

Abstract
Objective:
To examine the relationship of clinical and genetic features of patients with facioscapulohumeral muscular dystrophy (FSHD) with 7–10 residual D4Z4 repeats in a large genetically defined FSHD1 cohort.
Methods:
We performed a prospective cross-sectional observational study of 74 clinically affected patients with FSHD1. Measures of clinical severity were compared between patients with 1–6 D4Z4 repeats and 7–10 repeats, and included D4Z4 CpG methylation, age at diagnosis, age-adjusted clinical severity score, a muscle pathology grade of quadriceps biopsies (0 = normal, 12 = severe dystrophic changes), quantitative myometry of biopsied muscles, global manual muscle testing scores, and frequency of wheelchair use.
Results:
Twenty-eight (37.8%) participants had 7–10 D4Z4 repeats, and compared to participants with 1–6 repeats, were diagnosed 6.6 years older (p = 0.17); had lower CpG methylation than would be predicted by D4Z4 repeat size (p = 0.04); had age-adjusted clinical severity 39.8 points lower (p = 0.004); had muscle pathology grades that were 2.4 points less severe (p < 0.0001); had quantitative myometry 28.3% predicted of normal higher (p = 0.002); had global manual muscle testing scores 0.6 higher (p = 0.005); and did not require wheelchairs.
Conclusion:
Patients with FSHD with 7–10 D4Z4 repeats have milder disease than other genetically defined patients with FSHD1. The lower than predicted methylation in the 7–10 residual repeat group may suggest that additional epigenetic factors play a role in the severity of disease expression.
Facioscapulohumeral muscular dystrophy (FSHD) is a common muscular dystrophy due in its most prevalent form (FSHD1) to deletions in the D4Z4 macrosatellite repeat region on chromosome 4q35.1,2 While normal individuals have >10 repeats, patients with FSHD1 have between 1 and 10 repeats. The current model for disease proposes FSHD is an epigenetic disease: deletions lead to decreased CpG methylation and opening of chromatin structure.3 The open chromatin structure in combination with a polyadenylation signal polymorphism allow the transcription of a normally repressed gene located within the D4Z4 repeat, DUX4, which causes disease by a toxic gain of function.4
Patients with the smallest number of repeats demonstrate a more severe phenotype, including earlier wheelchair use and increased frequency of extramuscular manifestations (retinal vasculopathy and hearing loss).5–7 A number of articles have looked at the relationship of D4Z4 methylation levels to penetrance of disease expression, with decreased methylation in patients expressing disease.8,9 A linear relationship exists between the number of residual D4Z4 repeats and estimates of methylation in the D4Z4 region.9 So for most patients with FSHD1, the repeat contraction is sufficient to decrease methylation and cause disease. Symptomatically affected patients with 7–10 repeats, unlike patients with 1–6 repeats, have divergent methylation values from those predicted by D4Z4 contractions alone, suggesting that the contraction per se is insufficient to cause disease but requires additional (epigenetic) factors.9 Here we sought to determine if symptomatic patients with 7–10 D4Z4 repeats have differences in disease severity compared to patients with 1–6 repeats.
METHODS
We performed a prospective cross-sectional observational study of genetically confirmed and clinically affected patients with FSHD1 at the University of Rochester Medical Center conducted from 2002 to 2013.
Standard protocol approvals, registrations, and patient consents.
The study was approved by the institutional review board, and written and informed consent was obtained from all participants.
FSHD participants were between 18 and 75 years of age. D4Z4 residual fragment size was determined by Southern Blot after double digestion with EcoRI/BlnI restriction enzymes.2,10 CpG methylation measurements were determined by Southern Blot after cleavage with the methylation-sensitive endonuclease FseI.10 The Δ methylation value is the observed methylation value corrected for the repeat array size: Δ methylation = (observed CpG methylation) − (predicted methylation based on repeat size).9 The predicted methylation model was derived from a large cohort of patients and healthy controls (e-Methods on the Neurology® Web site at Neurology.org).9
Participants filled out a standardized self-reported medical history (https://www.urmc.rochester.edu/fields-center/protocols/). For each participant, clinical severity was determined based on pattern of muscle involvement (0–10, increasing from face and shoulders, to lower extremity) and adjusted for age: 1,000 × (2XCS)/age (table e-1).e1 Manual muscle testing was performed utilizing a modified 13-point Medical Research Council Scale (bilateral shoulder abductors, forward flexors, elbow flexors/extensors, wrist extensors, hip flexors, knee flexors/extensors, and ankle dorsiflexors).e2 Quadriceps muscle biopsy pathology was determined by one neuropathologist rater using an ordinal severity scale (0 = normal, 12 = severely dystrophic). Quantitative myometry of the quadriceps was performed prior to biopsy and normalized to percent predicted based on age, sex, and height (e-Methods).e2,e3
Descriptive statistics (mean, median, and frequency) were used to evaluate each outcome. Participants were divided into 2 groups based on residual D4Z4 repeat size for comparisons (1–6 vs 7–10 repeats). Differences between groups were determined by 2-sample t test for continuous measures, Wilcoxon rank-sum test for ordinal values, and χ2 for frequencies. Ninety-five percent confidence limits were calculated by standard procedures. The relationship between age-adjusted clinical severity and D4Z4 repeat was investigated visually first and mathematically by fitting trendlines (higher R2 values determining optimal fit). For box and whisker plots: boxes represent 50% of the distribution; thick line, median values; whiskers to nearest outlier, or 1.5 times box size; and dots, outliers. Missing data were assumed to be missing at random. All statistical testing was 2-sided with p < 0.05 used as a cutoff. Statistical testing was performed using SAS version 9.4 (SAS Institute Inc, Cary, NC) and STATA version 11.2 (StataCorp, College Station, TX).
RESULTS
Seventy-four participants with FSHD1 were included (table 1). Just over 1/3 of participants had 7–10 D4Z4 repeats. There were no differences based on sex or patient-reported age at diagnosis, although participants with 7–10 repeats were older at evaluation. No participants had documented extramuscular manifestations of FSHD1. After adjusting for age, participants with 7–10 repeats were less severely affected. The relationship of the age-adjusted clinical severity to residual D4Z4 repeat number was not linear (figure, A, left y-axis). For participants with 1–6 repeats, the residual repeat size appears to have a linear effect on clinical severity. But the relationship flattens out for participants with 7–10 repeats. Looking at the distribution of Δ methylation values for the 2 groups shows (1) a Δ methylation with a median value that approaches zero for participants with 1–6 repeats; and (2) a shift to negative values for participants with 7–10 repeats, which corresponds to the flatter portion of the age-adjusted clinical severity vs repeat size curve (figure, A, right y-axis). Overall, looking at a broad group of clinical severity measures, participants with 7–10 residual D4Z4 repeats were more mildly affected, including increased combined average manual muscle testing scores, increased quantitative myometry of quadriceps muscle (figure, B), and lower quadriceps muscle pathology scores. No participant with 7–10 residual repeats was using a wheelchair for mobility, despite the older age of the population (figure, C).
Table 1.
Clinical characteristics of patients with FSHD with 1–6 D4Z4 repeats compared to 7–10 repeats
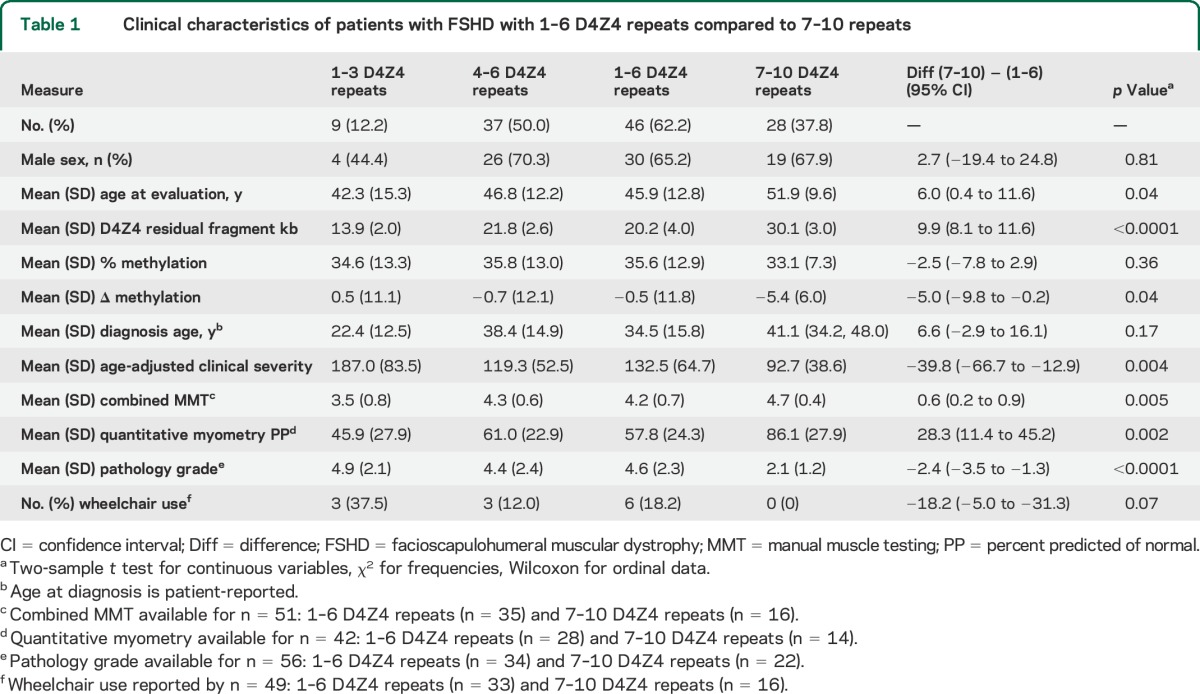
Figure. The relationship of facioscapulohumeral muscular dystrophy 1 residual D4Z4 repeats and Δ methylation to disease severity.

(A) The relationship of age-adjusted clinical severity to D4Z4 repeat size is not linear (left y-axis, n = 74). While the relationship of severity to repeat size is linear from 1 to 6 repeats, some other factor besides repeat size appears to determine severity for 7–10 repeats. Below box plots of the Δ methylation (right y-axis) for 1–6 repeats (left) and 7–10 repeats (right). Note the negative Δ methylation values for 7–10 repeats. (B) Participants with 7–10 repeats have increased quadriceps strength by quantitative myometry (1–3 repeats [n = 6], 4–6 repeats [n = 23], and 7–10 repeats [n = 14]). (C) No participants with 7–10 repeats reported using wheelchairs (1–3 repeats [n = 8], 4–6 repeats [n = 26], and 7–10 repeats [n = 17]).
DISCUSSION
FSHD1 is an epigenetic disease with considerable variability across the size of the D4Z4 repeat array.e1,e4 While attempts to split out groups for examination may be arbitrary, a number of points related to deletion size and methylation are becoming clear:
Patients with the smallest number of residual fragments1–3 have more severe disease. These patients are typically diagnosed younger, have higher penetrance by age,e4 are more likely to use a wheelchair,5,e5 and are more likely to experience extramuscular manifestations of FSHD, including symptomatic retinal vasculopathy, hearing loss requiring hearing aids, or seizures or cognitive changes.6,7,e6 These patients have decreased methylation determined by the number of residual repeats with Δ methylation values approximating zero.9 However, low methylation in itself does not completely explain severity, as patients with FSHD2, who have low methylation values, have not been described with severe extramuscular manifestations of disease or with early onset.10
Patients with FSHD with the largest number of residual D4Z4 fragments have a milder disease or remain nonpenetrant,5,e5,e7 with an older age at onset,e5 lower penetrance by age, lower frequency of wheelchair use, and typically do not have extramuscular manifestations. In this group, the methylation values do not appear to be completely driven by the number of residual repeats—the Δ methylation values are negative, suggesting some other factor, whether genetic, epigenetic, or environmental, is required for them to express disease.9 A genotype–phenotype cohort in Japan did not identify disease in this group.e8 Additionally, mutations in SMCHD1 are not only associated with FSHD2 but also modify FSHD1: an article described families in the 7–10 repeat range, with a mild phenotype, where certain individuals with more severe disease were found to have unexpectedly low methylation values, then subsequently found to also carry mutations in SMCHD1.e9
Ultimately, disease expression in FSHD1 appears to be a unique combination of factors, which includes deletion of repetitive elements that affects not only methylation, but may have additional physical properties related to size in the 1–3 repeat group, and a second group with 7–10 repeats where the size of the repeat is not sufficient to cause disease, but some other (epigenetic) factor is also required.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and family members.
GLOSSARY
- FSHD
facioscapulohumeral muscular dystrophy
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Jeffrey Statland and Rabi Tawil contributed to drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data, and statistical analysis. Stephen Tapscott, Richard Lemmers, and Silvère van der Maarel contributed to study concept or design, drafting/revising the manuscript for content, and acquisition of data. Colleen Donlin-Smith contributed to drafting/revising the manuscript for content and acquisition of data.
STUDY FUNDING
The Cellular and Molecular Pathophysiology Study in FSHD has been funded in whole or in part by the NIH (grant 1PO1NS069539-01) and the Fields Center for FSHD and Neuromuscular Research. The project described in this publication was supported by the University of Rochester CTSA award number UL1 RR024160 from the National Center for Research Resources and the National Center for Advancing Translational Sciences of the NIH. R.L. and S.M. are supported by funding from Spieren voor Spieren and from the Prinses Beatrix Spierfonds. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Dr. Statland's work on this project was supported by a NCATS grant awarded to the University of Kansas Medical Center for Frontiers: The Heartland Institute for Clinical and Translational Research #KL2TR000119.
DISCLOSURE
J. Statland's work on this project was supported by an NCATS grant awarded to the University of Kansas Medical Center for Frontiers: The Heartland Institute for Clinical and Translational Research #KL2TR000119. C. Donlin-Smith, S. Tapscott, R. Lemmers, S. van der Maarel, and R. Tawil report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.van Deutekom JC, Wijmenga C, van Tienhoven EA, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet 1993;2:2037–2042. [DOI] [PubMed] [Google Scholar]
- 2.Wijmenga C, Hewitt JE, Sandkuijl LA, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 1992;2:26–30. [DOI] [PubMed] [Google Scholar]
- 3.Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010;329:1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Maarel SM, Tawil R, Tapscott SJ. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol Med 2011;17:252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lunt PW, Jardine PE, Koch MC, et al. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD). Hum Mol Genet 1995;4:951–958. [DOI] [PubMed] [Google Scholar]
- 6.Lutz KL, Holte L, Kliethermes SA, Stephan C, Mathews KD. Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology 2013;81:1374–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Statland JM, Sacconi S, Farmakidis C, Donlin-Smith CM, Chung M, Tawil R. Coats syndrome in facioscapulohumeral dystrophy type 1: frequency and D4Z4 contraction size. Neurology 2013;80:1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaillard MC, Roche S, Dion C, et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology 2014;83:733–742. [DOI] [PubMed] [Google Scholar]
- 9.Lemmers RJ, Goeman JJ, van der Vliet PJ, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet 2014;24:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Greef JC, Lemmers RJ, Camano P, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 2010;75:1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.