

Abstract
Deficiencies of protein ion channels underlie many currently incurable human diseases. Robust networks of pumps and channels are usually responsible for the directional movement of specific ions in organisms ranging from microbes to humans. We thus questioned whether minimally selective small molecule mimics of missing protein channels might be capable of collaborating with the corresponding protein ion pumps to restore physiology. Here we report vigorous and sustainable restoration of yeast cell growth by replacing missing protein ion channels with imperfect small molecule mimics. We further provide evidence that this tolerance for imperfect mimicry is attributable to collaboration between the channel-forming small molecule and protein ion pumps. These results illuminate a mechanistic framework for pursuing small molecule replacements for deficient protein ion channels that underlie a range of challenging human diseases.
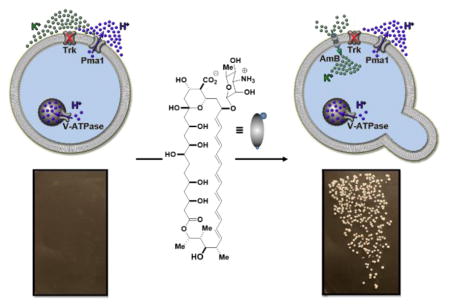
There are many currently incurable human diseases that are caused by missing protein ion channels, including cystic fibrosis, Bartter syndrome, Dravet syndrome, and Dent’s disease.1,2 Like many other human diseases caused by missing proteins, these diseases are difficult to treat, and new approaches are needed. Some small molecules can perform ion channel-like functions,3–11 suggesting the possibility of replacing missing protein ion channels with small molecule mimics. Closely replicating the functions of ion selective and tightly regulated protein channels with small molecules is challenging. However, robust protein networks comprised of pumps and channels drive targeted ions in targeted directions throughout the spectrum of living systems.12 We thus questioned whether relatively unselective and unregulated small molecule mimics of missing protein channels might be capable of collaborating with the corresponding protein ion pumps to restore physiology.
Yeast represent an excellent model system for studying eukaryotic physiology.13 Moreover, deficiencies of specific protein ion channels in yeast are known to lead to dramatic no growth phenotypes, thus providing a unique opportunity for using cell growth as a readout for physiology restoration. In yeast, ATP-driven V-ATPase and Pma1 proton pumps in the vacuolar and plasma membranes, respectively, collaborate with passive Trk potassium channels in the plasma membrane to achieve intracellular movement of potassium required for cell growth (Figure 1a, left).14 Loss of Trk channels impairs this uptake of environmental potassium and results in a no growth phenotype (Figure 1a, middle).15–17 Because the primary drivers of ion movement, the corresponding ATP-driven pumps, are still active in such yeast, we hypothesized that a small molecule ion channel permeable to potassium could collaborate with V-ATPase and Pma1 to restore cell growth (Figure 1a, right).
Figure 1.
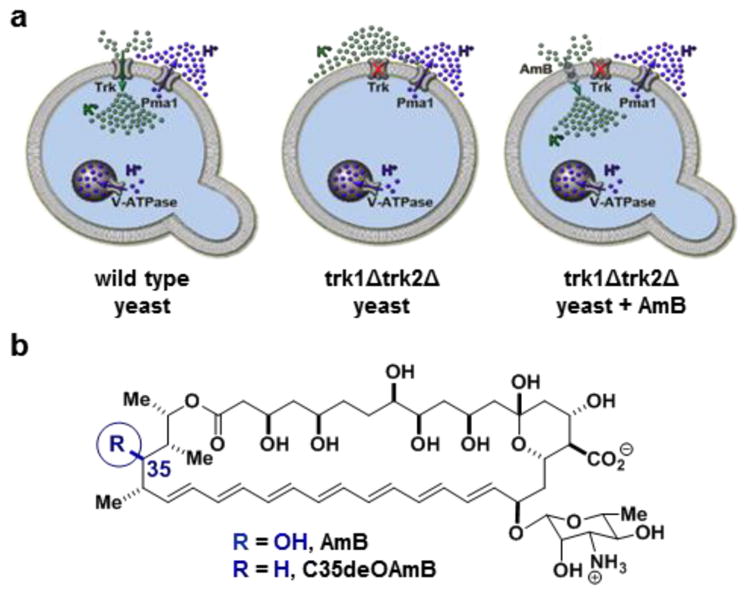
(a) The prospect of replacing missing protein channels with small molecule mimics. (b) Chemical structures of the archetypical ion channel-forming small molecule amphotericin B 7 and its single atom-deficient and channel-inactivated derivative C35-deoxy amphotericin B (C35deOAmB).
The ion channel-forming natural product amphotericin B (Figure 1b) was identified as a small molecule that could enable testing of this hypothesis.3 AmB can permeabilize yeast cells to potassium and other ions.18,19 AmB is also highly toxic to yeast, and this toxicity was thought to be inextricably linked to its membrane permeabilization. However, we found a synthesized derivative of AmB lacking a single oxygen atom at C35 (C35deOAmB) (Figure 1b) does not form ion channels, and yet still maintains potent fungicidal activity.20 Further studies revealed that AmB primarily kills yeast by binding and extracting sterols from membranes and is only cytotoxic when the amount of AmB exceeds that of ergosterol.21,22 This all suggested the channel activity of AmB might be separated from its cytocidal activity by simply adding this compound at low concentrations. Moreover, AmB and C35deOAmB, which differ via a single atom, represent a unique pair of probes for determining the impact of small molecule-mediated ion channel activity on organismal physiology.
We thus tested the hypothesis that AmB could restore cell growth in potassium channel-deficient yeast with a modified functional complementation experiment.23 Consistent with prior reports,15,16 growth was observed when wild type Saccharomyces cerevisiae were streaked onto agar plates containing normal concentrations of potassium (Figure 2a, left), and no growth was observed for the potassium channel-deficient strain (trk1Δtrk2Δ) (Figure 2a, middle). Strikingly, the addition of a low concentration of AmB (125 nM) to the agar plate vigorously restored growth of the trk1Δtrk2Δ mutant (Figure 2a. right).
Figure 2.

(a) Restoration of cell growth with a small molecule mimic of a missing protein channel. (b) Disc diffusion with AmB on a plate of trk1Δtrk2Δ cells. (c) AmB restores cell growth at concentrations below its minimum inhibitory concentration, while C35deOAmB does not restore growth. (d) Tetraethylammonium diminishes AmB-mediated growth restoration in trk1Δtrk2Δ cells but has no effect on trk1Δtrk2Δ cells grown under permissive conditions (100 mM KCl). (e) AmB restores uptake of extracellular 86Rb+, a surrogate for K+, in trk1Δtrk2Δ cells. (f) Vigorous restoration of cell growth is observed upon treating trk1Δtrk2Δ cells with AmB. (g) Similar to wild type cells, AmB-rescued trk1Δtrk2Δ cells show sustained growth over a period of >40 days. (h) A pre-formed AmB-ergosterol complex dramatically increases the range of concentrations over which physiology is restored. NS, not significant. *** P ≤ 0.0001. Graphs depict means ± SEM.
A series of additional experiments confirmed the observed restoration of cell growth is caused by the small molecule-based ion channel activity. A disc diffusion assay visually revealed the predicted dependence of this growth rescue on the concentration of AmB (Figure 2b). To quantify this concentration dependence and eliminate the potentially complicating issue of plating efficiency,24 we also measured trk1Δtrk2Δ yeast cell growth in a broth dilution assay (Figure 2c). Consistent with the disc diffusion results, no cell growth was observed in the absence of AmB, a dose-dependent increase of growth was observed at intermediate concentrations, and no growth was observed at or above the minimum inhibitory concentration of this antifungal agent. Further ruling out any type of generic hormetic effects,25 no growth stimulatory effects were observed when wild type cells were treated with AmB (Figure S1).
To directly probe the importance of the ion channel activity of AmB, we also tested the single atom variant C35deOAmB, which does not form ion channels (Figure 1b).20 This derivative failed to restore growth in trk1Δtrk2Δ cells at any tested concentration (Figure 2c). In a complementary experiment, we utilized the sterically bulky tetraethylammonium cation to block the AmB-based ion channel.26 This cation inhibited the functional complementation observed with AmB in a dose-dependent manner without causing general toxicity (Figure 2d). We also monitored uptake of radioactive 86Rb+ as a reporter of transmembrane potassium movement (Figure 3e).27 86Rb+ uptake was observed in wild type yeast but not in the trk1Δtrk2Δ mutant, and no uptake was observed when the trk1Δtrk2Δ mutant was treated with the channel-inactivated derivative C35deOAmB. In contrast, 86Rb+ uptake was restored when trk1Δtrk2Δ cells were treated with AmB.
Figure 3.
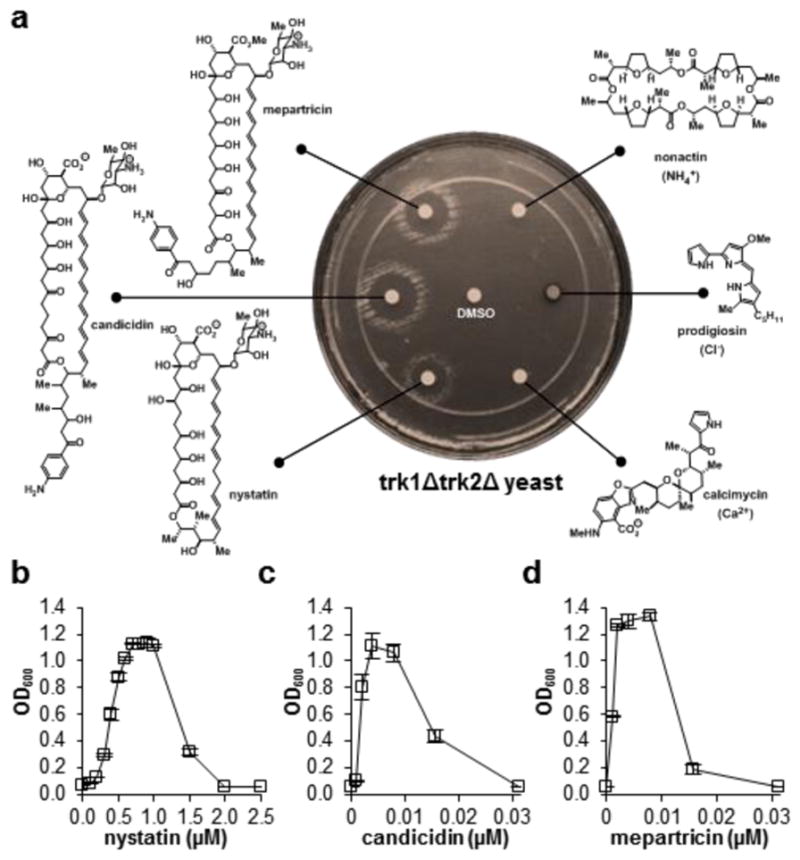
(a) A series of potassium-transporting polyene macrolide natural products restore vigorous cell growth in trk1Δtrk2Δ yeast, whereas no growth is observed upon treating with other small molecules that selectively transport other ions. (b–d) Nystatin, candicidin, and mepartricin were similarly able to restore physiology in a liquid broth dilution assay over a variety of concentrations. Graphs depict means ± SEM.
We further quantified the vigor and sustainability of this AmB ion channel-mediated restoration of yeast cell growth. AmB-treated trk1Δtrk2Δ cells reached a maximum cell density that matched that of the wild type (Figure 2f), and the doubling time for AmB-rescued trk1Δtrk2Δ cells was only 1.7 times longer (Figure S3). Equivalent levels of cell viability in wild type and AmB-rescued trk1Δtrk2Δ cells were also observed (Figure S4). To probe the sustainability of this rescue effect, we reiterated the max cell density and doubling time experiments for over a month. Like wild type cells, the AmB-rescued trk1Δtrk2Δ cells showed sustained vigorous cell growth throughout this entire period of time (Figure 2g). Removing AmB from the media at any point resulted in rapid loss of growth for the trk1Δtrk2Δ cells. We further confirmed the mechanism-based hypothesis that a pre-formed AmB-ergosterol complex should retain the capacity to permeabilize yeast cells (Figure S2) but show substantially decreased cell killing.21 This pre-formed complex dramatically extended the range of concentrations over which rescue is observed (Figure 3h).
To probe the scope and limitations of this tolerance for imperfect mimicry of missing protein channels with small molecules, a series of additional ion transporting natural products were evaluated. Vigorous restoration of trk1Δtrk2Δ cell growth was observed with other small molecules that transport potassium, including nystatin, candicidin, and mepartricin, but not with those that selectively transport NH4+ (nonactin), Cl− (prodigiosin), and Ca2+ (calcimycin) (Figure 3).
Finally, we tested the mechanistic hypothesis that these potassium channel-forming small molecules restore physiology by collaborating with the V-ATPase and Pma1 proton pumps. Such a model predicts selective sensitivity of the small molecule-rescued mutants to chemical inhibition of these pumps. As a negative control, AmB-treated wild type and AmB-rescued trk1Δtrk2Δ cells were equally sensitive to nocodazole, an off-pathway inhibitor of microtubule dynamics (Figure 4a). In contrast, AmB-rescued trk1Δtrk2Δ cells were exceptionally sensitive to inhibition of V-ATPase with bafilomycin (Figure 4b) and Pma1 with ebselen (Figure 4c). Similar results were observed with nystatin-, candicidin-, and mepartricin-rescued trk1Δtrk2Δ cells (Figure d–f).
Figure 4.
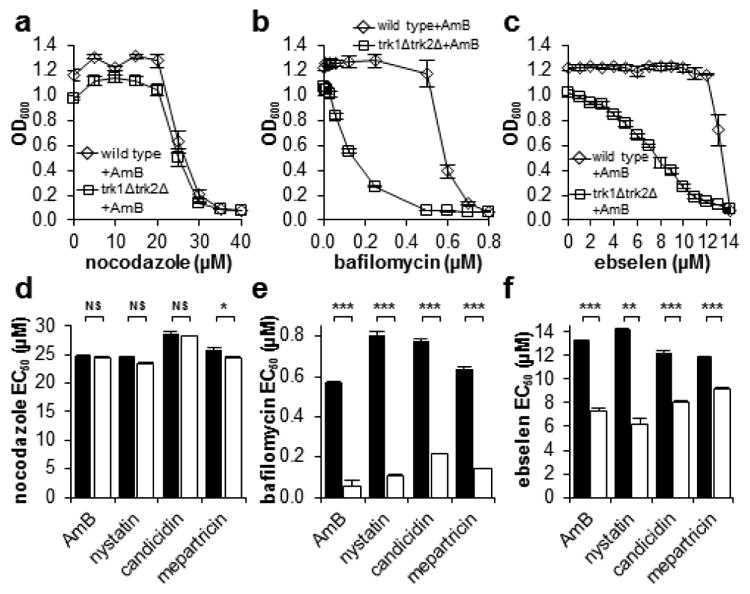
(a) AmB-treated wild type S. cerevisiae and trk1Δtrk2Δ cells are equally sensitive to the off-pathway microtubule inhibitor nocodazole. (b) AmB-rescued trk1Δtrk2Δ cells are substantially more sensitive to the V-ATPase inhibitor bafilomycin compared to wild type cells. (c) AmB-rescued trk1Δtrk2Δ cells are substantially more sensitive to the Pma1 inhibitor ebselen than wild type cells. (d–f) EC50 values for various inhibitors of cell growth against wild type (black bars) and trk1Δtrk2Δ cells (white bars) treated with optimum rescue concentrations of potassium-transporting polyene macrolide natural products AmB, nystatin, candicidin, and mepartricin. NS, not significant. * P ≤ 0.05. ** P ≤ 0.001. *** P ≤ 0.0001. Graphs depict means ± SEM.
Thus, imperfect small molecule mimics of mission protein ion channels can restore physiology in yeast, and evidence supports that this phenomenon is attributable to functional collaboration between small molecules channels and protein ion pumps. A common channels and pumps architecture is responsible for directional ion movement in organisms from yeast to humans,12 suggesting that the same type of functional collaboration observed herein might enable small molecule surrogates for missing protein ion channels to impact on human disease.
Supplementary Material
Acknowledgments
We gratefully acknowledge L. Jan for trk1Δtrk2Δ strain SGY1528, W. Dobrucki for help with 86Rb+ assays, and M. Sivaguru for help with cell viability assays. We also thank HHMI and the NIH (GM080436) for funding. M.D.B. is an HHMI Early Career Scientist. A.S.G. is an NSF graduate fellow.
Footnotes
Author Contributions
A.G.C., J.H., A.S.G., K.A.D., and M.D.B, designed the experiments and interpreted the data. A.G.C and J.H. performed the experiments. A.G.C, J.H., A.S.G., K.A.D., and M.D.B wrote the paper.
Notes
The authors declare no competing financial interests.
Materials, methods, and supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hubner CA, Jentsch TJ. Hum Mol Gen. 2002;11:2435. doi: 10.1093/hmg/11.20.2435. [DOI] [PubMed] [Google Scholar]
- 2.Rouleau G. Ion Channel Diseases. Elsevier; Amsterdam: 2008. [Google Scholar]
- 3.Ermishkin LN, Kasumov KM, Potzeluyev VM. Nature. 1976;262:698. doi: 10.1038/262698a0. [DOI] [PubMed] [Google Scholar]
- 4.El-Etri M, Cuppoletti J. Am J Physiol. 1996;270:L386. doi: 10.1152/ajplung.1996.270.3.L386. [DOI] [PubMed] [Google Scholar]
- 5.Busschaert N, Gale PA. Angew Chem Int Ed Engl. 2013;52:1374. doi: 10.1002/anie.201207535. [DOI] [PubMed] [Google Scholar]
- 6.Jiang C, Lee ER, Lane MB, Xiao YF, Harris DJ, Cheng SH. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1164. doi: 10.1152/ajplung.2001.281.5.L1164. [DOI] [PubMed] [Google Scholar]
- 7.Koulov AV, Lambert TN, Shukla R, Jain M, Boon JM, Smith BD, Li H, Sheppard DN, Joos JB, Clare JP, Davis AP. Angew Chem Int Ed Engl. 2003;42:4931. doi: 10.1002/anie.200351957. [DOI] [PubMed] [Google Scholar]
- 8.Shen B, Li X, Wang F, Yao X, Yang D. PloS One. 2012;7:e34694. doi: 10.1371/journal.pone.0034694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakai N, Matile S. Langmuir. 2013;29:9031. doi: 10.1021/la400716c. [DOI] [PubMed] [Google Scholar]
- 10.Fyles TM. Acc Chem Res. 2013;46:2847. doi: 10.1021/ar4000295. [DOI] [PubMed] [Google Scholar]
- 11.Gokel GW, Negin S. Acc Chem Res. 2013;46:2824. doi: 10.1021/ar400026x. [DOI] [PubMed] [Google Scholar]
- 12.Gouaux E, Mackinnon R. Science. 2005;310:1461. doi: 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- 13.Bolotin-Fukuhara M, Dumas B, Gaillardin C. FEMS Yeast Res. 2010;10:959. doi: 10.1111/j.1567-1364.2010.00693.x. [DOI] [PubMed] [Google Scholar]
- 14.Cyert MS, Philpott CC. Genetics. 2013;193:677. doi: 10.1534/genetics.112.147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaber RF, Styles CA, Fink GR. Mol Cell Biol. 1988;8:2848. doi: 10.1128/mcb.8.7.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko CH, Gaber RF. Mol Cell Biol. 1991;11:4266. doi: 10.1128/mcb.11.8.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minor DL, Jr, Masseling SJ, Jan YN, Jan LY. Cell. 1999;96:879. doi: 10.1016/s0092-8674(00)80597-8. [DOI] [PubMed] [Google Scholar]
- 18.Ermishkin LN, Kasumov KM, Potseluyev VM. Biochim Biophys Acta. 1977;470:357. doi: 10.1016/0005-2736(77)90127-4. [DOI] [PubMed] [Google Scholar]
- 19.Bolard J. Biochim Biophys Acta. 1986;864:257. doi: 10.1016/0304-4157(86)90002-x. [DOI] [PubMed] [Google Scholar]
- 20.Gray KC, Palacios DS, Dailey I, Endo MM, Uno BE, Wilcock BC, Burke MD. Proc Natl Acad Sci USA. 2012;109:2234. doi: 10.1073/pnas.1117280109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson TM, Clay MC, Cioffi AG, Diaz KA, Hisao GS, Tuttle MD, Nieuwkoop AJ, Comellas G, Maryum N, Wang S, Uno BE, Wildeman EL, Gonen T, Rienstra CM, Burke MD. Nature Chem Biol. 2014;10:400. doi: 10.1038/nchembio.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palacios DS, Dailey I, Siebert DM, Wilcock BC, Burke MD. Proc Natl Acad Sci USA. 2011;108:6733. doi: 10.1073/pnas.1015023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee MG, Nurse P. Nature. 1987;327:31. doi: 10.1038/327031a0. [DOI] [PubMed] [Google Scholar]
- 24.Brajtburg J, Elberg S, Medoff G, Kobayashi GS. Antimicrob Agents Chemother. 1981;19:199. doi: 10.1128/aac.19.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaiser J. Science. 2003;302:376. doi: 10.1126/science.302.5644.376. [DOI] [PubMed] [Google Scholar]
- 26.Borisova MP, Ermishkin LN, Silberstein AY. Biochim Biophys Acta. 1979;553:450. doi: 10.1016/0005-2736(79)90300-6. [DOI] [PubMed] [Google Scholar]
- 27.Sentenac H, Bonneaud N, Minet M, Lacroute F, Salmon JM, Gaymard F, Grignon C. Science. 1992;256:663. doi: 10.1126/science.1585180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.