

Abstract
NMDARs and ASIC1a both exist in central synapses and mediate important physiological and pathological conditions, but the functional relationship between them is unclear. Here we report several novel findings that may shed light on the functional relationship between these two ion channels in the excitatory postsynaptic membrane of mouse hippocampus. Firstly, NMDAR activation induced by either NMDA or OGD led to increased [Ca2+]i and greater apoptotic and necrotic cell deaths in cultured hippocampal neurons; these cell deaths were prevented by application of NMDAR antagonists. Secondly, ASIC1a activation induced by pH 6.0 extracellular solution (ECS) showed similar increases in apoptotic and necrotic cell deaths; these cell deaths were prevented by ASIC1a antagonists, and also by NMDAR antagonists. Since increased [Ca2+]i leads to increased cell deaths and since NMDAR exhibits much greater calcium permeability than ASIC1a, these data suggest that ASIC1a-induced neuronal death is mediated through activation of NMDARs. Thirdly, treatment of hippocampal cultures with both NMDA and acidic ECS induced greater degrees of cell deaths than either NMDA or acidic ECS treatment alone. These results suggest that ASIC1a activation up-regulates NMDAR function. Additional data supporting the functional relationship between ASIC1a and NMDAR are found in our electrophysiology experiments in hippocampal slices, where stimulation of ASIC1a induced a marked increase in NMDAR EPSC amplitude, and inhibition of ASIC1a resulted in a decrease in NMDAR EPSC amplitude. In summary, we present evidence that ASIC1a activity facilitates NMDAR function and exacerbates NMDAR-mediated neuronal death in pathological conditions. These findings are invaluable to the search for novel therapeutic targets in the treatment of brain ischemia.
Keywords: NMDA receptor, Acid-sensing ion channel 1a, Whole-cell patch-clamping recording, Oxygen-glucose deprivation, Neurotoxicity, Neuroprotection
Introduction
Ischemia-induced neuronal death is a major problem associated with stroke. Among various theories on the molecular mechanism of ischemic neuronal damage, hypotheses involving the overactivation of NMDARs have attracted more attention because of the unique properties of NMDARs. NMDAR activities are associated with various neuronal processes ranging from learning and memory to neuronal degeneration. Activation of NMDARs requires both glutamate and glycine binding to their respective sites, as well as membrane depolarization to remove the Mg2+ blockade (Mayer et al. 1984; Johnson and Ascher 1987). Once activated, NMDARs are highly permeable to Ca2+. It is this high Ca2+ permeability that makes them the ideal mediator of extracellular Ca2+ influx, which in turn activates Ca2+-dependent enzymes, including nNOS, protease, protein kinase, and DNase inneurons and damage cells (Schanne et al. 1979; Choi 1985; Mayer and Westbrook 1987; Lipton and Rosenberg 1994; Lee et al. 1999; Arundine and Tymianski 2004; Makhro etal. 2010; Szydlowska and Tymianski 2010). In addition, the function of NMDARs can be modified by many extracellular molecules, such as protons (H+), Zn2+, and polyamine (MacDermott et al. 1986; Mayer and Vyklicky 1989; Rock and Macdonald 1992; Williams 1997; Cull-Candy et al. 2001).
Recent studies have shown that the activation of homomeric acid-sensing ion channel 1a (ASIC1a) could also lead to Ca2+ influx into neurons and cause cell damage in cultured cortical and hippocampal neurons (Xiong et al. 2004; Gao et al. 2005).
Homotrimeric ASIC1a channels are a proton-gated and Ca2+-permeable ion channel that conducts Na+ and Ca2+ (Waldmann et al. 1997; Xiong et al. 2004; Yermolaieva et al. 2004). They are widely expressed in peripheral and central nervous systems (CNS) in mammals (Waldmann etal. 1997; Waldmann and Lazdunski 1998; Waldmann et al. 1999; Alvarez de la Rosa et al. 2002, 2003). ASIC1a channels are present at excitatory postsynaptic synaptic terminals (Wemmie et al. 2003, 2006; Zha et al. 2006), which could be activated by sudden pH drops from 7.4 to 6.9 and below, and show a half-maximal activation at pH 6.2 (Waldmann et al. 1997). After activation, ASIC1a rapidly goes into desensitization state (Waldmann et al. 1997; Xiong et al. 2004; Wemmie et al. 2006). In physiological condition, ASIC1a plays an important role in long-term potentiation (Wemmie et al. 2002) and dendritic structural plasticity in hippocampal neurons (Xiong et al. 2004; Zha et al. 2006). Behavior studies showed that disrupting ASIC1a impaired spatial learning, eye-blinking conditioning, and fear conditioning of the mouse (Wemmie et al. 2002, 2003). Conversely, over-expression of ASIC1a in transgenic mice increased fear conditioning (Wemmie et al. 2004).
In pathological conditions, such as brain ischemia, activation of ASIC1a induced Ca2+ overload in the neuron and caused cell death (Xiong et al. 2004; Gao et al. 2005). Pharmacologically blocking ASIC1a channels can also reduce ischemic neuronal death (Xiong et al. 2004; Gao et al. 2005).
Since NMDARs and ASIC1a show functional similarity in physiological and ischemic conditions, and since glutamate transmitter release is always accompanied by proton release from the presynaptic terminal, these two receptors may be co-activated (Krishtal et al. 1987; Zha et al. 2006). We ask whether the co-activation of these two ion channels in the postsynaptic site is obligatory or not. Since NMDAR is the main player in central synaptic transmission, completely blocking NMDARs is harmful for normal synaptic function, causes chronic neuronal degeneration, and does not benefit neuronal survival in brain ischemia (Hardingham and Bading 2003). Thus, we decided to examine whether inhibition of ASIC1a affects NMDAR function, whether inhibition of ASIC1a can prevent ischemic cell death, and what the underlying mechanism is.
Our previous electrophysiology studies in mouse brain slices have shown that electrical stimulation of Shaffer collateral fibers evoked excitatory postsynaptic currents in physiological pH (7.4) in low Mg2+ (0.1 mM) extracellular solution (ECS) under the blockade of AMPA, GABAA, and glycine receptors. In this condition, NMDAR activity should be mostly facilitated. We found that the excitatory currents were completely blocked by NMDAR antagonists, which suggests that the currents were mediated by NMDARs. Another set of patch-clamp recording data showed that NMDAR currents were distinctly suppressed when ASIC1a antagonists were added to the perfusion solution (pH 7.4), suggesting that NMDARs, not ASIC1a, are the main player in mediating excitatory synaptic transmission in the hippocampus.
In this study, we found that treatment of cultured hippocampal neurons with NMDA-induced neuronal deaths at physiological pH (7.4). Interestingly, inhibition of ASIC1a greatly reduced NMDA-induced neuronal death. However, stimulation of ASIC1a in pH 6.0 ECS greatly enhanced NMDA-induced neuronal death. We then further examined the involvement of ASIC1a in the NMDAR-mediated [Ca2+]i increase and apoptotic effector enzyme activation.
Materials and Methods
Primary Culture of Hippocampal Neurons
Dissociated cultures of hippocampal neurons were prepared from postnatal 0–1 day mice. All animal care and experimental procedures were complied with local and internal guidelines on ethical use of animals and were approved by the University animal welfare committee. The tissue was isolated by a standard enzyme treatment protocol. Briefly, shredded tissue was treated with 0.125 % trypsin (T1426, Sigma-Aldrich, USA) and incubated for 25 min at 37 °C. After centrifuging the mixture, the supernatant was discarded. The neurons were plated onto coverslips coated with poly-L-lysine (D0301, Sigma-Aldrich, USA) and then grown in DMEM with/F12 and 10 % fetal calf serum (21103-049, Gibco, USA) for the first 24 h. The cultures were maintained at 37 °C in a 5 % CO2 humidified atmosphere. The culture medium was replaced by neurobasal containing 2 % B27 on the second day. The culture medium was replaced every 3 days. Glial growth was suppressed by addition of 5-fluoro-2-deoxyuridine (10 μM) and uridine (10 μM) (Sigma-Aldrich, USA). Mature neurons (12–16 DIV) were used for experiments. To induce neuronal injury, hippocampal cultures were washed three times in Mg2+-free ECS containing (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 20 HEPES, and 3 glucose, with pH 7.4 and osmolarity 310–320 mOsm.
NMDA Treatment
To examine NMDA’s effect on cultured hippocampal neurons, specific blockade of synaptic AMPARs, glycine receptors, GABAA receptors, and voltage-gated calcium channels (VGCCs) were achieved by adding NBQX (5 μM), strychnine (1 μM), bicuculline (10 μM), and nifedipine (10 μM) into the Mg 2+-free ECS. The cultures were then exposed to Mg2+-free ECS containing NMDA (100 μM) and glycine (10 μM) for 25 min at room temperature (RT). Following a thorough wash with 1.3 mM Mg2+ and 15 mM glucose ECS (normal ECS), the cultures were returned to the normal culture medium and continued to grow for 24 h.
Acidic ECS Treatment
To test the effect of acidic ECS (pH 6.0) on cultured hippocampal neurons, the cultures were washed 3 times with Mg2+-free ECS, followed by exposure to Mg2+-free acidic ECS for 25 min at room temperature. The treatment was terminated by washing the cultures with normal ECS. The cultures were then returned to the normal medium and continued to grow for 24 h.
Oxygen-Glucose-Deprivation
To test the effect of OGD on cultured hippocampal neurons, the cultures were washed three times with Mg2+- and glucose-free ECS (deoxygenated in the anaerobic container for 30 min before use), and then the cultures were then transferred to an anaerobic container in Mg2+ and glucose-free ECS saturated by 5 % CO2/95 % N2 gases. The cultures remained in this anoxic condition for 90 min at 37 °C. OGD treatment was terminated by removing the cultures out of the container. The cultures were washed three times with normal ECS and were then returned to the normal culture medium, allowing to grow for 24 h. NBQX (5 μM), bicuculline (10 μM), strychnine (1 μM), and nifedipine (10 μM) were contained in the ECS to block AMPARs, GABAA receptors, glycine receptors, and VGCCs, respectively. Specific inhibitors of ASIC1a or NMDARs, such as amiloride and PcTX1, or Ketamine and APV were added to the ECS as needed.
Cell Viability Assay
Apoptotic neuronal death was determined by visualizing neurons stained with Hoechst-33342 (C1022, Beyotime Institute of Biotechnology, China). For visualizing apoptotic neurons, Hoechst-33342 (5 μg/ml) was added to the culture medium 24 h after treatments and incubated at 37 °C for 25 min. Images were taken with a fluorescence microscope (IX71SIF-3, Olympus, Japan). Cells with condensed or fragmented chromatin morphology were considered as apoptotic. These observations were quantified by double-blind counting of apoptotic and total neurons in each visual field and expressed as percentage. Necrotic cell death was quantified by measuring lactate dehydrogenase (LDH) released into the culture medium 24 h after treatments using a Cyto Tox 96 assay kit (Promega, Madison, WI). The absorbance readings were measured using a microplate reader (MULTISKAN MK3, Thermo Scientific, USA).
Ca2+ Imaging
The cultured hippocampal neurons grown on 8 × 8 mm glass coverslips were transferred to 12-well plates, washed twice with normal ECS, and then incubated with Fluo-4 AM (4 μM; F312, Dojindo Laboratories, Japan) in normal ECS for 60 min at 37 °C in the dark. The cultures were washed again and equilibrated for 30 min at room temperature. The cultures were then washed in modified ECS (Mg2+-free, 3 mM glucose, 2.9 mM Ca2+). After this, the coverslip was transferred into a container on the inverted confocal microscope (LEICA TCS SPE, Germany), and was incubated with modified ECS containing NBQX, bicuculline, strychnine, and nifedipine to block AMPARs, glycine receptors, GABAA receptors, and VGCCs, respectively, for 15 min at room temperature. Before scanning the images, the container was placed on the stage of the confocal microscope equipped with a 63 × oil-immersion objective lens and set up in xyt mode with a laser wavelength of Ex = 488 nm and Em = 530 nm. The resolution of frame scanned was set to 512 × 512 pixels.
Five minute baseline images followed by 30-min experimental treatment images were taken. The intensities of fluorescence were analyzed with the dynamic intensity analysis module of LAS AF. For normalizing the variations of Ca2+ loading, we used relative fluorescence intensity ΔFt/F0 to express the changes in the intracellular Ca2+ concentration (fold of Ca2+ increase). Ft is the averaged fluorescence intensity of ROI from each frame at the given time, and F0 is the averaged 5-min baseline fluorescence intensity form the same ROI immediately prior to experimental treatments. Background fluorescence (from a region within the same field lacking cells) was subtracted from ROI averages (Feldman et al. 2008).
Electrophysiology
Preparation of brain slices: Prior to decapitation, postnatal 5 to 6-week C57/BL6J mice were anesthetized with isoflurane. The brain was removed and placed in oxygenated (95 % O2 & 5 % CO2) ACSF at 4 °C containing (mM) 126 NaCl, 2.5 KCl, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, and 10 glucose. The osmolarity of the ACSF was adjusted to 300 mOsm and the pH to 7.2–7.4. Acute coronal brain slices (300 μm) containing the hippocampus were obtained with a vibrating microtome (Leica VT 1000S, Nassloch, Germany). The slices were then transferred to a chamber containing oxygenated ACSF and maintained at room temperature for at least 1 h prior to patch-clamp voltage recordings. Postsynaptic responses were evoked by electrical stimulation of the Schaffer collaterals (SCs) with a bipolar microelectrode positioned in the stratum radiatum of hippocampal slices. Stimulations were delivered every 12 s with the pulse duration of 100 μs. NMDAR-mediated excitatory postsynaptic currents (NMDAR EPSCs) were recorded from CA1 pyramidal neurons in voltage-clamp mode. The neuronal membrane potential was held at −30 mV. The recording electrode was filled with intracellular solution containing (in mM) 130 Cs+-methanesulfonate, 10 HEPES, 2 MgCl2, 2 ATP-Mg, 0.5 GTP, 1 lignocaine (lidocaine) N-ethyl bromide (QX-314), and 2 EGTA. The osmolarity was adjusted to 280 mOsm and the pH to 7.2–7.4. NMDAR EPSCs were pharmacologically isolated by perfusion of the brain slices with low Mg2+ ACSF (MgCl2 0.1 mM; CaCl2 2.9 mM) containing CNQX (5 μM), bicuculline (10 μM), and strychnine (1 μM), respectively, to block synaptic AMPAR, GABAA receptor, and glycine receptor responses.
Western Blot
Hippocampal cultures that were treated by NMDA for 30 min at room temperature were lysed in cell lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 % Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM EDTA, 1 % Na3VO4, 0.5 μg/ml leupeptin, and 1 mM phenylmethanesulfonyl fluoride). Protein samples (30 μg) were run in sodium dodecyl sulfate-polyacrylamide gels (12 %) for 1.5 h using Tris–glycine running buffer. Gels were then transferred onto a PVDF membrane in transfer buffer containing 25 mM Tris-base, 0.2 M glycine, and 20 % (v/v) methanol for 1 h at 70 V. The membrane was blocked with 5 % non-fat milk in 1 × TBS, 0.1% Tween-20 at 25 °C for 1 h and subsequently incubated overnight at 4 °C with primary antibodies for cleaved caspase-3 at Asp 175 (rabbit anti-mouse, 1:1000, Cell Signaling Technology, USA) and β-tubulin (rabbit anti-mouse, 1:1000, Cell Signaling Technology, USA). After washing with TBST, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody (Goat anti-rabbit IgG, ZSGB-BIO, China) for 1 h, and protein signals were detected with a chemiluminescence system (Clinx Science Instruments). Quantification of protein levels was achieved by densitometry analysis using Image J software and expressed as the ratio of the values of the detected protein band to the β-tubulin band.
Hippocampal slices were transferred from ice-cold oxygenated ACSF to room-temperature oxygenated ACSF after slicing, and were incubated for 1.5 h to allow recovery. The slices were then incubated in low glucose (3 mM)and Mg2+-free D-hanks solution (pH 7.4) containing bicuculline (10 μM), NBQX (5 μM), strychnine (1 μM), and nifedipine (10 μM) for 15 min at room temperature to block GABAA receptors, AMPARs, glycine receptors, and VGCCs, respectively. The pH value of the incubation solution was then dropped to 6.0 for 30 min to activate ASIC1a under continuous blockade of GABAA receptors, AMPARs, glycine receptors, and VGCCs. The slices were put into pre-cold lysis buffer immediately after the acidic solution treatment. The extracted protein samples (30 μg) from the slices were run in sodium dodecyl sulfate-polyacrylamide gels (8 %) for 1.5 h using Tris-glycine running buffer. Gels were then transferred onto a PVDF membrane in transfer buffer containing 25 mM Tris-base, 0.2 M glycine, and 20 % (v/v) methanol for 2.5 h at 70 V. The membrane was blocked with 5 % non-fat milk in 1× TBS, 0.1 % Tween-20 at room temperature (RT) for 1 h and subsequently incubated overnight at 4 °C with primary antibodies for probing phosphorylated NR2B (p-NR2B) (rabbit anti-mouse 1:1000, Cell Signaling Technology, USA). After washing, the membrane was incubated with secondary antibody (Goat anti-rabbit IgG, 1:5000, ZSGB-BIO, China) for 1 h at RT. The protein signals were detected and quantification of protein levels was achieved.
Data Analysis
The data are presented as mean ± SEM. ANOVA analysis was used for comparison among multiple groups, followed by the post hoc analysis for comparison between two groups. Student’s t test was also used. Statistical significance was defined as p ≤ 0.05.
Results
To explore the effect of activation of either NMDARs or ASIC1a alone, and the activation of both ASIC1a and NMDARs on neuronal damage, we used, respectively, NMDA, pH 6.0 acidic ECS, and OGD to challenge the hippocampal cultures. The parameters measured included neuronal viability, intracellular Ca2+ concentration increase, and apoptosis-related caspase-3 levels. Interestingly, we found that NMDARs played a pivotal role in neuronal death induced by activation of either NMDARs or ASIC1a, and even more so with activation of both.
Overactivation of NMDARs Induces Neuronal Death
It is documented that in brain ischemia, the ensuing neuronal death is due to mass glutamate transmitter release and overstimulation of NMDARs (Hardingham and Bading 2003). Using Hoechst-33342 staining, we observed that NMDA treatment of hippocampal cultures induced a 54 ± 4 % neuron death with the characteristic apoptotic morphological changes (Fig. 1a, b), including cell shrinkage, nuclear condensation, and fragmentation. This NMDA-induced apoptotic cell death was effectively prevented by pre-inhibition of NMDARs with their specific antagonists APV or Ketamine (Fig. 1a, b).
Fig. 1.
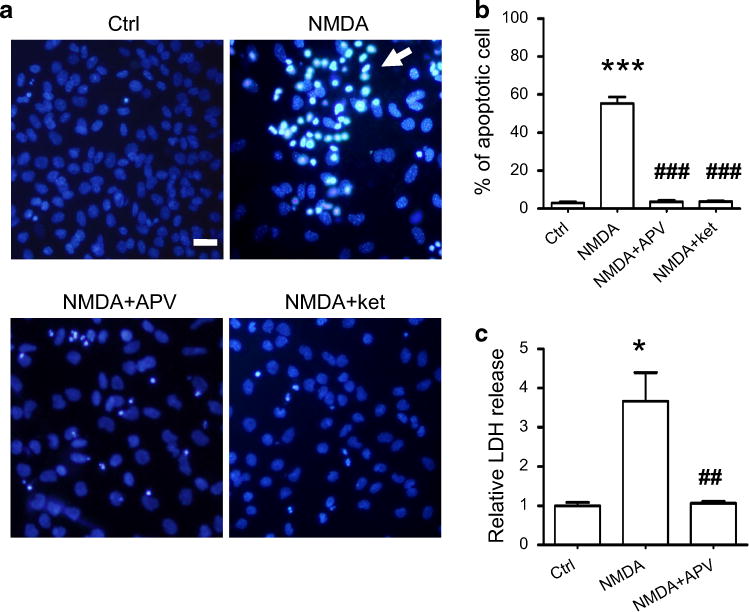
Overstimulation of NMDAR induces neuronal death. a Hippocampal cultures (16 DIV) stained with Hoechst-33342 did not show any obvious apoptotic cell death (upper left panel). NMDA treatment induced a significant increase in apoptotic neuronal death as evidenced by typical morphological features of nuclear condensation and DNA fragmentation (upper right panel). NMDAR antagonist APV (100 μM) or ketamine (30 μM) prevented NMDA-induced cell deaths (lower left and right panels). Scale bar is 20 μm. b Quantitative analysis of apoptotic death of cultured hippocampal neurons is shown in the histogram. There was a small percentage (3 %) of cell death in control cultures (n = 7), whereas NMDA exposure led to a 54 % cell death rate (n = 8). Antagonizing NMDARs markedly reduced neuronal death to ~6 % (n = 6). ***p < 0.005 compared with control; ###p < 0.005 compared with NMDA treatment. c LDH release in cultures representing the extent of necrotic neuronal death was measured and is shown in the histogram. LDH release was fourfold higher in the NMDA-treated cultures (n = 5) than in the control (n = 5). LDH release was greatly reduced by blocking NMDARs with APV (n = 3). *p < 0.05 compared with the control; ##p < 0.01 compared with NMDA treatment.
LDH released into extracellular environment has been a useful indicator for evaluating cell necrosis (Xiong et al. 2004). NMDA treatment of hippocampal cultures caused a fourfold increase of LDH concentration in the extracellular medium (Fig. 1c). This increase in LDH levels was effectively prevented by blocking NMDARs prior to NMDA treatment. These data suggest that overstimulation of NMDARs caused the cultured neurons to undergo both apoptotic and necrotic death.
Overactivation of ASIC1a also Induces Neuron Death
Excess glutamate release and acidosis often occur concurrently in brain ischemia. We treated the cultures with pH 6.0 ECS to mimic brain tissue acidosis under the blockade of AMPARs, glycine receptors, GABAA receptors, and VGCCs, respectively. We found that acidic ECS induced an increase in apoptotic neuron death (32 ± 4 %) (Fig. 2a, b), and also led to a significant increase in LDH release (~twofold) (Fig. 2c). The apoptotic cell death and LDH release evoked by acidic ECS were all prevented by the non-selective ASIC1a blocker amiloride or specific ASIC1a blocker PcTX1. These data suggest that over-stimulation of ASIC1a induced both apoptotic and necrotic neuronal deaths in cultured hippocampal neurons.
Fig. 2.
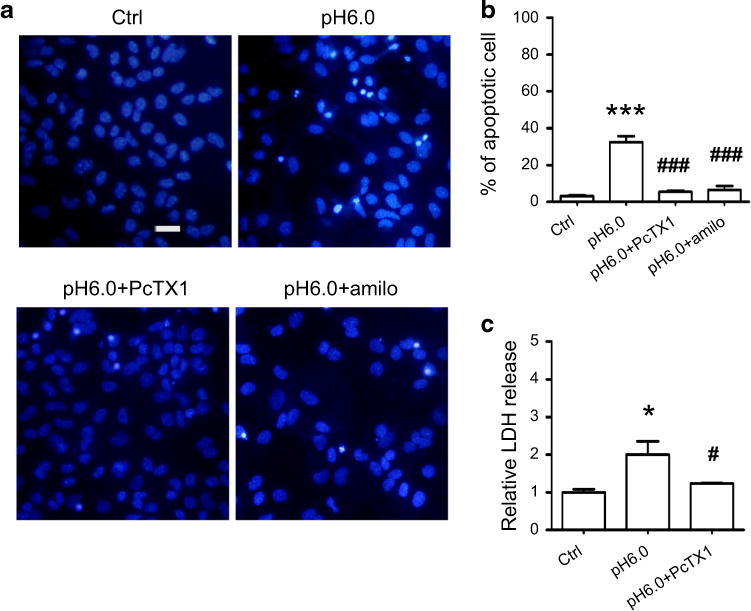
Acidic ECS induces neuronal death. a Representative images of hippocampal neuronal cultures showing that acidic ECS (pH 6.0) treatment induced more apoptotic cell deaths (upper right panel) than control (upper left panel). Blocking ASIC1a by pre-treatment with its antagonists PcTX1 (50 nM) or amiloride (100 μM) effectively prevented acid-induced apoptotic cell deaths (lower left and right panels). Scale bar is 20 μm. b Quantitative analysis of apoptotic death of cultured hippocampal neurons is shown in the histogram. Acidic ECS induced ~32 % apoptotic cell death (n = 4). Blocking ASIC1a by pre-treatment with PcTX1 (n = 3) or amiloride (n = 3) greatly reduced neuronal deaths to 5.5 and 6.6 %, respectively. ***p < 0.005 compared with the control (n = 7); ###p < 0.005 compared with pH 6.0 treated cultures. c The LDH release in acidic ECS-treated cultures is twofold higher (n = 3) than in control cultures (n = 5). Inhibition of ASIC1a reduced LDH release to 1.23 times that of control (n = 3). *p < 0.05 compared with the control; #p < 0.05 compared with pH 6.0 treated cultures.
Stimulation of ASIC1a Enhances NMDAR-Mediated Neuronal Death
The release of glutamate transmitter is commonly accompanied by the release of H+ during synaptic activity (Krishtal et al. 1987; Wemmie et al. 2003, 2006). Our electrophysiology data from mouse brain slices showed that stimulation of ASIC1a by pH 6.0 acidic ECS enhanced synaptic NMDAR activity in the early phase (within the first 2 min after the onset of pH 6.0 ECS perfusion), but exhibited an inhibitory effect on NMDAR activity thereafter (Fig. 3a–c). The observation that H+ inhibited NMDAR EPSCs was made by Traynelis two decades ago (Traynelis and Cull-Candy 1990, 1991). However, H+-enhancing NMDAR activity was observed for the first time. The most exciting founding was that H+ made NMDAR EPSCs rise time much faster and the decay time much slower (Fig. 3d–f). These data suggest that ASIC1a activity facilitates NMDAR channel open and hinder the channel close.
Fig. 3.
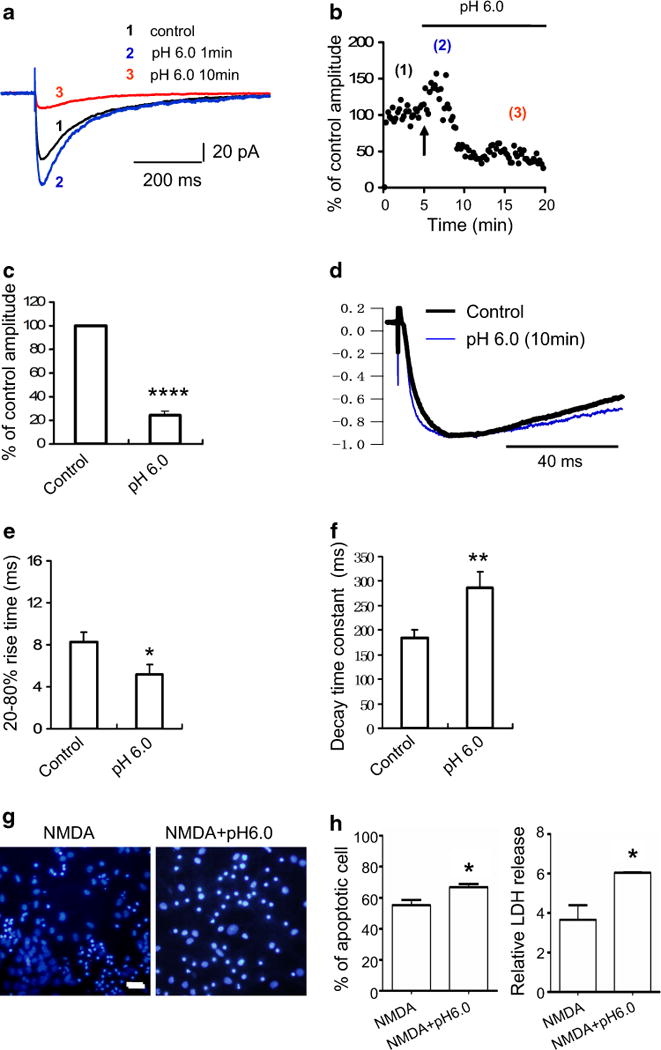
Stimulation of ASIC1a enhances NMDAR activity and exacerbates NMDAR-mediated neuronal death. a Whole-cell voltage-clamp recordings from mouse hippocampal slices showing the amplitudes of NMDAR EPSCs in normal ACSF, in the first 2 min after onset of pH 6.0 ACSF perfusion, and in the prolonged pH 6.0 ACSF perfusion phase, as indicated by normalized traces 1, 2, and 3. b Dynamic changes of the amplitudes of NMDAR EPSCs in brain slices before and during pH 6.0 ACSF perfusion. (1) The 5-min baseline recording of the control NMDAR EPSC amplitudes; (2) the amplitudes of NMDAR EPSCs transiently increased at the onset of pH 6.0 ACSF perfusion; (3) the amplitudes of NMDAR EPSCs decreased in the prolonged pH 6.0 ACSF perfusion phase. Arrow indicates the onset of pH 6.0 ACSF perfusion. c Summarized histogram showing that the NMDAR EPSC amplitudes were greatly suppressed by prolonged perfusion of brain slices in pH 6.0 ACSF (n = 5). ****p < 0.001. d Normalized traces showing that the perfusion of brain slices in pH 6.0 ACSF caused the rise time of NMDAR EPSCs to become faster and their decay time slower. e The 20–80 % rise time of NMDAR EPSCs in brain slices was significantly reduced by pH 6.0 ACSF perfusion compared to that of the control. *p < 0.05. f The decay time constant of NMDAR EPSCs in brain slices was significantly prolonged by pH 6.0 ACSF perfusion. **p < 0.01. g NMDA treatment-induced apoptotic neuronal death (left panel) was exacerbated by lowering the ACSF pH to 6.0 (right panel). Scale bar is 20 μm. h Histogram showing that the percentage of apoptotic neuronal death (left panel) induced by NMDA treatment (n = 8) was greatly enhanced by lowering the ACSF pH to 6.0 (n = 7). *p < 0.05. The LDH release induced by NMDA treatment (right panel) was markedly increased by lowering the ACSF pH to 6.0. *p < 0.05
The early phase of ASIC1a activation played a crucial role in the facilitation of NMDAR-mediated neuronal death, and the facilitation of the channel opening as well (Fig. 3g, h). NMDA induced a 54 ± 4 % neuronal death in normal physiological pH (7.3–7.4), whereas NMDA with acidic ECS at the pH 6.0 induced a much higher rate of apoptotic cell death (67 ± 2.42 %) and necrotic cell death (LDH release increased by more than sixfold).
Stimulation of ASIC1a Up-Regulates NMDAR Activity
To test the hypothesis that ASIC1a activation enhances NMDAR activity, we tried to inhibit ASIC1a activation by adding amiloride or PcTX1 prior to treating the cultures with NMDA or acidic ECS. We found that the apoptotic cell death was greatly reduced to 4.1 ± 0.8 or 4.23 ± 1.7 %, and that the necrotic cell death (as indicated by LDH release) was markedly reduced as well (Fig. 4a–c) to a level comparable to the control group. These data clearl show that activation of ASIC1a greatly enhanced NMDA-induced cell death.
Fig. 4.
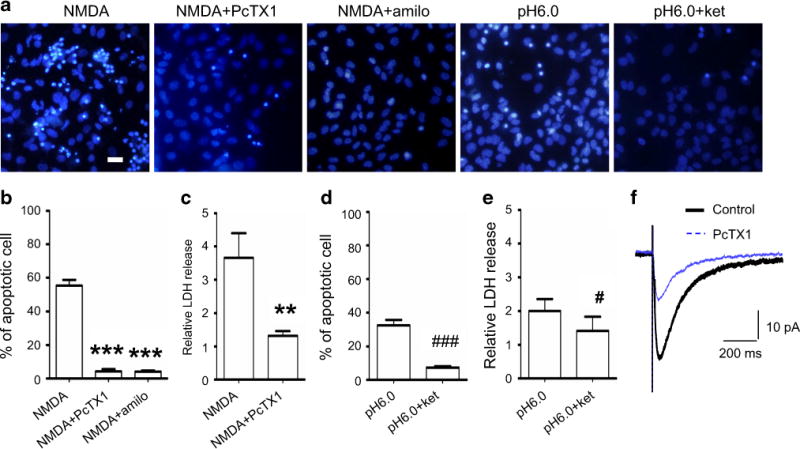
Antagonizing ASIC1a suppresses NMDA-induced neuronal death. a Representative images showing that NMDA treatment-induced apoptotic neuronal death was prevented by antagonizing ASIC1a with PcTX1 or amiloride. However, the pH 6.0-induced cell death was also prevented by blocking NMDAR with Ketamine. Scale bar is 20 μm. b Quantitative analysis showing that pre-treatment with either PcTX1 or amiloride effectively prevented NMDA treatment-induced apoptotic cell deaths. ***p < 0.005. c LDH release induced by NMDA (3.7-fold of the control; n = 5) was greatly reduced by pre-treatment with PcTX1 (1.3-fold of the control; n = 6). **p < 0.01 compared with NMDA-treated cultures. d Quantitative analysis of apoptosis showing that pH 6.0 induced 32 % (n = 4) apoptotic cell death, which was lowered to 7.4 % (n = 4) by NMDAR blocker ketamine. ###p < 0.005 compared with pH 6.0-treated cultures. e LDH release induced by pH 6.0 treatment was twofold (n = 4) that of the control (n = 6), whereas this increase in LDH release was brought down to 1.4-fold (n = 3) that of the control by blocking NMDARs with ketamine. #p < 0.05 compared with pH 6.0 treatment. f Whole-cell voltage-clamp recordings in brain slices showing that NMDAR EPSCs were markedly suppressed by PcTX1 (25 nM), which suggests that antagonizing ASIC1a prevented neuronal apoptosis via suppression of NMDAR activity.
In another experiment, we used NMDAR antagonist to block NMDARs and treated the culture with acidic ECS. Interestingly, we found that both apoptotic and necrotic cell deaths were distinctly reduced (7 ± 0.92 %) (Fig. 4a, d, e).
Since the phosphorylation of NR2B subunits of the NMDAR was well known to correlate with cell death (Tu et al. 2010; Lai et al. 2014), we examined whether activation of ASIC1a resulted in an increase in NR2B phosphorylation. We incubated mouse hippocampal slices with normal ECS, acidic ECS, and acidic ECS containing APV (50 μM), respectively. We found that the slices incubated in acidic ECS had a dramatic increase in NR2B phosphorylation compared to those slices incubated in normal ECS and in acidic ECS containing APV (supplementary Fig. 1a, b).
Together, these data suggest that activation of ASIC1a up-regulated NMDAR activity and exacerbated NMDAR-mediated cell death in acidic conditions, which is consistent with previous reports that ASIC1a channels conduct negligible Ca2+ entry to the cells (Samways et al. 2009). We speculate that ASIC1a potentiates and/or facilitates NMDAR function in both physiological and pathological conditions. This speculation is supported by our electro-physiology experimental results from brain slices (Fig. 4f), which showed a great reduction of NMDAR EPSC amplitudes after pharmacological inhibition of ASIC1a channel activity by PcTX1 (25 nM). It is also supported by Western blot analysis results, which showed that acidic ECS treatment of the hippocampal slices induced a marked increase in NR2B phosphorylation (supplementary Fig. 1). These data provide strong support for the idea that ASIC1a activity caused neuronal death through up-regulation of NMDARs. Without activation of NMDARs, ASIC1a alone could not induce significant neuronal death.
Stimulation of ASIC1a Facilitates NMDAR-Mediated Ca2+ Increase
NMDARs are the main Ca2+ permeable ion channels that mediate Ca2+ overload and result in neuronal death in brain ischemia. Using NMDA to challenge hippocampal cultures caused a great intracellular Ca2+ concentration ([Ca2+]i) increase in neurons when leaving both NMDARs and ASIC1a channels unblocked (Fig. 5a, c, f). It was logical to see that the [Ca2+]i increase was prevented after blocking NMDARs with ketamine, since NMDARs are the main Ca2+ permeable ion channels in the neurons (Mayer and Westbrook 1987; Iino et al. 1990). After inhibition of ASIC1a with amiloride or PcTX1, NMDA could not induce any appreciable [Ca2+]i increase, although NMDARs were left unblocked (Fig. 5b, c). Using pH 6.0 ECS to challenge hippocampal cultures caused a small amount of [Ca2+]i increase in neurons when both NMDARs and ASIC1a channels were left unblocked (Fig. 5d); however, even this small amount of [Ca2+]i increases was likely mediated by NMDARs because after blocking the NMDARs, stimulation of ASIC1a alone did not induce any appreciable [Ca2+]i increase (Fig. 5d). Thus, ASIC1a channel activity plays a powerful role in facilitating NMDAR-mediated intracellular Ca2+ overload and neuronal death, especially in conditions of tissue acidification (Fig. 5e). We also calculated the total [Ca2+]i accumulation in the neurons by measuring the area underneath the recorded curve and above the baseline. We found that [Ca2+]i accumulation in the cell was 6.5-fold greater after challenge with NMDA than after challenge with pH 6.0 (Fig. 5f); [Ca2+]i accumulation in the neurons was dramatically increased to tenfold after challenge with both NMDA and acidic ECS compared to challenge with pH 6.0 alone.
Fig. 5.
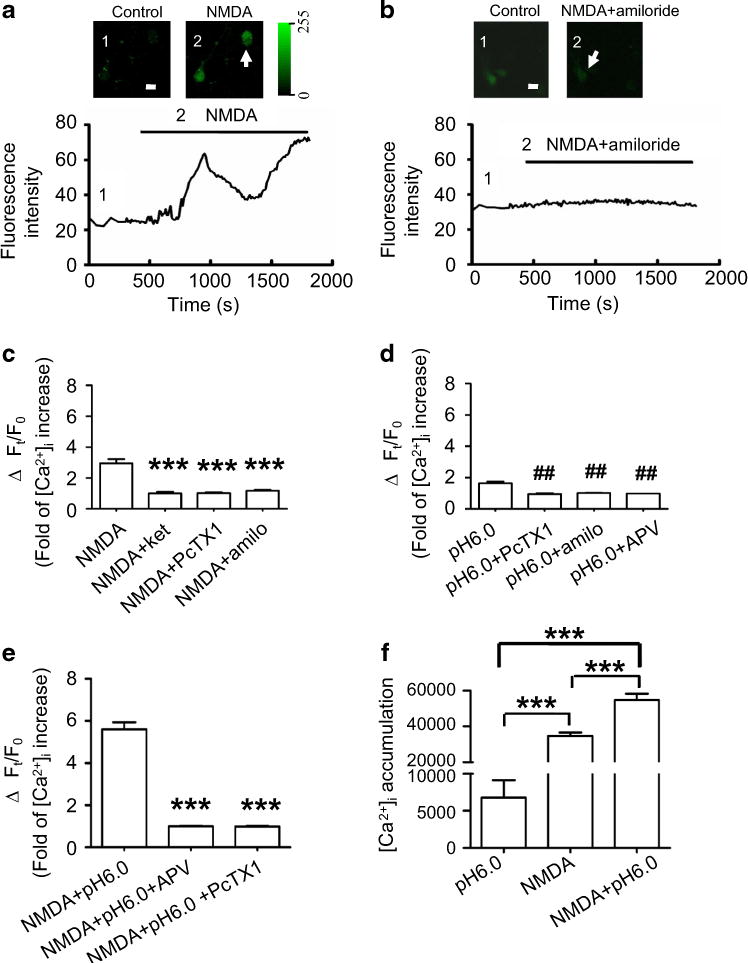
ASIC1a activity facilitates NMDAR-mediated intracellular Ca2+ increase in the neuron. a Representative images showing that over-stimulation of NMDARs evoked a sustained intracellular Ca2+ increase in cultured hippocampal neurons under the condition of blockade of AMPARs, GABAA receptors, glycine receptors, and VGCCs (inserted images on the top left and Ca2+ increase on the top right), as reflected by changes in fluorescence intensity within neurons over time (bottom trace). Scale bar is 10 μm. b NMDA could not induce an intracellular Ca2+ increase in the neuron after pre-inhibition of ASIC1a with amiloride (inserted images on the top left, top right and the trace at the bottom). Scale bar is 10 μm. c Summarized histogram showing that NMDA induced a threefold intracellular Ca2+ increase (n = 12) compared to control. Pre-treatment with ketamine prevented this NMDA-induced Ca2+ increase (n = 7). This NMDA-induced Ca2+ increase was also prevented by pre-treatment with PcTX1 (onefold; n = 21) and amiloride (1.7-fold; n = 12), respectively. ***p < 0.005 compared with NMDA treatment. d Summarized data showing that pH 6.0 treatment alone induced a 1.8-fold intracellular Ca2+ increase (n = 29). This effect was inhibited by ASIC1a antagonists PcTX1 (n = 25) and amiloride (n = 6), as well as the NMDAR antagonist APV (n = 10). ##p < 0.01 compared with pH 6.0 treatment. e Treatment of cultures with NMDA plus pH 6.0 induced a much higher increase in intracellular Ca2+ (5.6-fold; n = 13) compared to the treatment with NMDA or pH 6.0 ECS alone (see c, d). The enhanced intracellular Ca2+ increase was prevented by blocking either NMDARs with APV (n = 9) or ASIC1a with PcTX1 (n = 7). ***p < 0.005 compared with NMDA plus pH 6.0 ECS treatment. f Intracellular Ca2+ accumulation was measured and calculated by computing the area between the baseline and the fluorescence intensity traces for 25 min recordings. The area reflecting Ca2+ accumulation induced by NMDA treatment was 35,567 (n = 29), by pH 6.0 treatment was 5500 (n = 12), and by NMDA plus pH 6.0 treatment was 55,732 (n = 13). NMDA treatment alone induced a 6.5-fold higher intracellular Ca2+ accumulation in cultured neurons than pH 6.0 treatment alone (***p < 0.005), but NMDA pluspH 6.0 ECS treatment dramatically increased the intracellular Ca2+ accumulation to 1.57-fold that of NMDA treatment alone (***p < 0.005) and 10.1-fold that of pH 6.0 ECS treatment alone (###p < 0.005)
These data again suggest that direct activation of NMDARs allowed much more Ca2+ to enter neurons than the activation of ASIC1a that indirectly up-regulates NMDAR activity. Concurrent activation of both NMDARs and ASIC1a synergistically enhanced NMDAR-mediated intracellular Ca2+ overload in the cells and exacerbated neuronal death.
In some experiments we tried to determine if NMDAR activation could induce Ca2+ release from intracellular stores; to do so we used Ca2+-free ECS before and during NMDA treatment of the culture. As expected, we did not see any detectable [Ca2+]i concentration increase (data not shown). These results were consistent with the cell viability assay described above and further confirmed that the increased [Ca2+]i in the neuron is through NMDAR channels, not released from intracellular stores in our experimental conditions.
Stimulation of ASIC1a Enhances NMDAR-Associated Cleaved Caspase-3
A growing body of evidences has shown that activation of NMDARs induces remarkable intracellular Ca2+ concentration increases in neurons, and activation of ASIC1a channels at the same time enhances NMDAR-mediated [Ca2+]i increase (see Fig. 5). The excessive Ca2+ in the cytosol could activate Ca2+-dependent proteolytic enzymes, subsequently inducing necrotic cell death by degradation of intracellular proteins. Increased [Ca2+]i could also cause mitochondrial dysfunction, including loss of mitochondrial membrane potential, increased mitochondrial membrane permeability through opening mitochondrial permeability transition pore (MPTP), and leakage of apoptotic effectors such as SMACs (small mitochondria-derived activator of caspases) and cytochrome c (Mattson and Chan 2003; Halestrap et al. 2004). Those changes in mitochondria subsequently cause the activation of a group of caspases through a cascade of reactions (Fesik and Shi 2001), eventually producing a cleaved caspase-3 (the activate form of caspase-3), which is one of the main effector enzymes that carry out the cell death program. Although there exists a caspase-independent apoptotic cell death, but stroke-induced apoptosis occurs predominantly in neurons and is caspase-dependent (Broughton et al. 2009).
To investigate further the NMDAR-mediated intracellular Ca2+ increase that resulted in apoptotic neuronal death in hippocampal cultures, we measured the production of cleaved caspase-3 caused by excessive [Ca2+]i through performing Western blot analysis using a specific antibody to probe the cleaved caspase-3. We found that NMDA treatment in pH 7.4 ECS caused a greater increase in the production of cleaved caspase-3 in cultures compared to that of control cultures (no NMDA treatment; Fig. 6a, b). This increase in cleaved caspase-3 production was markedly reduced by pre-incubation of the cultures with NMDAR antagonist ketamine. As predicted, this increase in cleaved caspase-3 production was also remarkably reduced by pre-blocking ASIC1a channels with PcTX1 (Fig. 6a, b). This set of data is consistent with the results of Ca2+ imaging; it is also consistent with the results of cell viability assays and electrophysiology experiments. In contrast, activation of ASIC1a channel by pH 6.0 ECS greatly enhanced the increase in cleaved caspase-3 product (Fig. 6a, d). This ASIC1a-induced increase in cleaved caspase-3 is most likely mediated through NMDAR since this increase was remarkably suppressed by blocking NMDARs (Fig. 6a, c). Together, this set of experimental results clearly demonstrated that inhibition of ASIC1a channel activity suppressed NMDAR-associated cleaved caspase-3 production, and activation of ASIC1a channel activity enhanced NMDAR-associated cleaved caspase-3 production. These data provide further supportive evidence that ASIC1a up-regulates NMDAR function in cultured hippocampal neurons.
Fig. 6.
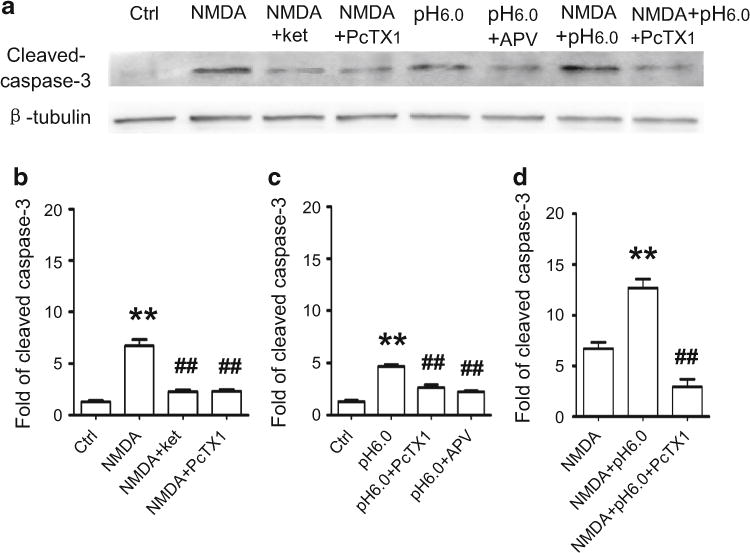
NMDA-induced cleaved caspase-3 increase is enhanced by acidic ECS. a Antibody specifically recognizing cleaved caspase-3 (activated caspase-3, ~7 kD) was used to probe the activated form of caspase-3. β-tubulin served as a loading control for various treatments. The protein bands show that NMDA treatment of hippocampal cultures induced a dramatic increase in cleaved caspase-3 production. Ketamine (30 μM) suppressed the NMDA-induced cleaved caspase-3 production, and PcTX1 (50 nM) also markedly suppressed cleaved caspase-3 production. Acidic ECS (pH 6.0) treatment also induced cleaved caspase-3 production. This increase was inhibited by NMDAR antagonist APV (100 μM). NMDA plus pH 6.0 treatment of the cultures induced a dramatic increase in cleaved caspase-3, and the increase was significantly reduced by antagonizing ASIC1a. b–d Quantitative analysis of the protein band density is shown in these histograms. b NMDA treatment of hippocampal neuronal cultures induced a dramatic 6.7-fold increase (n = 4) in cleaved caspase-3 compared to control (**p < 0.01). Blocking NMDARs with ketamine or PcTX1 distinctly reduced cleaved caspase-3 production to 2.29-fold (n = 4) and 2.32-fold (n = 4) that of the control. Either ketamine or PcTX1 significantly reduced the NMDA-induced cleaved caspase-3 production. ##p < 0.01. c pH 6.0 treatment of cultures increased the cleaved caspase-3 level to 4.7-fold that of the control (**p < 0.01). This large increase was lowered to twofold (n = 4) and 1.72-fold (n = 4) that of control, respectively, by either PcTX1 or APV. ##p < 0.01 compared with pH 6.0 ECS treatment. d NMDA plus pH 6.0 treatment of cultures induced a dramatic increase in cleaved caspase-3 level to 12.7-fold (n = 4) that of the control. This increase was distinctly higher than that of NMDA treatment alone (**p < 0.01). However, pre-treatment with PcTX1 effectively lowered this increase to only 2.93-fold of control (n = 4) (##p < 0.01)
Stimulation of ASIC1a Exacerbates NMDAR-Mediated Neuronal Death in OGD
Since excessive release of glutamate and acidosis is the central features of brain ischemia, we tried to determine whether ASIC1a channel activity exacerbates NMDAR-induced neuronal death in OGD condition (an accepted in vitro model for ischemia) (Xiong et al. 2004; Gao et al. 2005; Liu et al. 2007). We first verified if NMDAR mediated neuronal death by exposing cultures to OGD condition. Indeed, OGD treatment induced a huge increase in neuronal death which was demonstrated by cell viability assays and LDH measurements. This increase in neuronal death was prevented by pre-treatment of cultures with ketamine, a NMDAR blocker (Fig. 7a–c). This data confirmed that OGD induced increased glutamate release and caused neuronal death through overactivation of NMDARs. Next we tested if inhibition of ASIC1a activity could also prevent neuronal death—as we had observed in previous experiments—by treating the cultures with both NMDA and acidic ECS. As predicted, we did observe a great reduction in neuronal deaths in cultures when ASIC1a activity was inhibited by PcTX1. These results once again suggest that NMDAR-mediated neuronal death and ASIC1a activity up-regulated NMDAR activity in OGD condition.
Fig. 7.
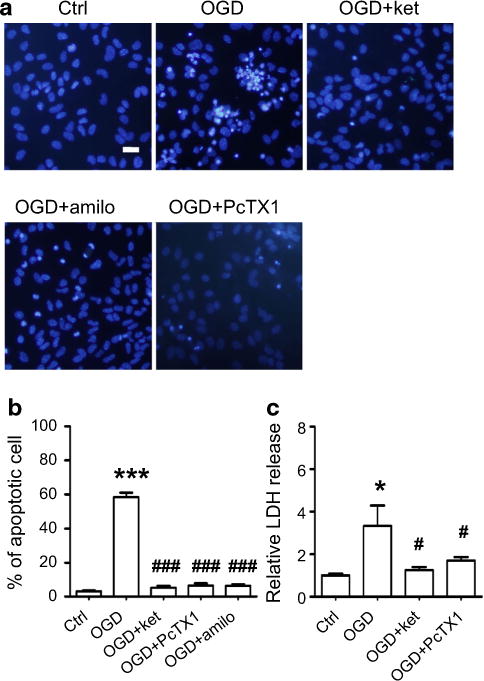
Inhibition of either NMDAR or ASIC1a protected neurons from OGD-evoked cell death. a Representative images of Hoechst-33342 stained neurons showing that OGD induced typical apoptotic cell death (upper middle panel). Cell death was largely prevented by inhibition of NMDARs with ketamine (upper right panel), or by inhibition of ASIC1a with amiloride (lower left panel) or PcTX1 (lower right panel). Scale bar is 20 μm. b Summarized data showing that OGD treatment of hippocampal neuronal cultures induced a large amount of apoptotic cell death (58.4 %; n = 4), which is much higher than that of control (3.1 %; n = 7; ***p < 0.005). However, when NMDARs were blocked with ketamine, this OGD-evoked cell death was reduced to 5.3 % (n = 4; ###p < 0.005). When ASIC1a was blocked with PcTX1 or amiloride, the cell death rate was reduced to 6.6 % (n = 4) and 6.5 % (n = 5), respectively. ASIC1a antagonists demonstrated very effective neuro-protective properties in the OGD treatment of hippocampal cultures (###p < 0.005). c LDH release is also measured in this set of experiments. Consistent with the data on apoptotic cell death, OGD induced high levels of LDH release (3.3-fold; n = 4) compared to control (*p < 0.05). As expected, NMDAR antagonists and ASIC1a antagonists suppressed LDH release in OGD-treated cultures to 1.3-fold (n = 4) and 1.7-fold (n = 6) that of control, respectively. #p < 0.05 compared with OGD treatment.
Discussion
Neurotransmitters glutamate and H+ are simultaneously released during excitatory synaptic transmission (DeVries 2001; Traynelis and Chesler 2001). The release of H+ from presynaptic terminals may bring about a rapid pH drop at synaptic cleft and activate ASIC1a (Waldmann et al. 1997; Xiong et al. 2004). On the other hand, glutamate molecules bind to and activate NMDARs, resulting in cell survival or cell death depending on the local environment and the amount of glutamate released (Hardingham and Bading 2003). NMDARs are highly Ca2+ permeable ion channels (Choi 1985, 1987; Mayer and Westbrook 1987) which can mediate large amounts of extracellular Ca2+ influx to neurons when they are overactivated (Manev et al. 1989; Marcoux et al. 1990). Ca2+ accumulates in neurons due to the inactivation of ATP-dependent ion pumps (Hardingham and Bading 2003; Brookes et al. 2004). Overstimulation of NMDARs induces excessive Ca2+ accumulation in neurons, which in turn triggers many downstream neurotoxic cascades. These include the uncoupling of mitochondrial electron transfer from ATP synthesis, as well as the activation and overstimulation of enzymes such as calpains and other proteases, protein kinases, nitric oxide synthase (NOS), calcineurins, and endonucleases (Brookes et al. 2004). Alterations in activity of these enzymes can lead to increased production of toxic reactive oxygen species (ROS) such as nitric oxide (NO), superoxide and hydrogen peroxide, proteins, and DNA degradation, leading to alterations in the organization of the cytoskeleton and activation of genetic signals that trigger the cell death.
In our cell viability assay experiments, we reproduced previous findings that overstimulation of NMDARs with agonist NMDA induced necrotic and apoptotic death in cultured hippocampal neurons. The NMDA-induced cell death was exacerbated when ECS’s pH dropped to 6.0, which would have activated ASIC1a in the postsynaptic membrane (Krishtal et al. 1987). In addition, reducing ECS pH to 6.0 alone without adding extra NMDA to the medium also induced necrotic and apoptotic neuronal death, which is consistent with previous reports that activation of ASIC1a with acidic ECS or ischemic condition resulted in neuronal death (Xiong et al. 2004; Gao et al. 2005). Our most exciting finding is that the NMDAR-mediated neuronal death can be prevented or greatly reduced by specifically blocking ASIC1a.
When using calcium imaging to study the relationship between receptor channel activity and the pattern of neuronal Ca2+ influx, we found that stimulation of ASIC1a alone induced only a small intracellular Ca2+ increase, whereas stimulation of NMDAR channels alone evoked a 6.5-fold greater Ca2+ influx into neurons. Moreover, activation of both NMDAR and ASIC1a evoked a huge intracellular Ca2+ increase (tenfold). Most interestingly, this increase in [Ca2+]i was greatly suppressed by inhibition of ASIC1a. These Ca2+ imaging data further support our cell viability assay finding that neuronal death due to NMDA-evoked intracellular Ca2+ increase can be prevented by blocking ASIC1a.
ASIC1a is Required for NMDAR Function
The release of H+ from presynaptic terminals brings about a pH drop at synaptic cleft and activates ASIC1a (Krishtal et al. 1987; Waldmann et al. 1997; Xiong et al. 2004). The data from our patch-clamp recordings in hippocampal brain slices showed that acidic pH enhanced NMDAR EPSC amplitudes shortly after the onset of pH drop in ECS, whereas the NMDAR EPSC amplitudes were greatly reduced after keeping perfusion of the brain slice in acidic pH. However, 20–80 % rise time of NMDAR EPSCs was always reduced no matter the EPSC amplitude increased or decreased induced by acidic ECS perfusion. These data suggest that ASIC1a activity evoked by acidic ECS do enhance NMDAR function through affecting NMDAR channel open. It might be the desensitization feature of ASIC1a and the inhibitory property of H+ on NMDAR EPSC that made NMDAR EPSC amplitude decrease after prolonged acidic ECS perfusion understandable.
Other evidences were when inhibition of ASIC1a greatly suppressed NMDAR EPSC amplitude and impeded NMDAR channel open at physiological pH (data in separate paper). Moreover, at basic pH ECS (pH 8.0), the NMDAR EPSC amplitude was greatly increased, but the 20–80 % rise time was markedly increased which means that NMDAR channel open was slowed down. These data again confirmed that ASIC1a activity plays a key role in facilitating NMDAR channel open.
In other set of experiments involving hippocampal cultures, we found that prolonged exposure to NMDA resulted in a great amount of neuronal death at pH 7.4, whereas exposure to pH 6.0 ECS alone without addition of NMDA resulted in a comparable amount of neuronal death. However, inhibition of ASIC1a abolished the NMDA-induced neuronal death. These data strongly suggest that direct overstimulation of NMDARs is responsible for the neuronal death. We hypothesized that activating ASIC1a through increasing the concentration of H+ in ECS can indirectly activate NMDARs and cause neuronal damage.
Taken together, our data strongly suggest that NMDAR-mediated synaptic transmission is the primary excitatory pathway, and that ASIC1a activity is essential for modulating the efficiency of this transmission process. In other words, ASIC1a plays an important role in maintaining the synaptic functional homeostasis in physiological condition.
ASIC1a Exacerbate NMDAR-Mediated Neuronal Death
Overstimulation of NMDARs causes the necrotic and apoptotic neuronal deaths. Acute Na+ influx mediates toxic neuronal swelling and largely contributes to necrotic neuronal death (Choi 1987). However, Ca2+ overload mediated by NMDARs is the primary factor that causes cell death (Choi 1985, 1987). This is because Ca2+entry through NMDARs is tightly linked to the overactivation of NOS, which is the most cytotoxic signaling pathway downstream from NMDARs (Sattler et al. 1999; Lau and Tymianski 2010). High [Ca2+]i is deleterious to mitochondrial function because the mitochondria is the primary intracellular buffer for [Ca2+]i. Mitochondrial dysfunction leads to not only apoptotic neuronal death due to the release of apoptotic inducers, but also reduces ATP production and causes necrotic cell death (Szydlowska and Tymianski 2010).
ASIC1a channelis activated by a sudden pH drop from 7.4 to 6.8 and below (Waldmann et al. 1997; Yermolaieva et al. 2004; Baron et al. 2008), and then rapidly undergoes a de-sensitization stage (Waldmann et al. 1997; Diochot et al. 2007). The activation phase of ASIC1a may be most crucial for initiation or enhancing NMDAR function. Our electro-physiology data showed that acidic stimulation of ASIC1a immediately increased not only NMDAR EPSC amplitudes, but also reduced their rise time as described above, which is linked to NMDAR channel open kinetics (Erreger et al. 2005) and the conductance (Erreger et al. 2005). In addition, our Western blot results added more evidence that activation of ASIC1a markedly increased NMDAR’s NR2B subunit phosphorylation. In contrast, blocking ASIC1a not only reduced the amplitudes of NMDAR EPSCs, but also increased their rise time. These electrophysiological data revealed for the first time the functional relationship between NMDAR and ASIC1a at the glutamatergic postsynaptic membrane. It is now easier to under why NMDAR-mediated neuronal death is exacerbated in acidic ECS.
It seems that NMDAR and ASIC1a do not induce cell death through parallel and separate pathways, because ASIC1a activation-induced cell death could be prevented by blocking either NMDARs or ASIC1a. Zha and co-workers proposed that ASIC1a induced intracellular Ca2+ influx largely through VGCCs (Zha et al. 2006; Samways et al. 2009). However, our data suggest that ASIC1a induces [Ca2+]i increase through activating NMDARs because we already blocked VGCCs in our experiments. It is also not very likely that ASIC1a itself mediated the large Ca2+ influx, since ASIC1a has negligible permeability to Ca2+ (Samways et al. 2009). This is illustrated in our experiments where, after blocking NMDARs, stimulation of ASIC1a could not induce either increased NR2B phosphorylation or greater neuronal deaths.
To determine whether the ASIC1a activation-induced [Ca2+]i overload and neuronal death is mediated through NMDAR channels, the activity of major downstream proteins in NMDAR-related intracellular signaling pathway needed to be further examined; for example, nNOS activation or CaMKII phosphorylation.
ASIC1a Facilitates NMDAR-Mediated Ca2+ Entering Neurons
Previous measurements estimate the Ca2+ permeability of ASIC1a (PCa/Na) to 0.02–0.4 (Kovalchuk Yu et al. 1990; Bassler et al. 2001; Wu et al. 2004; Neaga et al. 2005), which is higher than values obtained for other members of the ASICs. However, this Ca2+ permeability is several hundred times less than that of NMDARs (Mayer and Westbrook 1987). It is therefore debatable whether direct Ca2+ entry through these channels makes a substantial and direct contribution to [Ca2+]i homeostasis. Zha and co-workers reported a very small fraction of Ca2+ influx directly through ASIC1a receptors, most [Ca2+]i got into neurons was secondary through VGCCs (Zha et al. 2006). We need to point out here that when we measured [Ca2+]i changes, VGCCs were blocked by containing nifedipine in ECS. Samways and colleagues (2009) showed that native and recombinant homomeric ASIC1a did not show a measurable Ca2+ permeability. Most recent studies suggest that ASIC1a is not a primary mediator of [Ca2+]i increase in physiological or pathological conditions (Samways et al. 2009).
Our experimental findings strongly suggest that ASIC1a channel activation can directly influence the activity of NMDARs, thereby altering their Ca2+ permeability and indirectly inducing [Ca2+]i increases. Our novel findings may be helpful for explaining why loss of ASIC1a disrupts hippocampal-dependent LTP and spatial memory in the Morris water maze (Wemmie et al. 2002, 2003), why ASIC1a affects the density of dendritic spines development (Zha et al. 2006), and how ASIC1a causes neuronal death through disrupting [Ca2+]i homeostasis in brain ischemia (Xiong et al. 2004; Gao et al. 2005; Sherwood et al. 2011). The key to answering all these questions is the discovery of the close association between ASIC1a and NMDAR channel function. Although further studies need to be conducted to ascertain whether ASIC1a and NMDAR interact directly at the cell membrane level or through intracellular protein–protein interactions, but the most important conclusion from our study is that inhibition of ASIC1a can effectively inhibit NMDAR-mediated Ca2+ increase without fully suppressing NMDAR-mediated synaptic transmission.
Since neurons exposure to acidic ECS alone evoked a small amount of intracellular Ca2+ increased than exposure to NMDA alone, we theorize that in normal physiological condition, the activity of neurons in the neuronal network causes just enough glutamate release to meet the need for activation of NMDARs in normal physiological synaptic transmission. Extracellular pH drop may up-regulate NMDAR function by exciting ASIC1a, but the total number of activated NMDARs is not changed because there is no increased glutamate release; on the contrary, the released glutamate might be decreased (Traynelis and Cull-Candy 1990). The integrated output may be a slightly increase in intracellular Ca2+ as we observed in our experiments. However, adding NMDA to the medium directly activated more NMDARs and tipped off the balance toward inducing more Ca2+ influx into neurons. Thus, NMDA stimulation in an acidic environment can cause a much higher intracellular Ca2+ accumulation than does either stimulation alone. ASIC1a behaves like the gating controller for NMDARs, thereby influencing NMDAR-mediated Ca2+ influx.
Therapeutic Benefit of NMDAR-Associated Disease
Almost all NMDAR antagonists, except memantine (Areosa et al. 2005), have failed to pass clinical trials because of their poor therapeutic efficiency and poor tolerability (Ikonomidou et al. 1989; Ikonomidou and Turski 2002). This is understandable because NMDAR signaling plays important physiological roles in normal CNS synaptic transmission, as well as crucial adaptive processes such as synaptic plasticity, neuronal survival, and resistance to trauma (Forrest et al. 1994; Linden 1994; Kohara et al. 2001). The reason that memantine has shown the best clinical effect is because it is a low affinity and voltage-dependent unbinding NMDAR channel blocker that spares much of the synaptic NMDAR activity, but inhibits a large part of pathological chronic NMDAR activation (Parsons et al. 1999; Hardingham and Bading 2003; Areosa et al. 2005). Because brain ischemia is always accompanied by local acidosis, antagonists of ASIC1a may be ideal therapeutic agents for neuronal protection as they will not completely disrupt NMDAR-mediated synaptic transmission. When ASIC1a activity is inhibited, the CNS activates a compensate mechanism to maintain normal synaptic NMDAR function, thus minimizing disruption to the physiological function of NMDARs (Zha et al. 2006). Therefore, ASIC1a may be an attractive therapeutic target for the treatment of brain ischemia.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Natural Science Foundation of China (No. 81171233); the Natural Science Foundation of Shandong Province (No. ZR2010HM011); Foundation of Yantai Science and Technology (No. 2010252); and Taishan Scholar (tshw20110515).
Abbreviations
- NMDAR
N-methyl D-aspartate receptor
- ASIC1a
Acid-sensing ion channel 1a
- ACSF
Artificial cerebral spinal fluid
- OGD
Oxygen-glucose deprivation
- AMPA receptor
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- GABAA receptor
γ-Aminobutyric acid A-type receptor
- VGCC
Voltage-gated calcium channels
- PCs
Pyramidal cells
- Mg2+
Magnesium ion
- [Ca2+]i
Intracellular calcium concentration
- NBQX
1,2,3,4-Tetrahydro-6-nitro-2,3-dioxobenzo[f]quinoxaline-7-sulfonamide
- APV
2-Amino-5-phosphonopentanoic acid
- PcTX1
Psalmotoxin 1
- ECS
Extracellular solution
Footnotes
Conflict of interest We have no conflict of interest.
Electronic supplementary material The online version of this article (doi:10.1007/s12640-015-9530-3) contains supplementary material, which is available to authorized users.
References
- Alvarez de la Rosa D, Zhang P, et al. Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci USA. 2002;99(4):2326–2331. doi: 10.1073/pnas.042688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez de la Rosa D, Krueger SR, et al. Distribution, subcellular localization and ontogeny of ASIC1 in the mammalian central nervous system. J Physiol. 2003;546(Pt 1):77–87. doi: 10.1113/jphysiol.2002.030692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Areosa SA, Sherriff F, et al. Memantine for dementia. Cochrane Database Syst Rev. 2005;2006(2):CD003154. doi: 10.1002/14651858.CD003154.pub3. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61(6):657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron A, Voilley N, et al. Acid sensing ion channels in dorsal spinal cord neurons. J Neurosci. 2008;28(6):1498–1508. doi: 10.1523/JNEUROSCI.4975-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler EL, Ngo-Anh TJ, et al. Molecular and functional characterization of acid-sensing ion channel (ASIC) 1b. J Biol Chem. 2001;276(36):33782–33787. doi: 10.1074/jbc.M104030200. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, et al. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287(4):C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Broughton BR, Reutens DC, et al. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40(5):e331–e339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58(3):293–297. doi: 10.1016/0304-3940(85)90069-2. [DOI] [PubMed] [Google Scholar]
- Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7(2):369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, et al. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11(3):327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- DeVries SH. Exocytosed protons feedback to suppress the Ca2+ current in mammalian cone photoreceptors. Neuron. 2001;32(6):1107–1117. doi: 10.1016/s0896-6273(01)00535-9. [DOI] [PubMed] [Google Scholar]
- Diochot S, Salinas M, et al. Peptides inhibitors of acid-sensing ion channels. Toxicon. 2007;49(2):271–284. doi: 10.1016/j.toxicon.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, et al. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563(Pt 2):345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DH, Horiuchi M, et al. Characterization of acid-sensing ion channel expression in oligodendrocyte-lineage cells. Glia. 2008;56(11):1238–1249. doi: 10.1002/glia.20693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesik SW, Shi Y. Structural biology. Controlling the caspases. Science. 2001;294(5546):1477–1478. doi: 10.1126/science.1062236. [DOI] [PubMed] [Google Scholar]
- Forrest D, Yuzaki M, et al. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron. 1994;13(2):325–338. doi: 10.1016/0896-6273(94)90350-6. [DOI] [PubMed] [Google Scholar]
- Gao J, Duan B, et al. Coupling between NMDA receptor and acid-sensing ion channel contributes to ischemic neuronal death. Neuron. 2005;48(4):635–646. doi: 10.1016/j.neuron.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Clarke SJ, et al. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res. 2004;61(3):372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26(2):81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Iino M, Ozawa S, et al. Permeation of calcium through excitatory amino acid receptor channels in cultured rat hippocampal neurones. J Physiol. 1990;424:151–165. doi: 10.1113/jphysiol.1990.sp018060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1(6):383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Mosinger JL, et al. Sensitivity of the developing rat brain to hypobaric/ischemic damage parallels sensitivity to N-methyl-aspartate neurotoxicity. J Neurosci. 1989;9(8):2809–2818. doi: 10.1523/JNEUROSCI.09-08-02809.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325(6104):529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, et al. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291(5512):2419–2423. doi: 10.1126/science.1057415. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Yu N, Krishtal OA, et al. The proton-activated inward current of rat sensory neurons includes a calcium component. Neurosci Lett. 1990;115(2–3):237–242. doi: 10.1016/0304-3940(90)90461-h. [DOI] [PubMed] [Google Scholar]
- Krishtal OA, Osipchuk YV, et al. Rapid extracellular pH transients related to synaptic transmission in rat hippocampal slices. Brain Res. 1987;436(2):352–356. doi: 10.1016/0006-8993(87)91678-7. [DOI] [PubMed] [Google Scholar]
- Lai TW, Zhang S, et al. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–188. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460(2):525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Lee JM, et al. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399(6738 suppl):A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Linden R. The survival of developing neurons: a review of afferent control. Neuroscience. 1994;58(4):671–682. doi: 10.1016/0306-4522(94)90447-2. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330(9):613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27(11):2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott AB, Mayer ML, et al. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321(6069):519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- Makhro A, et al. Functional NMDA receptors in rat erythrocytes. Am J Physiol Cell Physiol. 2010;298(6):C1315–C1325. doi: 10.1152/ajpcell.00407.2009. [DOI] [PubMed] [Google Scholar]
- Manev H, Favaron M, et al. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol Pharmacol. 1989;36(1):106–112. [PubMed] [Google Scholar]
- Marcoux FW, Probert AW, Jr, et al. Hypoxic neuronal injury in tissue culture is associated with delayed calcium accumulation. Stroke. 1990;21(11 Suppl):III71–III74. [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003;5(12):1041–1043. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Vyklicky L., Jr The action of zinc on synaptic transmission and neuronal excitability in cultures of mouse hippocampus. J Physiol. 1989;415:351–365. doi: 10.1113/jphysiol.1989.sp017725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL. Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol. 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, et al. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309(5965):261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Neaga E, Amuzescu B, et al. Extracellular trypsin increases ASIC1a selectivity for monovalent versus divalent cations. J Neurosci Methods. 2005;144(2):241–248. doi: 10.1016/j.jneumeth.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, et al. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist—a review of preclinical data. Neuropharmacology. 1999;38(6):735–767. doi: 10.1016/s0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- Rock DM, Macdonald RL. The polyamine spermine has multiple actions on N-methyl-D-aspartate receptor single-channel currents in cultured cortical neurons. Mol Pharmacol. 1992;41(1):83–88. [PubMed] [Google Scholar]
- Samways DS, Harkins AB, et al. Native and recombinant ASIC1a receptors conduct negligible Ca2+ entry. Cell Calcium. 2009;45(4):319–325. doi: 10.1016/j.ceca.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, et al. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284(5421):1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Schanne FA, et al. Calcium dependence of toxic cell death: a final common pathway. Science. 1979;206(4419):700–702. doi: 10.1126/science.386513. [DOI] [PubMed] [Google Scholar]
- Sherwood TW, Lee KG, et al. Heteromeric acid-sensing ion channels (ASICs) composed of ASIC2b and ASIC1a display novel channel properties and contribute to acidosis-induced neuronal death. J Neurosci. 2011;31(26):9723–9734. doi: 10.1523/JNEUROSCI.1665-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47(2):122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Chesler M. Proton release as a modulator of presynaptic function. Neuron. 2001;32(6):960–962. doi: 10.1016/s0896-6273(01)00549-9. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature. 1990;345(6273):347–350. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. Pharmacological properties and H+ sensitivity of excitatory amino acid receptor channels in rat cerebellar granule neurones. J Physiol. 1991;433:727–763. doi: 10.1113/jphysiol.1991.sp018453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu W, Xu X, et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140(2):222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann R, Lazdunski M. H(+)-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr Opin Neurobiol. 1998;8(3):418–424. doi: 10.1016/s0959-4388(98)80070-6. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, et al. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386(6621):173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, et al. H(+)-gated cation channels. Ann New York Acad Sci. 1999;868:67–76. doi: 10.1111/j.1749-6632.1999.tb11274.x. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Chen J, et al. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34(3):463–477. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Askwith CC, et al. Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci. 2003;23(13):5496–5502. doi: 10.1523/JNEUROSCI.23-13-05496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmie JA, Coryell MW, et al. Overexpression of acid-sensing ion channel 1a in transgenic mice increases acquired fear-related behavior. Proc Natl Acad Sci USA. 2004;101(10):3621–3626. doi: 10.1073/pnas.0308753101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmie JA, Price MP, et al. Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci. 2006;29(10):578–586. doi: 10.1016/j.tins.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Williams K. Interactions of polyamines with ion channels. Biochem J. 1997;325(Pt 2):289–297. doi: 10.1042/bj3250289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LJ, Duan B, et al. Characterization of acid-sensing ion channels in dorsal horn neurons of rat spinal cord. J Biol Chem. 2004;279(42):43716–43724. doi: 10.1074/jbc.M403557200. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118(6):687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Yermolaieva O, Leonard AS, et al. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci USA. 2004;101(17):6752–6757. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha XM, Wemmie JA, et al. Acid-sensing ion channel 1a is a postsynaptic proton receptor that affects the density of dendritic spines. Proc Natl Acad Sci USA. 2006;103(44):16556–16561. doi: 10.1073/pnas.0608018103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.