

Abstract
Objective:
To determine the frequency and relative importance of the most life-affecting symptoms in myotonic dystrophy type 2 (DM2) and to identify the factors that have the strongest association with these symptoms.
Methods:
We conducted a cross-sectional study of adult patients with DM2 from a National Registry of DM2 Patients to assess the prevalence and relative importance of 310 symptoms and 21 symptomatic themes. Participant responses were compared by age categories, sex, educational attainment, employment status, and duration of symptoms.
Results:
The symptomatic themes with the highest prevalence in DM2 were the inability to do activities (94.4%), limitations with mobility or walking (89.2%), hip, thigh, or knee weakness (89.2%), fatigue (89.2%), and myotonia (82.6%). Participants identified the inability to do activities and fatigue as the symptomatic themes that have the greatest overall effect on their lives. Unemployment, a longer duration of symptoms, and less education were associated with a higher average prevalence of all symptomatic themes (p < 0.01). Unemployment, a longer duration of symptoms, sex, and increased age were associated with a higher average effect of all symptomatic themes among patients with DM2 (p < 0.01).
Conclusions:
The lives of patients with DM2 are affected by a variety of symptoms. These symptoms have different levels of significance and prevalence in this population and vary across DM2 subgroups in different demographic categories.
Myotonic dystrophy type 2 (DM2) is an autosomal dominant adult muscular dystrophy caused by abnormal CCTG repeat expansions in the cellular nucleic acid binding protein (CNBP) gene (also known as the zinc finger protein 9 [ZNF9] gene) on chromosome 3.1 Clinically, DM2 produces a multisystemic and diverse set of symptoms. Frequently reported DM2 symptoms include pain, progressive weakness, myotonia, cataracts before age 50, hypogonadism, cognitive impairment, cardiac arrhythmias, tremor, hypersomnia, and fatigue.2–5
While prior studies have identified the importance of pain,2,6–11 impaired sleep,12–17 and cognitive symptoms18–22 in myotonic dystrophy, the prevalence of each of these varied symptoms in DM2 is unknown. In addition, the relative burden that each symptom has on the DM2 population has not been defined. In preparation for DM2 clinical trials, and in order to optimize the clinical care and understanding of patients with DM2, it is important to know what symptoms and issues are perceived by this population as having the greatest effect on their daily life.
The Patient-Reported Impact of Symptoms in Myotonic Dystrophy Type 2 (PRISM-2) Study is a national cross-sectional evaluation of DM2-affected participants. Herein, we describe patient interviews and a large-scale survey initiative to (1) clinically define DM2 disease burden from the patient's point of view, and (2) identify factors potentially associated with reduced disease burden.
METHODS
Study participants.
Inclusion criteria included (1) age 21 years or older and (2) diagnosis of DM2 by genetic confirmation or clinical criteria. Genetic confirmation required a CCTG repeat size greater than 75 in the CNBP gene on chromosome 3. Participants without genetic testing were required to have (1) clinical signs of weakness and myotonia (clinical or electrodiagnostic) with one parent or child with clinically or genetically proven DM2, or (2) clinical DM2 based on clinical guidelines,3 the patient's family history, and a review of the patient's medical record by a physician expert in this disorder.
Study design.
Phase 1: DM2 qualitative interviews.
We conducted in-depth individual interviews with 15 genetically confirmed adult patients with DM2 known to the University of Rochester neuromuscular clinic. We used semistructured interviews and open-ended questions about clinical symptoms. During each interview, we asked participants to identify the symptoms and issues that have the greatest impact on their lives. Participant responses were audio-recorded, transcribed, coded into symptom clusters, and analyzed using a 3-investigator approach and a qualitative framework technique.23,24 Interview transcripts were reviewed by 3 independent coders to identify symptoms. Consensus meetings were held to identify discrepancies between reviewers and then to construct a final list of symptoms. Patient-reported symptoms were then grouped and tabulated. Symptoms were further classified into symptomatic themes to obtain an overall picture of DM2 health status.23,25
Phase 2: National cross-sectional study of participants with DM2.
Phase 2 utilized participants with DM2 from the National Registry of Myotonic Dystrophy and Facioscapulohumeral Muscular Dystrophy (FSHD) Patients and Family Members (http://www.urmc.rochester.edu/nihregistry/).26 Registry membership is based on written consent, patient-reported information (family history, symptoms, etc.), and review of each member's medical record including clinical examinations and laboratory results.26 Approximately 60% the DM2 members of the registry have genetic confirmation of their disease. Patients without DNA testing are classified through stringent review of medical records and based on current clinical guidelines and family history. The principal investigator of the National Registry (Richard T. Moxley III, MD) confirms the diagnosis obtained through medical record review for DM2.
We generated a survey of questions representing all of the DM2 symptoms and symptomatic themes identified in phase 1 and additional symptoms previously reported to be important in other adult muscular dystrophy populations (i.e., myotonic dystrophy type 1, and FSHD).23,25 For each symptom, participants were asked, “How much does the following impact your life now?” Participants responded using a Likert-type scale ranging from 1 to 6: (1) I don't experience this; (2) I experience this but it does not affect my life; (3) It affects my life a little; (4) It affects my life moderately; (5) It affects my life very much; and (6) It affects my life severely. Participants provided their age, race, sex, age of symptom onset, employment status, and level of education. Participants were also given an opportunity to list and describe symptoms that were important to them but not included in the survey.
Registry participants were mailed a recruitment letter and DM2 survey with instructions. Surveys were sent on March 8, 2011, and collected until May 26, 2011. In an effort to have a questionnaire of comfortable length and to reduce participant burden, 2 DM2 surveys were produced. Each included demographic questions, the 21 symptomatic themes identified in phase 1, and close to half of the 310 symptom questions. We randomly allocated each DM2 registry subject to one of the 2 surveys. Participants were offered the option to verbally answer survey questions over the phone if they thought that hand weakness, poor vision, or reading difficulties would interfere with their ability to complete the paper survey. A summary of the activities and analyses of phases 1 and 2 is provided in figure 1.
Figure 1. Flow diagram of activities and analyses.

Identifying the most critical symptoms and themes in myotonic dystrophy type 2 (DM2).
Statistical analysis.
We determined the frequency of each DM2 symptom and theme using input from participants in phase 2. In addition, we calculated the average importance of each symptom and theme (life impact score) to the DM2 phase 2 participants who experience the symptom or theme. For this metric, numerical values were assigned to each participant response as follows: the patient experiences the issue but it does not affect the patient's life = 0; the issue affects the patient's life a little = 1; the issue affects the patient's life moderately = 2; the issue affects the patient's life very much = 3; and the issue affects the patient's life severely = 4. In instances in which a participant marked between 2 choices, the response was scored as the average of the 2 choices. Average life impact scores for each symptom and theme were calculated using the average response of all participants who stated that they experienced the symptom or theme. In addition, a population impact score (percentage of participants in whom an issue was experienced multiplied by the average life impact score of the issue) was determined for each item. Lastly, composite prevalence scores and composite impact scores were calculated by averaging participant responses across all themes.23
Participant responses were further categorized based on (1) sex, (2) age (18–60 years; 61+ years of age), (3) employment status, (4) education level (those with a college degree; those without a college degree), and (5) duration of symptoms (0–20 years; 21+ years). Group categorization cutpoints were determined before analysis. We obtained descriptive statistics for the prevalence and impact score for each theme for the entire sample and for each subgroup. We compared the prevalence of each theme between the different subgroups using Fisher exact tests.24 We compared the distributions of relative impact scores for each theme, composite prevalence scores, and composite impact scores between the different subgroups using Kruskal-Wallis tests.24
Standard protocol approvals, registrations, and patient consents.
The University of Rochester institutional review board approved all aspects of this study. As directed by the local institutional review board, all interviewed participants and survey participants received and reviewed a detailed information letter before their involvement with this research.
RESULTS
Phase 1: DM2 qualitative interviews.
We conducted exploratory interviews with 7 woman and 8 men with DM2. We obtained 943 direct quotes. These quotes related to the symptoms and issues that, in the opinion of the participants, had the greatest impact on their everyday life. Recurring similar quotes were grouped to identify 310 symptoms and 21 symptomatic themes potentially relevant to the DM2 population.
Phase 2: National cross-sectional study of participants with DM2.
We sent surveys to all DM2 registry participants (n = 120). Seventy-four patients with DM2 (62%) chose to become participants in the cross-sectional study. Participants represented 31 different states and answered 12,713 of 13,008 (97.7%) DM2 symptom survey questions. Table 1 presents their demographic information.
Table 1.
Clinical and demographic information of phase 2 DM2 respondents from the National Registry

Prevalence of DM2 themes and symptoms.
The most frequently occurring symptomatic themes included the inability to do activities (94.4%), limitations with mobility or walking (89.2%), hip, thigh, or knee weakness (89.2%), and fatigue (89.2%).
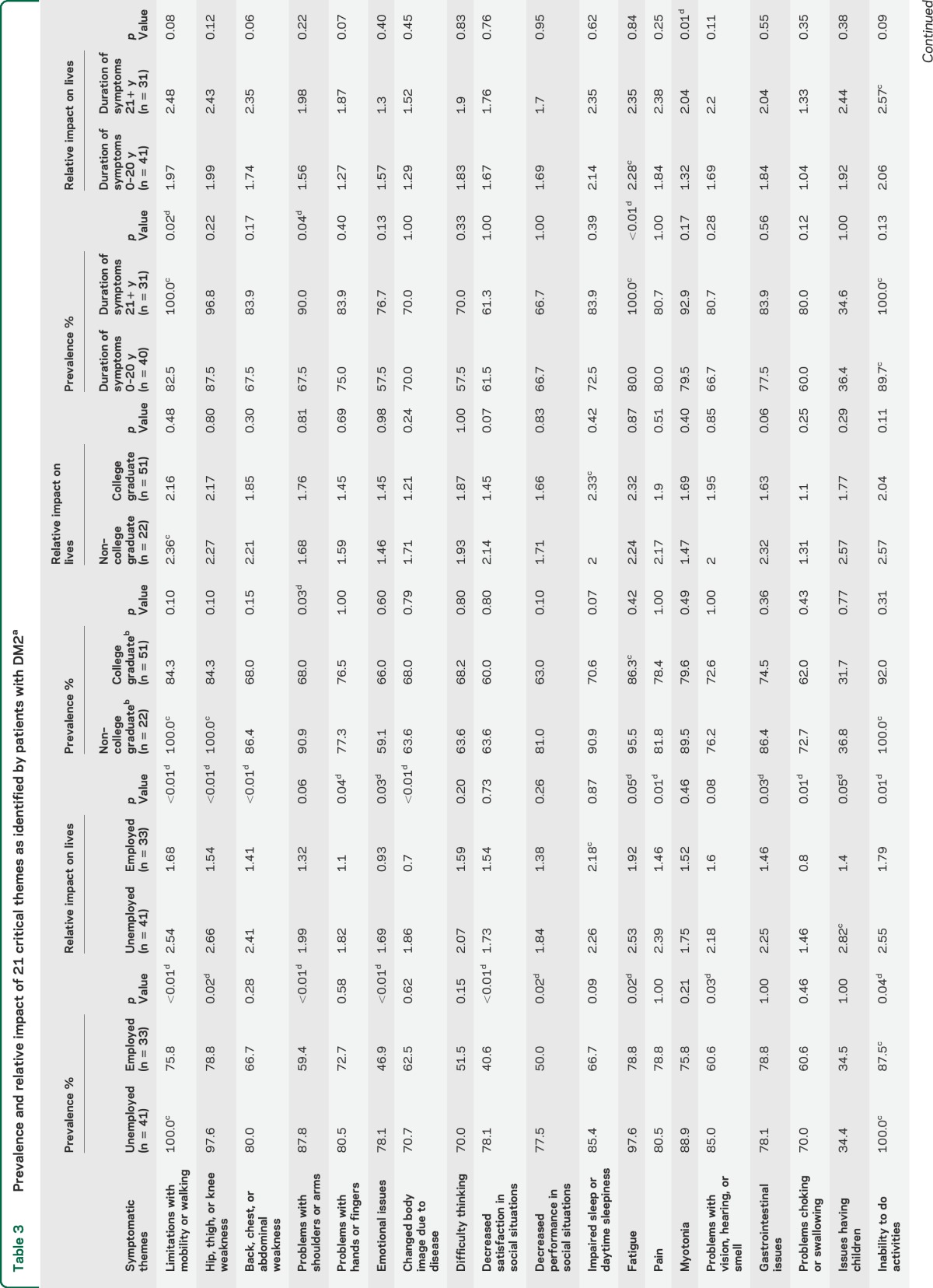
The frequency of each symptomatic theme is shown for sex and age subgroups in table 2 and for employment, education, and symptom duration subgroups in table 3. The relative prevalence of all 310 symptoms and 21 themes is shown in table e-1 on the Neurology® Web site at Neurology.org.
Table 2.
Prevalence and relative impact of 21 critical themes as identified by patients with DM2a
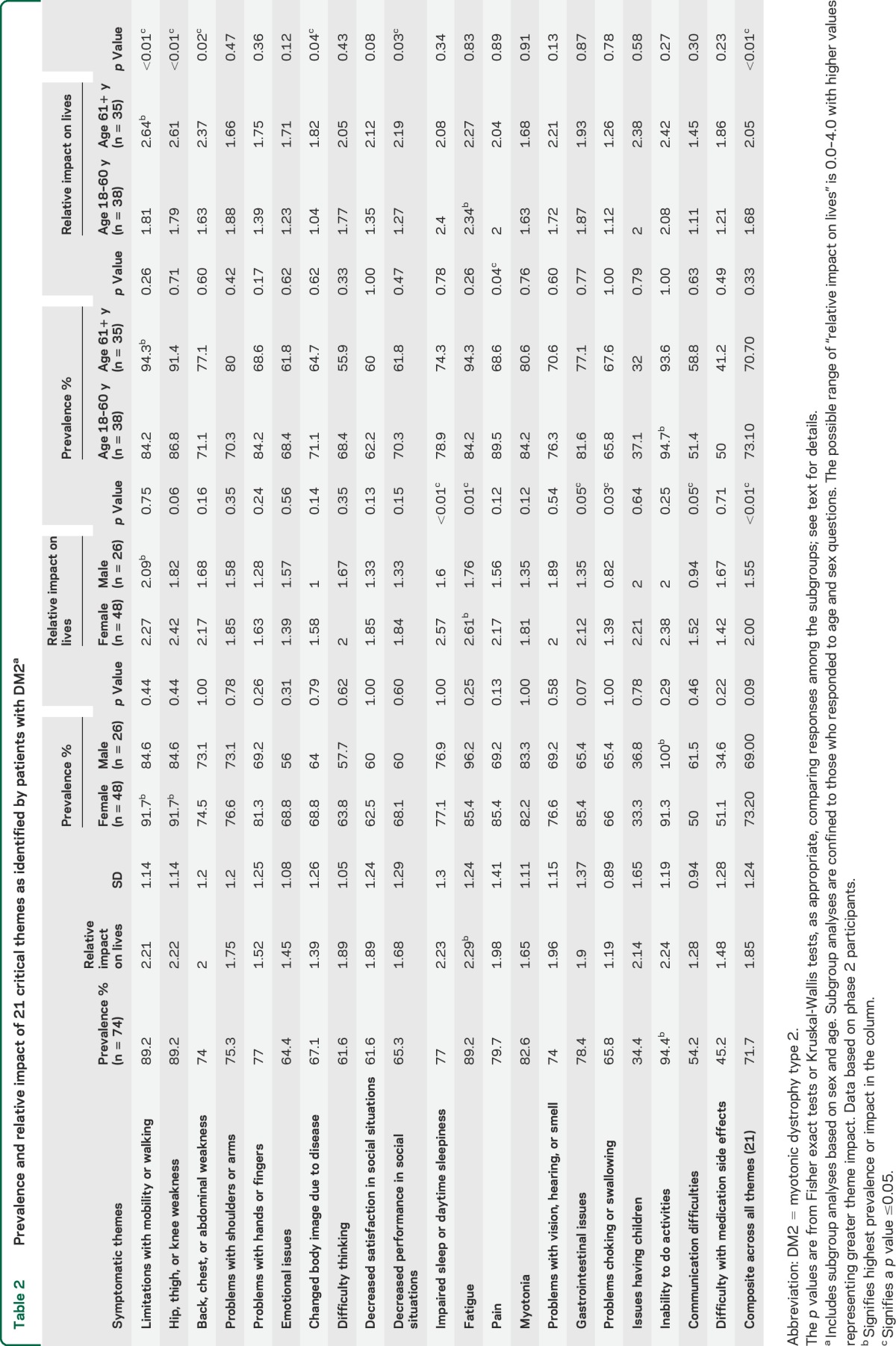
Table 3.
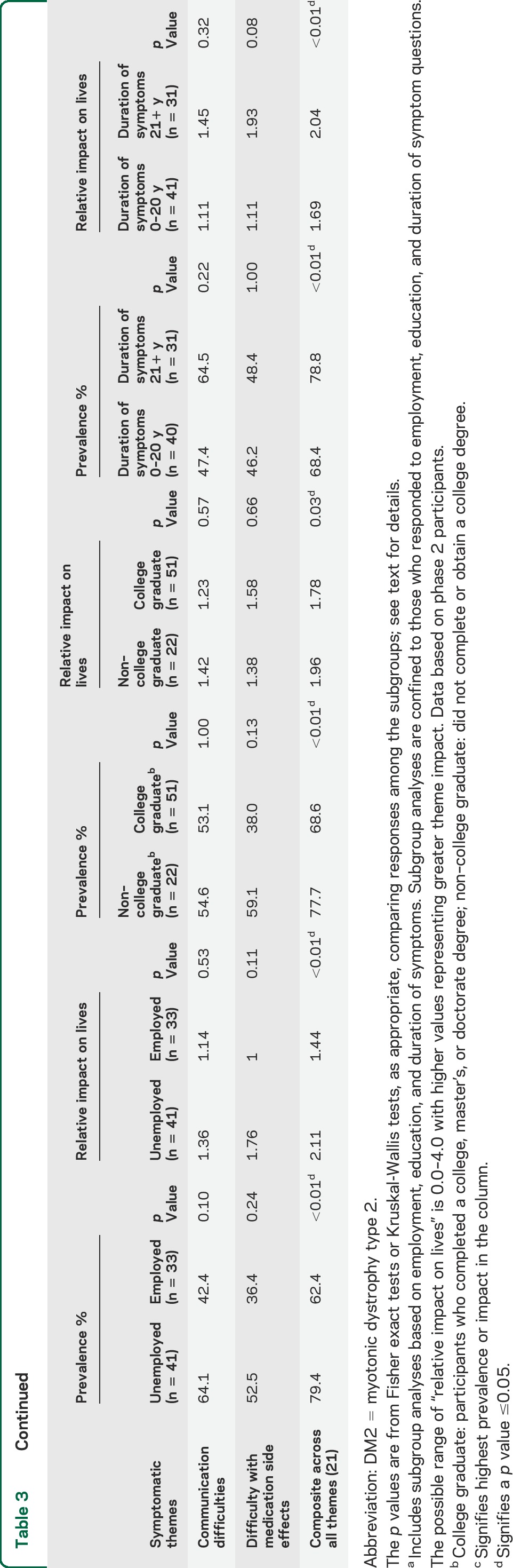
Prevalence and relative impact of 21 critical themes as identified by patients with DM2a
Of the 310 evaluated symptoms, 11 core symptoms were present in more than 95% of the respondents. These symptoms included difficulty getting up from the floor or ground (100%), leg weakness (100%), difficulty squatting down (100%), difficulty walking up hills or inclines (100%), difficulty rising from a seated position (97.4%), difficulty lifting objects because of leg weakness (97.4%), impaired walking (97.4%), difficulty with stairs (97.4%), the inability to run (97.4%), difficulty with balance (97.3%), and difficulty with rough ground (97.2%) (table e-1). Anger toward a parent who transmitted the disease was the only issue that was not reported by any participants.
The prevalence of select symptomatic themes was different between subgroups (tables 2 and 3). Participants older than 60 years had a higher prevalence of pain (p = 0.04) compared with younger participants. There were no differences in theme prevalence between female and male participants. Participants who had a college degree had problems with their shoulders and arms less frequently than those without a college degree (p = 0.03). Participants with a longer duration of symptoms were more likely to report fatigue (p < 0.01), limitations with mobility or walking (p = 0.02), or problems with shoulders or arms (p = 0.04).
Compared with employed participants, unemployed participants had a higher prevalence of limitations with mobility or walking (p < 0.01), problems with shoulders or arms (p < 0.01), emotional issues (p < 0.01), decreased satisfaction in social situations (p < 0.01), hip, thigh, or knee weakness (p = 0.0189), fatigue (p = 0.02), decreased performance in social situations (p = 0.02), problems with vision, hearing, or smell (p = 0.03), and activity limitation (p = 0.04) (figure 2, table 3).
Figure 2. Prevalence of symptomatic themes by employment status.

*Signifies p < 0.05.
The overall mean composite prevalence across all themes was associated with disease duration, education level, and employment status (p < 0.01) (table 3) but not with sex or age older than 60 years (table 2). The largest subgroup difference (in composite prevalence across all themes) occurred between employed (62.4%) and unemployed (79.4%) participants (table 3). Among individual symptomatic themes, the largest subgroup difference in prevalence occurred with “decreased satisfaction in social situations.” There was a 37.5% difference in the prevalence of this symptomatic theme between groups defined by employment status, with only 40.6% of employed participants with DM2 endorsing this issue and 78.1% of unemployed participants with DM2 endorsing this issue (figure 2).
Themes with the greatest life impact scores.
The themes that had the highest life impact score (range 0–4) in participants with DM2 were fatigue (2.29), inability to do activities (2.24), impaired sleep or daytime sleepiness (2.23), hip, thigh, or knee weakness (2.22), and limitations with mobility or walking (2.21) (table 2).
Of the 310 other symptoms evaluated, a lack of job secondary to disability (3.00), an inability to turn on a light with a pull cord (3.00), difficulty with stairs (2.88), difficulty getting up from the floor or ground (2.87), difficulty playing sports (2.85), difficulty squatting down (2.82), and a reliance on hand railings (2.80) had the highest life impact scores (table e-1).
In subgroup analyses of themes, female participants were more affected by impaired sleep or daytime sleepiness (p < 0.01), fatigue (p < 0.01), problems choking or swallowing (p = 0.03), and gastrointestinal issues (p = 0.0453) than male participants (table 2). Older participants were more affected than younger participants by mobility and walking (p < 0.01), hip, thigh, or knee weakness (p < 0.01), back, chest, or abdominal weakness (p = 0.02), changed body image (p = 0.04), and decreased performance in social situations (p = 0.03) (table 2).
Participants who were unemployed were more affected (life impact score) than employed participants with DM2 in the following areas: limitations with mobility and walking (p < 0.01), hip, thigh, or knee weakness (p < 0.01), back, chest, or abdominal weakness (p < 0.01), and changed body image due to disease (p < 0.01). There were no differences in life impact scores between participants with high vs low educational achievement. Participants with longer duration of symptoms were more affected by myotonia (p = 0.01) than those with symptom duration less than 21 years (table 3).
The average composite impact from all themes was worse in those who were unemployed, female, of older age, and had a longer duration of symptoms (p < 0.01) (tables 2 and 3). Having a college degree was also associated with a less severe composite burden from all themes (p = 0.03) (table 3).
Population impact scores.
The themes that had the greatest population impact scores (range 0–4) included inability to do activities (2.11), fatigue (2.04), hip, thigh, and knee weakness (1.98), and limitations with mobility or walking (1.97). The symptoms that had the greatest population impact scores were difficulty getting up from the floor or ground (2.87), difficulty squatting down (2.82), difficulty with stairs (2.80), and leg weakness (2.71) (table e-1).
DISCUSSION
This study utilizes direct patient input to identify the most prevalent and life-altering symptoms in DM2. These symptoms, including some that are clinically underrecognized, represent dysfunction in the physical, emotional, and social aspects of health. Our findings help define the diverse clinical phenotype of DM2 and potentially provide clues for the early identification and treatment of mildly symptomatic patients.
Patients with DM2 are often misdiagnosed early in the course of their disease. Prior work has identified an average delay of 14.4 years from a patient's first symptoms to their initial diagnosis.27 While additional research is needed, we think that the symptoms identified by our participants are similar to those symptoms that initially motivate a patient with DM2 to seek clinical care. Clinicians capable of identifying the early clinical features of DM2 have an opportunity to limit the diagnostic delay of their patients. A correct and efficient diagnosis of DM2 may lead to earlier treatment, appropriate monitoring for superimposed disease (e.g., cancer, hyperlipidemia, diabetes, cardiac conduction block, and hypothyroidism), and timely reproductive and medical counseling.
This study demonstrates that the symptoms that are most prevalent in a population are not always those that are most important to a population. While limitations with mobility and walking and hip, thigh, or knee weakness were the most prevalent symptomatic themes in participants with DM2, both were deemed to have less impact on participants' lives than fatigue. Both the prevalence and relative impact of individual symptoms will be worthwhile to consider when identifying the most appropriate therapeutic objectives in DM2. Of note, myotonia, one of the characteristic features by which DM2 is often recognized, ranked only 15th in relative impact among the 21 symptomatic themes.
The prevalence of different symptomatic themes varied among subgroups. One unexpected example was that participants aged 18 to 60 years had a higher prevalence of pain compared with those older than 60 years. While the basis for this finding is uncertain, it may be related to the higher amount of muscle mass in younger patients, increased coping mechanisms for pain among older individuals, or possibly that older participants were less severely affected (e.g., survival effect or other form of sampling bias). The higher prevalence of pain in the older group may also be related to age alone irrespective of DM2.
In some instances, the prevalence of a specific symptom was similar between subgroups while the average impact of the symptom on the patients in the subgroups differed. For instance, we found that women with DM2 were more severely affected by impaired sleep or daytime sleepiness, fatigue, problems choking or swallowing, and gastrointestinal issues than their male counterparts. While gastrointestinal issues are known to occur at higher rates in women,28 the association between these other symptoms and sex in DM2 warrants additional study.
The greatest difference in symptomatic theme prevalence and impact was seen between those patients who were employed vs those who were unemployed. We believe that employment status is highly dependent on a patient's overall disease burden. However, it is also possible that employment has specific effects on certain aspects of disease burden. For instance, we found that patients who were employed reported better satisfaction in social situations than those without jobs.
A limitation of our study is that our sample may not be a perfect representation of the general DM2 population. For example, we cannot exclude the possibility that the nonresponding registry participants have a different clinical profile than our responders, or that the symptoms of registry participants differ from the general DM2 population. Our sample was also predominantly female (64.9%). While we did not find any statistically significant difference in the prevalence of any symptomatic themes based on sex (table 2), our sample may overemphasize the impact of symptomatic themes that have a greater impact on the lives of women with DM2 (e.g., gastrointestinal issues and fatigue). In addition, because medication use and quality of care were not assessed, it is possible that the prevalence of treatable symptoms (e.g., pain, fatigue, daytime sleepiness, myotonia) was affected by symptomatic treatments.
The findings from PRISM-2 indicate that there is a distinct and diverse pattern of symptomatic disease in DM2. Knowledge from this study helps us to more clearly understand the complexities of the DM2 phenotype from a patient's perspective. In addition, knowledge of this study may ultimately prove useful in the clinical management of patients with DM2, in reducing DM2 diagnostic delay, and in the future design of disease-specific patient-reported outcome measures for this population.
Supplementary Material
GLOSSARY
- DM2
myotonic dystrophy type 2
- FSHD
facioscapulohumeral muscular dystrophy
- PRISM-2
Patient-Reported Impact of Symptoms in Myotonic Dystrophy Type 2 Study
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Chad Heatwole: study concept and design, acquisition of data, analysis and interpretation, critical revision of the manuscript for important intellectual content, study supervision. Dr. Nicholas Johnson: acquisition of data, analysis and interpretation, critical revision of the manuscript for important intellectual content. Dr. Rita Bode: analysis and interpretation, critical revision of the manuscript for important intellectual content. Jeanne Dekdebrun: acquisition of data. Nuran Dilek: analysis and interpretation. James E. Hilbert: acquisition of data. Elizabeth Luebbe: acquisition of data, analysis and interpretation, critical revision of the manuscript for important intellectual content. William Martens: analysis and interpretation. Dr. Michael P. McDermott: analysis and interpretation, critical revision of the manuscript for important intellectual content. Christine Quinn: acquisition of data. Dr. Nan Rothrock: analysis and interpretation, critical revision of the manuscript for important intellectual content. Dr. Charles Thornton: analysis and interpretation, critical revision of the manuscript for important intellectual content. Dr. Barbara G. Vickrey: analysis and interpretation, critical revision of the manuscript for important intellectual content. Dr. David Victorson: analysis and interpretation, critical revision of the manuscript for important intellectual content. Dr. Richard T. Moxley: analysis and interpretation, critical revision of the manuscript for important intellectual content.
STUDY FUNDING
Support for PRISM-2 was provided by the Goldberg Nathan Foundation for Myotonic Dystrophy Type-2 Research, the National Institute of Arthritis and Musculoskeletal and Skin Disorders (1K23AR055947), the Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center (U54NS48843-01), the Muscular Dystrophy Association, the New York State Empire Clinical Research Investigator Program, the Saunders Family Fund, and the University of Rochester Clinical Translational Science Institute.
DISCLOSURE
C. Heatwole receives grant funding from the NIH, FDA, and Cure SMA foundation. He is the founder and CEO of the Neuromuscular Quality of Life Institute. He receives royalties for the Myotonic Dystrophy Health Index. He reviews medical cases for Imedecs and Maximus. He has provided expert testimony for neuromuscular cases unrelated to this research. C. Heatwole has provided consultation to Biogen and aTyr Pharma. N. Johnson serves as an associate editor for Neurology: Genetics. He is funded by the NIH, grant 1K23NS091511-01. He has received research support from the Muscular Dystrophy Association, Valerion Therapeutics, Isis Pharmaceuticals, and Biogen Idec. R. Bode reports no disclosures relevant to the manuscript. J. Dekdebrun, N. Dilek, J. Hilbert, E. Luebbe, and W. Martens report no disclosures relevant to the manuscript. M. McDermott is a consultant for Cerebral Assessment Systems, Inc., AstraZeneca, and the New York State Department of Health and was a consultant for Asubio Pharmaceuticals. He serves or has served on data and safety monitoring boards for studies sponsored by Novartis Pharmaceuticals Corporation, AstraZeneca, aTyr Pharma, the ALS Association, and the Muscular Dystrophy Association. He receives research support from NIH, FDA, NYSTEM, the Spinal Muscular Atrophy Foundation, and Rhythm Pharmaceuticals, Inc. He serves on the editorial board of Movement Disorders and as an editor for Chance. C. Quinn reports no disclosures relevant to the manuscript. N. Rothrock is funded by NIH grants U2CCA186878, U54AR057943, and U54AR057951, receives research support from the AO Foundation and Coleman Foundation, and receives royalties from a publication within UpToDate. She was previously funded by NIH grants HHSN271201200036C, HHSN265200423601C, and RC4CA157236. The dates of her awards are as follows: U2CCA186878: 2014–2018, U54AR057943: 2009–2015, U54AR057951: 2009–2015, AO Foundation: 2011–2015, Coleman Foundation: 2011–2015, HHSN271201200036C: 2013–2014, HHSN265200423601C: 2012–2013, and RC4CA157236: 2010–2013. C. Thornton served as consultant to Isis Pharmaceuticals, received sponsored research support from Isis Pharmaceuticals and Biogen Idec, is funded by NIH grants U01NS072323 and U54NS48843, and received research support from the Muscular Dystrophy Association. B. Vickrey serves on the scientific advisory board for the Sports Concussion Institute. She receives research support from NINDS, the US Veterans Administration Health Services Research and Development Service, UniHealth Foundation, and California Community Foundation, and she is or has recently been a consultant to EMD Serono Canada and Genentech. D. Victorson and R. Moxley report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001;293:864–867. [DOI] [PubMed] [Google Scholar]
- 2.Day JW, Ricker K, Jacobsen JF, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 2003;60:657–664. [DOI] [PubMed] [Google Scholar]
- 3.Udd B, Meola G, Krahe R, et al. 140th ENMC International Workshop: myotonic dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management. Neuromuscul Disord 2006;16:403–413. [DOI] [PubMed] [Google Scholar]
- 4.Heatwole C, Johnson N, Goldberg B, Martens W, Moxley R., III Laboratory abnormalities in patients with myotonic dystrophy type 2. Arch Neurol 2011;68:1180–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson NE, Heatwole CR. Myotonic dystrophy: from bench to bedside. Semin Neurol 2012;32:246–254. [DOI] [PubMed] [Google Scholar]
- 6.Jensen MP, Hoffman AJ, Stoelb BL, Abresch RT, Carter GT, McDonald CM. Chronic pain in persons with myotonic dystrophy and facioscapulohumeral dystrophy. Arch Phys Med Rehabil 2008;89:320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen MP, Abresch RT, Carter GT. The reliability and validity of a self-report version of the FIM instrument in persons with neuromuscular disease and chronic pain. Arch Phys Med Rehabil 2005;86:116–122. [DOI] [PubMed] [Google Scholar]
- 8.Miro J, Raichle KA, Carter GT, et al. Impact of biopsychosocial factors on chronic pain in persons with myotonic and facioscapulohumeral muscular dystrophy. Am J Hosp Palliat Care 2009;26:308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nieto R, Raichle KA, Jensen MP, Miro J. Changes in pain-related beliefs, coping, and catastrophizing predict changes in pain intensity, pain interference, and psychological functioning in individuals with myotonic muscular dystrophy and facioscapulohumeral dystrophy. Clin J Pain 2012;28:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miro J, Gertz KJ, Carter GT, Jensen MP. Pain location and intensity impacts function in persons with myotonic dystrophy type 1 and facioscapulohumeral dystrophy with chronic pain. Muscle Nerve 2014;49:900–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suokas KI, Haanpaa M, Kautiainen H, Udd B, Hietaharju AJ. Pain in patients with myotonic dystrophy type 2: a postal survey in Finland. Muscle Nerve 2012;45:70–74. [DOI] [PubMed] [Google Scholar]
- 12.Tieleman AA, Knoop H, van de Logt AE, Bleijenberg G, van Engelen BG, Overeem S. Poor sleep quality and fatigue but no excessive daytime sleepiness in myotonic dystrophy type 2. J Neurol Neurosurg Psychiatry 2010;81:963–967. [DOI] [PubMed] [Google Scholar]
- 13.Chokroverty S, Bhat S, Rosen D, Farheen A. REM behavior disorder in myotonic dystrophy type 2. Neurology 2012;78:2004. [DOI] [PubMed] [Google Scholar]
- 14.Bhat S, Sander HW, Grewal RP, Chokroverty S. Sleep disordered breathing and other sleep dysfunction in myotonic dystrophy type 2. Sleep Med 2012;13:1207–1208. [DOI] [PubMed] [Google Scholar]
- 15.Shepard P, Lam EM, St Louis EK, Dominik J. Sleep disturbances in myotonic dystrophy type 2. Eur Neurol 2012;68:377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lam EM, Shepard PW, St Louis EK, et al. Restless legs syndrome and daytime sleepiness are prominent in myotonic dystrophy type 2. Neurology 2013;81:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romigi A, Albanese M, Placidi F, et al. Sleep disorders in myotonic dystrophy type 2: a controlled polysomnographic study and self-reported questionnaires. Eur J Neurol 2014;21:929–934. [DOI] [PubMed] [Google Scholar]
- 18.Minnerop M, Weber B, Schoene-Bake JC, et al. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 2011;134:3530–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaul C, Schmidt T, Windisch G, et al. Subtle cognitive dysfunction in adult onset myotonic dystrophy type 1 (DM1) and type 2 (DM2). Neurology 2006;67:350–352. [DOI] [PubMed] [Google Scholar]
- 20.Bugiardini E, Meola G; DM-CNS Group. Consensus on cerebral involvement in myotonic dystrophy: workshop report: May 24–27, 2013, Ferrere (AT), Italy. Neuromuscul Disord 2014;24:445–452. [DOI] [PubMed] [Google Scholar]
- 21.Meola G, Sansone V. Cerebral involvement in myotonic dystrophies. Muscle Nerve 2007;36:294–306. [DOI] [PubMed] [Google Scholar]
- 22.Axford MM, Pearson CE. Illuminating CNS and cognitive issues in myotonic dystrophy: workshop report. Neuromuscul Disord 2013;23:370–374. [DOI] [PubMed] [Google Scholar]
- 23.Heatwole C, Bode R, Johnson N, et al. Patient-reported impact of symptoms in myotonic dystrophy type 1 (PRISM-1). Neurology 2012;79:348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McColl E. Developing questionnaires. In: Fayers P, Hays R, editors. Assessing Quality of Life in Clinical Trials. Oxford, UK: Oxford Press; 2005:9–23. [Google Scholar]
- 25.Johnson NE, Quinn C, Eastwood E, Tawil R, Heatwole CR. Patient-identified disease burden in facioscapulohumeral muscular dystrophy. Muscle Nerve 2012;46:951–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hilbert JE, Kissel JT, Luebbe EA, et al. If you build a rare disease registry, will they enroll and will they use it? Methods and data from the National Registry of Myotonic Dystrophy (DM) and Facioscapulohumeral Muscular Dystrophy (FSHD). Contemp Clin Trials 2011;33:302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hilbert JE, Ashizawa T, Day JW, et al. Diagnostic odyssey of patients with myotonic dystrophy. J Neurol 2013;260:2497–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson S, Roberts L, Roalfe A, Bridge P, Singh S. Prevalence of irritable bowel syndrome: a community survey. Br J Gen Pract 2004;54:495–502. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.