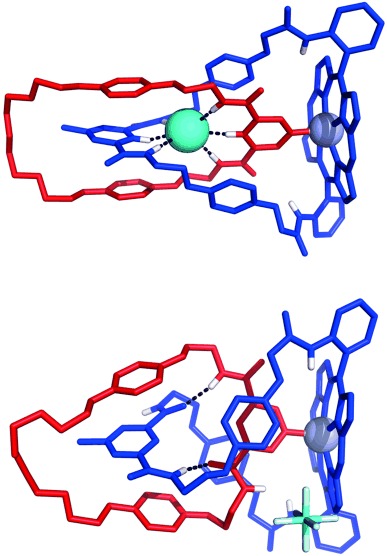
Solid-state structures of the [2]catenane 16⋅Cl (top) and the [2]catenane 16⋅PF6 (bottom). Solvent molecules and nonpolar hydrogen atoms have been omitted for clarity. Hydrogen bonds are represented as dashed lines.
The diffraction data obtained for the hexafluorophosphate catenane 16⋅PF6 were unusually weak and the structure was found to incorporate a significant degree of disorder, necessitating the use of extensive restraints during minimisation. Nevertheless, it is evident from the structure that the conformation of the hexafluorophosphate catenane 16⋅PF6 is largely unchanged from that of the chloride catenane 16⋅Cl in the solid state (Figure 6). In the absence of an encapsulated chloride anion, the pyridine–zinc coordinative bond and approximate interlocked co-conformation of the two macrocycles are preserved, but the 3,5-pyridinium bis(amide) and 3,5-pyridine bis(amide) groups adopt a syn–anti conformation, which is stabilised by two intercomponent NH⋅⋅⋅O amide–amide hydrogen bonds. The hexafluorophosphate counteranion is not involved in short contacts with the hydrogen-bond-donor groups from either of the bis(amide) motifs, which is in accordance with its assumed non-coordinating role.31
Halide recognition and sensing experiments
Encouraged by the crystallographic evidence that the chloride counteranion is encapsulated within the interlocked binding cavity of the [2]catenane 16⋅Cl in the solid state, we employed 1H NMR, UV/Vis and fluorescence spectroscopic titration experiments to investigate the ability of the [2]catenane host system to recognise and sense halide anions in solution.
1H NMR titration experiments
Addition of an increasing concentration of tetrabutylammonium (TBA) chloride to a 1.5 mM solution of compound 16⋅PF6 in [D6]DMSO induced progressive shifts in the 1H NMR signals for protons 14, 15 and c (Figure 7), which is consistent with fast-exchange complexation of the chloride anion within the [2]catenane’s interlocked binding domain. A 1:1 stoichiometric association constant of K=2144(149) M−1 was determined by analysis of the chloride-concentration-dependent shifts of the external pyridinium proton 16 with WinEQNMR232 software (Figure 8). In contrast, comparable titration experiments using TBAI and TBABr salts produced no convincing evidence of a binding interaction between the larger halide anions and the catenane’s interlocked cavity. Addition of up to 10 equivalents of TBABr and TBAI to a [D6]DMSO solution of the catenane resulted in only slight (Δδ≤0.11 ppm) perturbations in the 1H NMR signals for the cavity protons 14, 15 and c (Figure 8), which could not be definitively assigned to a binding event. The [2]catenane host system 16⋅PF6 therefore appears to display an impressive selectivity for chloride over bromide and iodide anions in DMSO, which may reflect an optimal host–guest complementarity relationship between the chloride anion and the catenane’s preorganised hydrogen-bond- donating binding pocket.33
Figure 7.
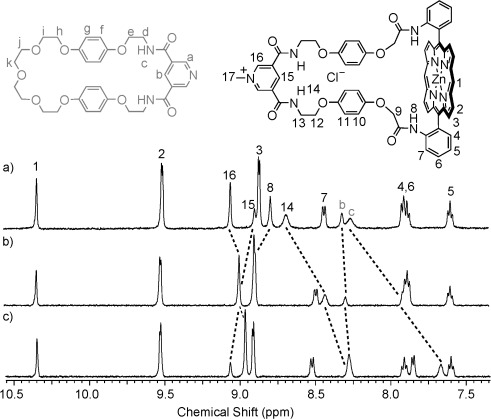
Partial 1H NMR spectra of a 1.5 mM solution of the [2]catenane 16⋅PF6 in [D6]DMSO at 293 K after addition of a) 0, b) 1 and c) 5 equivalents of TBACl.
Figure 8.
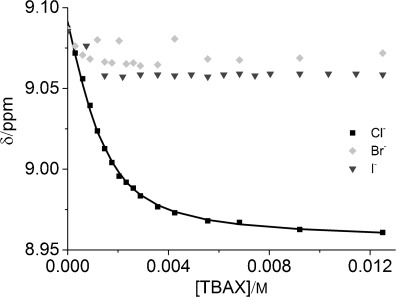
Changes in the chemical shift of the pyridinium proton 16 on addition of the TBAX salts (X=Cl−, Br− and I−) to 1.5 mM solutions of compound 16⋅PF6 in [D6]DMSO at 298 K. Square data points represent experimental data; continuous line represents the calculated binding curve for K=2144 M−1.
UV/Vis and fluorescence experiments
UV/Vis and fluorescence spectroscopic titration experiments revealed that the [2]catenane exhibits modest optical chloride-sensing capabilities: upon titration of TBACl into solutions of the catenane in DMSO a gradual hypsochromic shift of approximately 1 nm was observed in the Soret band absorbance, with the formation of a single isosbestic point at 418.5 nm,34 along with approximately 9 and 4 % increases in the intensities of the emission maxima at 595 nm and 650 nm, respectively (Figure 9). Analysis of the UV/Vis titration data by using Specfit35 software revealed a 1:1 stoichiometric association constant of log K=3.38±0.04 (K=2396 M−1), which is in good agreement with the value determined by 1H NMR spectroscopy. By comparison, addition of increasing concentrations of TBABr and TBAI to DMSO solutions of the catenane produced no discernible shifts in the maximum of the Soret band absorbance and only slight, random fluctuations in the intensities of the fluorescence emission spectra (see the Supporting Information, Figures S27 and S28), which corroborates the 1H NMR evidence that these larger halides do not significantly interact with the [2]catenane host system in DMSO.
Figure 9.
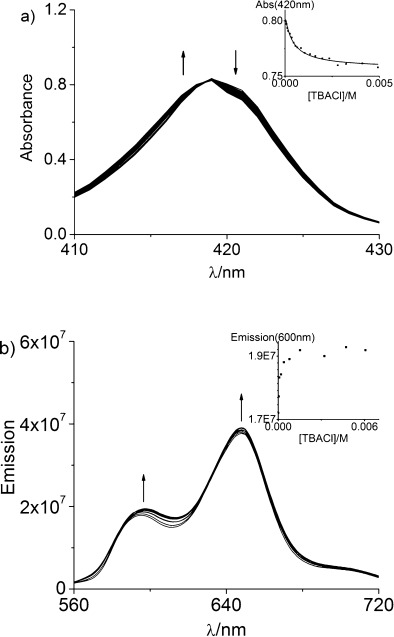
a) Changes in the Soret band absorbance of a 2 μM solution of the [2]catenane 16⋅PF6 in DMSO upon addition of an increasing concentration of TBACl (final TBACl concentration: 4.9 mM). Inset: change in absorbance at 420 nm as a function of [TBACl]; square data points represent experimental data; continuous line represents the calculated binding curve for K=2396 M−1. b) Changes in the fluorescence emission spectrum of a 40 μM solution of the the [2]catenane 16⋅PF6 in DMSO upon addition of an increasing concentration of TBACl (final TBACl concentration: 6.1 mM). λex=548 nm. Inset: change in emission at 600 nm as a function of [TBACl].
Conclusions
A new strapped-porphyrin-containing [2]catenane anion-host system was prepared through an amide-condensation-based clipping reaction, which was directed by chloride anion templation in combination with pyridine–zinc ligation and aromatic donor–acceptor interactions. Upon removal of the halide template, the catenane was shown to selectively recognise chloride (K=2144(149) M−1) over larger halide anions in the competitive solvent [D6]DMSO. The [2]catenane also exhibits a modest ability to optically sense chloride anions in DMSO through small but detectable changes in the absorption and emission spectra of the porphyrin chromophore.
Experimental Section
All solvents and reagents were purchased from commercial suppliers and used as received, unless otherwise stated. Dry solvents were obtained by purging with N2 and then passing through an MBraun MPSP-800 column. H2O was deionised and microfiltered by using a Milli-Q Millipore machine. Et3N was distilled and stored over KOH. TBA salts were stored in a vacuum desiccator containing P2O5 prior to use. 1H, 13C, 19F and 31P NMR spectra were recorded on a Varian Mercury-VX 300, a Varian Unity Plus 500, a Bruker AVD500 or a Bruker AVII500 with cryoprobe at 293 K. Chemical shifts are quoted in parts per million relative to the residual solvent peak. Mass spectra were obtained by using a Micromass LCT (ESMS) instrument or a MALDI Micro MX instrument. Electronic absorption spectra were recorded on a PG instruments T60U spectrometer.
X-ray crystallography
Single crystal diffraction data for compounds 3 b and Zn⋅3 a were collected at 150(2) K using graphite monochromated MoKα radiation (λ=0.71073 Å) on a Nonius Kappa CCD diffractometer. Cell parameter determination and refinement and raw frame data integration were carried out by using the DENZO-SMN package.36 Diffraction data for compounds 13, 14⋅Cl, 16⋅Cl and 16⋅PF6 were collected at 100(2) K by using silicon double crystal monochromated synchrotron radiation (λ=0.68890 Å) at Diamond Light Source, beamline I19,37 with a custom-built Rigaku diffractometer.38 Cell parameter determination and refinement and raw frame data integration were carried out by using the CrysAlisPro39 package. The structures were solved by charge-flipping methods using SUPERFLIP40 and refined by full matrix least squares on F2 using the CRYSTALS41 suite. All non-hydrogen atoms were refined with anisotropic displacement parameters. Where appropriate, disordered regions were modelled by using refined partial occupancies, geometric restraints were applied to ensure a physically reasonable model, and thermal and vibrational restraints were applied to maintain sensible ADPs; in addition, when present, diffuse disordered solvent and counteranions were modelled by treating the discrete Fourier transform of the void region as contributions to the calculated structure factors with PLATON/SQUEEZE.42 Hydrogen atoms were generally visible in the difference map and were treated in the conventional manner.43 CCDC 1412108–1412113 contain the supplementary crystallographic data (excluding structure factors) for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Synthetic procedures and characterisation data
The syntheses of the known compounds 218 and 4–7b13a, 44 are described in the supporting information. Compounds 1,19 1528 and 1729 were prepared by slight modification of previously reported procedures.
α,α- and α,β-Bis(amino)porphyrins 3 a and 3 b: 5,15-Bis-(2-nitrophenyl)porphyrin 2 (0.47 g, 0.84 mmol) was suspended in 37 % HCl(aq) (35 mL). The mixture was sonicated briefly and then stirred at room temperature for 20 min. SnCl2⋅2 H2O (0.10 g, 4.42 mmol) was added and the reaction mixture was stirred vigorously under N2 for 16 h. After cooling to 0 °C, saturated NH4OH(aq) was added cautiously until the pH reached 7. CH2Cl2 (30 mL) was added. The mixture was stirred vigorously at room temperature for 30 min and then transferred to a separating funnel. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3×25 mL). The combined CH2Cl2 extracts were washed with H2O (2×50 mL), dried over MgSO4 and concentrated on a rotary evaporator to give a purple solid, which contained a mixture of the α,α- and α,β-bis(amino)porphyrin derivatives 3 a and 3 b. This atropisomeric mixture was separated by column chromatography. The α,β-isomer 3 b was eluted with 98:2 CH2Cl2/EtOAc and obtained as a purple solid (0.165 g, 40 %). 9:1 CH2Cl2/EtOAc was then used to elute the α,α-isomer 3 a, which was also obtained as a purple solid (0.152 g, 37 %).
Characterisation data for α,α-5,15-bis(2-aminophenyl)porphyrin 3 a: 1H NMR (500 MHz; CDCl3): δ=10.31 (s, 2 H, porphyrin-meso-H), 9.41 (d, 3J=4.6 Hz, 4 H, β-pyrrole-H), 9.12 (d, 3J=4.6 Hz, 4 H, β-pyrrole-H), 7.97 (d, 3J=8.2 Hz, 2 H, Ar–H), 7.67–7.64 (m, 2 H, Ar–H), 7.25–7.22, (m, 2 H, Ar–H), 7.17 (d, 3J=8.2 Hz, 2 H, Ar–H), 3.56 (br s, 4 H, NH2), −3.15 ppm (br s, 2 H, pyrrole-NH); 13C NMR (75 MHz; CDCl3): δ=147.3, 146.9, 145.6, 134.8, 132.0, 130.8, 129.7, 126.3, 117.7, 115.3, 114.9, 105.0 ppm; UV/vis (CH2Cl2): λmax (ε)=405 (250 000), 501 (18 000), 536 (5000), 574 (6000), 628 nm (1500 mol−1 m3 cm−1); ESMS m/z: 493.21 ([M+H]+; C32H25N6 requires 493.21); HRMS m/z: 493.2133 ([M+H]+; C32H25N6 requires 493.2135).
Characterisation data for α,β-5,15-bis(2-aminophenyl)porphyrin 3 b: 1H NMR (500 MHz; CDCl3): δ=10.31 (s, 2 H, porphyrin-meso-H), 9.40 (d, 3J=3.8 Hz, 4 H, β-pyrrole-H), 9.12 (d, 3J=3.8 Hz, 4 H, β-pyrrole-H), 7.92 (d, 3J=7.6 Hz, 2 H, Ar–H), 7.67–7.64 (m, 2 H, Ar–H), 7.24–7.21, (m, 2 H, Ar–H), 7.18 (d, 3J=8.2 Hz, 2 H, Ar–H), 3.59 (br s, 4 H, NH2), −3.16 ppm (br s, 2 H, pyrrole-NH); ESMS m/z: 493.22 ([M+H]+; C32H25N6 requires 493.21; UV/vis (CH2Cl2): λmax (ε)=405 (268 000), 502 (19 000), 536 (5500), 574 (6000), 629 nm (1000 mol−1 m3 cm−1); HRMS m/z: 493.2134 ([M+H]+; C32H25N6 requires 493.2135). Single crystals suitable for X-ray structural determination were grown by slow evaporation from CH2Cl2. Single crystal data: C32H24N6, Mr=492.57; monoclinic, P21/c; a=8.5216(2), b=10.9750(3), c=13.7764(4) Å; α=γ=90°, β=102.6958(14)°; V=1256.96(6) Å3; data/restraints/parameters: 2399/0/172; Rint=0.024; final R1=0.047 (I>2σ(I)); wR2=0.118 (I>2σ(I)); Δρmin,max=−0.24, +0.35 e Å−3.
ZnII α,α-bis(amino)porphyrin Zn⋅3 a: The α,α-bis(amino)porphyrin 3 a (0.15 g, 0.30 mmol) was dissolved in CH2Cl2 (25 mL) and a solution of Zn(OAc)2⋅2H2O (0.33 g, 1.52 mmol) in MeOH (25 mL) was added. The solution was stirred at room temperature under nitrogen for 18 h, before being concentrated on a rotary evaporator without applying heat. The residual solid was dissolved in DMF (2 mL) and H2O (40 mL) was added. The resulting precipitate was collected by filtration, washed with H2O (8×15 mL) followed by MeOH (2×2.5 mL) and dried under high vacuum. After recrystallization (CH2Cl2/hexane), the product was obtained as a pink/purple solid (0.15 g, 91 %). 1H NMR (300 MHz; [D6]DMSO): δ=10.29 (s, 2 H, meso-H), 9.46 (d, 3J=4.7 Hz, 4 H, β-pyrrole-H), 8.93 (d, 3J=4.7 Hz, 4 H, β-pyrrole-H), 7.77 (dd, 3J=7.3 Hz, 4J=1.5 Hz, 2 H, meso-Ar–H), 7.58–7.53 (m, 2 H, meso-Ar–H), 7.16 (d, 3J=7.6 Hz, meso-Ar–H), 7.09–7.04 (m, 2 H, meso-Ar–H), 4.36 ppm (s, 4 H, NH2); 13C NMR (75 MHz; [D6]DMSO): δ=149.5, 149.0, 148.2, 148.1, 134.3, 132.2, 131.4, 129.0, 126.7, 115.6, 115.4, 114.6, 105.6 ppm; ESMS m/z: 555.15 ([M+H]+; C32H23N6Zn requires 555.13); HRMS m/z 555.1280 ([M+H]+; C32H23N6Zn requires 555.1270); UV/vis (CH2Cl2): λmax (ε)=406 (107 000), 535 (7000), 570 nm (2000 mol−1 m3 cm−1). Single crystals of the methanol solvate suitable for X-ray structural determination were grown by slow evaporation from CH2Cl2/MeOH. Single crystal data: C33H26N6OZn, Mr=587.99; triclinic, P$\bar 1$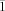 , a=10.3352(2), b=11.5808(2), c=12.3227(3) Å; α=75.5368(8), β=69.8504(9), γ=88.4691(10)°; V=1337.91(5) Å3; data/restraints/parameters: 5470/0/388; Rint=0.022; final R1=0.032 (I>2σ(I)); wR2=0.066 (I>2σ(I)); Δρmin,max=−0.52, +0.50 e Å−3.
, a=10.3352(2), b=11.5808(2), c=12.3227(3) Å; α=75.5368(8), β=69.8504(9), γ=88.4691(10)°; V=1337.91(5) Å3; data/restraints/parameters: 5470/0/388; Rint=0.022; final R1=0.032 (I>2σ(I)); wR2=0.066 (I>2σ(I)); Δρmin,max=−0.52, +0.50 e Å−3.
Ethyl 2-(4-{2-[(tert-butoxycarbonyl)amino]ethoxy}phenoxy)acetate 8: tert-Butyl[2-(4-hydroxyphenoxy)ethyl]carbamate 7 (0.75 g, 2.96 mmol) was dissolved in dry THF (100 mL) and NaH (0.148 g of a 60 % dispersion in mineral oil, 3.70 mmol) was added. The mixture was stirred at room temperature under N2 for 20 min. Ethyl bromoacetate (0.99 g, 0.66 mL, 5.92 mmol) was added and the reaction mixture was heated to 50 °C, and maintained at this temperature under N2 for 18 h, before being cooled to room temperature, diluted with H2O (30 mL) and extracted with CH2Cl2 (4×30 mL). The combined organic extracts were washed with brine (50 mL), dried over MgSO4 and concentrated under reduced pressure. Purification of the residue by column chromatography (2 % MeOH in CH2Cl2) afforded the product as a viscous, pale yellow oil (0.95 g, 94 %). 1H NMR (300 MHz; [D6]acetone): δ=6.87–6.83 (m, 8 H, hydroquinone-Ar–H), 6.14 (br s, 1 H, NH), 4.63 (s, 2 H, OCH2CO2Et), 4.19 (quartet, 3J=7.0 Hz, 2 H, CH2CH3), 3.98 (t, 3J=5.58 Hz, 2 H, OCH2CH2NH) 3.45–3.39 (m, 2 H, OCH2CH2NH), 1.40 (s, 9 H, tBu-CH3), 1.24 ppm (t, 3J=7.0 Hz, 3 H, CH2CH3); 13C NMR (75 MHz; [D6]acetone): δ=169.7, 156.8, 154.5, 153.4, 116.5, 116.2, 78.8, 68.2, 66.5, 61.4, 40.8, 28.7, 14.5 ppm; ESMS m/z: 362.15 ([M+Na]+; C17H25NNaO6 requires 362.16); 701.32 ([2 M+Na]+; C34H50N2NaO12 requires 701.33); HRMS m/z: 362.1569. ([M+Na]+; C17H25NNaO6 requires 362.1572).
Ethyl 2-[4-(2-aminoethoxy)phenoxy]acetate hydrochloride 9⋅HCl: Ethyl 2-(4-{2-[(tert-butoxycarbonyl)amino]ethoxy}phenoxy)acetate 8 was dissolved in Et2O (25 mL). Gaseous HCl was bubbled slowly through the solution for a period of 2 h, during which time a white precipitate formed. The mixture was then stirred at room temperature under N2 for an additional 2 h. The precipitate was collected by filtration, washed with Et2O (5×5 mL) and dried under vacuum to afford the product as a white solid (0.75 g, 97 %). 1H NMR (300 MHz; [D6]DMSO): δ=8.12 (br s, 3 H, NH3⋅Cl), 6.94–6.87 (m, 4 H, hydroquinone-Ar–H), 4.71 (s, 2 H, OCH2CO2Et), 4.15 (quartet, 3J=7.0 Hz, 2 H, CH2CH3), 4.10 (t, 3J=5.3 Hz, 2 H, OCH2CH2NH), 3.19–3.15 (br m, 2 H, OCH2CH2NH), 1.20 ppm (t, 3J=7.0 Hz, 3 H, CH2CH3); 13C NMR (75 MHz; [D6]DMSO): δ=168.9, 152.4, 152.1, 115.7, 115.5, 65.2, 64.8, 60.6, 38.3, 14.1 ppm; ESMS m/z: 240.12 ([M−Cl]+; C12H18NO4 requires 240.12); 262.10 ([M−HCl+Na]+; C12H17NaNO4 requires 262.11); HRMS m/z: 240.1236 ([M−Cl]+; C12H18NO4 requires 240.1230).
Compound 11: 3,5-Pyridinedicarboxylic acid (0.197 g, 1.18 mmol) was suspended in dry CH2Cl2 (30 mL). Oxalyl chloride (0.75 g, 0.50 mL, 5.89 mmol) and DMF (1 drop) were added. The mixture was stirred at room temperature under N2 for 18 h, by which time it had formed a homogenous solution. The solvent was removed on a rotary evaporator and the residue dried under high vacuum for 60 min to afford 3, 5-bis(chlorocarbonyl) pyridine 10 as a waxy, off-white solid, which was redissolved in dry CH2Cl2 (20 mL). The solution was cooled to 0 °C in an ice bath and a solution of 9⋅HCl in dry CH2Cl2 (20 mL) and dry Et3N (2.5 mL) was added dropwise by syringe. After addition was complete, the reaction mixture was stirred at 0 °C under N2 for 15 min. The ice bath was then removed and the reaction mixture was allowed to stir at room temperature for 18 h. The solution was washed with 10 % citric acid(aq) (2×50 mL), followed by saturated NaHCO3(aq) (2×50 mL) and H2O (50 mL). The organic layer was dried over MgSO4 and concentrated under reduced pressure. Purification of the residue by column chromatography (2–4 % MeOH in CH2Cl2 afforded the product as a white solid (0.52 g, 73 %). 1H NMR (500 MHz; CDCl3): δ=9.14 (d, 4J=2.2 Hz, 2 H, py-Ar–H), 8.48 (t, 4J=2.2 Hz, 1 H, py-Ar–H), 6.80 (t, 3J=5.6 Hz, 2 H, amide-NH), 6.87–6.83 (m, 8 H, Ar–H), 4.55 (s, 4 H, OCH2CO2Et), 4.25 (quartet, 3J=7.1 Hz, 4 H, CH2CH3), 4.10 (t, 3J=5.1 Hz, 4 H, NHCH2CH2O), 3.88–3.85 (m, 4 H, NHCH2CH2O), 1.26 ppm (t, 3J=7.1 Hz, 6 H, CH2CH3); 13C NMR (75 MHz; CDCl3): δ=169.2, 165.0, 153.2, 152.4, 150.7, 133.6, 129.7, 115.8, 115.4, 66.9, 66.1, 61.4, 39.8, 14.1 ppm; ESMS m/z: 632.21 ([M+Na]+; C31H35N3NaO10+ requires 632.22); 1241.47 ([2 M+Na]+; C62H70N6NaO20 requires 1241.45); HRMS m/z: 632.2216 ([M+Na]+; C31H35N3NaO10 requires 632.2215).
Compound 12: Compound 11 (0.50 g, 0.82 mmol) was dissolved in CH2Cl2 (40 mL) and MeOH (40 mL). A solution of KOH (0.18 g, 3.28 mmol) in H2O (7 mL) was added. The solution was stirred at RT under N2 for 60 h, during which time a white precipitate formed. H2O (40 mL) was added and the organic solvents were removed on a rotary evaporator. The pH of the remaining aqueous suspension was adjusted to 7 by addition of 10 % citric acid(aq) and the solid was collected by filtration, washed with H2O (4×15 mL), MeOH (3×5 mL) and CH2Cl2 (3×10 mL) and dried under high vacuum to afford the product as a white solid (0.40 g, 88 %). 1H NMR (300 MHz; [D6]DMSO): δ=9.06 (d, 4J=2.2 Hz, 2 H, py-Ar–H), 8.98 (t, 3J=5.3 Hz, 2 H, amide-NH), 8.57 (t, 4J=2.2 Hz, 1 H, py-Ar–H), 6.85 (d, 3J=9.1 Hz, 4 H, hydroquinone-Ar–H), 6.75 (d, 3J=9.1 Hz, 4 H, hydroquinone-Ar–H), 4.38 (s, 4 H, OCH2CO2H), 4.07 (t, 3J=5.3 Hz, 4 H, OCH2CH2NH), 3.64–3.59 ppm (m, 4 H, OCH2CH2NH); ESMS m/z: 554.16 (M+H]+; C27H28N3O10 requires 554.18); 576.15 ([M+Na]+; C27H27N3NaO10 requires 576.16); HRMS m/z: 576.1641 ([M−H]−; C27H26N3O10 requires 552.1624).
3,5-Pyridine bis(amide)-strapped porphyrin macrocycle 13: Dry, de-gassed DMF was added to a flask containing Zn⋅3 a (0.14 g, 0.25 mmol), compound 12 (0.14 g, 0.25 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (0.12 g, 0.63 mmol), 1-hydroxybenzotriazole hydrate (0.077 g, ca. 0.57 mmol) and 4-(dimethylamino)pyridine (0.030 g, 0.25 mmol). The mixture was sonicated briefly and then stirred vigorously at room temperature under N2 for 60 h. After removal of the solvent under reduced pressure, the residual solid was dissolved in 9:1 CH2Cl2/MeOH mixture (100 mL), dry-loaded onto silica and purified by column chromatography (1–5 % MeOH in CH2Cl2) to afford the product as a purple solid (0.12 g, 44 %). 1H NMR (500 MHz; [D6]DMSO): δ=10.43 (s, 2 H, porphyrin-meso-H), 9.51 (d, 3J=4.4 Hz, 4 H, porphyrin-β-pyrrole-H), 8.83 (d, 3J=4.4 Hz, 4 H, porphyrin-β-pyrrole-H), 8.70 (s, 2 H, amide-NH), 8.60 (d, 3J=8.3 Hz, 2 H, Ar–H), 8.46 (s, 2 H, py-Ar–H), 8.43 (br t, 2 H, amide-NH), 8.15 (s, 1 H, py-Ar–H), 8.04 (dd, 3J=7.3 Hz, 4J=1.5 Hz, 2 H, Ar–H), 7.93–7.89 (m, 2 H, Ar–H), 7.65–7.61 (m, 2 H, Ar–H), 6.21 (d, 3J=9.0 Hz, 4 H, hydroquinone-Ar–H), 5.28 (d, 3J=9.0 Hz, 4 H, hydroquinone-Ar–H), 3.92 (t, 3J=4.9 Hz, 4 H, OCH2CH2NH), 3.76 (s, 4 H, OCH2CONH), 3.56–3.53 ppm (m, 4 H, OCH2CH2NH); 13C NMR (75 MHz, [D6]DMSO): δ=166.5, 164.6, 152.7, 150.2, 150.1, 149.4, 149.3, 137.6, 135.3, 133.2, 133.1, 132.8, 131.0, 129.1, 128.9, 123.1, 121.1, 114.9, 114.5, 113.2, 106.3, 66.7, 66.6 ppm, one 13C signal is coincident with [D6]DMSO; ESMS m/z: 1094.29 ([M+Na]+; C59H45N9NaO8Zn requires 1094.26); HRMS m/z: 1094.2452 ([M+Na]+; C59H45N9NaO8Zn requires 1094.2575). Single crystals suitable for X-ray structural determination were grown by layered diffusion of hexane into a CH2Cl2/MeOH solution of the macrocycle. Single crystal data: C59H45N9O8Zn⋅CH2Cl2, Mr=1158.37; triclinic, P$\bar 1$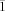 ; a=14.0873(8), b=14.2958(7), c=15.5922(9) Å; α=63.513(5), β=71.193(5), γ=82.978(4)°; V=2659.6(3) Å3; data/restraints/parameters: 7853/0/721; Rint=0.085; final R1=0.057 (I>2σ(I)); wR2=0.147 (I>2σ(I)); Δρmin,max=−0.77, +0.86 e Å−3.
; a=14.0873(8), b=14.2958(7), c=15.5922(9) Å; α=63.513(5), β=71.193(5), γ=82.978(4)°; V=2659.6(3) Å3; data/restraints/parameters: 7853/0/721; Rint=0.085; final R1=0.057 (I>2σ(I)); wR2=0.147 (I>2σ(I)); Δρmin,max=−0.77, +0.86 e Å−3.
3,5-Pyridinium bis(amide)-strapped porphyrin macrocycle 14⋅Cl: Macrocycle 13 (0.105 g, 0.098 mmol) was dissolved in dry, de-gassed DMF (6 mL). NaHCO3 (0.058 g, 0.68 mmol) was added, followed by MeI (0.34 g, 0.15 mL, 2.41 mmol). The reaction mixture was heated at 80 °C under N2 for 2 h, using a reflux condenser to prevent loss of MeI. After cooling to room temperature, the excess MeI was removed on a rotary evaporator. The remaining DMF solution was diluted with CH2Cl2 (100 mL) and then washed with 1 M NH4Cl(aq) (6×75 mL) followed by H2O (2×75 mL). After concentration of the organic layer under reduced pressure, and recrystallisation of the residual solid (CH2Cl2/MeOH/hexane), the product was obtained as a purple solid (0.11 g, 99 %). 1H NMR (500 MHz, [D6]DMSO): δ=10.42 (s, 2 H, porphyrin-meso-H), 9.48 (d, 3J=4.4 Hz, 4 H, porphyrin-β-pyrrole-H), 9.40 (s, 2 H, pyridinium-Ar–H), 9.14 (br s, 2 H, amide-NH), 8.99 (s, 1 H, pyridinium-Ar–H), 8.75 (d, 3J=4.4 Hz, 4 H, porphyrin-β-pyrrole-H), 8.57 (d, 3J=9.2 Hz, 2 H, porphyrin-meso-Ar–H), 8.34 (s, 2 H, amide-NH), 8.03 (d, 3J=7.6 Hz, 2 H, porphyrin-meso-Ar–H), 7.90–7.86 (m, 2 H, porphyrin-meso-Ar–H), 7.62–7.58 (m, 2 H, porphyrin-meso-Ar–H), 6.30 (d, 3J=9.0 Hz, 4 H, hydroquinone-Ar–H), 5.29 (d, 3J=9.0 Hz, 4 H, hydroquinone-Ar–H), 4.30 (s, 3 H, methylpyridinium-CH3), 3.98 (t, 3J=4.8 Hz, 4 H, CH2), 3.80 (s, 4 H, CH2), 3.69–3.66 ppm (m, 4 H, CH2); 13C NMR (125 MHz; [D6]DMSO): δ=166.5, 161.3, 152.6, 150.1, 149.4, 149.2, 147.1, 140.3, 137.6, 135.2, 133.2, 132.8, 132.7, 131.0, 128.9, 123.1, 121.0, 114.9, 114.4, 113.1, 106.3, 66.6, 66.4, 48.4 ppm, one 13C signal is coincident with [D6]DMSO; ESMS m/z: 1086.32 ([M−Cl]+; C60H48N9O8Zn requires 1086.29); HRMS m/z: 1086.2929 ([M−Cl]+; C60H48N9O8Zn requires 1086.2912). Single crystals of the pyridine solvate suitable for X-ray structural determination were grown by layered diffusion of hexane into a CH2Cl2/MeOH/pyridine solution of the macrocycle. Single crystal data: C65H53N10O8Zn⋅Cl, Mr=1203.03; monoclinic, P21/c; a=10.1084(3), b=21.8767(7), c=31.8505(12) Å; α=γ=90°, β=90,349(3)°; V=7043.2(4) Å3; data/restraints/parameters: 11 350/0/766; Rint=0.100; final R1=0.077 (I>2σ(I)); wR2=0.182 (I>2σ(I)); Δρmin,max=−0.61, +0.51 e Å−3.
Chloride catenane 16⋅Cl: 3,5-Pyridinedicarboxylic acid (0.014 g, 0.081 mmol) was suspended in dry CH2Cl2 (3 mL). Oxalyl chloride (0.051 g, 0.034 mL, 0.41 mmol) and DMF (1 drop) were added and the mixture was stirred at room temperature under N2 until it had formed a homogenous solution (2.5 h). After removal of the solvent on a rotary evaporator, the residual off-white solid was dried under high vacuum for 3 h and then re-dissolved in dry CH2Cl2 (2.5 mL). Et3N (0.041 g, 0.057 mL, 0.41 mmol) was added and the solution was added dropwise to a solution of compound 15 (0.038 g, 0.081 mmol) and macrocycle 14⋅Cl (0.040 g, 0.032 mmol) in dry CH2Cl2 (10 mL). The reaction mixture was stirred at room temperature under N2 for 90 min, then diluted with CH2Cl2 (5 mL). The solution was washed sequentially with H2O (10 mL), saturated NaHCO3(aq) (10 mL) and brine (10 mL), dried over MgSO4 and concentrated on a rotary evaporator. The residue was purified by preparative thin layer chromatography (SiO2; 8 % MeOH in CH2Cl2, then 5 % MeOH in EtOAc) and recrystallisation (CH2Cl2/MeOH, then CH2Cl2/MeOH/hexane) to give the product as a purple solid (0.017 g, 30 %). 1H NMR (500 MHz; [D6]DMSO): δ=10.34 (s, 2 H, porphyrin-meso-H), 9.52 (d, 3J=4.6 Hz, 4 H, porphyrin-ß-pyrrole-H), 9.04 (s, 1 H, pyridinium-Ar–H), 8.97 (s, 2 H, pyridinium-Ar–H), 8.96 (s, 2 H, amide-NH), 8.91 (d, 4 H, 3J=4.6 Hz, porphyrin-ß-pyrrole-H), 8.52 (d, 3J=8.0 Hz, 2 H, Ar–H), 8.31 (br s, 2 H, amide-NH), 8.28 (s, 1 H, py-Ar–H), 7.91 (t, 3J=8.0 Hz, 2 H, Ar–H), 7.86 (d, 3J=7.4 Hz, 2 H, Ar–H), 7.72 (br s, 2 H, amide-NH), 7.59 (t, 3J=7.4 Hz, 2 H, Ar–H), 6.28 (d, 3J=9.0 Hz, 4 H, hydroquinone-Ar–H), 6.14 (d, 3J=8.2 Hz, 4 H, hydroquinone-Ar–H), 6.03 (d, 3J=9.0 Hz, 4 H, hydroquinone-Ar–H), 5.45 (d, 3J=8.2 Hz, 4 H, hydroquinone-Ar–H), 4.47 (s, 3 H, methylpyridinium-CH3), 3.76 (br t, 4 H, CH2), 3.72 (s, 4 H, CH2), 3.56 (br t, 4 H, CH2), 3.50–3.46 (m, 16 H, CH2), 3.44–3.42 (m, 4 H, CH2), 3.02–2.99 ppm (m, 4 H, CH2), signal corresponding to the external pyridinium proton 16 is not observed in [D6]DMSO owing to signal broadening; 13C NMR (125 MHz; [D6]DMSO): δ=166.4, 162.4, 159.9, 152.3, 152.2, 151.5, 150.3, 149.4, 149.2, 147.2, 145.8, 137.2, 136.0, 134.2, 132.8, 132.1, 131.7, 131.2, 128.7, 126.7, 123.6, 122.9, 114.6, 114.4, 114.3, 114.0, 113.8, 106.2, 69.9, 69.8, 69.0, 67.3, 65.0, 48.6 ppm, one 13C signal is coincident with [D6]DMSO; ESMS m/z: 1681.49 ([M−Cl]+; C91H85N12O17Zn requires 1681.54); HRMS m/z: 1683.5339 ([M−Cl]+; C91H85N12O17Zn requires 1683.5419). Single crystals suitable for X-ray structural determination were grown by layered diffusion of hexane into a CH2Cl2/MeOH solution of the catenane. Single crystal data: C91H85N12O17Zn⋅2(CH2Cl2)⋅Cl, Mr=1889.44; triclinic, P$\bar 1$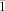 ; a=14.5199(2), b=16.4604(2), c=21.6127(4) Å; α=99.0338(12), β=104.2010(14), γ=92.0154(11)°; V=4930.86(13) Å3; data/restraints/parameters: 15 424/0/1153; Rint=0.025; final R1=0.037 (I>2σ(I)); wR2=0.087 (I>2σ(I)); Δρmin,max=−0.69, +0.90 e Å−3.
; a=14.5199(2), b=16.4604(2), c=21.6127(4) Å; α=99.0338(12), β=104.2010(14), γ=92.0154(11)°; V=4930.86(13) Å3; data/restraints/parameters: 15 424/0/1153; Rint=0.025; final R1=0.037 (I>2σ(I)); wR2=0.087 (I>2σ(I)); Δρmin,max=−0.69, +0.90 e Å−3.
Hexafluorophosphate catenane 16⋅PF6: The chloride catenane 16⋅Cl (0.009 g, 0.005 mmol) was dissolved in DMSO (4 mL). The flask was wrapped with foil to protect the contents from light and a solution of AgNO3 (0.004 g, 0.026 mmol) in H2O (0.16 mL) was added. After stirring at room temperature under N2 for 45 min, the solution was filtered through a plug of celite and then added dropwise to 0.05 M NH4PF6(aq) (60 mL), which resulted in the formation of a purple precipitate. The precipitate was collected by filtration, washed with H2O (6×7.5 mL), EtOH (3×2 mL) and hexane (3×5 mL) and dried under vacuum to yield the product as a purple solid (0.007 g, 73 %). 1H NMR (500 MHz, [D6]DMSO): δ=10.33 (s, 2 H, porphyrin-meso-H), 9.49 (d, 3J=4.2 Hz, 4 H, porphyrin-β-pyrrole-H), 9.08 (s, 2 H, pyridinium-Ar–H), 8.84–8.81 (m, 5 H, 4 porphyrin-β-pyrrole-H+1 pyridinium-Ar–H), 8.72 (s, 2 H, amide-NH), 8.63 (br s, 2 H, amide-NH), 8.49 (br s, 2 H, amide-NH), 8.39 (d, 3J=8.0 Hz, 2 H, porphyrin-meso-Ar–H), 8.32 (s, 1 H, py-Ar–H), 7.93 (d, 3J=6.9 Hz, 2 H, porphyrin-meso-Ar–H), 7.88–7.85 (m, 2 H, porphyrin-meso-Ar–H), 7.61–7.58 (m, 2 H, porphyrin-meso-Ar–H), 6.39 (d, 3J=7.8 Hz, 4 H, hydroquinone-Ar–H), 6.28 (d, 3J=8.0 Hz, 4 H, hydroquinone-Ar–H), 6.00 (d, 3J=8.0 Hz, 4 H, hydroquinone-Ar–H), 5.87 (d, 3J=7.8 Hz, 4 H, hydroquinone-Ar–H), 4.23 (s, 3 H, methylpyridinium-CH3), 3.83–3.81 (m, 4 H, CH2), 3.71–3.69 (m, 4 H, CH2), 3.64 (s, 4 H, CH2), 3.55–3.51 (m, 4 H, CH2), 3.42–3.39 (m, 4 H, CH2), 3.08–3.02 (m, 8 H, CH2), 2.99–2.97 3.83–3.81 ppm (m, 4 H, CH2); 19F NMR (470 MHz, [D6]DMSO): δ=−70.1 ppm (d, 2J(F,P)=711 Hz), 31P NMR (203 MHz, [D6]DMSO): δ=−144.2 ppm (septet, 2J(F,P)=711 Hz); ESMS m/z: 1683.51 ([M−PF6]+; C91H85N12O17Zn requires 1683.54). Single crystals suitable for X-ray structural determination were grown by layered diffusion of di-isopropyl ether into an acetone solution of the catenane. Single crystal data: C91H85N12O17Zn⋅0.5(PF6), Mr=1756.59; tetragonal, P4/ncc; a=41.5009(6), b=41.5009(6), c=24.4007(5) Å; α=β=γ=90°, V=42 025.9(15) Å3; data/restraints/parameters: 10 933/3740/1415; Rint=0.110; final R1=0.091 (I>2σ(I)); wR2=0.237 (I>2σ(I)); Δρmin,max=−0.51, +0.87 e Å−3.
Acknowledgments
We thank the Wellcome Trust and the European Research Council (under the European Union’s 7th Framework Program; FP7/2007-2013), ERC Advanced Grant Agreement (no. 267426) for postdoctoral funding (A.B.) and the Engineering and Physical Sciences Research Council for studentships (A.B., M.J.L.). N.L.K. thanks the Royal Commission for the Exhibition of 1851 for a Research Fellowship. We also thank Diamond Light Source for an award of beam time on I19 (MT9981), the beamline scientists (Drs. David Allan, Harriott Nowell, Sarah Barnett and Mark Warren) for technical support, Dr. Richard Knighton for collection of a preliminary single crystal data set and Prof. Stephen Faulkner for the use of spectrometers.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Sauvage JP, editor; Dietrich-Buchecker C, editor. Molecular Catenanes, Rotaxanes and Knots: A Journey through the World of Molecular Topology. Weinheim: Wiley-VCH; 1999. [Google Scholar]
- 1b.Evans NH, Beer PD. Supramolecular Chemistry: From Molecules to Nanomaterials. Hoboken: Wiley; 2012. (Eds.: J. W. Steed, P. A. Gale) [Google Scholar]
- 2.For examples, see
- 2a.Leigh DA, Wong JKY, Dehez F, Zerbetto F. Nature. 2003;424:174–179. doi: 10.1038/nature01758. [DOI] [PubMed] [Google Scholar]
- 2b.Hernandez JV, Kay ER, Leigh DA. Science. 2004;306:1532–1537. doi: 10.1126/science.1103949. for a review of synthetic molecular motors and machines, see: [DOI] [PubMed] [Google Scholar]
- 2c.Kay ER, Leigh DA, Zerbetto F. Angew. Chem. Int. Ed. 46:72–191. doi: 10.1002/anie.200504313. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007;119 [Google Scholar]
- 3.Lee J-J, White AG, Rice DR, Smith BD. Chem. Commun. 2013;49:3016–3018. doi: 10.1039/c3cc40630j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a.Barin G, Frasconi M, Dyar SM, Iehl J, Buyukcakir O, Sarjeant AA, Carmieli R, Coskun A, Wasielewski MR, Stoddart JF. J. Am. Chem. Soc. 2013;135:2466–2469. doi: 10.1021/ja3125004. [DOI] [PubMed] [Google Scholar]
- 4b.Andrievsky A, Ahuis F, Sessler JL, Vogtle F, Gudat D, Moini M. J. Am. Chem. Soc. 1998;120:9712–9713. [Google Scholar]
- 5a.Collier CP, Mattersteig G, Wong EW, Luo Y, Beverly K, Sampaio J, Raymo FM, Stoddart JF, Heath JR. Science. 2000;289:1172–1175. doi: 10.1126/science.289.5482.1172. [DOI] [PubMed] [Google Scholar]
- 5b.Klajn R, Fang L, Coskun A, Olson MA, Wesson PJ, Stoddart JF, Grzybowski BA. J. Am. Chem. Soc. 2009;131:4233–4235. doi: 10.1021/ja9001585. [DOI] [PubMed] [Google Scholar]
- 5c.Olson MA, Braunschweig AB, Fang L, Ikeda T, Klajn R, Trabolsi A, Wesson PJ, Benítez D, Mirkin CA, Grzybowski BA, Stoddart JF. Angew. Chem. Int. Ed. 48:1792–1797. doi: 10.1002/anie.200804558. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009;121 [Google Scholar]
- 5d.Li Q, Sue C-H, Basu S, Shveyd AK, Zhang W, Barin G, Fang L, Sarjeant AA, Stoddart JF, Yaghi OM. Angew. Chem. Int. Ed. 49:6751–6755. doi: 10.1002/anie.201003221. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010;122 [Google Scholar]
- 6a.Dietrich-Buchecker CO, Sauvage JP, Kintzinger JP. Tetrahedron Lett. 1983;24:5095–5098. [Google Scholar]
- 6b.Dietrichbuchecker CO, Sauvage JP, Kern JM. J. Am. Chem. Soc. 1984;106:3043–3045. [Google Scholar]
- 7.For examples, see
- 7a.Leigh DA, Lusby PJ, Teat SJ, Wilson AJ, Wong JKY. Angew. Chem. Int. Ed. 40:1538–1543. doi: 10.1002/1521-3773(20010417)40:8<1538::AID-ANIE1538>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001;113 [Google Scholar]
- 7b.Goldup SM, Leigh DA, Lusby PJ, McBurney RT, Slawin AMZ. Angew. Chem. Int. Ed. 47:6999–7003. doi: 10.1002/anie.200801904. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008;120 [Google Scholar]
- 7c.Leigh DA, Lusby PJ, McBurney RT, Morelli A, Slawin AMZ, Thomson AR, Walker DB. J. Am. Chem. Soc. 2009;131:3762–3771. doi: 10.1021/ja809627j. [DOI] [PubMed] [Google Scholar]
- 7d.Goldup SM, Leigh DA, Long T, McGonigal PR, Symes MD, Wu J. J. Am. Chem. Soc. 2009;131:15924–15929. doi: 10.1021/ja9070317. [DOI] [PubMed] [Google Scholar]
- 7e.Prakasam T, Lusi M, Elhabiri M, Platas-Iglesias C, Olsen J-C, Asfari Z, Cianférani-Sanglier S, Debaene F, Charbonnière LJ, Trabolsi A. Angew. Chem. Int. Ed. 52:9956–9960. doi: 10.1002/anie.201302425. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 for a review of strategies for the metal-directed syntheses of mechanically bonded molecules, see: [Google Scholar]
- 7f.Beves JE, Blight BA, Campbell CJ, Leigh DA, McBurney RT. Angew. Chem. Int. Ed. 50:9260–9327. doi: 10.1002/anie.201007963. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011;123 [Google Scholar]
- 8a.Amabilino DB, Ashton PR, Balzani V, Boyd SE, Credi A, Lee JY, Menzer S, Stoddart JF, Venturi M, Williams DJ. J. Am. Chem. Soc. 1998;120:4295–4307. [Google Scholar]
- 8b.Lu J, Turner DR, Harding LP, Byrne LT, Baker MV, Batten SR. J. Am. Chem. Soc. 2009;131:10372–10373. doi: 10.1021/ja9041912. [DOI] [PubMed] [Google Scholar]
- 8c.Chang C-F, Chuang C-J, Lai C-C, Liu Y-H, Peng S-M, Chiu S-H. Angew. Chem. Int. Ed. 51:10094–10098. doi: 10.1002/anie.201205498. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- 8d.Loots L, Barbour LJ. Chem. Commun. 2013;49:671–673. doi: 10.1039/c2cc37953h. [DOI] [PubMed] [Google Scholar]
- 9a.Hunter CA. J. Am. Chem. Soc. 1992;114:5303–5311. [Google Scholar]
- 9b.Vögtle F, Meier S, Hoss R. Angew. Chem. Int. Ed. Engl. 31:1619–1622. [Google Scholar]; Angew. Chem. 1992;104 [Google Scholar]
- 9c.Johnston AG, Leigh DA, Nezhat L, Smart JP, Deegan MD. Angew. Chem. Int. Ed. Engl. 34:1212–1216. [Google Scholar]; Angew. Chem. 1995;107 [Google Scholar]
- 9d.Lee J-J, Baumes JM, Connell RD, Oliver AG, Smith BD. Chem. Commun. 2011;47:7188–7190. doi: 10.1039/c1cc10946d. [DOI] [PubMed] [Google Scholar]
- 10.Barnes JC, Fahrenbach AC, Cao D, Dyar SM, Frasconi M, Giesener MA, Benítez D, Tkatchouk E, Chernyashevskyy O, Shin WH, Li H, Sampath S, Stern CL, Sarjeant AA, Hartlieb KJ, Liu Z, Carmieli R, Botros YY, Choi JW, Slawin AMZ, Ketterson JB, Wasielewski MR, Goddard WA, Stoddart JF. Science. 2013;339:429–433. doi: 10.1126/science.1228429. [DOI] [PubMed] [Google Scholar]
- 11.Gilday LC, Lang T, Caballero A, Costa PJ, Felix V, Beer PD. Angew. Chem. Int. Ed. 52:4356–4360. doi: 10.1002/anie.201300464. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 [Google Scholar]
- 12a.Fujita M, Ibukuro F, Hagihara H, Ogura K. Nature. 1994;367:720–723. [Google Scholar]
- 12b.Armspach D, Ashton PR, Ballardini R, Balzani V, Godi A, Moore CP, Prodi L, Spencer N, Stoddart JF, Tolley MS, Wear TJ, Williams DJ. Chem. Eur. J. 1995;1:33–55. [Google Scholar]
- 12c.Fang L, Basu S, Sue C-H, Fahrenbach AC, Stoddart JF. J. Am. Chem. Soc. 2011;133:396–399. doi: 10.1021/ja1087562. [DOI] [PubMed] [Google Scholar]
- 12d.Cougnon FBL, Ponnuswamy N, Jenkins NA, Pantoş GD, Sanders JKM. J. Am. Chem. Soc. 2012;134:19129–19135. doi: 10.1021/ja3075727. [DOI] [PubMed] [Google Scholar]
- 13a.Sambrook MR, Beer PD, Wisner JA, Paul RL, Cowley AR. J. Am. Chem. Soc. 2004;126:15364–15365. doi: 10.1021/ja045080b. [DOI] [PubMed] [Google Scholar]
- 13b.Huang BQ, Santos SM, Felix V, Beer PD. Chem. Commun. 2008:4610–4612. doi: 10.1039/b808094a. [DOI] [PubMed] [Google Scholar]
- 13c.Evans NH, Allinson ESH, Lankshear MD, Ng KY, Cowley AR, Serpell CJ, Santos SM, Costa PJ, Felix V, Beer PD. RSC Adv. 2011;1:995–1003. [Google Scholar]
- 13d.Hancock LM, Gilday LC, Kilah NL, Serpell CJ, Beer PD. Chem. Commun. 2011;47:1725–1727. doi: 10.1039/c0cc04683c. [DOI] [PubMed] [Google Scholar]
- 13e.White NG, Beer PD. Chem. Commun. 2012;48:8499–8501. doi: 10.1039/c2cc34210c. [DOI] [PubMed] [Google Scholar]
- 14a.Zhao YJ, Li YL, Li YJ, Zheng HY, Yin XD, Liu HB. Chem. Commun. 2010;46:5698–5700. doi: 10.1039/c0cc01120g. [DOI] [PubMed] [Google Scholar]
- 14b.Chae MK, Suk JM, Jeong KS. Tetrahedron Lett. 2010;51:4240–4242. [Google Scholar]
- 15.Caballero A, Zapata F, Beer PD. Coord. Chem. Rev. 2013;257:2434–2455. For a review of anion templation strategies for the synthesis of anion host systems and sensors, see. [Google Scholar]
- 16. For previous examples of catenane-based optical and electrochemical anion sensors, see.
- 16a.Evans NH, Rahman H, Leontiev AV, Greenham ND, Orlowski GA, Zeng Q, Jacobs RMJ, Serpell CJ, Kilah NL, Davis JJ, Beer PD. Chem. Sci. 2012;3:1080–1089. [Google Scholar]
- 16b.Caballero A, Zapata F, White NG, Costa PJ, Félix V, Beer PD. Angew. Chem. Int. Ed. 51:1876–1880. doi: 10.1002/anie.201108404. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- 17. For previous examples of porphyrin-containing catenanes, see.
- 17a.Gunter MJ, Hockless DCR, Johnston MR, Skelton BW, White AH. J. Am. Chem. Soc. 1994;116:4810–4823. [Google Scholar]
- 17b.Gunter MJ, Farquhar SM. Org. Biomol. Chem. 2003;1:3450–3457. doi: 10.1039/b307383a. [DOI] [PubMed] [Google Scholar]
- 17c.Gunter MJ, Farquhar SM, Mullen KM. New J. Chem. 2004;28:1443–1449. [Google Scholar]
- 17d.Beyler M, Heitz V, Sauvage JP. Chem. Commun. 2008:5396–5398. doi: 10.1039/b811013a. [DOI] [PubMed] [Google Scholar]
- 17e.Langton MJ, Matichak JD, Thompson AL, Anderson HL. Chem. Sci. 2011;2:1897–1901. [Google Scholar]
- 17f.Beyler M, Heitz V, Sauvage JP. New J. Chem. 2011;35:1751–1757. [Google Scholar]
- 17g.Coumans RGE, Elemans J, Rowan AE, Nolte RJM. Chem. Eur. J. 2013;19:7758–7770. doi: 10.1002/chem.201203983. [DOI] [PubMed] [Google Scholar]
- 17h.Aleman Garcia MA, Bampos N. Org. Biol. Chem. 2013;11:27–30. doi: 10.1039/c2ob26587g. [DOI] [PubMed] [Google Scholar]
- 18.Manka JS, Lawrence DS. Tetrahedron Lett. 1989;30:6989–6992. [Google Scholar]
- 19.Balaz M, Collins HA, Dahlstedt E, Anderson HL. Org. Biol. Chem. 2009;7:874–888. doi: 10.1039/b814789b. [DOI] [PubMed] [Google Scholar]
- 20. When TFA was used as the catalyst, compound 22was isolated in lower yields of 9–15 %. Despite numerous attempts to optimise the reaction conditions, we were unable to reproduce the very high yield reported by Manka and Lawrence for compound (ref. [18])
- 21. Compounds 3 a3 b1meso161G−1and can be distinguished by slight differences in the H NMR chemical shift of their -aryl proton signals. Variable temperature H NMR experiments indicated that the two compounds are stable to interconversion in [D ]DMSO at room temperature but begin to isomerise rapidly when the temperature is raised above 70 °C. The two atropisomers were observed to remain in slow exchange on the H NMR timescale at 500 MHz up to temperatures of at least 150 °C, from which the lower limit of the kinetic barrier to rotation can be estimated to be Δ ≥94 kJ mol.
- 22a.Gunter MJ, Mander LN. J. Org. Chem. 1981;46:4792–4795. [Google Scholar]
- 22b.Young R, Chang CK. J. Am. Chem. Soc. 1985;107:898–909. [Google Scholar]
- 23.The α,β-isomer 3 b3 a3 b223 a. could subsequently be converted into the desired α,α-isomer by using the equilibrium displacement technique described by Lindsey (ref. [45]). After heating a toluene solution of compound in the presence of silica gel, and separation of the resultant isomeric mixture by column chromatography (CH Cl /EtOAC), the α,α-isomer was isolated in yields of 54–75 %.
- 24.Andrews PR, Munro SLA, Sadek M, Wong MG. J. Chem. Soc. Perkin Trans. 2. 1988:711–718. [Google Scholar]
- 25.The axially coordinated methanol molecule was found to be disordered over two positions, with fractional occupancies of 62 and 38 %. The Zn⋅⋅⋅O distance measurement is based upon the higher occupancy position, which is assumed to be the more thermodynamically stable
- 26.Reaction of the equivalent α,β-isomer of the zinc(II) bis(amino)porphyrin Zn⋅3 b1213131613. with the bis-acid afforded the corresponding α,β-isomeric form of macrocycle in 24 % yield. However, although this experiment proved that the strap component of macrocycle is sufficiently long and flexible to allow formation of this α,β-macrocyclic species, variable-temperature H NMR experiments in [D ]DMSO confirmed that the α,α- and α,β-isomeric forms of macrocycle are kinetically stable to interconversion up to temperatures of at least 100 °C.
- 27.When the reaction was carried out in the absence of NaHCO3. demetallation of the zinc(II) porphyrin took place, presumably owing to a build-up of HI in the reaction mixture, and the product was isolated in its free base form. A similar observation was recently made by Flood and co-workers (ref. [46])
- 28.Hancock LM, Gilday LC, Carvalho S, Costa PJ, Felix V, Serpell CJ, Kilah NL, Beer PD. Chem. Eur. J. 2010;16:13082–13094. doi: 10.1002/chem.201002076. [DOI] [PubMed] [Google Scholar]
- 29.Hancock LM, Beer PD. Chem. Eur. J. 2009;15:42–44. doi: 10.1002/chem.200802029. [DOI] [PubMed] [Google Scholar]
- 30.Unfortunately, it was not possible to obtain a comparative 11422161314. H NMR spectrum of the strapped porphyrin macrocycle ⋅Cl in CD Cl owing to the macrocycle’s extremely low solubility in this solvent. The hydroquinone protons 10 and 11 are unusually shielded in the spectrum of the [2]catenane ⋅Cl, but similar shifts were observed for these protons in the spectra of the non-interlocked porphyrin-containing macrocycles and ⋅Cl, which was primarily attributed to the shielding effect of the porphyrin’s aromatic ring currents.
- 31.The PF6−6. counteranion is thought to be disordered over two positions, each with 50 % occupancy. Whereas one of the two partially occupied PF counteranions was clearly visible in the difference map, and was modelled accordingly, the other was considerably more difficult to locate as it was highly disordered around symmetry operators. The residual electron density corresponding to this highly disordered anion was therefore modelled by using Platon SQUEEZE (ref. [42])
- 32.Hynes MJ. J. Chem. Soc. Dalton Trans. 1993:311–312. [Google Scholar]
- 33.When the TBA salts of AcO−24−624133133. and H PO were added to [D ]DMSO solutions of the catenane, perturbations in the signals for both the cavity protons and the porphyrin ring protons were observed, along with significant spectral broadening. The observed changes were tentatively assigned to the existence of two separate oxoanion binding modes: the dominant one involving an interaction between the guest anion and the catenane’s interlocked binding cavity and a second, weaker binding mode involving direct coordination of the anion to the zinc(II) cation, which presumably results in displacement of the pyridine group. Further evidence for this direct zinc–anion interaction was provided by the observation of gradual bathochromic shifts in the Soret band absorbance upon addition of increasing concentrations of TBAAcO and TBAH PO to DMSO solutions of the catenane. Unfortunately however, owing to problems with H NMR spectral broadening, in combination with the complicated nature of the competing equilibrium binding processes, we were unable to obtain reliable quantitative binding data for these oxoanions. The titration experiments were also attempted in 1:1 CDCl /CD OD. In this solvent mixture we did not observe any evidence of direct oxoanion coordination to the zinc(II) centre by H NMR or UV/Vis spectroscopy. However, we were unable to calculate accurate association constants for anion binding in 1:1 CDCl /CD OD as a result of the catenane’s low solubility and tendency towards aggregation in this solvent mixture.
- 34.To investigate the solvent dependency of the catenane’s optical chloride sensing properties, we also carried out the UV/Vis titration experiments in acetone and acetonitrile. Addition of increasing concentrations of TBACl to 2 mM. solutions of the catenane in these solvents produced gradual hypsochromic shifts of approximately 2 nm in the Soret band absorbance, comparable to but marginally greater in magnitude than the spectral changes observed in DMSO.
- 35.Specfit v. 2.02, Spectrum Software Associates, Chapel Hill, NC, USA
- 36.Otwinowski Z, Minor W. In: Methods in Enzymology, Vol.276: Macromolecular Crystallography, part A. Carter CW Jr, Sweet RM, editors. Waltham, MA: Academic Press; 1997. pp. 307–326. [Google Scholar]
- 37.Nowell H, Barnett SA, Christensen KE, Teat SJ, Allan DR. J. Synchrotron Radiat. 2012;19:435–441. doi: 10.1107/S0909049512008801. [DOI] [PubMed] [Google Scholar]
- 38.Cosier J, Glazer AM. J. Appl. Crystallogr. 1986;19:105–107. [Google Scholar]
- 39.CrysAlisPro, Agilent Technologies, Yarnton, Oxfordshire, UK
- 40.Palatinus L, Chapuis G. J. Appl. Crystallogr. 2007;40:786–790. [Google Scholar]
- 41.Betteridge PW, Carruthers JR, Cooper RI, Prout K, Watkin DJ. J. Appl. Crystallogr. 2003;36:1487–1487. [Google Scholar]
- 42a.Vandersluis P, Spek AL. Acta Crystallogr. Sect. A. 1990;46:194–201. [Google Scholar]
- 42b.Spek AL. J. Appl. Crystallogr. 2003;36:7–13. [Google Scholar]
- 43.Cooper RI, Thompson AL, Watkin DJ. J. Appl. Crystallogr. 2010;43:1100–1107. [Google Scholar]
- 44.Goldenberg C, Wandestrick R, Binon F, Charlier R. Chim. Ther. 1973;8:259–270. [Google Scholar]
- 45.Lindsey J. J. Org. Chem. 1980;45:5215–5215. [Google Scholar]
- 46.Li Y-j, Zhao Y-j, Flood AH, Liu C, Liu H-b, Li Y-l. Chem. Eur. J. 2011;17:7499–7505. doi: 10.1002/chem.201100633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information