

Abstract
Knowledge of the genome-wide rate and spectrum of mutations is necessary to understand the origin of disease and the genetic variation driving all evolutionary processes. Here, we provide a genome-wide analysis of the rate and spectrum of mutations obtained in two Daphnia pulex genotypes via separate mutation-accumulation (MA) experiments. Unlike most MA studies that utilize haploid, homozygous, or self-fertilizing lines, D. pulex can be propagated ameiotically while maintaining a naturally heterozygous, diploid genome, allowing the capture of the full spectrum of genomic changes that arise in a heterozygous state. While base-substitution mutation rates are similar to those in other multicellular eukaryotes (about 4 × 10−9 per site per generation), we find that the rates of large-scale (>100 kb) de novo copy-number variants (CNVs) are significantly elevated relative to those seen in previous MA studies. The heterozygosity maintained in this experiment allowed for estimates of gene-conversion processes. While most of the conversion tract lengths we report are similar to those generated by meiotic processes, we also find larger tract lengths that are indicative of mitotic processes. Comparison of MA lines to natural isolates reveals that a majority of large-scale CNVs in natural populations are removed by purifying selection. The mutations observed here share similarities with disease-causing, complex, large-scale CNVs, thereby demonstrating that MA studies in D. pulex serve as a system for studying the processes leading to such alterations.
All aspects of evolutionary change ultimately depend on the rate of origin and molecular composition of mutations. An accurate characterization of the different types of mutations is therefore critical to our understanding of evolutionary rates, the origin of genetic disorders, and how signatures of natural selection are interpreted (Halligan and Keightley 2009). Mutation-accumulation (MA) experiments combined with whole-genome sequencing have been shown to provide a powerful tool for surveying genome-wide mutation patterns. In an MA experiment, repeated bottlenecking of initially genetically identical lines reduces the efficiency of selection to a minimum, thereby enabling the capture of nearly all de novo mutations, with the exception of the small minority of mutations that cause immediate lethality and/or sterility (Kibota and Lynch 1996).
Although this type of experiment has enhanced our understanding of the rate and spectrum of spontaneous mutational changes across the tree of life (Denver et al. 2004, 2009; Haag-Liautard et al. 2008; Lynch et al. 2008b; Lynch 2010b; Conrad et al. 2011; Ness et al. 2012; Sung et al. 2012a), previous MA studies are limited by an inability to characterize the de novo rates of large-scale insertions and deletions that have mechanisms of origin and repair distinct from base-substitution mutations (Hastings et al. 2009). Furthermore, most MA experiments in eukaryotes have been confined to either haploid asexuals or obligately sexual species maintained as homozygous lines by extreme inbreeding, conditions that might influence various aspects of genome stability. For example, mutation assays involving inbred sexual lines reduce or eliminate the possibility of capturing genomic changes that are extremely deleterious or lethal in the homozygous state. Inbred lines of normally outcrossing species may also exhibit unusual mutational features owing to the exposure of segregating recessive factors influencing replication fidelity (Lynch 2008, 2010a). Finally, although the mutational processes from an asexual, diploid, homozygous MA experiment have been investigated (Zhu et al. 2014), the lack of heterozygosity limited the ability to detect gene-conversion and/or internal genome homogenizing effects and the influence of these processes on genome composition (Nagylaki 1983; Lamb 1984).
One way to avoid these issues is to perform MA experiments with natural isolates of diploid species capable of asexual reproduction that can therefore maintain normal levels of heterozygosity. Lines propagated by single, clonally produced offspring allow recessive deleterious mutations to accumulate in a heterozygous background. To this end, we performed deep-coverage, genome-wide, next-generation sequencing on two long-term MA experiments using genotypes of the microcrustacean Daphnia pulex directly derived from natural environments and permanently harboring naturally occurring levels of heterozygosity. Most D. pulex alternate between bouts of sexual and asexual reproduction in nature (cyclical parthenogenic reproduction) but can still be maintained in culture in an exclusively clonal fashion (Hebert and Ward 1972; Lynch and Gabriel 1983). As a consequence of annual phases of sex, cyclical parthenogenic populations are generally in Hardy-Weinberg equilibrium like most other obligately sexual species (Lynch 1983).
A second unique aspect of D. pulex biology convenient for mutation analyses is that some natural isolates have completely lost the ability to reproduce sexually, hereafter referred to as obligate asexuals (Innes and Hebert 1988; Paland et al. 2005; Lynch et al. 2008a; Tucker et al. 2013). Because such genotypes accumulate mutations in nature in a perpetually clonal manner, a direct comparison between MA experiments and patterns of variation in natural asexual lineages has the potential to yield insight into the role of selection in either eliminating or promoting various classes of mutation (Tucker et al. 2013; So et al. 2015).
In this study, we provide joint, genome-wide estimates of the rates and spectra of base-substitution mutations, gene-conversion-like processes, and de novo copy-number variants (CNVs) for both cyclically parthenogenetic and obligately asexual D. pulex genotypes. By comparing the asexual MA experiment to patterns of polymorphism found in asexual populations in a previous study, we also give insight into how selection is operating on de novo CNVs.
Results
To estimate mutation rates in D. pulex, we performed deep-coverage, next-generation sequencing on two separate MA experiments (average sequencing coverage of 36×) (Supplemental Table S1), whereby successive genetic bottlenecks of isogenic clonal sublines were performed in a naturally occurring, heterozygous state: one experiment founded by an obligately asexual genotype from Linwood Lake, Ontario (ASEX), consisting of four sublines propagated for an average of 170 generations, and the other founded by a cyclically parthenogenetic genotype from Slimy Log Pond, Oregon (CYC), consisting of three sublines propagated for an average of 85 generations.
Base-substitution mutation
After filtering low coverage, repeat, and gap regions from the whole-genome alignments and applying strict criteria for base quality and alignment scores (Methods), we were able to screen 30.1% and 37.2% of the D. pulex genome for base-substitution mutations in the ASEX and CYC MA lines, respectively. We identified 733 base-substitution mutations, yielding genome-wide base-substitution mutation rates of 1.02 × 10−8 (SE = 0.80) per site per generation for ASEX and 4.33 × 10−9 (SE = 0.34) per site per generation for CYC (Table 1; Fig. 1).
Table 1.
Base-substitution mutation statistics for D. pulex and previous MA studies
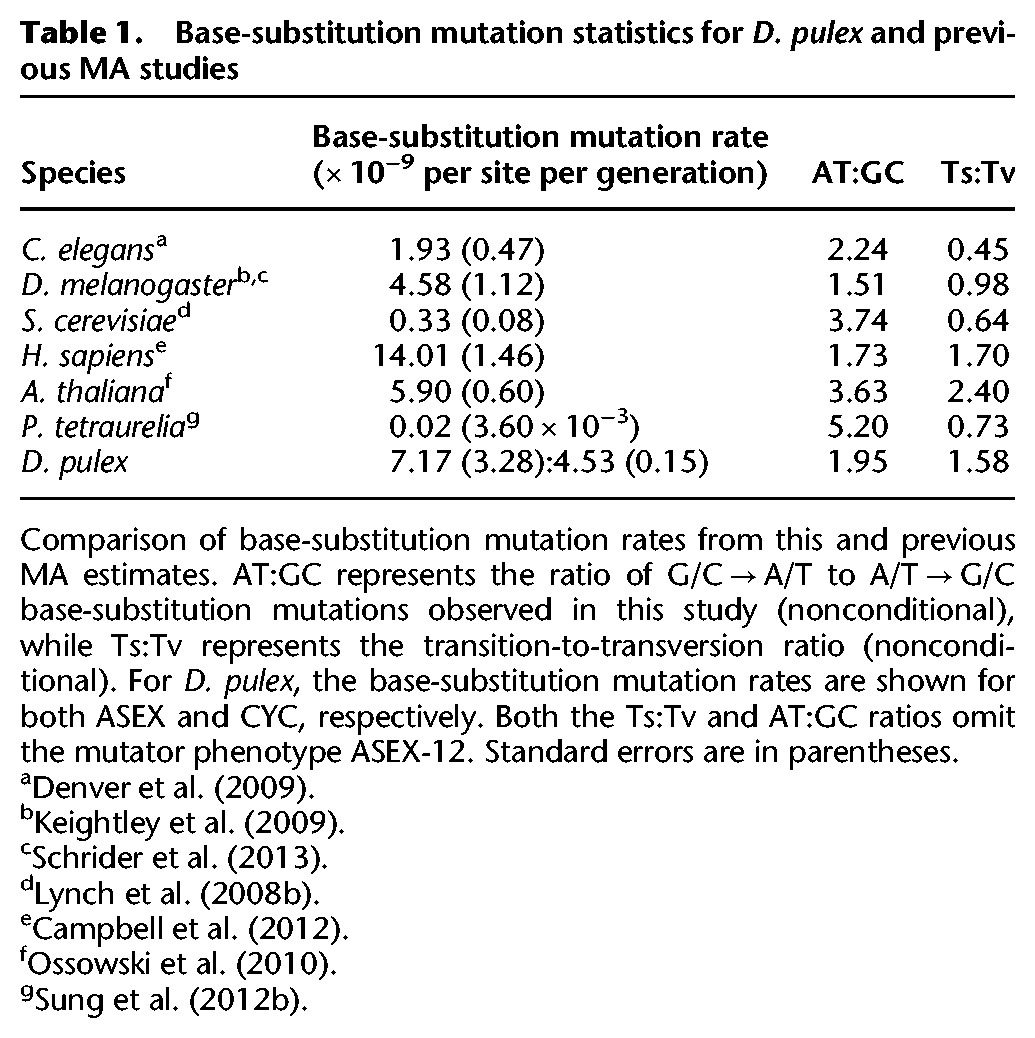
Figure 1.

Conditional base-substitution rates for both the ASEX and CYC experiments (per site per generation). For each type of base-substitution mutation, X → Y denotes the conditional mutation rate per site per generation (× 10−9, y-axis) from nucleotide X to Y.
It is important to note the higher rate of base-substitution mutation in the ASEX experiment is driven by a possible mutator phenotype in one of the four ASEX sublines (ASEX-12) (Supplemental Table S1). The base-substitution mutation rate in ASEX-12 was 9.2× greater than the average of the other ASEX sublines. For ASEX-12, we analyzed the functional categories (COG database) (Tatusov et al. 2003) for genes in which nonsynonymous mutations occurred in the experiment, with the reasoning that a subline-specific mutation(s) may be driving the increased base-substitution rate. After surveying 27 genes with nonsynonymous mutations, only one gene was regarded as having a possible influence in DNA replication and repair, the replication factor C (RFC) large subunit (Supplemental Table S3; Supplemental Fig. S1). RFC, a DNA clamp-loader protein essential for DNA replication and repair, catalyzes the opening of the DNA clamp, PCNA, allowing PCNA to encircle the DNA strand and recruit mismatch repair (MMR)–associated proteins (McAlear et al. 1996). We observe two nonsynonymous mutations in the large subunit of RFC.
A knockout study of the RFC large subunit in yeast resulted in mutator phenotypes (Xie et al. 1999). Although these two mutations are not in the DNA or PCNA binding domains (Supplemental Fig. S1), the observed hydrophilic (P) → hydrophobic (Q) replacement and a hydrophilic (E) → positively charged (K) replacement occurring in close proximity (11 amino acids) could lead to loss or reduced function, thereby providing an explanation for the increased mutation rate of ASEX-12. If we remove this outlier subline, the ASEX rate of mutation is 3.34 (SE = 0.49) × 10−9 per site per generation, which is lower than the observed rate in CYC (4.53 × 10−9 per site per generation).
As in numerous previous experiments directly measuring mutation rates in various eukaryotes, we observed an appreciable number of single nucleotide mutations separated by <50 bp (Schrider et al. 2011, 2013; Campbell et al. 2012; Michaelson et al. 2012; Zhu et al. 2014; Peters et al. 2015). In particular, nine mutations within the CYC lines were found within 50 bp of another mutation (representing 13% of base changes in these lines), and 76 such mutations were found in the ASEX lines (32% of ASEX mutations). These clusters represent multinucleotide mutations (MNMs) (Schrider et al. 2011), in which multiple bases are changed simultaneously, perhaps by an error-prone translesion polymerase (Harris and Nielsen 2014). The majority of these clusters (80%) are found in ASEX-12, suggesting they may be another consequence of the mutator allele we propose is present in that line. In ASEX, only 16% of mutations are MNMs when excluding ASEX-12. We also note that while the most commonly observed MNM is a pair of adjacent mutations, only one such pair is present in our data, and therefore, most MNMs we observe are similar to what we observed in the RFC large subunit of ASEX-12 (Supplemental Fig. S1; Supplemental Table S22). Thus, it is unclear if these putative MNMs were created by the same process as in other species or if a different mechanism is at play.
Gene-conversion-like processes
We report gene-conversion processes, which we define as all genome-homogenizing processes with the exception of deletions that result in loss of heterozygosity (LOH). We estimated the rates of LOH via gene-conversion-like processes to be 1.18 (SE = 0.39) × 10−5 and 1.14 (0.010) × 10−6 per site per generation in the ASEX and CYC lines, respectively (Supplemental Table S4). We assigned transmission to either A/T or G/C at each converted AT/GC heterozygous position and observed that converted sites were significantly biased toward GC alleles (one-sample proportion test) (for description, see Lesecque et al. 2013). Genome-wide, 54.2% (P < 10−6, 95% CI [0.525, 0.560]) of AT/GC heterozygous sites were converted to G or C, which is slightly higher than a genome-wide de novo estimate in yeast (51.3% GC-bias, P < 10−3) (Mancera et al. 2008). We utilized a maximum likelihood model to estimate average conversion tract lengths and found average tract lengths for ASEX were 1358 bp (95% CI [1358, 1590]), 236 bp (95% CI [187, 302]), 197 bp (95% CI [166, 234]), and 28.3 bp (95% CI [28.2, 28.4]), for ASEX-2, ASEX-12, ASEX-41, and ASEX-48, respectively (Fig. 2A,B; Supplemental Fig. S2). For the CYC experiment, tract length estimates were 84.2 bp (95% CI [83.5, 85.0]), 48.1 bp (95% CI [48.0, 48.4]), and 65.6 bp (95% CI [65.1, 66.0]) for CYC-17, CYC-237, and CYC-97, respectively (Supplemental Table S9).
Figure 2.

(A) Parallel coordinate visualization of the minimum and maximum lengths for conversion tracts. The minimum (min) and maximum (max) possible lengths for each event are shown on the x-axis, with the tract length (bp) shown along the y-axis. (B) Frequency distribution of tract lengths. Visualization of the size distribution of the midpoint of the minimum and maximum tract lengths for all 879 conversion tracts in the ASEX experiment (see Supplemental Materials and Methods). Vertical bars group conversion-like tracts by size in 100-bp increments. The red vertical dashed line denotes the median tract length (221 bp). (C) Complex region of large-scale duplication directly flanking a concentrated region of gene-conversion region. Black dots (top track) represent −log10P for each consecutive sliding window on ∼900 kb of scaffold 51 in ASEX-2 and denote the probability the normalized depth of sequencing coverage deviates from 1:1 by chance between the MA subline where large-scale duplication occurred and the normalized average coverage of the other sublines (see Methods). On the middle track (red and blue), the normalized, log2 coverage ratio of the subline where the de novo CNV mutation versus a composite of the other three subline comparisons is plotted in 500-bp windows, with sliding 250-bp increments. The “LOH sites” tract represents a density plot of LOH positions (i.e., individual nucleotide positions of LOH), while the bottom track (gene conversion tracts) shows density of gene-conversion-like tracts within the region. The conversion region begins within 500 bp of the large-scale duplication. Note how the far 3′ end of the conversion region has intervening regions of deletion, starting ∼730 kb, indicating further mutational complexity.
De novo CNV analysis
We also observed many genomic duplication and deletion mutations. The rates of LOH (per heterozygous “site”) resulting from spontaneous deletion were 4.06 (SE = 1.33) × 10−5 and 1.16 (SE = 0.99) × 10−5 per site per generation for ASEX and CYC, respectively, comparable to a previous study in D. pulex (2–30 kb = 6.7 × 10−5 per site per generation) (Xu et al. 2011). We also observed many large-scale de novo CNVs (which we define as indels >0.1 Mb) (Figs. 3, 4; Supplemental Tables S5). In ASEX there were seven large-scale duplications, ranging from 0.10 to 1.35 Mb, and 13 large-scale deletions, ranging from 0.10 to 1.01 Mb. In contrast, in the CYC MA lines we observed only one large-scale deletion of 0.40 Mb resulting from a highly complex mutational event (Fig. 5), and a single large-scale duplication of 0.35 Mb. It is important to note that CYC was propagated for fewer generations, which results in only a fourfold difference in deletion rate between ASEX and CYC despite the presence of 12 more large-scale CNVs in ASEX. We also found a significant enrichment of these mutations on asexual-conferring Chromosomes XIII and IX in the ASEX MA experiment (eight/20 de novo CNVs >100 kb on 13 scaffolds that comprise Chromosomes XIII and IX, out of 200 scaffolds total; P < 4.0 × 10−5, Fisher's exact test). Twelve large-scale CNVs were randomly selected for validation by real-time quantitative PCR, and all were subsequently verified (Supplemental Tables S6, S7).
Figure 3.

Large-scale duplication and deletion. (A) Large-scale duplication; (B) large-scale deletion. Black dots (top track on each figure) represent −log10P for each consecutive sliding window on scaffold 8 (A) and scaffold 17 (B) from the D. pulex reference assembly, where P denotes the probability the depth of sequencing coverage deviates from 1:1 by chance between the MA subline where the de novo CNV occurred and other sublines (see Methods). On the middle track in A (blue, duplication) and B (red, deletion), the normalized, log2 coverage ratio of the subline where the de novo CNV mutation versus a composite of the other three subline comparisons is plotted for both scaffolds in 500-bp windows, with sliding 250-bp increments (see Methods). The black arrow below the normalized coverage tracts denotes all D. pulex full-gene models nested within each duplication and deletion. Note that the separate scaffolds of these two events are not the same length, so the deletion (310 kb) and duplication (1 Mb) are not to scale. Large-scale CNVs were subsequently validated by RT-qPCR.
Figure 4.

Size spectrum of de novo duplications and deletions (indels) >3000 bp. (A) All indel sizes for both the ASEX and CYC experiments >3 kb; (B) only large-scale de novo indels >100 kb, split into duplications and deletions.
Figure 5.

Complex mutation event leading to multiple duplications and deletions in the MA subline CYC-17. Arrows denote the left and right Illumina read pairs (with arrows not signifying left or right, specifically) and provide evidence for complex de novo events on scaffold 18 in CYC-17, including a large-scale deletion (center of figure) and multiple smaller duplications and deletions. While distance between read pairs should only be a few hundred base pairs, the arrows show read pairs with drastic differences in mapping distance and are indicative of indel activity. The sequence coverage track represents the normalized depth of sequencing coverage, relative to the other CYC MA sublines, where lighter color represents lower relative coverage (e.g., the 401-kb deletion). Also, −logP is plotted for each consecutive sliding window (i.e., the probability of a region deviating from 1:1 coverage ratios by chance, or the probability of an indel), and higher values relative to lower values represent increasing probability of CNV (see Methods).
De novo CNV rate analysis
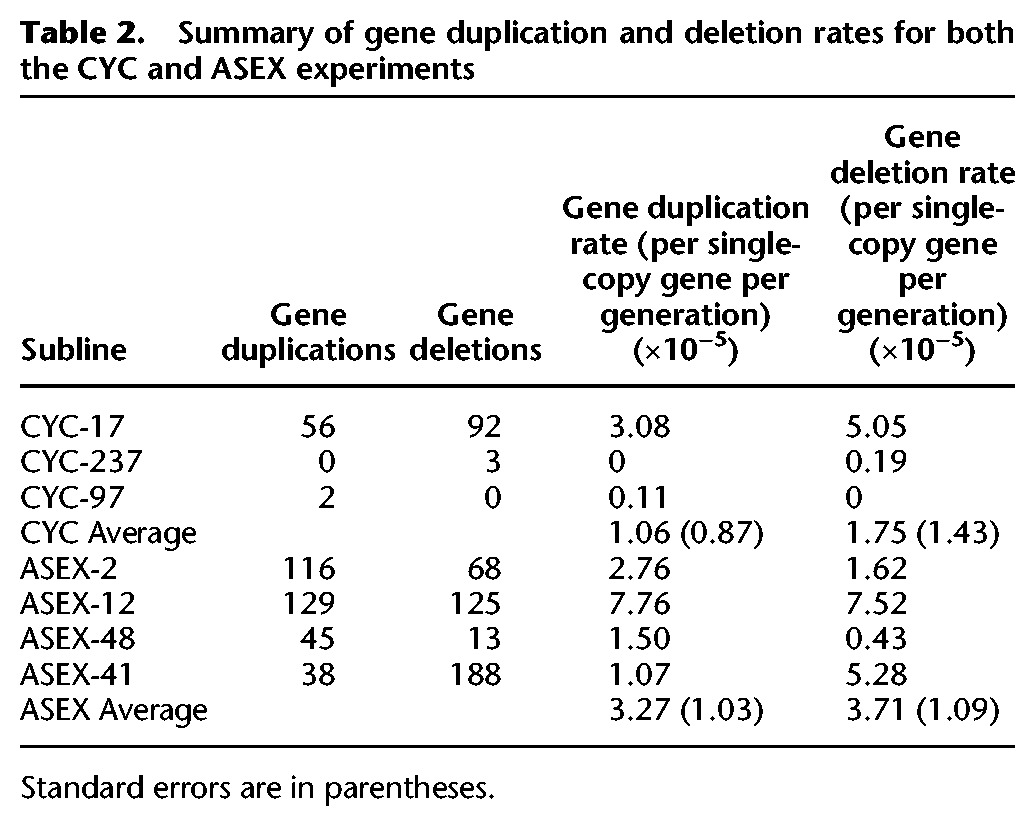
In the ASEX MA experiment, copy-number change was observed for 722 single-copy genes, via 328 gene duplications and 394 gene deletions, yielding gene duplication and deletion rates of 3.27 (SE = 1.03) × 10−5 and 3.71 (SE = 1.09) × 10−5 per single-copy gene per generation, respectively (see Methods). In the CYC MA experiment, copy-number change was observed for 153 single copy genes, via 58 duplications and 95 deletions, leading to gene duplication and deletion rates of 1.06 (SE = 0.87) × 10−5 and 1.75 (1.43) × 10−5 per single-copy gene per generation, respectively (Table 2). We also looked at the rates of duplication and deletion for indels >3 kb (duplication or deletion per bp or “site” per generation). The rates of spontaneous deletion are 7.6× higher in ASEX, and the rate of duplication is 5.0× higher in ASEX relative to CYC (Supplemental Table S10).
Table 2.
Summary of gene duplication and deletion rates for both the CYC and ASEX experiments
Copy-number variation and selection
Asexual D. pulex lineages provide an opportunity for studying genomic consequences of natural selection because a specific, asexual-linked Chromosome XIII and IX haplotype confers obligate asexuality when introgressed into a cyclical parthenogenic background by asexual males that undergo sporadic meiosis (Tucker et al. 2013). Therefore, as this shared haplotype that confers the origin of asexual lineages accumulates mutations in a lineage-specific fashion, it provides an estimate of the ages of asexual lineages (Tucker et al. 2013). We applied the same method (see Supplemental Materials and Methods) to re-estimate the ages of extant asexual lineages from across North America with our revised mutation rate estimates, and used these revised estimates of lineage age to estimate the total number of base pairs we would expect to undergo CNV in the absence of selection. We then compared this null estimate to the observed number of base pairs subject to CNV on these chromosomes in natural populations. Tucker et al. (2013) concluded that base substitutions were accumulating in a neutral fashion on the asexual haplotype (dN/dS, or the ratio of nonsynonymous base-substitutions per site to the synonymous base substitutions per site = 0.94, with a ratio of 1.0 implying neutral evolution). However, after comparing the total accumulation of de novo CNVs (>3 kb) in natural populations to the null expectation derived from the MA experiments (CNVs > 3 kb), there are 19× fewer base pairs subject to de novo CNVs than would be expected in the absence of selection (Supplemental Table S8). Although the disparity in the rates that CNVs are accumulating between natural populations and ASEX could be explained by a single ASEX subline with many large-scale CNVs (and none in the sublines), it is important to note that we observed these mutations in all sublines for ASEX (Supplemental Table S5). These results suggest that selection is acting differently on these two different classes of mutations.
We further found that while large-scale de novo CNVs (i.e., indels >0.1 Mb) commonly accumulate on XIII and IX in the ASEX MA experiment, no large-scale de novo CNVs were found on this haplotype in natural populations (Supplemental Methods; Supplemental Table S4), suggesting that purifying selection against these large-scale mutations is in part leading to the discrepancy between observed and expected de novo CNV.
Discussion
MA studies have substantially increased our knowledge of the rate and molecular spectrum of mutations across cellular life, but the de novo rates of certain mutational classes still remain a mystery. This study allows the ascertainment of classes of mutations that until now have been unobservable, most notably the de novo rates of large-scale CNV and gene-conversion-like processes. While the base-substitution mutation rates in Daphnia are similar to those reported for other species, we discovered specific mutations that potentially underlie a mutator phenotype. We observe high rates of gene duplication and deletion driven by large-scale de novo CNVs (>100 kb). We also report average conversion-like tract lengths for all sublines. Finally, in regions where lineage-specific de novo CNVs can be studied in asexual populations, we find that CNVs are accumulating at lower rates in natural populations compared with estimates derived from this MA experiment.
The D. pulex base-substitution mutation rate (avg. ≈ 3.8 × 10−9 per site per generation) is similar to those reported for Caenorhabditis elegans (Denver et al. 2009), Arabidopsis thaliana (Ossowski et al. 2010), and Drosophila melanogaster (Keightley et al. 2009; Schrider et al. 2013) but is higher than estimates in unicellular eukaryotes (Lynch et al. 2008b; Ness et al. 2012; Sung et al. 2012b; Schrider et al. 2013; Zhu et al. 2014) and lower than estimates in humans (Table 1; Campbell et al. 2012). Transitions arise more frequently than transversions (1.52:1), as observed in previous MA studies. If we consider the conditional mutation rates, i.e., the number of each type of base-substitution mutation normalized by the genome-wide G+C and A+T composition (Fig. 1; Supplemental Table S2; Lynch 2007), the rate of G/C → A/T mutations is 2.5-fold higher than that of A/T → G/C mutations, a bias observed in previous studies (Denver et al. 2004; Lynch et al. 2008b; Keightley et al. 2009; Campbell et al. 2012; Sung et al. 2012b; Zhu et al. 2014). Based on this A+T bias, we would expect an equilibrium A+T composition of 72%. With an observed A+T composition of 59%, the genome composition of D. pulex cannot be explained by base-substitution forces alone, and opposing forces such as GC-biased gene conversion may be countering the observed mutational pressure to A+T. Taking into account the GC-bias of conversion we report (54.2%, P < 10−6, 95% CI [0.525, 0.560]), the expected equilibrium A+T composition in D. pulex is actually 2% A+T. Even if we consider the lower bound of the confidence interval for G/C bias, we would still expect an equilibrium A+T composition of 11%.
In previous MA studies, the A/T bias we report has been attributed to the oxidation of 8-oxo-guanine (Duncan and Miller 1980) or to the spontaneous deamination of 5-methylcytosine, which is a main source of C:G → T:A transitions (Friedberg 2003). As 5-methylcytosine is often found in CpG islands, we investigated whether CpG context is influencing the GC/AT bias via C:G → T:A transitions by comparing CpG context mutations versus all other genomic contexts (Supplemental Tables S13–S21; for details, see Sung et al. 2015). If methylation at CpG islands was driving the observed mutational bias via deamination, we would expect an enrichment of 5′-X[C/G → X]G/C-3′ and 5′-G/C[C/G → X]X-3′ mutations (i.e., CpG context mutations) relative to the contexts of other mutation triplets (where X is either A,T, G, or C). Overall, we observe that both ASEX and CYC are not significantly enriched for CpG context mutations (Fisher's exact text, P < 0.05) (Supplemental Tables S13, S19). However, the ASEX experiment was only marginally nonsignificant (P = 0.06 for ASEX compared with P = 0.46 for CYC). We therefore investigated CpG context in each of the seven ASEX and CYC sublines and determined that there was one outlier that was significantly enriched with CpG context mutations, the mutator phenotype ASEX-12 (P = 0.04) (Supplemental Table S15).
We also found that the enrichment of CpG context mutations in ASEX-12 is driven by a significant enrichment of C:G → T:A transitions relative to the other mutational outcomes. We discovered that 45/71 mutations are C:G → T:A transitions, and these transitions represent one-third of the possible mutations from a CpG context (χ2 = 27.5, N = 71, P = 1.56 × 10−7). Although, taken together, these results suggest germline methylation of cytosine at CpG contexts results in C:G → T:A mismatches that are normally repaired by MMR processes, future studies are needed to investigate the germline state of methylation in D. pulex.
Previous D. pulex studies have suggested that LOH reduces genomic diversity at much higher rates than the input of variation by base-substitution mutation possibly contributing to the demise of obligately asexual lineages (Omilian et al. 2006; Tucker et al. 2013). In ASEX, which has a level of heterozygosity that is representative of North American D. pulex (Tucker et al. 2013), LOH via gene-conversion processes occurs more than 17-fold more often than gain of heterozygosity via base substitutions. When also considering LOH due to deletion, LOH arises about 75-fold more often than gain of heterozygosity, supporting previous claims that LOH generates substantially more genomic input than base-substitution mutations in D. pulex.
We also report average tract lengths for both the ASEX and CYC experiment. Meiotic conversion tracts are relatively small (>1 kb) compared with mitotic conversion tracts, which are generally 1 kb to many kb (Jeffreys and May 2004; Chen et al. 2007). In Daphnia, parthenogenesis occurs via abortive meiosis (Hiruta et al. 2010), and conversion-like processes could be expected to show a mixture of tract lengths due to both meiotic and mitotic processes. We provide evidence suggesting both meiotic-like and mitotic-like conversion processes are operating. While for the ASEX experiment, the average tract length is similar to that expected from meiotic conversion processes (median = 221 bp) (Fig. 2B), ASEX-2 has a significantly higher average tract length (∼1.5 kb) (Supplemental Fig. S2; Supplemental Table S9) compared with the other ASEX and CYC sublines. The majority of ASEX-2 tracts (293 out of 339 gene-conversion-like events) reside in three large-scale regions of concentrated conversion. These concentrated regions of conversion tracts are ∼300 kb (scaffold 13), ∼125 kb (scaffold 14), and ∼400 kb (scaffold 51) and are directly flanked by large-scale duplications (e.g., Fig. 2C). These regions are not observed in the other ASEX or CYC sublines. While in the ASEX experiment the overall median tract length is 221 bp, the median tract lengths in these regions are 314 bp (scaffold 13), 1381 bp (scaffold 14), and 1446 bp (scaffold 51), suggesting these complex regions of larger conversion tracts are the result of mitotic and/or replication-based repair processes that are driving the increased average tract length in ASEX-2.
The combined rate of gene copy-number change by either duplication or deletion in D. pulex (average of ∼5.4 × 10−5/single-copy gene/generation) is higher than direct estimates from previous MA experiments: D. melanogaster (1.1 × 10−6 per gene per generation) (Schrider et al. 2013), C. elegans (3.4 × 10−7 per gene per generation) (Lipinski et al. 2011), and Saccharomyces cerevisiae (5.5 × 10−6 dup per gene per generation [no gene deletion estimate]) (Lynch et al. 2008b). It is important to note that our study is limited to duplication and deletions of single-copy genes to eliminate biases that arise when reads map to multiple locations and to control for genetic differences between isolates. Genes residing in clusters of tandem duplicates, which account for half of the reported gene duplicates in the D. pulex assembly (Colbourne et al. 2011), may undergo higher rates of copy-number change due to an increased probability of unequal crossing over during recombination (Read et al. 2004). Thus, the total rate of gene duplication and deletion in D. pulex may be higher than the estimates we provide.
The difference in mutation rates between ASEX and CYC is also of interest. Relative to the CYC lines, in the ASEX experiment, the base-substitution mutation rate is 2.4-fold greater (although the rates are similar if the candidate mutator phenotype, ASEX-12, is removed from the mutation-rate calculation), the combined rate of gene duplication and deletion is approximately threefold higher, duplications (>3 kb) arise 5× more often, deletions (>3 kb) arise 7× more often, and the rate of gene-conversion processes is about 10.4-fold greater. However, we are hesitant to conclude that there are differences in mutation rates between all asexual and cyclically parthenogenetic D. pulex, as the small sample size of this experiment (N = 1 for asexuals and sexuals) does not justify such a conclusion and as the differences we observe are well within the variation observed among individuals within the same species (Conrad et al. 2011; Schrider et al. 2013). Future studies are needed to determine if mutation rates are higher in asexual D. pulex populations.
As observed in a previous yeast MA experiment (Zhu et al. 2014), by maintaining D. pulex ameiotically and in a heterozygous state, we are able to study large-scale CNVs that are typically lost due to complementation of recessive lethal mutations in experiments that utilize sexual reproduction. We find that the rate of large-scale CNVs is 1.5 × 10−2 events per haploid genome per generation, higher than the rates observed in an ameiotically propagated yeast MA study (3.2 × 10−6 events per haploid genome per generation, Zhu et al. 2014), the only previous MA study reporting such de novo mutations. When we consider the number of nucleotides in the two genomes (events per base pair per generation), large-scale de novo CNVs arise about 5000× more often in D. pulex.
The overrepresentation of CNVs on the asexual-conferring haplotype (Chromosomes XIII and IX) is intriguing, as this haplotype was originally introgressed from a divergent sister-species, D. pulicaria (origin of asexuality ∼1250 yr before present) (Tucker et al. 2013). Because of this foreign origin, there is 2× the silent-site heterozygosity on Chromosomes XIII and IX compared with all other genomic regions in asexuals (synonymous-site heterozygosity, or Πs = 0.0442 vs. 0.0223 per site) (Tucker et al. 2013), suggesting that chromosomal divergence is contributing to the increased mutation rates in this region. It has been observed that mutations accumulate at higher rates in F1 hybrids (Scavetta and Tautz 2010; Yang et al. 2015), and repeat regions are more likely to change in length if the homologous sequence has a drastically divergent length (Amos et al. 1996). However, perhaps most pertinent to this study is that mutations have been shown to arise at a higher rate in heterozygous regions of genomes that have interspersed homozygous and heterozygous domains (Yang et al. 2015). Future studies are needed to fully understand the mechanistic phenomena leading to the observed changes on D. pulex Chromosomes XIII and IX in asexual lines.
Because large changes in the order, orientation, and copy number of genomic elements can affect transcriptional regulators and gene dosage, large-scale CNVs can be highly deleterious and may be subject to higher levels of purifying selection than single-base mutations (Itsara et al. 2010). Furthermore, large-scale CNVs have been associated with debilitating neurodevelopmental disorders and genetic diseases in humans (Zhang 2003; Sebat et al. 2007; Stone et al. 2008; Girirajan et al. 2011). Thus, while Tucker et al. (2013) reported no purifying selection operating on base substitutions in young asexual lineages (based on the ratio of nonsynonymous base-substitution mutations/nonsynonymous site to synonymous base substitutions/synonymous site, dN/dS = 0.94; with a ratio of 1.0 corresponding to neutral base-substitution accumulation), we find that large-scale CNVs are being removed in asexual Daphnia populations (Supplemental Table S8). If we consider that on average each large-scale CNV (>100 kb) reported in this MA experiment changes the copy number of 15.87 genes, their observed removal in the natural populations we examined is consistent with the idea that large-scale CNVs have a detrimental effect on the fitness of an organism and that they are therefore often removed by purifying selection. Taken together, these findings show the importance of large-scale CNVs on genome stability and that Daphnia is an ideal model to study the rate and consequences of all mutational classes.
Methods
MA line propagation, and DNA sequencing procedures
D. pulex MA lines were initiated from two independent genotypes, one from Linwood, Ontario (ASEX), and the other from Slimy Log Pond, Oregon (CYC). For both experiments, a single individual was grown up into an isogenic (clonal) population and separated into sublines, each maintained under the following conditions: 100 mL of filtered lake water, stored at 20°C, 12 h on:12 h off, light:dark environment, and fed ad libitum with Scenedesmus obliquus. Every generation, a randomly chosen daughter was selected to propagate the next generation, and back-ups were maintained to restore lineages in the event of mortality, production of males only, overproduction of ephippia, or operator error. Over the course of the experiment, each replicate MA line was propagated through single-progeny descent, reducing the effective population size to approximately one, minimizing the effect of natural selection, and accumulating all but the most deleterious mutations (Kibota and Lynch 1996). At the end of the experiment, a total of eight randomly selected MA lines (four from each of the two genotypes) were chosen for whole-genome sequencing from the remaining sublines.
DNA was extracted using CTAB buffer and purified using phenol/chloroform in phase-lock tubes. In order to collect enough DNA for sequencing (1–10 µg), a single female from each of the randomly chosen MA lines was clonally propagated to hundreds of individuals. Standard Illumina paired-end libraries were prepared at the Center for Genomic and Bioinformatics at Indiana University (CYC) and BGI (ASEX), sequenced, and mapped to the D. pulex genome reference assembly using Novoalign (Novocraft Technologies). The reference genome alignments from each of the eight lines were converted to SAM format and analyzed using SAMtools (Li et al. 2009). Final sequence-coverage depth for mapped reads ranged from 18 to 55× (Supplemental Table S5) across the eight lines.
Identifying de novo copy-number variation and gene duplication and deletion
Our study is limited to duplications and deletions of single-copy genes to eliminate biases that arise when reads map to multiple locations and to control for genetic differences between isolates. Genes residing in clusters of tandem duplicates, which account for half of the reported gene duplicates in the D. pulex assembly (Colbourne et al. 2011), may undergo higher rates of copy-number change due to an increased probability of unequal crossing over during recombination (Read et al. 2004). Therefore, we restricted our analysis of the rates of gene duplication and deletion to uniquely mapped sequences within the 200 largest scaffolds of the reference. This approach is able to analyze duplication and deletion across 10,521 single-copy (Colbourne et al. 2011) genes in the reference.
To detect de novo CNVs, defined as regions that had either been duplicated or deleted throughout the MA experiment, a sliding window approach via CNV-seq software (Xie and Tammi 2009) was utilized for all pairwise comparisons of the ASEX and CYC MA lines, independently. To identify CNVs, we required a minimum of four consecutive 500-bp windows (sliding in 250-bp increments to have a significantly different depth of coverage deviating from a 1:1 ratio; P-value = 0.001, log2-threshold = 0.45). After genome-wide CNV regions were identified for all pairwise comparisons, we then identified annotated D. pulex genes located within these CNV regions. Each gene was classified as either a deletion or duplication and compared against the average depth of coverage across all other sublines for each identified gene and for each experiment (ASEX and CYC), independently. If a gene within a CNV region for a given line had an increased depth of coverage relative to the other lines, this gene was categorized as a gene-duplication candidate, while a relative decrease in the depth of coverage was categorized as a gene deletion candidate because of its co-occurrence with a CNV region. We then filtered out duplication and deletion candidate genes without depth of coverage ratios supporting a duplication or deletion (deletion depth of coverage ratio ≤0.6, duplication depth of coverage ratio ≥1.4, as using strict ratios of 0.5 for deletion and 1.5 for duplication cause many duplications and deletions to be filtered out), leaving us with high-confidence gene duplication and deletions located within CNV regions >1000 bp. Although some previous studies (Alkan et al. 2011; Mills et al. 2011) have defined CNVs as indels >50 bp, we did not use a read-pair approach (as described in Fig. 2; Alkan et al. 2011; Mills et al. 2011) because of the divergence of the ASEX progenitor from the D. pulex reference genome. Thus, our CNV minimum length cutoff was chosen to return high-confidence CNV calls with a depth of coverage method by joining multiple 500-bp windows, with each window having a significant difference in average depth of coverage.
For each line, duplication and deletion rates were estimated with µg = n/(2i × T), where n is the number of gene duplications or deletions within a line, i is the total number of single-copy genes that were analyzed for copy-number change, and T is the total number of generations the line was propagated. Due to differences in mapping efficiency (given the genetic divergence between ASEX and the reference), we reasoned that i could differ between ASEX and CYC, and we filtered single-copy genes with low coverage for each experiment, independently. The standard errors (SE) of the gene duplication and deletion rates were calculated for individual sublines independently, with the formula where µg is the rate of gene duplication or deletion, ng is the number of analyzed single-copy genes, and T is the number of generations (Lynch et al. 2008b; Denver et al. 2009; Ossowski et al. 2010). The overall SE across all sublines was estimated with where (s) is the standard deviation of the gene duplication or deletion rates, and N is the number of sublines.
We also calculated rates of duplication and deletion based on events large enough to duplicate or delete a Daphnia gene (duplications and deletions >3 kb). The new formula is µ = n/(2i × T), where n is the number of de novo CNVs (duplication or deletion >3 kb), i is the total number of bps analyzed, and T is the total number of generations for each subline, arriving at a rate of duplication or deletion/bp/generation.
To address the issues of nonunique and low-coverage genomic regions, which hinder our ability to accurately estimate CNV lengths based on our sliding window approach by interrupting CNV events, we utilized an agglomerative, hierarchical clustering algorithm for all CNV-seq pairwise comparisons. We then merged CNV calls <10 kb apart, which accounts for the lengths of 95% of assembly gaps and low complexity (repeat) regions in the D. pulex reference genome. This method was applied to natural asexual populations and the ASEX and CYC experiment to assign CNV lengths for all pairwise comparisons. To assign lineage-specific or MA subline-specific CNVs, we used the output CNV regions from our pairwise-comparison after clustering and computed the intersection of all pairwise CNV comparisons using BEDOPS (Neph et al. 2012).
CNV validations
We performed RT-PCR validation for 10 randomly selected de novo CNVs for ASEX sublines where genomic DNA was available after sequencing. RT-PCR was performed using Roche LightCycler 480 DNA SYBR green I master kit on the LightCycler 480 (Roche) hardware, and reactions included 10 ng of DNA, 0.5 mM of each primer, and 10 mL of 1X SYBR green I master mix, which includes reaction buffer, MgCl2, dNTP mix, FastStart Taq DNA polymerase, and SYBR green I dye.
Each Roche LightCycler 384-well clear qPCR plate (catalog no. 05102430001) contained the following: First, three technical replicates of two randomly selected genes residing within a candidate de novo CNV for the line where the CNV occurred and three technical replicates for these two genes in a “control” line where no candidate de novo CNV was observed. We used ASEX-48 as control for all CNVs, as only two de novo CNVs were observed, with no CNV overlap with other sublines. Second, a standard curve, replicated three times per gene, with one standard curve per gene, dilution series from 0.78–50 ng; and third, negative controls that consisted of no template control, no master mix control, and no primer control.
Samples, standards, and controls were then randomly assigned to wells (http://www.random.org/) to avoid any spatial biases that may arise on the LightCycler plate. RT-PCR cycling conditions followed the manufacturer's recommendations and included an initial denaturation at 95°C followed by 40 cycles of denaturing for 10 sec at 95°C, annealing for 20 sec, and extension for 30 sec at 72°C. Melting curve analysis was generated by running the RT-PCR amplicons through one cycle of 5 sec at 95°C and then 1 min at 65°C, followed by a ramp up to 97°C.
Data access
The data from this study have been submitted to the NCBI BioProject (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA275628.
Supplementary Material
Acknowledgments
We thank J.F. Gout, S. Reynolds, S. Xu, S. Glaholt, and M. Hahn for helpful discussions and G. Fox for computational support and expertise. E. Williams and M. Westphal maintained the MA lines. Funding was provided by grants from National Institutes of Health to M.L. (R01GM036827 and R01GM101672-01A1), from the Murdock Charitable Trust and the National Science Foundation to S.S. (MCB-1150213), and from the National Institute of Environmental Health Sciences to J.R.S. (R01ES019324).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.191338.115.
References
- Alkan C, Coe BP, Eichler EE. 2011. Genome structural variation discovery and genotyping. Nat Rev Genet 12: 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos W, Sawcer SJ, Feakes RW, Rubinsztein DC. 1996. Microsatellites show mutational bias and heterozygote instability. Nat Genet 13: 390–391. [DOI] [PubMed] [Google Scholar]
- Campbell CD, Chong JX, Malig M, Ko A, Dumont BL, Han LD, Vives L, O'Roak BJ, Sudmant PH, Shendure J, et al. 2012. Estimating the human mutation rate using autozygosity in a founder population. Nat Genet 44: 1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JM, Cooper DN, Chuzhanova N, Ferec C, Patrinos GP. 2007. Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet 8: 762–775. [DOI] [PubMed] [Google Scholar]
- Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, et al. 2011. The ecoresponsive genome of Daphnia pulex. Science 331: 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JEM, DePristo MA, Lindsay SJ, Zhang YJ, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Morris K, Lynch M, Thomas WK. 2004. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430: 679–682. [DOI] [PubMed] [Google Scholar]
- Denver DR, Dolan PC, Wilhelm LJ, Sung W, Lucas-Lledo JI, Howe DK, Lewis SC, Okamoto K, Thomas WK, Lynch M, et al. 2009. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci 106: 16310–16314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan BK, Miller JH. 1980. Mutagenic deamination of cytosine residues in DNA. Nature 287: 560–561. [DOI] [PubMed] [Google Scholar]
- Friedberg EC. 2003. DNA damage and repair. Nature 421: 436–440. [DOI] [PubMed] [Google Scholar]
- Girirajan S, Campbell CD, Eichler EE. 2011. Human copy number variation and complex genetic disease. Annu Rev Genet 45: 203–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag-Liautard C, Dorris M, Maside X, Macaskill S, Halligan DL, Houle D, Charlesworth B, Keightley PD. 2008. Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature 453: 128. [DOI] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD. 2009. Spontaneous mutation accumulation studies in evolutionary genetics. Annu Rev Ecol Evol Syst 40: 151–172. [Google Scholar]
- Harris K, Nielsen R. 2014. Error-prone polymerase activity causes multinucleotide mutations in humans. Genome Res 24: 1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. 2009. Mechanisms of change in gene copy number. Nat Rev Genet 10: 551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PDN, Ward RD. 1972. Inheritance during parthenogenesis in Daphnia magna. Genetics 71: 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiruta C, Nishida C, Tochinai S. 2010. Abortive meiosis in the oogenesis of parthenogenetic Daphnia pulex. Chromosome Res 18: 833–840. [DOI] [PubMed] [Google Scholar]
- Innes DJ, Hebert PDN. 1988. The origin and genetic-basis of obligate parthenogenesis in Daphnia pulex. Evolution 42: 1024–1035. [DOI] [PubMed] [Google Scholar]
- Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ, Eichler EE. 2010. De novo rates and selection of large copy number variation. Genome Res 20: 1469–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, May CA. 2004. Intense and highly localized gene conversion activity in human meiotic crossover hot spots. Nat Genet 36: 151–156. [DOI] [PubMed] [Google Scholar]
- Keightley PD, Trivedi U, Thomson M, Oliver F, Kumar S, Blaxter ML. 2009. Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genome Res 19: 1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibota TT, Lynch M. 1996. Estimate of the genomic mutation rate deleterious to overall fitness in E. coli. Nature 381: 694–696. [DOI] [PubMed] [Google Scholar]
- Lamb BC. 1984. The properties of meiotic gene conversion important in its effects on evolution. Heredity (Edinb) 53(Pt 1): 113–138. [DOI] [PubMed] [Google Scholar]
- Lesecque Y, Mouchiroud D, Duret L. 2013. GC-biased gene conversion in yeast is specifically associated with crossovers: molecular mechanisms and evolutionary significance. Mol Biol Evol 30: 1409–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski KJ, Farslow JC, Fitzpatrick KA, Lynch M, Katju V, Bergthorsson U. 2011. High spontaneous rate of gene duplication in Caenorhabditis elegans. Curr Biol 21: 306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 1983. Ecological genetics of Daphnia pulex. Evolution 37: 358–374. [DOI] [PubMed] [Google Scholar]
- Lynch M. 2007. The origins of genome architecture. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Lynch M. 2008. The cellular, developmental and population-genetic determinants of mutation-rate evolution. Genetics 180: 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010a. Evolution of the mutation rate. Trends Genet 26: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010b. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci 107: 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Gabriel W. 1983. Phenotypic evolution and parthenogenesis. Am Nat 122: 745–764. [Google Scholar]
- Lynch M, Seyfert A, Eads B, Williams E. 2008a. Localization of the genetic determinants of meiosis suppression in Daphnia pulex. Genetics 180: 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, Dickinson WJ, Okamoto K, Kulkarni S, Hartl DL, et al. 2008b. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci 105: 9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM. 2008. High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlear MA, Tuffo KM, Holm C. 1996. The large subunit of replication factor C (Rfc1p/Cdc44p) is required for DNA replication and DNA repair in Saccharomyces cerevisiae. Genetics 142: 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, et al. 2012. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151: 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagylaki T. 1983. Evolution of a finite population under gene conversion. Proc Natl Acad Sci 80: 6278–6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, et al. 2012. BEDOPS: high-performance genomic feature operations. Bioinformatics 28: 1919–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Colegrave N, Keightley PD. 2012. Estimate of the spontaneous mutation rate in Chlamydomonas reinhardtii. Genetics 192: 1447–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omilian AR, Cristescu MEA, Dudycha JL, Lynch M. 2006. Ameiotic recombination in asexual lineages of Daphnia. Proc Natl Acad Sci 103: 18638–18643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Lucas-Lledo JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M. 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327: 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paland S, Colbourne JK, Lynch M. 2005. Evolutionary history of contagious asexuality in Daphnia pulex. Evolution 59: 800–813. [PubMed] [Google Scholar]
- Peters BA, Kermani BG, Alferov O, Agarwal MR, McElwain MA, Gulbahce N, Hayden DM, Tang YT, Zhang RY, Tearle R, et al. 2015. Detection and phasing of single base de novo mutations in biopsies from human in vitro fertilized embryos by advanced whole-genome sequencing. Genome Res 25: 426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read LR, Raynard SJ, Ruksc A, Baker MD. 2004. Gene repeat expansion and contraction by spontaneous intrachromosomal homologous recombination in mammalian cells. Nucleic Acids Res 32: 1184–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scavetta RJ, Tautz D. 2010. Copy number changes of CNV regions in intersubspecific crosses of the house mouse. Mol Biol Evol 27: 1845–1856. [DOI] [PubMed] [Google Scholar]
- Schrider DR, Hourmozdi JN, Hahn MW. 2011. Pervasive multinucleotide mutational events in eukaryotes. Curr Biol 21: 1051–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Houle D, Lynch M, Hahn MW. 2013. Rates and genomic consequences of spontaneous mutational events in Drosophila melanogaster. Genetics 194: 937–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. 2007. Strong association of de novo copy number mutations with autism. Science 316: 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So M, Ohtsuki H, Makino W, Ishida S, Kumagai H, Yamaki KG, Urabe J. 2015. Invasion and molecular evolution of Daphnia pulex in Japan. Limnol Oceanogr 60: 1129–1138. [Google Scholar]
- Stone JL, O'Donovan MC, Gurling H, Kirov GK, Blackwood DHR, Corvin A, Craddock NJ, Gill M, Hultman CM, Lichtenstein P, et al. 2008. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455: 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M. 2012a. Drift-barrier hypothesis and mutation-rate evolution. Proc Natl Acad Sci 109: 18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Tucker AE, Doak TG, Choi E, Thomas WK, Lynch M. 2012b. Extraordinary genome stability in the ciliate Paramecium tetraurelia. Proc Natl Acad Sci 109: 19339–19344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Gout JF, Miller SF, Williams E, Foster PL, Lynch M. 2015. Asymmetric context-dependent mutation patterns revealed through mutation–accumulation experiments. Mol Biol Evol 32: 1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. 2003. The COG database: An updated version includes eukaryotes. BMC Bioinformatics 4: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker AE, Ackerman MS, Eads BD, Xu S, Lynch M. 2013. Population-genomic insights into the evolutionary origin and fate of obligately asexual Daphnia pulex. Proc Natl Acad Sci 110: 15740–15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Tammi MT. 2009. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics 10: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Counter C, Alani E. 1999. Characterization of the repeat-tract instability and mutator phenotypes conferred by a Tn3 insertion in RFC1, the large subunit of the yeast clamp loader. Genetics 151: 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Omilian AR, Cristescu ME. 2011. High rate of large-scale hemizygous deletions in asexually propagating Daphnia: implications for the evolution of sex. Mol Biol Evol 28: 335–342. [DOI] [PubMed] [Google Scholar]
- Yang S, Wang L, Huang J, Zhang X, Yuan Y, Chen JQ, Hurst LD, Tian D. 2015. Parent–progeny sequencing indicates higher mutation rates in heterozygotes. Nature 523: 463–467. [DOI] [PubMed] [Google Scholar]
- Zhang JZ. 2003. Evolution by gene duplication: an update. Trends Ecol Evol 18: 292–298. [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA. 2014. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci 111: E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.