

Abstract
Objective:
To identify the genetic defects present in 3 families with muscular dystrophy, contractures, and calpain 3 deficiency.
Methods:
We performed targeted exome sequencing on one patient presenting a deficiency in calpain 3 on Western blot but for which mutations in the gene had been excluded. The identification of a homozygous truncating mutation in the M-line part of titin prompted us to sequence this region in 2 additional patients presenting similar clinical and biochemical characteristics.
Results:
The 3 patients shared similar features: coexistence of limb-girdle weakness and early-onset diffuse joint contractures without cardiomyopathy. The biopsies showed rimmed vacuoles, a dystrophic pattern, and secondary reduction in calpain 3. We identified a novel homozygous mutation in the exon Mex3 of the TTN gene in the first patient. At protein level, this mutation introduces a stop codon at the level of Mex3. Interestingly, we identified truncating mutations in both alleles in the same region of the TTN gene in patients from 2 additional families. Molecular protein analyses confirm loss of the C-ter part of titin.
Conclusions:
Our study broadens the phenotype of titinopathies with the report of a new clinical entity with prominent contractures and no cardiac abnormality and where the recessive mutations lead to truncation of the M-line titin and secondary calpain 3 deficiency.
Identification of the precise genetic cause of muscular dystrophies (MD) is a complex task, especially since they are characterized by genetic heterogeneity and variable clinical expressivity, as with limb-girdle muscular dystrophy type 2A (LGMD2A).1 Indeed, about a third of mutations in the LGMD2A gene that encodes the protease calpain 3 have no quantitative consequences at protein level but only on its activity2 and secondary deficiencies in calpain 3 have been observed in a number of pathologic conditions including defects in dysferlin, anoctamin5, or titin.3–5
Titin (TTN, OMIM 188840) is the largest known protein (33,000 amino acids) and is encoded by 363 exons. It spans half a sarcomere and is anchored at the Z-disc and M-line.6 It plays an essential role in the skeletal and heart muscles, development, structure, extensibility, elasticity, and signaling of the sarcomeres.7 Mutations in the titin gene have been implicated in a range of disorders involving exclusively the cardiac muscle,8–12 exclusively the skeletal muscle,3,13 or both tissues.4,14
We report the identification of new truncating mutations in the C-terminal part of titin in 3 families presenting MD and in whom a calpain 3 deficiency was observed on Western blot (WB). The deficiency was qualified as being likely secondary since no mutations in the calpain 3 gene were identified. The clinical presentation of the patients resembles Emery-Dreifuss MD (EDMD), albeit with no cardiac abnormality. EDMD has until now been associated with mutations in genes coding for 3 major proteins: emerin (EMD), lamin A/C (LMNA), and FHL1.15–17 Other minor genes have been reported in EDMD-like patients including SYNE1 (nesprin-1), SYNE2 (nesprine2), TMEM43 (LUMA), SUN1 (SUN1), and SUN2 (SUN2).18–20 Our report extends the clinical phenotype spectrum of titinopathies.
METHODS
Standard protocol approvals, registrations, and patient consents.
All biological samples (blood and muscle biopsies) were obtained after informed consent was received from patients and their relatives.
Sequencing.
Targeted sequencing was performed using a custom-made SureSelect library on a HiSeq2000 instrument (Illumina; San Diego, CA). Details are indicated in the e-Methods on the Neurology® Web site at Neurology.org. Sanger sequencing was used to confirm variants in family 1 and to sequence the M-line TTN exons in families 2 and 3 (figure e-1). PCR products were amplified with primers shown in table e-1 and sequenced through Beckman Coulter Genomics (Hertfordshire, UK) services.
RT-PCR analysis.
Analysis of splicing in the C-ter part of titin was performed by reverse transcription (RT)-PCR on 1 μg of RNA extracted using Trizol and reverse transcribed using RevertAid H-first strand cDNA synthesis kit (ThermoScientific; Waltham, MA) and primers localized in Mex1 and Mex5. Nonsense-mediated decay (NMD) was evaluated by real-time quantitative RT-PCR analysis using primers located in Mex1 in a SyBer green system on a real-time thermocycler (LC480; Roche, Basel, Switzerland). Results were expressed in 2ΔΔCT normalized by P0. Primer sequences are detailed in table e-1.
Morphologic and immunofluorescence analyses.
Cryosections of fresh frozen deltoid or quadriceps femoris biopsies were histochemically and immunocytochemically analyzed by standard techniques.21 The following antibodies were used: desmin (D33, DAKO; Carpinteria, CA), α-β crystallin (G2JF, Novocastra; Newcastle upon Tyne, UK), myotilin (RSO34, Novocastra), titin N-ter staining (Millipore MAB1553; Billerica, MA), rabbit anti-Titin M10-1 polyclonal (1:100), and mouse anti-α-actinin monoclonal (1:1000, Sigma-Aldrich A7811; St. Louis, MO). Details are indicated in the e-Methods.
Western blot.
Multiplex WB was performed as described by Anderson and Davison22 using the following monoclonal antibodies (Novocastra): N-ter calpain3 (Calp3d/2C4; dilution 1:19), C-ter calpain3 (Calp3c/12A2; dilution 1:15), dystrophin (NCL-DYS1 and NCL-DYS2; dilution 1:150), dysferlin (NCL-Hamlet; dilution 1:1500), α-sarcoglycan (NCL-L-a-SARC; dilution 1:150), and γ-sarcoglycan (NCL-L-g-SARC; dilution 1:150). WB for titin was performed as previously described with the M10-1 titin antibody.23
RESULTS
Clinical and imaging findings.
Patient 1 is a man born from a consanguineous Algerian family with asymptomatic sibs and parents (table 1 and figure 1A). Disease onset was at 10 years with lower limb girdle weakness, waddling gait, and Achilles tendon contractures. The clinical course remained relatively stable from 10 to 17 years, but then progressed rapidly with difficulties climbing stairs rising from a chair, upper girdle weakness, and contractures extending to knees, elbows, and fingers at age 20 years. At age 23 years, the patient was partially wheelchair bound. Examination showed severe symmetrical proximal weakness, scapular winging, and weak abdominal muscles. Distal muscles were much less affected (table e-2). Joint contractures were marked in the 4 limbs while neck and spine were spared (figure 1B and table e-3). Tongue was hypertrophic. Creatine kinase (CK) measurements were 4–10 times the upper normal values. There was no facial, oculomotor, or pharyngeal weakness. There were neither respiratory (vital capacity 89%) nor cardiac abnormalities as evaluated by ECG and echocardiography. Lower limb muscle MRI showed a moderate to severe fatty degeneration in all pelvic, thigh, and lower leg muscles with some sparing of sartorius and, in particular, tibialis posterior and deep toe flexors (figure 1C).
Table 1.
Clinical data of the patients
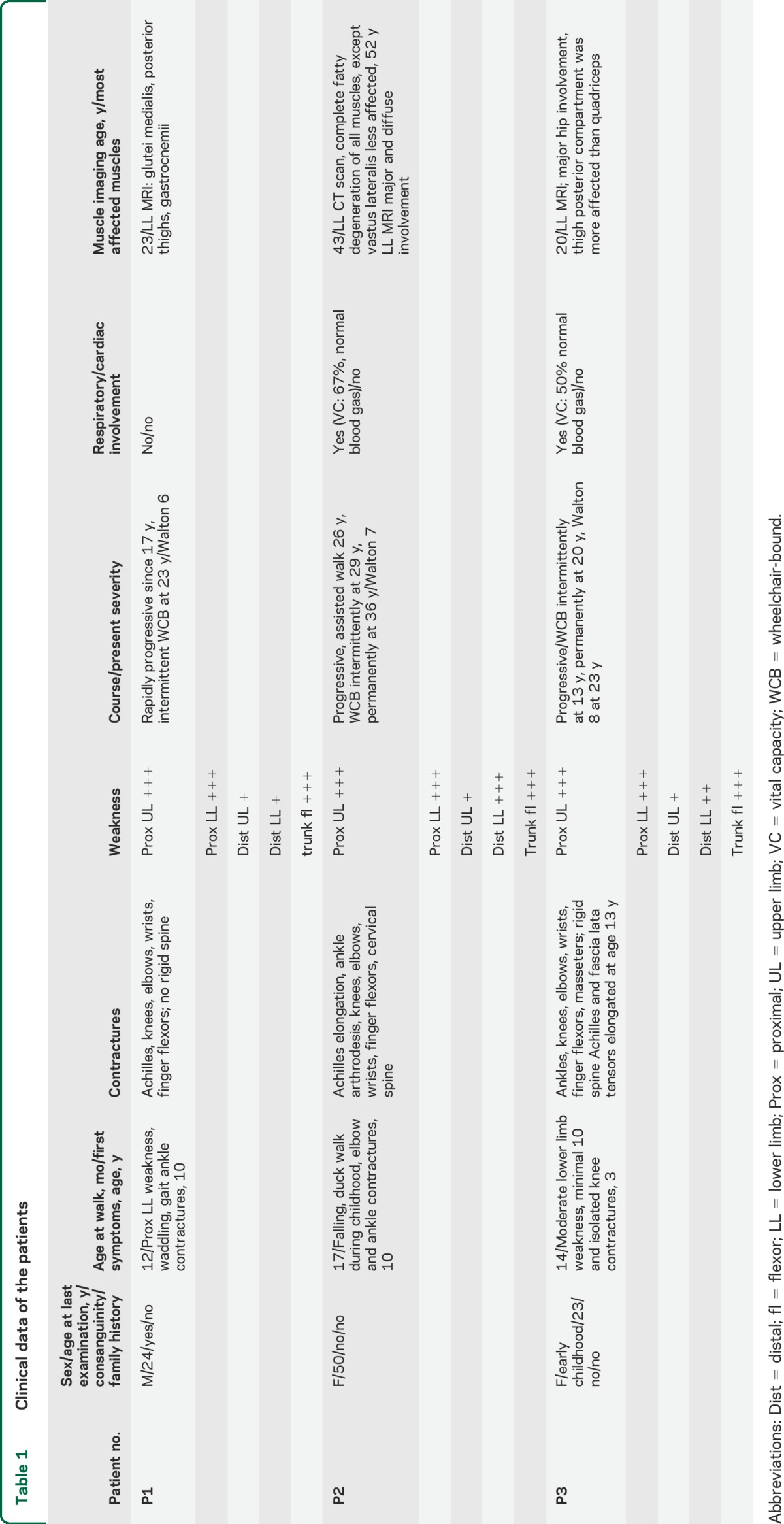
Figure 1. Pedigree of the families and clinical presentation and imaging of the patients.

(A) Pedigree of the families and clinical presentation and imaging of the patients. Filled-in symbols indicate the affected individual. Empty symbols indicate unaffected individuals. Numbers indicate individuals whose DNA was used for this study. The mutations identified are indicated for each tested individual. (B) Photographs of patients showing prominent contractures. Patient 1 (P1): P1-a: severe upper girdle involvement with scapular winging; P1-b: arm atrophy and elbow contractures. Patient 2 (P2): P2-a: finger flexor contractures; P2-b: severe proximal involvement and elbow contractures; P2-c: severe ankle contracture. Patient 3 (P3): P3-a: severe upper involvement with scapular winging; P3-b: rigid spine; P3-c, P3-d: finger and elbow contractures. (C) Muscle MRI of the patients at 3 different levels (axial T2 images through pelvis, thigh, and calf). P1 at 23 years: diffuse fatty degeneration in gluteal muscles with partial sparing of the sartorius and severe involvement of other femoral muscles and of the posterior and lateral compartments as well as tibialis anterior of the lower legs. P2 at 52 years: severe and diffuse involvement with fatty replacement of all muscles. P3 at 20 years: diffuse moderate fatty degenerative changes in all muscle groups with atrophy more marked in the hamstrings.
Patient 2 is a woman from a nonconsanguineous French Caucasian family with no other affected sibs (figure 1A). During childhood, she experienced falling, presented waddling gait, and had difficulties in sport activities. From age 10 years, she developed ankle and elbow contractures, underwent a surgical elongation of the Achilles tendons at 17 years, and underwent ankle arthrodesis at 23. The disease gradually worsened, with no running after the surgery, and difficulties rising from a chair and climbing stairs. She needed assistance for walking at 26 years, intermittent wheelchair at 29 years, and permanent wheelchair at 36 years. Examination at age 52 years showed a severe and diffuse weakness and atrophy involving upper and lower limbs and axial muscles. Wrists and fingers were less weak (table e-2). She presented marked contractures affecting spine and limbs (figure 1B). There was no facial, oculomotor, or pharyngeal weakness. Vital capacity was mildly reduced (67%). Cardiac echography and ECG were normal (table 1). CK was 1.5 times the upper normal value. Lower limb muscle imaging showed that thigh anterior compartment was clearly less affected than posterior at age 42 years as shown by CT scan (data not shown) and progressed to complete fatty degeneration in all compartments at age 52 years (figure 1C).
Patient 3 is the only affected member of a nonconsanguineous French Caucasian family (figure 1A). Since walking at age 19 months, there were difficulties rising from squatting position and climbing stairs, and running was impossible. At age 3 years, the first clinical investigation showed moderate lower limb proximal weakness with waddling gait, Gowers sign, and calf hypertrophy. CK level was 20 times the upper normal level. EMG was myogenic and muscle biopsy showed a dystrophic pattern. Isolated mild knee contractures were detected at 3.5 years old. From age 6 years, the knee contractures worsened, and contractures in the elbows, ankles, and spine occurred, whereas the weakness remained moderate and stable until age 11. Intermittent wheelchair use was required at age 13 years when bilateral Achilles and tensor fascia-latae tenotomy was performed resulting in increased weakness. Weakness of proximal upper limbs appeared from age 13 years with mild progression. A scoliosis required a corset since age 13 years, but no spinal arthrodesis was performed. At 18 years, she was wheelchair-bound but could stand alone. Her walking 10 meters with assistance was lost 2 years later. At the last examination, at age 23 years, weakness was marked in both girdles, in ankle dorsiflexion, and abdominal muscles (table e-2). Contractures were massive, affecting spine, proximal and distal limbs, and jaws (figure 1B and table e-3). No facial, ocular, or bulbar muscle weakness was found. Vital capacity was decreased by 50%, blood gas measurements were normal, as were cardiac echography and EGC. CK was 1.5 times the upper normal value. MRI performed at age 20 years showed moderate fatty degenerative changes diffusely in all muscle groups with marked atrophic change in the hamstring muscle (figure 1C).
Muscle biopsy findings.
Morphologic investigations by light and electron microscopy of muscle revealed a range of alterations (figure 2). The most frequent finding was the presence of rimmed vacuoles in about 10%–30% of muscle fibers (figure 2, B and C). Eosinophilic inclusions appeared as cytoplasmic bodies and rods (figure 2, B and C). Nuclear internalization was also a prominent feature. Oxidative reactions revealed variable abnormalities of the myofibrillar network. However, immunostaining for myofibrillar proteins (desmin, myotillin, and αB-crystallin) did not show any protein aggregates. Overall, there was marked fiber size variability and different degrees of necrotic or regenerating processes.
Figure 2. Morphologic alterations at histologic and ultrastructural level of skeletal muscle of the patients.

(A) Patient 1. Deltoid muscle biopsy, performed at 17 years, shows dystrophic pattern. (A.a) Increase of internalized nuclei and fiber size variation (hematoxylin & eosin [H&E]). (A.b) Clusters of regenerating fibers (H&E). (A.c) NADH reductase staining shows minor irregularities of the myofibrillar network. (B) Patient 2. (B.a) Muscle section shows great fiber size variation, increased connective tissues, and rimmed vacuoles (H&E). (B.b) Subsarcolemmal rimmed vacuoles (Gomori trichrome). (B.c) Fuchsinophilic inclusion corresponding to cytoplasmic bodies and rods (Gomori trichrome). (C) Patient 3. First biopsy. (C.a) Presence of rimmed vacuoles in several muscle fibers associated with necrotic/regenerating fibers and increased connective tissue (H&E). (C.b) Multiple rimmed vacuoles in the same fibers (Gomori trichrome). (C.c) Second biopsy. Red inclusions correspond to nemaline bodies (Gomori trichrome). Scale bars are indicated on the images. (D) Patient 2. First biopsy. (D.a) Rimmed vacuoles contain degradation products and lamellate myeloid structures. (D.d) Increased autophagic material in both subsarcolemmal and central areas of the myofiber. Patient 2. Second biopsy. (D.b) Nemaline bodies dispersed in areas containing thin filament accumulations, organelles, and debris. (D.e) Areas of focal myofibrillar disorganization correspond to dissolution of M-band structure by subsequent disintegration of myosin filament with almost intact Z-disc. Patient 3. (D.c) Electron microscopy demonstrates large tubulo-filamentous inclusions disrupting the myofibrillar network; filamentous inclusions also were found inside the nucleus. (D.f) Electrondense inclusions correspond to cytoplasmic bodies surrounded by amorphous material containing cellular debris and filaments. Scale bars are indicated on the images.
Electron microscopy demonstrated structural alterations corresponding to rimmed vacuoles containing degradation products and lamellate myeloid structures (figure 2, D.a and D.d). Nemaline bodies were found surrounded by amorphous material and cellular debris in the second biopsy from patient 2 (figure 2D.b). We also observed M-line disruption with Z-disc preservation in some places (figure 2D.e). Large tubulo-filamentous inclusions were found both disrupting the myofibrillar network and inside nuclei (figure 2D.c). Electron-dense inclusions corresponding to cytoplasmic bodies were also found (figure 2D.f).
Molecular pathology and genetics.
For all these patients, a number of immunofluorescence and molecular genetic analyses were performed previously without any mutations being identified (table e-4). However, a complete or massive deficiency in calpain 3 by WB was evidenced in all of them (figure 3A). We performed targeted DNA capture focused on an 820 gene panel related to muscular function and high-throughput sequencing of DNA sample of patient 1. The average mean read depth of bases was 350× with 96.4% of the target regions having at least 25×. The sample contained more than 3,952 variants with 3,834 already present in reference databases and 118 new variants. They included 502 indel with 328 already referenced in dbSNP. After filtering out heterozygous variants, common single nucleotide variations, and synonymous changes, and then filtering in variants present in exons or splice sites, 2 indels in AMOT and COL18A1 and one nonsense mutant in TTN (Chr2: 179393519; NM_001267550.1c.107184T>A; NP_001254479.1 p.Y35653*) remained. The 2 indels were in-frame and discarded as potential causative variants. Through Sanger sequencing of family members, we confirmed that the TTN variant segregated with the disease in the family with both parents being heterozygotes. Interestingly, this mutation is located on the 360th exon of titin, also called Mex3. This exon codes for a part of titin located in the M-line, a region where other MD-causing mutants were previously identified.3,4,13,14 This nonsense variant was the best candidate and the most likely cause of the disease.
Figure 3. Molecular analyses.

(A) Western blot analysis of calpain 3 in muscle biopsies of the 3 index cases (P1, P2, and P3) and one control (C). An antibody against a N-ter calpain 3 epitope was used in combination with dystrophin (Dysf; NCL-DYS1, Novocastra) and α-sarcoglycan (α-SG; NCL-L-α-SARC), allowing detection of the full length of 94 kDa and the autoproteolytic fragment of 30 kDa and an antibody against a calpain 3 C-ter epitope of with dysferlin (Dysf; NCL-Hamlet) and γ-sarcoglycan (γ-SG; NCL-L-γ-SARC) antibodies, allowing detection of the full length of 94 kDa and the autoproteolytic fragment of 60 kDa. All patients showed a complete or massive deficiency in calpain 3. (B) Western blot of the M-line titin using M10-1 titin antibody shows the absence of labeling compared to control. The control for P1 was loaded from the same gel but come from a nonadjacent line. Loading was normalized to Coomassie blue stained myosin heavy chain (lower panel). (C) Immunofluorescence analysis of titin using the N-terminus and C-terminus antibodies shows that the titin molecules are correctly incorporated in the sarcomere and are lacking the C-terminus. Scale bars = 20 μm.
Following this finding, we hypothesized that this region of titin could also be involved in the 2 other families. Sanger sequencing on the M-line titin (table e-1) identified 2 truncating mutations in patient 2: one single base deletion at the end of Mex1 (Chr2: 179395292; c.106275delT; p.E35351Nfs*54) and one nonsense mutation in Mex3 (Chr2: 179393500; c.107203C>T; p.Q35660*) and 2 truncating mutations in patient 3: a 5-base deletion at the end of Mex1 (Chr2: 179395428-432; c.106135_106139delACCTG; p.T35304Cfs*3) and a single base deletion in Mex2 (Chr2: 179394796; c.106647delG; p.F35475Sfs*4). The correct segregation of the mutations was confirmed in both families.
Molecular titin analyses.
We investigated the consequences of the mutations on the titin molecules by PCR, qPCR, WB, and immunofluorescence. First, we performed a RT-PCR across Mex1 and Mex6 exons (for patients 2 and 3) to define whether any cryptic splice site would have occurred and restored the frame. The correct size of the PCR product indicates that no splicing has occurred (figure e-2A). Second, we analyzed by real-time quantitative RT-PCR the level of titin transcript and observed a decrease, indicating that NMD has occurred due to the mutations (figure e-2B). Third, WB analysis performed with a C-ter titin antibody showed total absence of staining (figure 3B). Forth, immunofluorescence analysis was performed on muscles sections using N-ter and C-ter titin antibodies (figure 3C). The absence of staining with the C-ter antibody indicates the loss of the corresponding part of titin while the positive staining obtained with the N-ter antibody shows that the truncated titin molecules are nonetheless correctly inserted in the sarcomeres.
DISCUSSION
We report the identification of a new recessive titinopathy phenotype caused by novel truncating mutations in the C-ter part of titin segregating with the disease in 3 unrelated families. The patients shared common phenotypic features that include (1) coexistence of both limb-girdle weakness and early-onset contractures, preceding or accompanying initial weakness; (2) early-onset MD with normal neonatal period, proximal weakness in infancy-childhood, and progressive course in adolescence and adulthood, with permanent loss of ambulation from age 13 to 36 years; (3) unaffected facial, bulbar, and oculomotor muscles; (4) high CK levels, decreasing in the late stages of the disease; and (5) no identified cardiomyopathy to date. Finally, muscle biopsies showed rimmed vacuoles, increase of internal nuclei, cytoplasmic bodies, and dystrophic pattern. A key feature was massive secondary reduction of calpain 3 on WB.
Of special interest are the other titinopathy patients where mutations were identified in the vicinity of the mutations presented here. M-line titin homozygous truncations in Mex1 and Mex3 (figure 4) were reported in 2 consanguineous families of Moroccan and Sudanese origins with an early-onset myopathy with fatal cardiomyopathy with conduction disturbance (EOMFC).4 Several aspects are similar to our cases: autosomal recessive transmission, combination of a myopathy with lower limb predominant involvement, calf hypertrophy, joint and spine contractures, and secondary depletion of calpain 3. However, major differences in these patients are to be underlined: earlier onset, presence of a ptosis, fatal arrhythmic dilated cardiomyopathy developing from childhood, and the histology including minicore-like-lesions but without rimmed vacuoles.4 Recessive truncating titin mutations located in Mex1 have been reported in 5 unrelated individuals with congenital centronuclear myopathy (CNM).3 As in our cases, no cardiomyopathy was reported, but several major features differ: earlier onset, no contractures, and a distinctive histopathologic pattern without rimmed vacuoles. Simultaneously, recessive TTN truncating mutations located in Mex1, Mex2, and Mex3 were reported in a set of 4 families with congenital core myopathy combined with primary heart disease.14 The clinical spectrum varied from severe early-onset cases with arthrogryposis and neonatal cardiac failure to Emery-Dreifuss-like presentation. The patient harboring the Emery-Dreifuss-like phenotype was similar to our cases regarding the muscle phenotype combining weakness and contractures, but had neonatal onset and a severe arrhythmic dilated cardiomyopathy. Moreover, the histopathologic signature differed, with minicores as the main finding. It should be noted that calpain 3 deficiency was not investigated in those cases. The adult-onset dominant titinopathies, tibial muscular dystrophy (TMD) and hereditary myopathy with early respiratory failure, share the absent cardiomyopathy and the presence of rimmed vacuoles but otherwise are not differential diagnostic alternatives. The Mex6 (exon 363) TMD-mutation homozygous LGMD2J is childhood onset and shows a secondary calpain deficiency, but the clinical presentation is different, without contractures.24–27 Overall, the presentation described here is unique among titinopathies.
Figure 4. Summary of all reported mutations in the M-line titin and consequences.

Each box represents a patient or a group of patients with similar genotype. When colored, the mutations are dominant. The position of the TMD/LGMD2 mutations is expanded. The numbering is based on the titin sequences NM_001267550.1 and NP_001254479.1. Each frame corresponds to a specific patient or a group in case of the FINMaj mutation. ARVC = arrhythmogenic right ventricular cardiomyopathy; CNM = centronuclear myopathy; EOMFC = early-onset myopathy with fatal cardiomyopathy; LGMD2J = limb-girdle muscular dystrophy type 2J; MmD-HD = multi-minicore disease with heart disease; TMD = tibial muscular dystrophy.
The frameshift mutations reported here are all located close to each other between the M6 and M8 domains of C-terminal titin in the sarcomeric M-band. As evidenced by WB and immunofluorescence analyses, the molecular consequences of these mutations are primary truncations of the titin protein without impact on their sarcomere incorporation. Of interest, dissolution of M-band structure in some myofibrils with disintegration of myosin filament and without major Z-disc alteration is an associated characteristic ultrastructural finding. Another consequence of the truncation of titin is the absence of the calpain 3 binding site in C-ter titin, which is probably the cause of its secondary defect. This particular feature is also present in other recessive C-ter titinopathies including LGMD2J, EOMFC, and CNM,3–5 all with primary or secondary titin truncations.23
Whereas the proximity of all the reported mutations is coherent with the similarity in clinical features presented by the patients, it is intriguing that other patients, in whom the truncations affect the same region, present a fatal cardiac phenotype.4 The potential correlation of the heart phenotype severity with the localization of the TTN mutations has been discussed,14 suggesting that the closer a mutation is to the C-terminus, the less severe is the heart involvement. However, the factors determining the presence of heart involvement associated with M-line TTN mutations are probably more complex since some of the mutations we describe are located 5′ of the mutations previously described with cardiomyopathy.14 Possible explanations for the association of cardiomyopathy could be related to differences in the proportion of mutated titin isoform incorporated in the sarcomere, degree of NMD, degree of read-through of stop codons, protein stability, partner interactions, or other modifying mutations in the whole TTN gene, but this needs further studies to be clarified.
Supplementary Material
ACKNOWLEDGMENT
The authors thank J.F. Deleuze, A. Ferreiro, A. Behin, M. Fardeau, P. Laforet, T. Stojkovic, R.Y. Carlier, and P. Carlier for their contribution.
GLOSSARY
- CK
creatine kinase
- CNM
congenital centronuclear myopathy
- EDMD
Emery-Dreifuss muscular dystrophy
- EOMFC
early-onset myopathy with fatal cardiomyopathy with conduction disturbance
- LGMD2A
limb-girdle muscular dystrophy type 2A
- MD
muscular dystrophy
- NMD
nonsense-mediated decay
- RT-PCR
reverse transcription-PCR
- TMD
tibial muscular dystrophy
- WB
Western blot
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
R.D.C. planned and performed experiments for the genetic part of the study and participated in the redaction of the manuscript. R.B.Y. performed clinical evaluation of the patients and participated in the redaction of the manuscript. K.C. analyzed the data and wrote the manuscript. C.R., S.B., A.C., I.N., J.N., L.B.A., C.C., and R.O. performed experiments for the genetic part of the study. N.B.R. and E.M. performed experiments for the histopathology part of the study and contributed to the writing of the manuscript. M.B. performed the histoimmunofluorescence analyses. F.L. and A.V. performed experiments for the biochemical part of the study. B.U. analyzed the data and contributed to the writing of the manuscript. G.B. supervised the study, analyzed the data, and contributed to the writing of the manuscript. B.E. performed clinical evaluation of the patients and wrote the manuscript. I.R. supervised the study, analyzed the data, and wrote the manuscript.
STUDY FUNDING
This work was financially supported by Association Française contre les Myopathies (AFM-Telethon), the Assistance Publique-Hôpitaux de Paris, the Institut National de la Santé et de la Recherche Médicale, the Sorbonne Universités-Université Pierre et Marie Curie Paris 06, the Centre National de la Recherche Scientifique, and the NMD-CHIP Consortium, a FP7 HEALTH project of the European Commission (Development of Targeted DNAChips for High Throughput Diagnosis of Neuromuscular Disorders–Collaborative Project–FP7 Grant Agreement Number: HEALTH-F5-2008-223026; to R.D.C., S.B., I.N., G.B., and I.R.) and the Sigrid Juselius Foundation and the Finnish Academy Research Funds (B.U.).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Richard I, Broux O, Allamand V, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995;81:27–40. [DOI] [PubMed] [Google Scholar]
- 2.Milic A, Daniele N, Lochmuller H, et al. A third of LGMD2A biopsies have normal calpain 3 proteolytic activity as determined by an in vitro assay. Neuromuscul Disord 2007;17:148–156. [DOI] [PubMed] [Google Scholar]
- 3.Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 2013;81:1205–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carmignac V, Salih MA, Quijano-Roy S, et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol 2007;61:340–351. [DOI] [PubMed] [Google Scholar]
- 5.Haravuori H, Vihola A, Straub V, et al. Secondary calpain3 deficiency in 2q-linked muscular dystrophy: titin is the candidate gene. Neurology 2001;56:869–877. [DOI] [PubMed] [Google Scholar]
- 6.Linke WA, Hamdani N. Gigantic business: titin properties and function through thick and thin. Circ Research 2014;114:1052–1068. [DOI] [PubMed] [Google Scholar]
- 7.Fukuda N, Granzier HL, Ishiwata S, Kurihara S. Physiological functions of the giant elastic protein titin in mammalian striated muscle. J Physiol Sci 2008;58:151–159. [DOI] [PubMed] [Google Scholar]
- 8.Itoh-Satoh M, Hayashi T, Nishi H, et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun 2002;291:385–393. [DOI] [PubMed] [Google Scholar]
- 9.Taylor M, Graw S, Sinagra G, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011;124:876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun 1999;262:411–417. [DOI] [PubMed] [Google Scholar]
- 11.Frey N, Luedde M, Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol 2012;9:91–100. [DOI] [PubMed] [Google Scholar]
- 12.Peled Y, Gramlich M, Yoskovitz G, et al. Titin mutation in familial restrictive cardiomyopathy. Int J Cardiol 2014;171:24–30. [DOI] [PubMed] [Google Scholar]
- 13.Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet 2002;71:492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chauveau C, Bonnemann CG, Julien C, et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet 2014;23:980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 1999;21:285–288. [DOI] [PubMed] [Google Scholar]
- 16.Bione S, Maestrini E, Rivella S, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet 1994;8:323–327. [DOI] [PubMed] [Google Scholar]
- 17.Gueneau L, Bertrand AT, Jais JP, et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet 2009;85:338–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang WC, Mitsuhashi H, Keduka E, et al. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol 2011;69:1005–1013. [DOI] [PubMed] [Google Scholar]
- 19.Meinke P, Mattioli E, Haque F, et al. Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet 2014;10:e1004605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Q, Bethmann C, Worth NF, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery-Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet 2007;16:2816–2833. [DOI] [PubMed] [Google Scholar]
- 21.Malfatti E, Olive M, Taratuto AL, et al. Skeletal muscle biopsy analysis in reducing body myopathy and other FHL1-related disorders. J Neuropathol Exp Neurol 2013;72:833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson LV, Davison K. Multiplex Western blotting system for the analysis of muscular dystrophy proteins. Am J Pathol 1999;154:1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charton K, Sarparanta J, Vihola A, et al. CAPN3-mediated processing of C-terminal titin replaced by pathological cleavage in titinopathy. Hum Mol Genet 2015;24:3718–3731. [DOI] [PubMed] [Google Scholar]
- 24.Ohlsson M, Hedberg C, Bradvik B, et al. Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain 2012;135:1682–1694. [DOI] [PubMed] [Google Scholar]
- 25.Pfeffer G, Elliott HR, Griffin H, et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain 2012;135:1695–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palmio J, Evila A, Chapon F, et al. Hereditary myopathy with early respiratory failure: occurrence in various populations. J Neurol Neurosurg Psychiatry 2014;85:345–353. [DOI] [PubMed] [Google Scholar]
- 27.Udd B, Partanen J, Halonen P, et al. Tibial muscular dystrophy. Late adult-onset distal myopathy in 66 Finnish patients. Arch Neurol 1993;50:604–608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.