

Abstract
Hepatitis B virus (HBV) infection is a major public health problem worldwide. HBV is not directly cytotoxic to infected hepatocytes; the clinical outcome of infection results from complicated interactions between the virus and the host immune system. In acute HBV infection, initiation of a broad, vigorous immune response is responsible for viral clearance and self-limited inflammatory liver disease. Effective and coordinated innate and adaptive immune responses are critical for viral clearance and the development of long-lasting immunity. Chronic hepatitis B patients fail to mount efficient innate and adaptive immune responses to the virus. In particular, HBV-specific cytotoxic T cells, which are crucial for HBV clearance, are hyporesponsiveness to HBV infection. Accumulating experimental evidence obtained from the development of animal and cell line models has highlighted the importance of innate immunity in the early control of HBV spread. The virus has evolved immune escape strategies, with higher HBV loads and HBV protein concentrations associated with increasing impairment of immune function. Therefore, treatment of HBV infection requires inhibition of HBV replication and protein expression to restore the suppressed host immunity. Complicated interactions exist not only between innate and adaptive responses, but also among innate immune cells and different components of adaptive responses. Improved insight into these complex interactions are important in designing new therapeutic strategies for the treatment HBV infection. In this review, we summarize the current knowledge regarding the cross-talk between the innate and adaptive immune responses and among different immunocytes in HBV infection.
Keywords: Crosstalk, Hepatitis B virus, Innate immune, Adapative immune
Core tip: Hepatitis B virus (HBV) is poorly sensed by the innate immune system and can escape innate immune recognition at the early stage of infection. HBV-specific T-cell responses are timely and efficiently induced in acute self-limited infections but are deeply exhausted in chronic hepatitis B. The tolerogenic effect of the liver environment and the persistent exposure of T cells to high antigen loads play a key role in the pathogenesis of T-cell inhibition in chronic HBV infection. Combination of reduction of HBV and virus antigen loads and restoration of the anti-viral T-cell function may represent a strategy to cure chronic HBV infections.
INTRODUCTION
Hepatitis B virus (HBV) infection is a major public health problem worldwide. Approximately 30% of the world’s population show serological evidence of current or past HBV infection and 350 million people are chronically infected[1]. The outcome of HBV infection varies widely among infected patients from resolved acute infection, chronic hepatitis, and liver cirrhosis to hepatocellular carcinoma. Infections in approximately 5% of adults and 95% of neonates become persistent[2]. HBV itself is not directly cytotoxic to infected hepatocytes and the clinical outcome of infection results from complicated interactions between the virus and the host immune system[3-5]. The immune responses to HBV antigens, which are mediated through complex interactions between the innate immune and adaptive immune systems, are responsible both for viral clearance and disease pathogenesis. In acute HBV infection, a broad, vigorous immune response results in viral clearance associated with acute, self-limited inflammatory liver disease[6]. In contrast, chronic hepatitis B (CHB) patients fail to mount efficient innate and adaptive immune responses to the virus, with HBV-specific cytotoxic T cells (CTLs) in particular, being hyporesponsiveness to HBV infection[7,8]. The role of adaptive immune responses in the control of HBV infection is widely accepted, with HBV-specific T cell responses being essential for the termination of HBV infection. Furthermore, CD4+ T cells serve as the chief regulators of the adaptive immune response to HBV[5]. The innate immune system is the first line of active host defense against viral infection, and once activated, is linked to a favorable clinical outcome and subsequent robust adaptive immune responses[8]. The induction of innate immune responses by HBV during the phase of early infection is a longstanding controversy. The development of animal and cell culture models has yielded great improvements in our understanding of the innate immune responses during HBV infection. Furthermore, the strategies employed by HBV to counteract the innate antiviral pathways are being gradually recognized. It is known that effective recognition of viral infection and successive activation of antiviral innate immune responses are vital for host antiviral defense and largely depend on multiple regulators, including Toll-like receptors (TLRs)[9,10] and cytokines[11]. Efficient control of virus infections requires the coordinated actions of both innate and adaptive immune responses. Mounting effective innate and adaptive immune responses is critical for viral clearance and the development of long-lasting immunity. Complicated interactions exist not only between the innate and adaptive systems, but also among innate immune cells and among different components of adaptive responses.
A better understanding of the interplay between innate and adaptive immune responses and between the host immune response and the virus is crucial for the development of new antiviral therapeutic strategies aimed at eradicating chronic infections.
In this review, we summarize the current knowledge regarding the interactions between the innate and adaptive immune systems and among different immunocytes during HBV infection.
TLRS
TLRs are a group of highly conserved molecules that sense pathogen-associated molecular patterns (PAMPs). So far in humans and mice, TLR1 to 13 have been identified, which are extensively expressed in various immune and non-immune cells. Stimulation by their ligands initiates the activation of complex intracellular signal transduction networks and innate and adaptive immune-related cells, including natural killer (NK) cells, NK-T cells, monocytes, dendritic cells (DCs), T cells, B cells, and Tregs, as well as the production of antiviral effector interferons (IFNs) and proinflammatory cytokines[12]. TLRs play important roles in innate immune responses[13] to viral infections, including HBV. TLRs can activate DCs, improve antigen presentation, and initiate T cell immune responses. In vivo, TLRs also directly modulate HBV-specific T and B cell responses, which are essential for the termination of HBV infection[14]. Therefore, TLR responses are cell type-specific.
TLRs and innate immunity
Innate immunity is important in controlling infection immediately after contact with the pathogen and to initiate efficient development of an adaptive immune response. TLRs play a key role in the activation of innate immune responses to infectious agents[13]. The TLR family consists of intracellular and cell surface subgroups. The intracellular subgroup (TLR3, TLR7, TLR8 and TLR9) is localized in endosomes and recognizes nucleic acids, such as viral DNA or RNA, while the cell surface subgroup (TLR1, TLR2, TLR4/MD-2, TLR5 and TLR6) recognizes extracellular bacterial and fungal cell wall components, as well as some viral proteins[13-17]. Binding of TLR agonists to their receptors initiates the activation of complex networks of intracellular signal transduction pathways that leads to the induction of type I IFNs (IFNα/β), proinflammatory cytokines, and costimulatory molecules, which are involved in antiviral responses[18,19]. The importance of TLR receptor signaling in controlling HBV replication was confirmed by a study in which a single intravenous injection of ligands specific for TLR3, TLR4, TLR5, TLR7 and TLR9 provided efficient inhibition of HBV replication in a non-cytolytic and IFNα/β-dependent manner in HBV transgenic mice[20].
TLRs and DCs and peripheral blood mononuclear cells
TLRs are abundantly expressed on the surface of DCs, especially peripheral blood monocyte-derived DCs (moDCs). Plasmacytoid dendritic cells (pDCs) play a crucial role in triggering antiviral immunity through their ability to capture and process viral antigens and subsequently induce adaptive immune responses and the production of type I IFNs. pDCs are the key sensors of viral infections through expression of both TLR7 and TLR9[21]. Myeloid DCs (mDCs) respond to TLR1, -2, -4 and -9 ligands resulting in upregulation of CD40 and activation of allogeneic T cells[22]. TLR9 detects intracellular viral double-stranded (ds)DNA, which leads to the activation of nuclear factor κB (NF-κB) via the myeloid differentiation primary response 88 (MyD88) pathway, resulting in the activation of immune responses against HBV. However, expression of TLR9, MyD88, IRAK1, TRAF6, and NF-κB in peripheral blood mononuclear cells (PBMCs) of CHB patients is significantly decreased in comparison with healthy controls[23,24], which may result in an attenuated responses that ultimately lead to long-lasting HBV infection[25]. Reduced TLR9 expression in pDCs of CHB patients is associated with impaired IFNα production[26]. TLR2 and TLR4 mediate the activation of the same signaling pathways downstream of MyD88, including NF-κB, MAPK, and PI-3k/Akt pathways to inhibit hepadnaviral replication. One study indicated that expressions of TLR2 and TLR4 were downregulated in PBMCs during HBV infection[27], while another study showed that expression of TLR2 and TLR-4 in moDCs was significantly increased with disease progression[28]. The role of TLR2 and TLR-4 in the pathogenesis of requires further evaluation.
TLRs and NK cells
NK cells possess receptors allowing them to sense and respond to viral and bacterial patterns, including TLRs. Upon TLR activation (mainly TLR3 and TLR7), NK cells produce IFNγ[29-31], which also contributes to deleterious inflammation if produced in excessive amounts[29]. NK cells in CHB patients have an impaired IFNγ response to TLR9 stimulation compared to healthy controls although no differences have been observed in responses to the other TLR ligands. This suggests that multiple mechanisms may be involved in NK activation[32] and although viral clearance is suppressed in chronic HBV infection, the potential to mediate tissue injury is maintained.
TLRs and non-parenchymal cells
Non-parenchymal cells (NPCs), like Kupffer cells (KCs) and liver sinusoidal endothelial cells (LSECs), also participate in innate immune responses by producing various cytokines, including tumor necrosis factor-α (TNF-α) and IFNβ[28] in response to TLR signaling. Isogawa et al[20] demonstrated the involvement of NPCs rather than hepatocytes in antiviral activation induced by TLR ligands. HBV is recognized by the NPCs of the liver, mainly macrophages (KCs), although they are not infected. KCs respond to all TLR ligands by producing TNF-α or interleukin-6 (IL-6), to TLR3 and TLR4 ligands by producing IFNβ[22], to TLR1 and TLR8 ligands by upregulating major histocompatibility complex (MHC) class II and costimulatory molecules, and to TLR1, -2, -4 and -6 ligands by inducing high levels of T cell proliferation and IFNλ production in the mixed lymphocyte reaction[22].
LSECs are liver-resident antigen-presenting cells that are capable of antigen cross-presentation and induction of CD8+ T cell tolerance or immunity under different conditions[33,34]. Liu et al[35] demonstrated that pretreatment of LSECs with a TLR1/2 ligand or LPS (TLR4 ligand) relieved their suppressive functions to induce T cell immunity, while Wu et al[22] suggested that, on stimulation by TLR ligands, LSECs have similar responses to KCs. Another study that demonstrated that, among different TLR ligands, hepatic NPCs show significant production of IFNβ only in response to TLR3 stimulation[36]. However, in the presence of HBsAg, TLR-induced expression of IFNγ, interferon sensitive genes and proinflammatory cytokines in murine KCs and LSECs was efficiently suppressed, whereas the expression of anti-inflammatory cytokines was enhanced[37].
Although regarded as a type of antigen-presenting cell (APC), NPCs display a restricted TLR-mediated activation profile compared with “classical” APCs. Therefore, antiviral effects induced by TLR receptor activation should be carefully evaluated in therapeutic design to maintain the balance between viral control and liver injury. Furthermore, coordination of innate and adaptive immune responses may be highly important for the control of viral infection[19].
TLRs and adaptive immunity
Several studies have demonstrated that TLR2 is expressed by activated and memory CD4+ and CD8+ T cells and serves as a costimulatory molecule[38,39]. In some studies, TLR3 and TLR9 expression on human CD8+ T cells was also demonstrated to promote IFNγ production upon stimulation[40,41]. However, one study showed that, although all TLRs were able to induce CD8+ T cell activation in vitro, there were profound differences in their CD8+ T cell activation capacity in vivo. TLR3 and TLR9 induced CD8+ T cell activation, while, TLR2 and TLR4 were not only incapable of inducing CD8+ T cell priming, but also inhibiting CD8+ T cell expansion[42].
B cells represent an important link between the adaptive and innate immune systems in that they express both antigen-specific B cell receptors (BCRs) as well as various TLRs[43]. Conventionally, signaling through the BCR initiates a sequence of events that are necessary for B cell activation and differentiation of. In combination with BCR signaling, TLR signaling plays multiple roles in B cell differentiation and activation and the outcome is largely context-dependent[44]. However, activation of resting B cells by simultaneous involvement of TLR-2 and the costimulatory molecules CD40 and CD86 could be BCR-independent[45,46]. Expressions and activation of TLRs in immune cells in HBV infection are illustrated in Figure 1.
Figure 1.
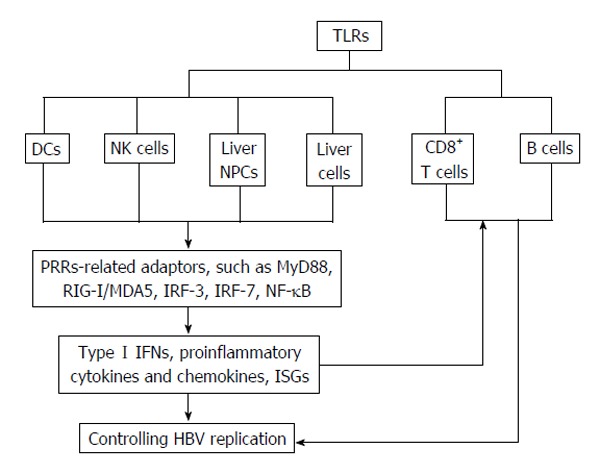
Expressions and activation of toll-like receptors in innate and adaptive immune cells in controlling hepatitis B virus infection. HBV: Hepatitis B virus; NPCs: Non-parenchymal cells; NK: Natural killer; IFN: Interferon; TLRs: Toll-like receptors; ISG: Interferon-stimulated genes; RIG-I: Retinoic acid inducible gene I; IRF: Interferon-regulatory factors; NF-κB: Nuclear factor κB; PBMCs: Periperal blood mononuclear cells; MDA5: Melanoma differentiation associated gene 5; DCs: Dendritic cells.
CYTIKINES
Cytokines and chemokines play a crucial role in initiating, maintaining, and regulating immunological homeostasis and inflammatory processes. Cytokines are released by many different cell types and activate cells of both the innate and adaptive immune system[47]. Cytokine-mediated immune responses play a pivotal role in determining the clinical outcome of HBV infection. Different patterns of serum cytokines and chemokines are associated with different phases of HBV infection. Non-cytolytic intracellular viral inactivation by IFNγ and TNF-α play an important role in the clearance of HBV in resolved acute HBV infection without killing infected cells. The recognition of PAMPs by PRRs such as TLRs, RIG-I like receptors, NOD-like receptors results in activation of intracellular pathways and leads to the production of antiviral, immunoregulatory and proinflammatory molecules[48].
IFNs
IFNs represent one of the first lines of host defense against invading pathogens. As key components of the innate immune system, IFNs have been demonstrated to restrict HBV replication by affecting multiple steps in the viral life cycle, including HBV RNA synthesis, pgRNA encapsulation, the turnover rate of viral proteins, and modulation of covalently closed circular (ccc)DNA formation[49,50] by inducing numerous IFN-stimulated genes[51]. IFNs are classified into three groups, types I, II and III, based on the structure of their receptors on the cell surface[47]. The early phase of viral infection is characterized mainly by the production of type I IFNα/β, and NK cell activation. The production of type I IFNs can be triggered directly by virus replication through cellular mechanisms that detect the presence of viral RNA or DNA. The main sources of IFNα/β are infected cells and pDCs, whereas IFNγ is produced primarily by NK and NKT cells[52]. IFNβ has also been identified as a major antiviral factor produced by NPCs in response to TLR3[36]. Recombinant IFN (rIFN)-α has been approved and successfully used as a standard treatment for chronic HBV infection[48]. Furthermore, treatment of the HBV-producing hepatocytes with rIFN-γ and rTNF-α efficiently suppresses HBV replication without cytolysis[53]. In addition, IFNs have immunomodulatory functions as indicated by the ability of IFNα treatment to recover HBV-impaired hepatocyte-intrinsic innate immunity[54].
TNF-α
TNF-α is another major antiviral cytokine which, like IFNγ, also stimulates adaptive immunity and the antiviral effects of CTLs[53,55]. The absence of TNF-α or early treatment with a TNF receptor blocker reduces viral clearance, persistently maintains elevated HBV viral load and increases expression of the inhibitory receptor, programmed death-1 (PD-1) in CD8+ T cells in a mouse model[56,57]. These results suggest that HBV is reactivated during therapy with TNF-α-blocking agents in clinical practice. In addition to the induction of non-cytopathic suppression and clearance of HBV in animal models, TNF-α rapidly blocks HBV replication by promoting destabilization of pre-existing cytoplasmic viral nucleocapsids containing viral RNA and DNA, as well as of empty nucleocapsids[57].
IL-6
Sodium-taurocholate cotransporting polypeptide (NTCP) has been identified as an HBV-specific receptor. Studies have shown that NTCP-mediates HBV entry is markedly inhibited by IL-6, with a strong inhibition of long-term HBsAg secretion and a profound reduction in intracellular HBV cccDNA[58]. Hösel et al[59] demonstrated that recognition of HBV patterns by liver NPCs results in IL-6-mediated control of HBV infection at the transcriptional level. In the early phase of infection, IL-6 rather than IFN mediates control of the virus, limiting activation of the adaptive immune response and preventing death of HBV-infected hepatocytes[59].
IL-12
IL-12 is an immunomodulatory cytokine that promotes cellular immunity. Research suggests that IL-12-based vaccination therapy strongly enhances hepatic HBV-specific CD8+ T cell responses, restores systemic HBV-specific CD4+ T cell responses and activates HBsAg-specific follicular Th-germinal center B cell responses, resulting in IFNγ secretion and anti-HBs antibody production[55]. Studies have also shown that IL-12 initiates LSEC-mediated CD8+ T cell immunity[35].
IL-18
IL-18 is produced mainly by activated macrophages, and like IL-12, induces IFNγ and TNF-α. It has been shown that IL-18 inhibits HBV replication in hepatoma cell lines and in the liver through induction of IFN-γ production by NK cells and T cells. HBeAg protein may suppress IL-18-mediated NF-κB signaling in NK and hepatoma cells and inhibit expression of IFNγ[60], which contributes to the establishment of HBV persistent infection. Studies have shown that IL-18 gene polymorphisms affect susceptibility to HBV infection and are associated with different outcomes of HBV infection. However, the results from other studies are conflicting. Motavaf suggested that the IL-18 genotype -607 A/A is associated with susceptibility to chronic HBV infection[61], while Karra indicated that it may be protective against HBV infection and associated with spontaneous clearance[62]. Thus, the effects of this IL-18 genotype on HBV infection remain to be fully elucidated.
IL-22
Despite hepatoprotective and anti-fibrotic functions in acute liver injury models, IL-22 exacerbates liver inflammation and fibrosis in chronic HBV-infected patients and HBV transgenic (Tg) mice by recruiting Th17 cells into the liver. IL-22 also induces upregulation of numerous IL-22 pathway-associated proinflammatory genes in HBV-infected liver tissues and exerts mitogenic and anti-apoptotic effects on hepatocytes[63]. Furthermore, IL-22 depletion was shown to significantly inhibit recruitment of antigen-non-specific inflammatory cells into the liver in HBV Tg mice, while, IFNγ mediated non-cytopathic inhibition of virus replication initiated by HBV-specific cytotoxic T cells was not affected[64]. This indicates that IL-22 has no direct inhibitory effects on virus replication.
Transforming growth factor-β and IL-10
Transforming growth factor (TGF)-β is an important cytokine for the maturation and differentiation of many different immune cells in the liver. This cytokine mediates dual immunoregulatory functions involving induction of proinflammatory or anti-inflammatory responses in cooperation with other soluble factors. It suppresses differentiation of Th1 and Th2 cells and promotes development of the Th17, Th9, and the Treg phenotypes[64]. Thus, TGF-β plays a dual role in HBV infection by suppressing immune responses against viral infection and inhibiting viral replication. TGF-β1 suppresses HBV replication primarily through transcriptional inhibition of pre-genomic RNA[65]. KCs in HBV-carrier mice express high levels of IL-10 and mediate the induction of systemic tolerance in an IL-10-dependent manner[66]. Blockade of IL-10 restores NK cell effector function in acute HBV infection, indicating that the immunosuppressive cytokine environment in chronic HBV infection may inhibit the ability of NK cells to produce IFNγ and subsequent activation of CD8+ T cells[67]. NK cells and regulatory B (Breg) cells also produce elevated IL-10 in CHB[68].
Other cytokines
IL-21, derived from HBV-specific CD4+ T cells plays key roles in sustaining CD8+ T cells and promoting B cell responses that are essential for effective HBV control[69]. IL-21 is not only mediates direct and effective suppression of HBV replication, but also reduces HBV replication by inhibiting IL-10 secretion[70]. However, as a mediator of inflammation, IL-21 is also involved in the development of HBV-induced liver cirrhosis and exacerbating liver injury[71].
IL-35 is a recently identified potent immunosuppressive cytokine of the IL-12 family, which is secreted by regulatory T (Treg) cells and the newly reported Breg cells. IL-35 suppresses the proliferation of HBV antigen-specific cytotoxic T-lymphocytes and IFNγ production in vitro and decreases the proliferation of CD4+CD45RA+ naïve T cells and the expansion of CD11c+ DCs ex vivo. High expression of IL-35 in CD4+ T cells may be one of the factors involved in the inhibition of cellular immune responses in chronic HBV infection[71-73].
DCS AND OTHER IMMUNE CELLS
DCs are the most efficient professional APCs, which stimulate the initial T cell activation and proliferation. Typically, immature DCs capture and process antigens to peptides which are then presented in the context of MHC class II or class I molecules. It is generally accepted that the function of DCs of patients with chronic HBV infection is impaired, resulting in more tolerogenic rather than immunogenic responses, which may contribute to viral persistence. However, whether DCs in chronic HBV patients are phenotypically and functionally equal to DCs from healthy donors is still open to discussion. A few studies have shown that the frequency and function of ex vivo-analyzed mDCs and pDCs are largely intact in patients with HBV infection and similar to those of healthy donor DCs, with the exception of reduced IFNα production by pDC from CHB patients[74]. Treatment of MoDCs with HBsAg resulted in enhanced cell surface expression of CD80, CD83, CD86 and MHC class II, and increased production of IL-12 p40, IL-12 p70, and IL-10[75]. Nevertheless, other studies showed that the pDCs isolated from CHB patients have lower expression of HLA-DR and the costimulatory molecules CD80 and CD40, leading to low allo-stimulatory function, and lower levels of IFN-α and IL-12 production[76-78]. The major role of DCs in CHB immunopathogenesis mainly involves their interaction with other cells of the innate or adaptive immune systems.
DC s and NK cells
NK cell functions are closely related to those of DCs. DCs play a crucial role in the NK cell activation and a reciprocal functional interaction between NK cells and either pDCs or mDCs may play an important physiological role in the regulation of both innate and adaptive immune responses[79-81]. DCs efficiently enhances NK cell expression of CD69, proliferation, IFNγ secretion and cytotoxic activity. Studies have suggested that membrane-associated molecules, as well as soluble factors such as IL-12, TNF-α and type I IFNs, contribute to DC-mediated NK cell activation[82] and subsequent adaptive immune responses. CHB patients display a diminished functional interaction between poly(I:C)/IFNγ activated mDC and NK cells due to impaired mDC function and reduced IFNγ production compared to those of healthy individuals. Furthermore, restoration of TLR3-activated mDC activity leads to improved NK cell function, which underlies the impaired DC-induced NK cell dysfunction in CHB[83].
NK cells also promote the DCs maturation and markedly augment their capacity to produce proinflammatory cytokines and to stimulate T cell responses. The NK cell-mediated effects on DCs are dependent on cell membrane-associated molecules, such as NKp30 and soluble factors, such as TNF-α and IFNγ[82]. The intrahepatic pool of NK cells also plays a key role in the regulation of DC function in CHB patients[80]. Therefore, it can be speculated that enhancing this reciprocal interaction will reinforce the innate and thus, the adaptive immune response, which may contribute significantly to achieving effective antiviral immunity[81].
DCs and HBV-specific CD8+ T cells
Experimental evidence has shown that HBV-specific T cell responses are essential for the control of HBV infection. In chronic HBV infection, virus-specific CD8+ T cells are recruited to the liver, but are functionally or quantitatively impaired[84]. Typically, DCs activate resting T cells to initiate immune responses. Impaired DC function in patients with CHB may lead to insufficient T cell responses to HBV, which may be associated with persistent viral infection. HBV particles and purified HBsAg both contribute to the mDC dysfunction[85,86] and inhibit the antiviral function of autologous lymphocytes manifested by decreased IFNγ and IL-2 production and increased IL-10 secretion. A recent study demonstrated that HBcAg-pulsed DCs derived from CHB patients exhibited a stronger capacity to stimulate autologous CD4+ and CD8+ T cells to release IFNγ and induce HBV core 18-27 specific CTLs[87]. Furthermore, CpG-activated pDCs act synergistically in vitro with HBcAg-pulsed moDCs (core-DC) in inducing autologous HBV-specific CD8+ T cell proliferation and IFNγ production[88]. Thus, mature DCs efficiently induce Th1 polarization of T cells and generate HBcAg-specific CTLs. In addition, liver-resident CD103+ DCs are also highly immunogenic in hepatotropic viral infections and serve as a major APC to support the local CD8+ T cell responses[89].
DCs and Treg cells
Circulating CD4+ CD25+ Tregs have been demonstrated to maintain immunotolerance and suppress antigen-specific or antigen-non-specific T cell responses. In CHB patients, the frequency of CD4+ CD25 (high) Tregs is increased and correlates positively with serum viral load and has been shown to suppress HBV antigen-stimulated autologous PBMC proliferation and IFNγ production in vitro[90]. In CHB patients, DCs induce the expansion of Tregs, which continue to express high levels of forkhead box P3 (Foxp3) protein[91]. Furthermore, Tregs induced by NK-primed DCs are capable of inducing a suppressor effect via the negative co-stimulation of PD-1[92]. On the other hand, when triggered by a specific antigen, Tregs act on immature DCs via a feedback mechanism to block the upregulation of the costimulatory molecules, CD80 and CD86[91].
NK CELLS, NKT CELLS AND ADAPTIVE CELLS
NK cells represent the main effector cell population involved in innate immune responses against intracellular pathogens and tumor cells through their cytolytic activity and production of cytokines. NK cells are enriched in the liver, with a frequency of 30%-50% of intrahepatic lymphocytes in humans, which is 10-12-fold higher in CHB patients compared to healthy controls[93]. NKT cells share characteristics with innate lymphocytes and classic NK cell markers that link innate and adaptive immunity[94,95]. CD1d-restricted invariant NKT (iNKT) cells are a group of innate-like regulatory T cells, which play a central role in the regulation of the liver environment[96]. In addition to the direct killing of viral-infected cells without antigen-specific priming, NK cells regulate adaptive immune responses by producing interferon IFNγ, TNF-α and immunoregulatory cytokines[97]. The ability of NK cells to modulate T cell responses can be mediated through direct T-NK interactions, cytokine production, or indirectly through DCs and other cell types. Early NK cell interactions with other immune cells can have long-lasting effects on the number and quality of memory T cells, as well as impacting the exhaustion of T cells during chronic infections[98]. Evidence supporting the role of NK cells in acute HBV infection is conflicting. One study demonstrated that the activation and cytokine-producing function of NK cells was impaired in acute HBV patients[69], while another study demonstrated that NK cell activation and the development of NK and NKT cell responses is earlier than that of HBV-specific T cells, which may contribute to limiting the spread of HBV and lead to the timely induction of adaptive responses[99].
It is becoming increasingly apparent that NK cells exert a detrimental effect on the host during chronic HBV infection. As reviewed by Schuch et al[100], NK cells regulate adaptive immune responses by exhaustion of HBV-specific CD8+ T cells, probably by producing IL-10 and TGF-β on activation[101-104], upregulation of tumor necrosis factor-related apoptosis-inducing ligand[105] and by diminishing APC function during persistent virus infection[106]. NK cell depletion can improve memory T cell formation[107] and control persistent infection[108]. The absence of the inhibitory receptor 2B4 on NK cells resulted in a reduced virus-specific CD8+ T cell response that led to prolonged viral persistence[109]. Human regulatory NK cells (NKreg), which are a subgroup of NK cells, have been shown to produce IL-10 and reduce the proliferation of antigen-specific CD4+ T cells in vitro[110]. NKreg cells can also limit virus-specific CD8+ T cell immunity and promote chronic virus infection or immune pathology[92]. Furthermore, in a mouse model of acute infection, NK cells have also been shown to inhibit the generation of virus-specific memory T- and B-cells as well as virus-specific antibody production in a perforin-dependent manner[111]. iNKT cells play a central role in the regulation of the liver environment. Upon activation, iNKT cells secret large amounts of both Th1 and Th2 cytokines and play key regulatory roles in antimicrobial immunity[96]. One report showed that the number and cytokine-producing function of iNKT cells were comparable in CHB patients and healthy controls, while another study showed that iNKT cell frequency decreased with disease progression in CHB patients[112]. When activated by the ligand, alpha-galactosylceramide (alpha-GalCer), Vα14-positive NKT cells strongly enhance the induction and proliferation of HBsAg-specific CTLs in mouse models and promote the disruption of tolerance to HBV-specific CD8+ T cell antigens[113].
ADAPTIVE IMMUNE CELLS
B cells and T cells
Anti-HBs antibodies play an important role in the clearance of HBV particles in the blood and protection against reinfection of hepatocytes[114]. Memory B cell responses are indicative of a resolved previous infection[115] because the appearance of anti-HBs antibodies occurs relatively late after HBV exposure, and are usually absent in the clinical symptomatic phase of infection as well as in the chronic stage. The role of anti-HBs-positive B cells in the resolution or the pathogenesis of infection has been underestimated. In addition to anti-HBs production, B cells can act as APCs for antiviral CD4+ T cells[116]. A number of studies have yielded contradictory findings. Xu et al[117] suggested that expression of CD80, serum HBs antibody levels and the frequency of HBsAg-specific B cells were significantly decreased in CHB patients compared with healthy control subjects. In contrast, another study showed that there were no differences in the frequencies of B-lymphocytes expressing CD80 and CD86 between CHB patients and healthy controls[118]. Some data indicated that interactions between B and T cells may contribute to immunotolerance in mouse models with B cells as predominant APCs[119]. Sustained exposure to viral antigens can lead to an increase in the frequency of B cells with an exhausted phenotype in the liver[120] as well as the induction of negative costimulatory molecules, such as PD-1 and CTLA-4[121]. In contrast, some evidence demonstrates that an overwhelming B cell response plays a key role in HBV-associated acute liver failure[122,123]. Therefore, the function of B cells in HBV infection requires further investigation.
CD4+ T cells and CD8+ T cells
Evidence of the role of CD4+ T cells in the control of HBV infection is conflicting. Some data show that, similar to CD8+ T cells, the CD4+ T cell response in the acute phase of self-limiting infection is significantly greater and multi-specific than in the chronic phase. Furthermore, the induction of functional HBV-specific CD8+ T cell responses is dependent on early CD4+ T cell priming prior to HBV spread[114]. While CD4+ T cell depletion at the peak of HBV infection had no effect on viral replication in infected chimpanzees[124], depletion prior to HBV infection resulted in quantitatively and functionally impaired HBV-specific CD8+ T cell responses[125]. In the absence of early CD4+ T cell responses, intrahepatic CD8+ T cell priming results in T cell inactivation, tolerance or apoptosis[126,127]. Functional impairment of T cells may also contribute to hyperactivation of regulatory CD4+ FoxP3+ T cells that suppress virus-specific T cells, thereby affecting the quality and intensity of antiviral responses. In CHB patients, the frequency of circulating CD4+CD25+ Treg cells correlates significantly with serum viral load and liver injury[128]. Th17 cells, another CD4+ T cell subset, may contribute to disease progression and the pathogenesis of liver injury in HBV-infected patients. An increased Treg/Th17 ratio and the Th17 frequency at onset have significant predictive value for survival of patients with HBV-related acute-on-chronic liver failure[129,130].
LIVER CELLS AND ADAPTIVE IMMUNE
Liver cells include NPCs and hepatocytes. Under normal conditions, resident liver LSECs and KCs secrete IL-10 and TGF-β, maintaining a tolerogenic environment and restraining inflammatory responses to foreign antigens, such as HBV[67,131]. LSECs, as one type of local APC, are capable of antigen cross-presentation and subsequent tolerization of naive CD8+ T cells. Under certain conditions, LSECs can switch from a tolerogenic to an immunogenic state and promote the development of T cell immunity[131]. As in the setting of acute HBV infection, liver cells might be able to sense HBV infection and mount antiviral effects via an IFN response[132]. Furthermore, LSEC-mediated cross-presentation of soluble, circulating or hepatocyte-derived antigens to naïve CD8+ T cells results in the development of antigen-experienced memory-like T cells[133]. LSECs and KCs can reduce TLR expression leading to the inactivation of innate immunity[134]. LSECs are the major liver cell type responsible for the induction of TGF-β-dependent hepatic CD4+ CD25+ Foxp3+ Treg cells, which contribute to the tolerogenic features of the intrahepatic microenvironment[135,136]. In contrast to activated professional APCs, intrahepatic antigen presentation by HBV-positive hepatocytes suppresses HBV-specific CD8+ T cell responses or mediates T cell apoptosis via the PD-1/ PD-L1 pathway[137]. This may in part, explain the development of the tolerogenic hepatic microenvironment and the occurrence of persistent HBV infection in the liver. Thus, precise quantitative and qualitative regulation of CD4+ T responses is required to initiate the activation of CD8+ T cells to control the infection.
CONCLUSION
The innate immune system is the first line of host defense against infection immediately after the pathogen invasion. Its functions depend largely on multiple regulators, including TLRs and cytokines, mainly type I IFN and subsequent activation of adaptive immune responses. Initiation of effective adaptive immune responses, especially HBV-specific CD8+ T cell responses, is central to the control of HBV infection. Efficient clearance of viral infections requires the synergistic interaction of both innate and adaptive immune responses, which is vital for the development of long-lasting immunity. A better understanding of these complex interactions and their role in HBV infection is essential for designing effective immunotherapeutic regimens for CHB and designing new combination treatment strategies for the eradication HBV[138].
Footnotes
P- Reviewer: Ito H, Rodriguez-Frias F, Tai DI, Vaughan G S- Editor: Wang JL L- Editor: A E- Editor: Liu SQ
Supported by Grants of Yantai Science and Technology Plan Project, No. 2012116.
Conflict-of-interest statement: All the authors of the manuscript declare that they have no conflict of interest in connection with this paper
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: September 26, 2015
First decision: October 21, 2015
Article in press: December 11, 2015
References
- 1.Trépo C, Chan HL, Lok A. Hepatitis B virus infection. Lancet. 2014;384:2053–2063. doi: 10.1016/S0140-6736(14)60220-8. [DOI] [PubMed] [Google Scholar]
- 2.Lai CL, Ratziu V, Yuen MF, Poynard T. Viral hepatitis B. Lancet. 2003;362:2089–2094. doi: 10.1016/S0140-6736(03)15108-2. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, Yuan Z. Interplay between hepatitis B virus and the innate immune responses: implications for new therapeutic strategies. Virol Sin. 2014;29:17–24. doi: 10.1007/s12250-014-3412-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang FS, Zhang Z. Host immunity influences disease progression and antiviral efficacy in humans infected with hepatitis B virus. Expert Rev Gastroenterol Hepatol. 2009;3:499–512. doi: 10.1586/egh.09.50. [DOI] [PubMed] [Google Scholar]
- 5.Yang PL, Althage A, Chung J, Maier H, Wieland S, Isogawa M, Chisari FV. Immune effectors required for hepatitis B virus clearance. Proc Natl Acad Sci USA. 2010;107:798–802. doi: 10.1073/pnas.0913498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertoletti A, Tan AT, Gehring AJ. HBV-Specific Adaptive Immunity. Viruses. 2009;1:91–103. doi: 10.3390/v1020091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kondo Y, Kobayashi K, Asabe S, Shiina M, Niitsuma H, Ueno Y, Kobayashi T, Shimosegawa T. Vigorous response of cytotoxic T lymphocytes associated with systemic activation of CD8 T lymphocytes in fulminant hepatitis B. Liver Int. 2004;24:561–567. doi: 10.1111/j.1478-3231.2004.0982.x. [DOI] [PubMed] [Google Scholar]
- 8.Busca A, Kumar A. Innate immune responses in hepatitis B virus (HBV) infection. Virol J. 2014;11:22. doi: 10.1186/1743-422X-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarkar N, Panigrahi R, Pal A, Biswas A, Singh SP, Kar SK, Bandopadhyay M, Das D, Saha D, Kanda T, et al. Expression of microRNA-155 correlates positively with the expression of Toll-like receptor 7 and modulates hepatitis B virus via C/EBP-β in hepatocytes. J Viral Hepat. 2015;22:817–827. doi: 10.1111/jvh.12390. [DOI] [PubMed] [Google Scholar]
- 10.Wu JF, Chen CH, Ni YH, Lin YT, Chen HL, Hsu HY, Chang MH. Toll-like receptor and hepatitis B virus clearance in chronic infected patients: a long-term prospective cohort study in Taiwan. J Infect Dis. 2012;206:662–668. doi: 10.1093/infdis/jis420. [DOI] [PubMed] [Google Scholar]
- 11.Wu JF, Hsu HY, Chiu YC, Chen HL, Ni YH, Chang MH. The effects of cytokines on spontaneous hepatitis B surface antigen seroconversion in chronic hepatitis B virus infection. J Immunol. 2015;194:690–696. doi: 10.4049/jimmunol.1401659. [DOI] [PubMed] [Google Scholar]
- 12.Kondo Y, Ueno Y, Shimosegawa T. Toll-like receptors signaling contributes to immunopathogenesis of HBV infection. Gastroenterol Res Pract. 2011;2011:810939. doi: 10.1155/2011/810939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 14.Ma Z, Zhang E, Yang D, Lu M. Contribution of Toll-like receptors to the control of hepatitis B virus infection by initiating antiviral innate responses and promoting specific adaptive immune responses. Cell Mol Immunol. 2015;12:273–282. doi: 10.1038/cmi.2014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gay NJ, Gangloff M. Structure and function of Toll receptors and their ligands. Annu Rev Biochem. 2007;76:141–165. doi: 10.1146/annurev.biochem.76.060305.151318. [DOI] [PubMed] [Google Scholar]
- 16.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 17.Miyake K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol. 2007;19:3–10. doi: 10.1016/j.smim.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 19.Zhang E, Lu M. Toll-like receptor (TLR)-mediated innate immune responses in the control of hepatitis B virus (HBV) infection. Med Microbiol Immunol. 2015;204:11–20. doi: 10.1007/s00430-014-0370-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol. 2005;79:7269–7272. doi: 10.1128/JVI.79.11.7269-7272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzgerald-Bocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008;19:3–19. doi: 10.1016/j.cytogfr.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu J, Meng Z, Jiang M, Zhang E, Trippler M, Broering R, Bucchi A, Krux F, Dittmer U, Yang D, et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology. 2010;129:363–374. doi: 10.1111/j.1365-2567.2009.03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sajadi SM, Mirzaei V, Hassanshahi G, Khorramdelazad H, Daredor HY, Hosseini SM, Moogooi M, Ravary A, Arababadi MK, Kennedy D. Decreased expressions of Toll-like receptor 9 and its signaling molecules in chronic hepatitis B virus-infected patients. Arch Pathol Lab Med. 2013;137:1674–1679. doi: 10.5858/arpa.2012-0415-OA. [DOI] [PubMed] [Google Scholar]
- 24.Momeni M, Zainodini N, Bidaki R, Hassanshahi G, Daneshvar H, Khaleghinia M, Ebrahim M, Karimi-Googheri M, Askari A, Arababadi MK, et al. Decreased expression of toll like receptor signaling molecules in chronic HBV infected patients. Hum Immunol. 2014;75:15–19. doi: 10.1016/j.humimm.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 25.Shahrakyvahed A, Sanchooli J, Sanadgol N, Arababadi MK, Kennedy D. TLR9: an important molecule in the fight against hepatitis B virus. Postgrad Med J. 2014;90:396–401. doi: 10.1136/postgradmedj-2013-132309. [DOI] [PubMed] [Google Scholar]
- 26.Xie Q, Shen HC, Jia NN, Wang H, Lin LY, An BY, Gui HL, Guo SM, Cai W, Yu H, et al. Patients with chronic hepatitis B infection display deficiency of plasmacytoid dendritic cells with reduced expression of TLR9. Microbes Infect. 2009;11:515–523. doi: 10.1016/j.micinf.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z, Cheng Y, Xu Y, Liao J, Zhang X, Hu Y, Zhang Q, Wang J, Zhang Z, Shen F, et al. Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin Immunol. 2008;128:400–408. doi: 10.1016/j.clim.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 28.Lu X, Xu Q, Bu X, Ma X, Zhang F, Deng Q, Zhang Y, Ding J. Relationship between expression of toll-like receptors 2/4 in dendritic cells and chronic hepatitis B virus infection. Int J Clin Exp Pathol. 2014;7:6048–6055. [PMC free article] [PubMed] [Google Scholar]
- 29.Adib-Conquy M, Scott-Algara D, Cavaillon JM, Souza-Fonseca-Guimaraes F. TLR-mediated activation of NK cells and their role in bacterial/viral immune responses in mammals. Immunol Cell Biol. 2014;92:256–262. doi: 10.1038/icb.2013.99. [DOI] [PubMed] [Google Scholar]
- 30.Hart OM, Athie-Morales V, O’Connor GM, Gardiner CM. TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J Immunol. 2005;175:1636–1642. doi: 10.4049/jimmunol.175.3.1636. [DOI] [PubMed] [Google Scholar]
- 31.Girart MV, Fuertes MB, Domaica CI, Rossi LE, Zwirner NW. Engagement of TLR3, TLR7, and NKG2D regulate IFN-gamma secretion but not NKG2D-mediated cytotoxicity by human NK cells stimulated with suboptimal doses of IL-12. J Immunol. 2007;179:3472–3479. doi: 10.4049/jimmunol.179.6.3472. [DOI] [PubMed] [Google Scholar]
- 32.Ratnam DT, Sievert W, Visvanathan K. Natural killer cells display impaired responses to toll like receptor 9 that support viral persistence in chronic hepatitis B. Cell Immunol. 2012;279:109–115. doi: 10.1016/j.cellimm.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, Momburg F, Arnold B, Knolle PA. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med. 2000;6:1348–1354. doi: 10.1038/82161. [DOI] [PubMed] [Google Scholar]
- 34.Kern M, Popov A, Scholz K, Schumak B, Djandji D, Limmer A, Eggle D, Sacher T, Zawatzky R, Holtappels R, et al. Virally infected mouse liver endothelial cells trigger CD8+ T-cell immunity. Gastroenterology. 2010;138:336–346. doi: 10.1053/j.gastro.2009.08.057. [DOI] [PubMed] [Google Scholar]
- 35.Liu J, Jiang M, Ma Z, Dietze KK, Zelinskyy G, Yang D, Dittmer U, Schlaak JF, Roggendorf M, Lu M. TLR1/2 ligand-stimulated mouse liver endothelial cells secrete IL-12 and trigger CD8+ T cell immunity in vitro. J Immunol. 2013;191:6178–6190. doi: 10.4049/jimmunol.1301262. [DOI] [PubMed] [Google Scholar]
- 36.Wu J, Lu M, Meng Z, Trippler M, Broering R, Szczeponek A, Krux F, Dittmer U, Roggendorf M, Gerken G, et al. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology. 2007;46:1769–1778. doi: 10.1002/hep.21897. [DOI] [PubMed] [Google Scholar]
- 37.Jiang M, Broering R, Trippler M, Poggenpohl L, Fiedler M, Gerken G, Lu M, Schlaak JF. Toll-like receptor-mediated immune responses are attenuated in the presence of high levels of hepatitis B virus surface antigen. J Viral Hepat. 2014;21:860–872. doi: 10.1111/jvh.12216. [DOI] [PubMed] [Google Scholar]
- 38.Reynolds JM, Dong C. Toll-like receptor regulation of effector T lymphocyte function. Trends Immunol. 2013;34:511–519. doi: 10.1016/j.it.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Geng D, Zheng L, Srivastava R, Asprodites N, Velasco-Gonzalez C, Davila E. When Toll-like receptor and T-cell receptor signals collide: a mechanism for enhanced CD8 T-cell effector function. Blood. 2010;116:3494–3504. doi: 10.1182/blood-2010-02-268169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabiasco J, Devêvre E, Rufer N, Salaun B, Cerottini JC, Speiser D, Romero P. Human effector CD8+ T lymphocytes express TLR3 as a functional coreceptor. J Immunol. 2006;177:8708–8713. doi: 10.4049/jimmunol.177.12.8708. [DOI] [PubMed] [Google Scholar]
- 41.Babu S, Blauvelt CP, Kumaraswami V, Nutman TB. Cutting edge: diminished T cell TLR expression and function modulates the immune response in human filarial infection. J Immunol. 2006;176:3885–3889. doi: 10.4049/jimmunol.176.7.3885. [DOI] [PubMed] [Google Scholar]
- 42.Mandraju R, Murray S, Forman J, Pasare C. Differential ability of surface and endosomal TLRs to induce CD8 T cell responses in vivo. J Immunol. 2014;192:4303–4315. doi: 10.4049/jimmunol.1302244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crampton SP, Voynova E, Bolland S. Innate pathways to B-cell activation and tolerance. Ann N Y Acad Sci. 2010;1183:58–68. doi: 10.1111/j.1749-6632.2009.05123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hua Z, Hou B. TLR signaling in B-cell development and activation. Cell Mol Immunol. 2013;10:103–106. doi: 10.1038/cmi.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jain S, Chodisetti SB, Agrewala JN. Combinatorial signaling through TLR-2 and CD86 augments activation and differentiation of resting B cells. PLoS One. 2013;8:e54392. doi: 10.1371/journal.pone.0054392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jain S, Chodisetti SB, Agrewala JN. CD40 signaling synergizes with TLR-2 in the BCR independent activation of resting B cells. PLoS One. 2011;6:e20651. doi: 10.1371/journal.pone.0020651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin FC, Young HA. Interferons: Success in anti-viral immunotherapy. Cytokine Growth Factor Rev. 2014;25:369–376. doi: 10.1016/j.cytogfr.2014.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pei RJ, Chen XW, Lu MJ. Control of hepatitis B virus replication by interferons and Toll-like receptor signaling pathways. World J Gastroenterol. 2014;20:11618–11629. doi: 10.3748/wjg.v20.i33.11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guidotti LG, Morris A, Mendez H, Koch R, Silverman RH, Williams BR, Chisari FV. Interferon-regulated pathways that control hepatitis B virus replication in transgenic mice. J Virol. 2002;76:2617–2621. doi: 10.1128/JVI.76.6.2617-2621.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, Petersen J, Raimondo G, Dandri M, Levrero M. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest. 2012;122:529–537. doi: 10.1172/JCI58847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809, table of contents. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katze MG, He Y, Gale M. Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 53.Phillips S, Chokshi S, Riva A, Evans A, Williams R, Naoumov NV. CD8(+) T cell control of hepatitis B virus replication: direct comparison between cytolytic and noncytolytic functions. J Immunol. 2010;184:287–295. doi: 10.4049/jimmunol.0902761. [DOI] [PubMed] [Google Scholar]
- 54.Lan P, Zhang C, Han Q, Zhang J, Tian Z. Therapeutic recovery of hepatitis B virus (HBV)-induced hepatocyte-intrinsic immune defect reverses systemic adaptive immune tolerance. Hepatology. 2013;58:73–85. doi: 10.1002/hep.26339. [DOI] [PubMed] [Google Scholar]
- 55.Bertoletti A, Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Postgrad Med J. 2013;89:294–304. doi: 10.1136/postgradmedj-2011-301073rep. [DOI] [PubMed] [Google Scholar]
- 56.Tzeng HT, Tsai HF, Chyuan IT, Liao HJ, Chen CJ, Chen PJ, Hsu PN. Tumor necrosis factor-alpha induced by hepatitis B virus core mediating the immune response for hepatitis B viral clearance in mice model. PLoS One. 2014;9:e103008. doi: 10.1371/journal.pone.0103008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chyuan IT, Tsai HF, Tzeng HT, Sung CC, Wu CS, Chen PJ, Hsu PN. Tumor necrosis factor-alpha blockage therapy impairs hepatitis B viral clearance and enhances T-cell exhaustion in a mouse model. Cell Mol Immunol. 2015;12:317–325. doi: 10.1038/cmi.2015.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bouezzedine F, Fardel O, Gripon P. Interleukin 6 inhibits HBV entry through NTCP down regulation. Virology. 2015;481:34–42. doi: 10.1016/j.virol.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 59.Hösel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M, Tedjokusumo R, Esser K, Arzberger S, Kirschning CJ, et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology. 2009;50:1773–1782. doi: 10.1002/hep.23226. [DOI] [PubMed] [Google Scholar]
- 60.Jegaskanda S, Ahn SH, Skinner N, Thompson AJ, Ngyuen T, Holmes J, De Rose R, Navis M, Winnall WR, Kramski M, et al. Downregulation of interleukin-18-mediated cell signaling and interferon gamma expression by the hepatitis B virus e antigen. J Virol. 2014;88:10412–10420. doi: 10.1128/JVI.00111-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Motavaf M, Safari S, Alavian SM. Interleukin 18 gene promoter polymorphisms and susceptibility to chronic hepatitis B infection: a review study. Hepat Mon. 2014;14:e19879. doi: 10.5812/hepatmon.19879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karra VK, Gumma PK, Chowdhury SJ, Ruttala R, Polipalli SK, Chakravarti A, Kar P. IL-18 polymorphisms in hepatitis B virus related liver disease. Cytokine. 2015;73:277–282. doi: 10.1016/j.cyto.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y, Cobleigh MA, Lian JQ, Huang CX, Booth CJ, Bai XF, Robek MD. A proinflammatory role for interleukin-22 in the immune response to hepatitis B virus. Gastroenterology. 2011;141:1897–1906. doi: 10.1053/j.gastro.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schon HT, Weiskirchen R. Immunomodulatory effects of transforming growth factor-β in the liver. Hepatobiliary Surg Nutr. 2014;3:386–406. doi: 10.3978/j.issn.2304-3881.2014.11.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chou YC, Chen ML, Hu CP, Chen YL, Chong CL, Tsai YL, Liu TL, Jeng KS, Chang C. Transforming growth factor-beta1 suppresses hepatitis B virus replication primarily through transcriptional inhibition of pregenomic RNA. Hepatology. 2007;46:672–681. doi: 10.1002/hep.21726. [DOI] [PubMed] [Google Scholar]
- 66.Xu L, Yin W, Sun R, Wei H, Tian Z. Kupffer cell-derived IL-10 plays a key role in maintaining humoral immune tolerance in hepatitis B virus-persistent mice. Hepatology. 2014;59:443–452. doi: 10.1002/hep.26668. [DOI] [PubMed] [Google Scholar]
- 67.Dunn C, Peppa D, Khanna P, Nebbia G, Jones M, Brendish N, Lascar RM, Brown D, Gilson RJ, Tedder RJ, et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology. 2009;137:1289–1300. doi: 10.1053/j.gastro.2009.06.054. [DOI] [PubMed] [Google Scholar]
- 68.Gong Y, Zhao C, Zhao P, Wang M, Zhou G, Han F, Cui Y, Qian J, Zhang H, Xiong H, et al. Role of IL-10-Producing Regulatory B Cells in Chronic Hepatitis B Virus Infection. Dig Dis Sci. 2015;60:1308–1314. doi: 10.1007/s10620-014-3358-1. [DOI] [PubMed] [Google Scholar]
- 69.Li Y, Tang L, Hou J. Role of interleukin-21 in HBV infection: friend or foe? Cell Mol Immunol. 2015;12:303–308. doi: 10.1038/cmi.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li HJ, Kang FB, Li BS, Yang XY, Zhang YG, Sun DX. Interleukin-21 inhibits HBV replication in vitro. Antivir Ther. 2015;20:583–590. doi: 10.3851/IMP2950. [DOI] [PubMed] [Google Scholar]
- 71.Xiang XG, Xie Q. IL-35: a potential therapeutic target for controlling hepatitis B virus infection. J Dig Dis. 2015;16:1–6. doi: 10.1111/1751-2980.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li X, Tian L, Dong Y, Zhu Q, Wang Y, Han W, Liu X, Ni Q, Chen Y, Li L. IL-35 inhibits HBV antigen-specific IFN-γ-producing CTLs in vitro. Clin Sci (Lond) 2015;129:395–404. doi: 10.1042/CS20140511. [DOI] [PubMed] [Google Scholar]
- 73.Zhou Y, Zhang H, Li Y. IL-35 expression in peripheral blood CD4(+) T cells from chronic hepatitis B virus-infected patients directly correlates with virus load. Cytokine. 2015;73:169–175. doi: 10.1016/j.cyto.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 74.Tavakoli S, Mederacke I, Herzog-Hauff S, Glebe D, Grün S, Strand D, Urban S, Gehring A, Galle PR, Böcher WO. Peripheral blood dendritic cells are phenotypically and functionally intact in chronic hepatitis B virus (HBV) infection. Clin Exp Immunol. 2008;151:61–70. doi: 10.1111/j.1365-2249.2007.03547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jan RH, Lin YL, Chen CJ, Lin TY, Hsu YC, Chen LK, Chiang BL. Hepatitis B virus surface antigen can activate human monocyte-derived dendritic cells by nuclear factor kappa B and p38 mitogen-activated protein kinase mediated signaling. Microbiol Immunol. 2012;56:719–727. doi: 10.1111/j.1348-0421.2012.00496.x. [DOI] [PubMed] [Google Scholar]
- 76.Wang FS, Xing LH, Liu MX, Zhu CL, Liu HG, Wang HF, Lei ZY. Dysfunction of peripheral blood dendritic cells from patients with chronic hepatitis B virus infection. World J Gastroenterol. 2001;7:537–541. doi: 10.3748/wjg.v7.i4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gehring AJ, Ann D’Angelo J. Dissecting the dendritic cell controversy in chronic hepatitis B virus infection. Cell Mol Immunol. 2015;12:283–291. doi: 10.1038/cmi.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin C, Zou H, Wang S. Hepatitis B e Antigen Seroconversion Is Related with the Function of Dendritic Cells in Chronic Hepatitis B Virus Infection. Gastroenterol Res Pract. 2014;2014:413952. doi: 10.1155/2014/413952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gerosa F, Gobbi A, Zorzi P, Burg S, Briere F, Carra G, Trinchieri G. The reciprocal interaction of NK cells with plasmacytoid or myeloid dendritic cells profoundly affects innate resistance functions. J Immunol. 2005;174:727–734. doi: 10.4049/jimmunol.174.2.727. [DOI] [PubMed] [Google Scholar]
- 80.Gerosa F, Baldani-Guerra B, Nisii C, Marchesini V, Carra G, Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med. 2002;195:327–333. doi: 10.1084/jem.20010938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Degli-Esposti MA, Smyth MJ. Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol. 2005;5:112–124. doi: 10.1038/nri1549. [DOI] [PubMed] [Google Scholar]
- 82.Wehner R, Dietze K, Bachmann M, Schmitz M. The bidirectional crosstalk between human dendritic cells and natural killer cells. J Innate Immun. 2011;3:258–263. doi: 10.1159/000323923. [DOI] [PubMed] [Google Scholar]
- 83.Tjwa ET, van Oord GW, Biesta PJ, Boonstra A, Janssen HL, Woltman AM. Restoration of TLR3-activated myeloid dendritic cell activity leads to improved natural killer cell function in chronic hepatitis B virus infection. J Virol. 2012;86:4102–4109. doi: 10.1128/JVI.07000-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maini MK, Boni C, Lee CK, Larrubia JR, Reignat S, Ogg GS, King AS, Herberg J, Gilson R, Alisa A, et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med. 2000;191:1269–1280. doi: 10.1084/jem.191.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Apostolopoulos V, Thalhammer T, Tzakos AG, Stojanovska L. Targeting antigens to dendritic cell receptors for vaccine development. J Drug Deliv. 2013;2013:869718. doi: 10.1155/2013/869718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Op den Brouw ML, Binda RS, van Roosmalen MH, Protzer U, Janssen HL, van der Molen RG, Woltman AM. Hepatitis B virus surface antigen impairs myeloid dendritic cell function: a possible immune escape mechanism of hepatitis B virus. Immunology. 2009;126:280–289. doi: 10.1111/j.1365-2567.2008.02896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen W, Shi M, Shi F, Mao Y, Tang Z, Zhang B, Zhang H, Chen L, Chen L, Xin S, et al. HBcAg-pulsed dendritic cell vaccine induces Th1 polarization and production of hepatitis B virus-specific cytotoxic T lymphocytes. Hepatol Res. 2009;39:355–365. doi: 10.1111/j.1872-034X.2008.00468.x. [DOI] [PubMed] [Google Scholar]
- 88.Chen W, Zhang Z, Shi M, Chen L, Fu J, Shi F, Zhang B, Zhang H, Jin L, Wang FS. Activated plasmacytoid dendritic cells act synergistically with hepatitis B core antigen-pulsed monocyte-derived dendritic cells in the induction of hepatitis B virus-specific CD8 T-cell response. Clin Immunol. 2008;129:295–303. doi: 10.1016/j.clim.2008.07.026. [DOI] [PubMed] [Google Scholar]
- 89.Krueger PD, Kim TS, Sung SS, Braciale TJ, Hahn YS. Liver-resident CD103+ dendritic cells prime antiviral CD8+ T cells in situ. J Immunol. 2015;194:3213–3222. doi: 10.4049/jimmunol.1402622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peng G, Li S, Wu W, Sun Z, Chen Y, Chen Z. Circulating CD4+ CD25+ regulatory T cells correlate with chronic hepatitis B infection. Immunology. 2008;123:57–65. doi: 10.1111/j.1365-2567.2007.02691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yamazaki S, Inaba K, Tarbell KV, Steinman RM. Dendritic cells expand antigen-specific Foxp3+ CD25+ CD4+ regulatory T cells including suppressors of alloreactivity. Immunol Rev. 2006;212:314–329. doi: 10.1111/j.0105-2896.2006.00422.x. [DOI] [PubMed] [Google Scholar]
- 92.Lang PA, Lang KS, Xu HC, Grusdat M, Parish IA, Recher M, Elford AR, Dhanji S, Shaabani N, Tran CW, et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc Natl Acad Sci USA. 2012;109:1210–1215. doi: 10.1073/pnas.1118834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sitia G, Isogawa M, Iannacone M, Campbell IL, Chisari FV, Guidotti LG. MMPs are required for recruitment of antigen-nonspecific mononuclear cells into the liver by CTLs. J Clin Invest. 2004;113:1158–1167. doi: 10.1172/JCI21087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, Laccabue D, Zerbini A, Cavalli A, Missale G, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. 2007;81:4215–4225. doi: 10.1128/JVI.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Villanueva AI, Haeryfar SM, Mallard BA, Kulkarni RR, Sharif S. Functions of invariant NK T cells are modulated by TLR ligands and IFN-α. Innate Immun. 2015;21:275–288. doi: 10.1177/1753425914527327. [DOI] [PubMed] [Google Scholar]
- 96.Lawrenczyk A, Kim S, Wen X, Xiong R, Yuan W. Exploring the Therapeutic Potentials of iNKT Cells for Anti-HBV Treatment. Pathogens. 2014;3:563–576. doi: 10.3390/pathogens3030563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pollicino T, Koumbi L. Role natural killer group 2D-ligand interactions in hepatitis B infection. World J Hepatol. 2015;7:819–824. doi: 10.4254/wjh.v7.i6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cook KD, Waggoner SN, Whitmire JK. NK cells and their ability to modulate T cells during virus infections. Crit Rev Immunol. 2014;34:359–388. doi: 10.1615/critrevimmunol.2014010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fisicaro P, Valdatta C, Boni C, Massari M, Mori C, Zerbini A, Orlandini A, Sacchelli L, Missale G, Ferrari C. Early kinetics of innate and adaptive immune responses during hepatitis B virus infection. Gut. 2009;58:974–982. doi: 10.1136/gut.2008.163600. [DOI] [PubMed] [Google Scholar]
- 100.Schuch A, Hoh A, Thimme R. The role of natural killer cells and CD8(+) T cells in hepatitis B virus infection. Front Immunol. 2014;5:258. doi: 10.3389/fimmu.2014.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, Biron CA. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J Exp Med. 2009;206:2235–2251. doi: 10.1084/jem.20082387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gray JD, Hirokawa M, Ohtsuka K, Horwitz DA. Generation of an inhibitory circuit involving CD8+ T cells, IL-2, and NK cell-derived TGF-beta: contrasting effects of anti-CD2 and anti-CD3. J Immunol. 1998;160:2248–2254. [PubMed] [Google Scholar]
- 104.Conroy MJ, Mac Nicholas R, Grealy R, Taylor M, Otegbayo JA, O’Dea S, Mulcahy F, Ryan T, Norris S, Doherty DG. Circulating CD56dim natural killer cells and CD56+ T cells that produce interferon-γ or interleukin-10 are expanded in asymptomatic, E antigen-negative patients with persistent hepatitis B virus infection. J Viral Hepat. 2015;22:335–345. doi: 10.1111/jvh.12299. [DOI] [PubMed] [Google Scholar]
- 105.Peppa D, Gill US, Reynolds G, Easom NJ, Pallett LJ, Schurich A, Micco L, Nebbia G, Singh HD, Adams DH, Kennedy PT, Maini MK. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell-mediated deletion. J Exp Med. 2013;210:99–114. doi: 10.1084/jem.20121172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cook KD, Whitmire JK. The depletion of NK cells prevents T cell exhaustion to efficiently control disseminating virus infection. J Immunol. 2013;190:641–649. doi: 10.4049/jimmunol.1202448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Soderquest K, Walzer T, Zafirova B, Klavinskis LS, Polić B, Vivier E, Lord GM, Martín-Fontecha A. Cutting edge: CD8+ T cell priming in the absence of NK cells leads to enhanced memory responses. J Immunol. 2011;186:3304–3308. doi: 10.4049/jimmunol.1004122. [DOI] [PubMed] [Google Scholar]
- 108.Waggoner SN, Daniels KA, Welsh RM. Therapeutic depletion of natural killer cells controls persistent infection. J Virol. 2014;88:1953–1960. doi: 10.1128/JVI.03002-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sun C, Fu B, Gao Y, Liao X, Sun R, Tian Z, Wei H. TGF-β1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PLoS Pathog. 2012;8:e1002594. doi: 10.1371/journal.ppat.1002594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Deniz G, Erten G, Kücüksezer UC, Kocacik D, Karagiannidis C, Aktas E, Akdis CA, Akdis M. Regulatory NK cells suppress antigen-specific T cell responses. J Immunol. 2008;180:850–857. doi: 10.4049/jimmunol.180.2.850. [DOI] [PubMed] [Google Scholar]
- 111.Rydyznski C, Daniels KA, Karmele EP, Brooks TR, Mahl SE, Moran MT, Li C, Sutiwisesak R, Welsh RM, Waggoner SN. Generation of cellular immune memory and B-cell immunity is impaired by natural killer cells. Nat Commun. 2015;6:6375. doi: 10.1038/ncomms7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhu H, Zhang Y, Liu H, Zhang Y, Kang Y, Mao R, Yang F, Zhou D, Zhang J. Preserved Function of Circulating Invariant Natural Killer T Cells in Patients With Chronic Hepatitis B Virus Infection. Medicine (Baltimore) 2015;94:e961. doi: 10.1097/MD.0000000000000961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ito H, Ando K, Ishikawa T, Nakayama T, Taniguchi M, Saito K, Imawari M, Moriwaki H, Yokochi T, Kakumu S, et al. Role of Valpha14+ NKT cells in the development of Hepatitis B virus-specific CTL: activation of Valpha14+ NKT cells promotes the breakage of CTL tolerance. Int Immunol. 2008;20:869–879. doi: 10.1093/intimm/dxn046. [DOI] [PubMed] [Google Scholar]
- 114.Isogawa M, Tanaka Y. Immunobiology of hepatitis B virus infection. Hepatol Res. 2015;45:179–189. doi: 10.1111/hepr.12439. [DOI] [PubMed] [Google Scholar]
- 115.Wang Q, Sachse P, Semmo M, Lokhande M, Montani M, Dufour JF, Zoulim F, Klenerman P, Semmo N. T- and B-cell responses and previous exposure to hepatitis B virus in ‘anti-HBc alone’ patients. J Viral Hepat. 2015;22:1068–1078. doi: 10.1111/jvh.12428. [DOI] [PubMed] [Google Scholar]
- 116.Mitchison NA. T-cell-B-cell cooperation. Nat Rev Immunol. 2004;4:308–312. doi: 10.1038/nri1334. [DOI] [PubMed] [Google Scholar]
- 117.Xu X, Shang Q, Chen X, Nie W, Zou Z, Huang A, Meng M, Jin L, Xu R, Zhang JY, et al. Reversal of B-cell hyperactivation and functional impairment is associated with HBsAg seroconversion in chronic hepatitis B patients. Cell Mol Immunol. 2015;12:309–316. doi: 10.1038/cmi.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wongjitrat C, Sukwit S, Chuenchitra T, Seangjaruk P, Rojanasang P, Romputtan P, Srisurapanon S. CTLA-4 and its ligands on the surface of T- and B-lymphocyte subsets in chronic hepatitis B virus infection. J Med Assoc Thai. 2013;96 Suppl 1:S54–S59. [PubMed] [Google Scholar]
- 119.Raimondi G, Zanoni I, Citterio S, Ricciardi-Castagnoli P, Granucci F. Induction of peripheral T cell tolerance by antigen-presenting B cells. II. Chronic antigen presentation overrules antigen-presenting B cell activation. J Immunol. 2006;176:4021–4028. doi: 10.4049/jimmunol.176.7.4021. [DOI] [PubMed] [Google Scholar]
- 120.Mohamadkhani A, Naderi E, Sotoudeh M, Katoonizadeh A, Montazeri G, Poustchi H. Clinical feature of intrahepatic B-lymphocytes in chronic hepatitis B. Int J Inflam. 2014;2014:896864. doi: 10.1155/2014/896864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Frommer F, Heinen TJ, Wunderlich FT, Yogev N, Buch T, Roers A, Bettelli E, Müller W, Anderton SM, Waisman A. Tolerance without clonal expansion: self-antigen-expressing B cells program self-reactive T cells for future deletion. J Immunol. 2008;181:5748–5759. doi: 10.4049/jimmunol.181.8.5748. [DOI] [PubMed] [Google Scholar]
- 122.Farci P, Diaz G, Chen Z, Govindarajan S, Tice A, Agulto L, Pittaluga S, Boon D, Yu C, Engle RE, et al. B cell gene signature with massive intrahepatic production of antibodies to hepatitis B core antigen in hepatitis B virus-associated acute liver failure. Proc Natl Acad Sci USA. 2010;107:8766–8771. doi: 10.1073/pnas.1003854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dao DY, Hynan LS, Yuan HJ, Sanders C, Balko J, Attar N, Lok AS, Word RA, Lee WM. Two distinct subtypes of hepatitis B virus-related acute liver failure are separable by quantitative serum immunoglobulin M anti-hepatitis B core antibody and hepatitis B virus DNA levels. Hepatology. 2012;55:676–684. doi: 10.1002/hep.24732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003;77:68–76. doi: 10.1128/JVI.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Asabe S, Wieland SF, Chattopadhyay PK, Roederer M, Engle RE, Purcell RH, Chisari FV. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J Virol. 2009;83:9652–9662. doi: 10.1128/JVI.00867-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Isogawa M, Chung J, Murata Y, Kakimi K, Chisari FV. CD40 activation rescues antiviral CD8+ T cells from PD-1-mediated exhaustion. PLoS Pathog. 2013;9:e1003490. doi: 10.1371/journal.ppat.1003490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bertolino P, Bowen DG, McCaughan GW, Fazekas de St Groth B. Antigen-specific primary activation of CD8+ T cells within the liver. J Immunol. 2001;166:5430–5438. doi: 10.4049/jimmunol.166.9.5430. [DOI] [PubMed] [Google Scholar]
- 128.Xu D, Fu J, Jin L, Zhang H, Zhou C, Zou Z, Zhao JM, Zhang B, Shi M, Ding X, et al. Circulating and liver resident CD4+CD25+ regulatory T cells actively influence the antiviral immune response and disease progression in patients with hepatitis B. J Immunol. 2006;177:739–747. doi: 10.4049/jimmunol.177.1.739. [DOI] [PubMed] [Google Scholar]
- 129.Xue-Song L, Cheng-Zhong L, Ying Z, Mo-Bin W. Changes of Treg and Th17 cells balance in the development of acute and chronic hepatitis B virus infection. BMC Gastroenterol. 2012;12:43. doi: 10.1186/1471-230X-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liang XS, Li CZ, Zhou Y, Yin W, Liu YY, Fan WH. Changes in circulating Foxp3(+) regulatory T cells and interleukin-17-producing T helper cells during HBV-related acute-on-chronic liver failure. World J Gastroenterol. 2014;20:8558–8571. doi: 10.3748/wjg.v20.i26.8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.You Q, Cheng L, Kedl RM, Ju C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology. 2008;48:978–990. doi: 10.1002/hep.22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schurich A, Berg M, Stabenow D, Böttcher J, Kern M, Schild HJ, Kurts C, Schuette V, Burgdorf S, Diehl L, et al. Dynamic regulation of CD8 T cell tolerance induction by liver sinusoidal endothelial cells. J Immunol. 2010;184:4107–4114. doi: 10.4049/jimmunol.0902580. [DOI] [PubMed] [Google Scholar]
- 133.Ait-Goughoulte M, Lucifora J, Zoulim F, Durantel D. Innate antiviral immune responses to hepatitis B virus. Viruses. 2010;2:1394–1410. doi: 10.3390/v2071394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Knolle PA, Böttcher J, Huang LR. The role of hepatic immune regulation in systemic immunity to viral infection. Med Microbiol Immunol. 2015;204:21–27. doi: 10.1007/s00430-014-0371-0. [DOI] [PubMed] [Google Scholar]
- 135.Chang JJ, Lewin SR. Immunopathogenesis of hepatitis B virus infection. Immunol Cell Biol. 2007;85:16–23. doi: 10.1038/sj.icb.7100009. [DOI] [PubMed] [Google Scholar]
- 136.Carambia A, Freund B, Schwinge D, Heine M, Laschtowitz A, Huber S, Wraith DC, Korn T, Schramm C, Lohse AW, et al. TGF-β-dependent induction of CD4+CD25+Foxp3+ Tregs by liver sinusoidal endothelial cells. J Hepatol. 2014;61:594–599. doi: 10.1016/j.jhep.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 137.Mühlbauer M, Fleck M, Schütz C, Weiss T, Froh M, Blank C, Schölmerich J, Hellerbrand C. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J Hepatol. 2006;45:520–528. doi: 10.1016/j.jhep.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 138.Fazle Akbar SM, Al-Mahtab M, Hiasa Y. Designing immune therapy for chronic hepatitis B. J Clin Exp Hepatol. 2014;4:241–246. doi: 10.1016/j.jceh.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]