

Abstract
The functions of the major mammalian cytoplasmic poly(A) binding protein, PABPC1, have been characterized predominantly in the context of its binding to the 3′ poly(A) tails of mRNAs. These interactions play important roles in post-transcriptional gene regulation by enhancing translation and mRNA stability. Here, we performed transcriptome-wide CLIP-seq analysis to identify additional PABPC1 binding sites within genomically encoded mRNA sequences that may impact on gene regulation. From this analysis, we found that PABPC1 binds directly to the canonical polyadenylation signal in thousands of mRNAs in the mouse transcriptome. PABPC1 binding also maps to translation initiation and termination sites bracketing open reading frames, exemplified most dramatically in replication-dependent histone mRNAs. Additionally, a more restricted subset of PABPC1 interaction sites comprised A-rich sequences within the 5′ UTRs of mRNAs, including Pabpc1 mRNA itself. Functional analyses revealed that these PABPC1 interactions in the 5′ UTR mediate both auto- and trans-regulatory translational control. In total, these findings reveal a repertoire of PABPC1 binding that is substantially broader than previously recognized with a corresponding potential to impact and coordinate post-transcriptional controls critical to a broad array of cellular functions.
Keywords: CLIP-seq, poly(A) binding protein, polyadenylation signal, translational control
INTRODUCTION
The biogenesis of eukaryotic messenger RNAs (mRNAs) is tightly linked to the post-transcriptional addition of polyadenylate [poly(A)] tails to their 3′ ends. These poly(A) tails contribute to regulation of mRNA transcription, transport, stability, and translation (Mangus et al. 2003; Goss and Kleiman 2013). Poly(A) tail-dependent functions are mediated in large part via the association of one or more poly(A) binding proteins (PABPs). In mammals, there are six defined PABP isoforms; a single nuclear isoform, PABPN1, that impacts on the addition of poly(A) tails in the nucleus and five cytoplasmic PABPs, ePAB, PABPC1, PABPC2, PABPC4, and PABPC5, that are thought to play roles in regulating mRNA stability and translation in the cytoplasm (Wahle 1991; Mangus et al. 2003; Good et al. 2004). The overall structures and RNA-binding specificities of the five cytoplasmic PABPs are highly conserved (Adam et al. 1986; Burd and Dreyfuss 1994). They each contain four RNA recognition motifs (RRMs). RRMs 1 and 2 are primarily responsible for the high-affinity binding to homopolymeric adenosines (Kd = 1.8 nM), while RRMs 3 and 4 can bind to nonhomopolymeric AU sequences (Kd = 2.9 nM) (Sladic et al. 2004). With the exception of non-redundant impacts of PABPC4 in erythroid maturation (Kini et al. 2014) and vertebrate development (Gorgoni et al. 2011), the levels of functional specificity and/or redundancy of the mammalian cytoplasmic PABPs remain unexplored.
PABPC1 is the major cytoplasmic PABP isoform in adult mouse somatic cells and is abundantly expressed in all tissues (Kleene et al. 1994). The interaction of PABPC1 with poly(A) tails is well documented and defined in multiple contexts (Blobel 1973; Mangus et al. 2003). The corresponding functions of the PABPC1/poly(A) tail complex are primarily mediated in pathways of mRNA stabilization and translation enhancement (Wang and Kiledjian 2000; Wilusz et al. 2001; Kahvejian et al. 2005). These functions may be linked to the interactions of PABPC1 with the 5′ cap-binding complex via heterodimerization with eIF4G (Tarun and Sachs 1995; Wells et al. 1998; Peixeiro et al. 2012) Limited evidence points to additional binding sites and functions for PABPC1 within the eukaryotic mRNA transcriptome. For example, PABPC1 has been shown to bind to an A-rich element in the 5′ untranslated region (UTR) of its own mRNA (mouse and human), establishing an autoregulatory translation control circuit (de Melo Neto et al. 1995; Bag and Wu 1996; Hornstein et al. 1999). A recent study in Saccharomyces cerevisiae using a photoactivatable-ribonucleoside-enhanced crosslinking immunoprecipitation approach (PAR-CLIP) demonstrated in vivo binding of yeast poly(A) binding protein Pab1 to AU-rich elements in mRNAs (Baejen et al. 2014), including binding to the efficiency element (UAUAUA) of the yeast polyadenylation signal (Guo and Sherman 1995; Tuck and Tollervey 2013). Importantly, the PAR-CLIP approach used in this yeast study only identifies crosslinking events at uridines, which may bias the overall mapping results. Furthermore, the impact of Pab1 binding to the polyadenylation efficiency element in yeast remains undefined, as does any generalization of these findings to higher eukaryotic organisms.
The extent to which PABPC1 binds to genomically encoded sequences in the mammalian transcriptome remains undetermined. The presence of such interactions could have broad implications to the understanding of post-transcriptional gene regulation. To address this gap, we comprehensively mapped PABPC1 binding to sites throughout the mouse transcriptome. This analysis revealed robust PABPC1 occupancy within the 3′ untranslated region (3′ UTR) that is predominantly localized to the canonical polyadenylation signal (PAS). A distinct set of PABPC1 interactions, lacking a defined binding site motif, were mapped to 5′ and 3′ boundaries of the coding open reading frame (ORF), exemplified most clearly in the replication-dependent histone mRNAs. A third, and more restricted subset of PABPC1 binding sites, was identified at AU-rich sites within the 5′ UTRs of a select group of mRNAs and was demonstrated to affect translation. These studies substantially expand the known repertoire of PABPC1 interactions within the eukaryotic transcriptome and link a subset of these interactions to pathways of post-transcriptional control.
RESULTS
CLIP-seq identifies genomically encoded PABPC1 binding sites in the mouse transcriptome
We performed crosslinking immunoprecipitation coupled to high-throughput sequencing (CLIP-seq) to map PABPC1 binding sites within the transcriptome of mouse erythroleukemia (MEL) cells (see work flow; Fig. 1A). PABPC1 RNP complexes were captured by in vivo UV-crosslinking, followed by limited RNase I digestion, 32P-labeling of RNA in the complexes, and immunoprecipitation with an isotype-specific anti-PABPC1 antibody. The immunoprecipitated PABPC1 RNP complexes were resolved on an SDS-PAGE gel and complexes migrating in close proximity to the PABPC1 band (blue line, Fig. 1B) were excised for analysis. The slower running complexes were excluded from library preparation as they were assumed to represent PABPC1 multimers bound to poly(A) (red line, Fig. 1B). RNA fragments were isolated from the PABPC1 RNP complexes and used as templates for the construction of high-throughput sequencing libraries (see Materials and Methods).
FIGURE 1.

PABPC1 CLIP-seq analysis reveals binding to genomically encoded sequences within murine mRNAs. (A) Schematic of PABPC1 CLIP workflow showing immunoprecipitation and library preparation of PABPC1-bound RNAs for sequencing (see Materials and Methods). (B) Isolation of 32P-labeled PABPC1–mRNP complexes. RNase I trimmed and 32P-labeled RNP complexes were immunoprecipitated with an antibody to PABPC1 and the products were resolved on a denaturing gel. The left lane is an autoradiograph of the immunoprecipitated 32P-labeled PABPC1-RNA complexes and the right lane is an immunoblot of the same gel incubated with antibody to PABPC1. Blue and Red lines indicate PABPC1 monomers and multimers, respectively. (C) Correlation of PABPC1 CLIP-seq replicates. PABPC1 CLIP-seq tags per gene are plotted for three independent biological replicates (Spearman correlation coefficient, R > 0.96 for all comparisons). (D) Correlation of PABPC1 CLIP-seq and RNA-seq from MEL cells. RPKM per gene is plotted for CLIP-seq and RNA-seq (Spearman correlation coefficient, R = 0.792). (E) Pie chart of the distribution of PABPC1 CLIP tags within the transcriptome. (F) Relative distribution of PABPC1 CLIP tags along spliced mRNA transcripts. Gencode mRNAs were binned into 100 evenly sized regions and the coverage at each bin was used to create a composite profile. (G) Screenshots of the UCSC genome browser for two representative mRNAs (Slc25a1 and Pcbp1), showing distribution of PABPC1 CLIP tags along the length of the primary transcript. Note that Pcbp1 is encoded by an intronless gene.
Sequencing of the libraries generated from the PABPC1-bound RNA fragments yielded 14.8 million unique sequences across three biological replicates (Table 1). The mean size of the unique sequences protected from the RNase I treatment was 24 nt. These unique sequences were mapped to the mouse genome (mm10) with Novoalign, resulting in 7.5 million uniquely mapping CLIP tags that were used for downstream analysis (see Materials and Methods) (Table 1). Only 2.6% of sequencing reads corresponded to pure poly(A) sequences, which is likely a consequences of selecting RNP complexes migrating close to the PABPC1 protein bands (Supplemental Fig. S1). These were assumed to represent PABPC1 monomers, whereas the longer migrating bands were likely to be enriched for PABPC1 multimers bound to the poly(A) tail.
TABLE 1.
Summary of sequencing libraries and binding site identification
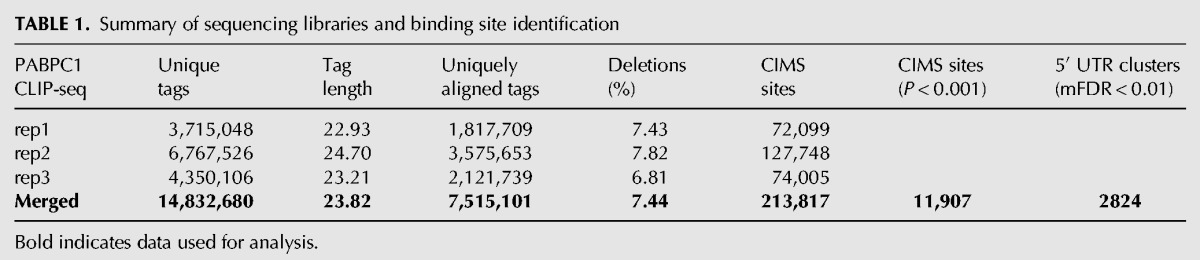
We first examined the correlation of CLIP tags per gene between biological replicates and found a high level of reproducibility between experiments (Spearman correlation coefficient; R > 0.96 for all comparisons) (Fig. 1C). This consistency between replicates allowed us to merge the biological samples for subsequent analyses. We also determined the correlation between PABPC1 CLIP-seq and mRNA-seq data that we generated from MEL cells (see Materials and Methods; Fig. 1D). The relatively high correlation coefficient between this CLIP-seq and mRNA-seq comparison (Spearman correlation coefficient; R2 = 0.79) suggested that the PABPC1 may recognize genomically encoded sequences shared by most mRNAs in the transcriptome. Overall, the high reproducibility of CLIP-seq replicates supported successful enrichment of PABPC1-bound fragments and warranted further investigation.
PABPC1 binds predominantly to the 3′ UTR of mRNAs
We next examined the distribution of PABPC1 CLIP tags across the mouse transcriptome. The great majority (73.89%) of CLIP tags mapped to the 3′ UTR, with the next highest amount of tags (19.63%) mapping to the coding sequence (CDS) (Fig. 1E). Since the average genomic length of 3′ UTRs is shorter than that of the CDS, this distribution indicates a strong enrichment of PABPC1 binding in 3′ UTRs. The remainder of the CLIP tags mapped to annotated 5′ UTRs, introns, long intergenic noncoding RNAs (lincRNAs), and microRNAs (miRNAs). A meta-analysis of PABPC1 CLIP tags across mature mRNA transcripts demonstrated a marked enrichment of CLIP tag density in proximity to annotated 3′ termini of mRNAs (Fig. 1F). This is in agreement with the majority of CLIP tags mapping to the 3′ UTR. Visual inspection of specific mRNAs revealed numerous CLIP tags clustering along the 3′ UTR with the most prominent peak occurring close to the 3′ termini (examples in Fig. 1G). Together, these data indicate that PABPC1 binds to genomically encoded sequences in numerous mRNAs, that most binding events occur in the 3′ UTR, and that the preponderant localization of the PABPC1 binding occurs in close proximity to the 3′ terminus of mRNAs.
PABPC1 binding is enriched at the termini of 3′ UTR
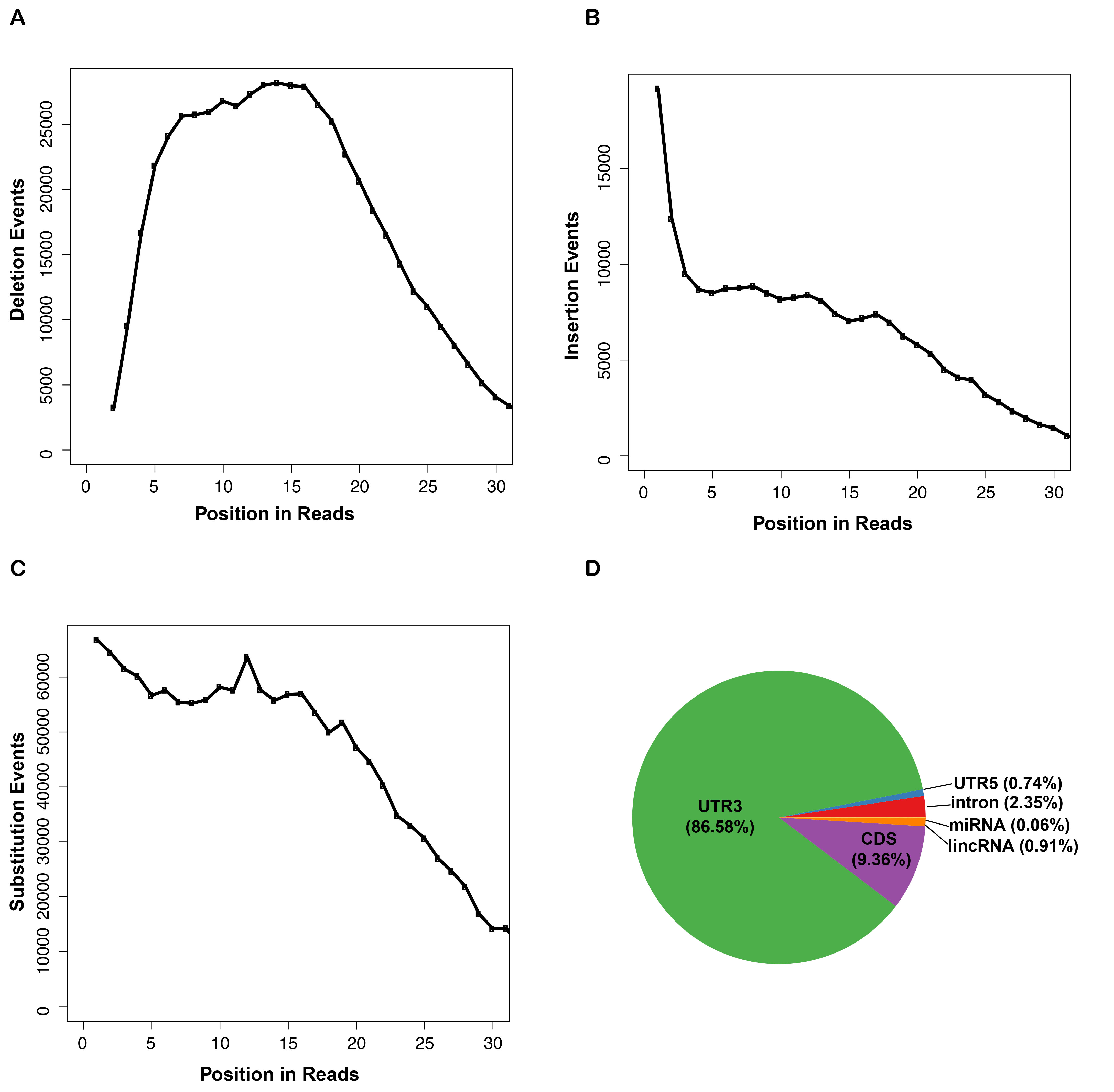
To enable closer inspection of PABPC1 binding sites, we mapped direct binding events at single-nucleotide resolution by crosslink induced mutation site (CIMS) analysis (Zhang and Darnell 2011; Moore et al. 2014). This analysis takes advantage of the propensity for reverse transcriptase to skip nucleotides with protein adducts that remain after proteinase K treatment of RNP complexes. In agreement with previous studies, we found that deletions, but not insertions or substitutions were enriched within the body of CLIP tags (Supplemental Fig. S2A–C). CIMS analysis of PABPC1 CLIP tags identified 11,907 significant (false discovery rate [FDR] < 0.001) direct binding sites within the mouse genome (Table 1; Supplemental Table S1). Within the transcriptome, 86% of CIMS sites were located in 3′ UTRs, 9% in the CDS, and the remaining 5% distributed across other regions (Supplemental Fig. S2D). To examine the distribution of CIMS sites in more detail, regions of mature mRNAs (5′ UTR, CDS, and 3′ UTR) were binned into 100 discrete units, and CIMS coverage across each bin was calculated (Fig. 2A). We did not observe any positional enrichment within the 5′ UTR, whereas an increase in binding events was observed toward the 3′ end of the CDS leading into the beginning of the 3′ UTR. While numerous binding sites were distributed throughout the 3′ UTR, the most robust sites of enrichment for PABPC1 binding localized to the terminal segments of 3′ UTRs. This distribution of CIMS sites is consistent with the distribution of CLIP tags toward the 3′ terminus of mRNAs (Fig. 1F) and is indicative of direct PABPC1 binding to these regions.
FIGURE 2.

CIMS analysis of PABPC1 CLIP tags reveals direct binding of PABPC1 to the cleavage and polyadenylation signal. (A) Relative distribution profile of CIMS sites along mRNAs. Position-specific coverage was calculated by parsing each region (UTRs and CDS) into 100 distinct bins and calculating CIMS coverage per bin. (B) Logo representing the average nucleotide sequence ±10 nt proximal to the CIMS sites (position 11 represents the CIMS site). (C) Z-score distribution of hexanucleotide analysis of CIMS sites (±15 nt) and a logo representing the 20 most enriched hexanucleotides. (D) Motif logo uncovered by MEME analysis of CIMS sites ±15-nt flanking sequence. (E) Motif logo uncovered by MEME analysis on CIMS sites ±15-nt flanking sequence after removing all possible PAS signal sequences. (F) Absolute distribution of CIMS relative to the end of annotated 3′ UTRs. (G) Absolute distribution of CIMS sites relative to the PAS signal sequence (AAUAAA). (H) Absolute distribution of experimentally determined poly(A) addition sites relative to CIMS sites.
PABPC1 binding sites are enriched for A/U-rich and A-rich motifs
We next determined the binding site sequence preference for PABPC1 within the mRNA population using multiple approaches. First, we analyzed the sequence content at each position surrounding CIMS sites. To do this, we anchored the analysis at CIMS sites and identified the base composition at each position ±10 nt from the CIMS site (Fig. 2B). This approach revealed a strong preference for adenosines interspersed with less frequent uridines (denoted as T's on the logo). Uridine was the most commonly cross-linked base (position 11), consistent with analyses of UV-induced cross-links for other RBPs (Williams and Konigsberg 1991) and most likely reflecting preferential formation of UV-induced crosslinking of proteins with uridine over other ribonucleosides (Sugimoto et al. 2012). We also calculated the enrichment of hexanucleotide sequences in the region ±15 nt from each CIMS sites relative to all mRNA sequences. We selected the top 20 most enriched hexanucleotides and created a position weight matrix and motif logo to represent the sequence content (Fig. 2C). A strong enrichment for adenosine and uridine was observed using this approach. Finally, using a de novo motif discovery algorithm (MEME) (Bailey et al. 2009), we identified an A/U-rich sequence that had a striking resemblance to the canonical mammalian polyadenylation signal sequence (AAUAAA) (Fig. 2D). The presence of this abundant and conserved sequence element may overshadow the identification of other true motifs. Therefore, to search for a secondary motif, we eliminated all sequences that contained the top 10 mammalian PAS sequences (corresponding to 55% of all CIMS sequences) and re-ran the MEME analysis (Tian et al. 2005). This secondary search revealed a purely A-rich sequence which was derived from ∼170 CIMS sites (Fig. 2E). Together, these orthogonal approaches led us to conclude that PABPC1 binds directly to both the PAS-like as well as to purely A-rich sequences within the mammalian transcriptome.
PABPC1 binds directly to the cleavage and polyadenylation signal of mammalian mRNAs
Due to the predominant binding of PABPC1 to 3′ terminal poly(A) tails, we considered the possibility that the observation of enriched binding at the PAS might reflect “bleed-over” from canonical poly(A) tail binding. Positional analysis of CIMS sites revealed that a preponderance of the PABPC1 CIMS sites mapped 20 to 25-nt upstream of the annotated 3′ terminus of mRNAs (Fig. 2F). This location coincides with the approximate positioning of the PAS and argues against “bleed-over” from the poly(A) tail. Furthermore, alignment of the CIMS relative to the canonical PAS sequence (AAUAAA) revealed a sharp enrichment for CIMS sites at this element (Fig. 2G). To confirm that CIMS sites were localized to active PAS elements, we used our mRNA-seq data generated from MEL cells to identify functional poly(A) addition sites in the MEL cell transcriptome (see Materials and Methods). We examined the distribution of active poly(A) addition sites relative to CIMS sites (Fig. 2H). This analysis revealed that active poly(A) addition sites were located 20–25 nt downstream from CIMS sites. These various approaches were internally consistent in demonstrating that PABPC1 binds directly to bona fide mRNA PAS elements. This binding to the PAS on mRNAs throughout the transcriptome likely explains the high level of correlation between CLIP-seq and RNA-seq data sets (Fig. 1D) and the predominance of CLIP tags mapping to the 3′ terminus of the 3′ UTR (Fig. 1E–G). In summary, these data support the conclusion that PABPC1 binds directly to PAS elements and to genomically encoded A-rich mRNA sequences, in addition to its well documented binding to mRNA poly(A) tails.
PABPC1 clusters are enriched in close proximity to the translation initiation and termination codons
As expected based on the correlation between mRNA abundance and PABPC1 CLIP tags, we found that the RNAs with the most CLIP tags were highly expressed mRNAs encoding the protein components of the ribosome and proteins that comprise the translation machinery (Supplemental Table S2). Surprisingly, we also observed that a number of replication-dependent histone mRNAs were represented among the top 1000 transcripts with highest CLIP tag density. Given that this class of mRNAs is unique in lacking a PAS and poly(A) tail, we chose to examine the corresponding pattern of PABPC1 binding in detail. The distribution of CLIP tags across histone mRNAs (Fig. 3A; as in Fig. 1F) was prominently enriched at the 5′ and 3′ ends of transcripts. This distribution contrasts strongly with the 3′ enrichment observed in the remainder of the mRNA transcriptome (cf. Figs. 3A and 1F, examples in Figs. 3B and 1G). Detailed mapping of PABPC1 CLIP tags in replication-dependent histone mRNAs revealed that they were highly enriched over the translation initiation and termination sites (Fig. 3C,D). Multiple analytic approaches failed to reveal any corresponding enriched primary sequence motif corresponding to these binding events. The pattern of binding within the replication-dependent histone mRNAs at start codons was similar to that of polyadenylated mRNAs with short 5′ UTRs (Fig. 3E). Importantly, we observed a similar enrichment over the start codon in genes with longer 5′ UTRs, although to a lesser extent. Similarly, PABPC1 binding in the vicinity of the stop codon of histone mRNAs displayed a sharp peak (Fig. 3D), as did the stop codon of polyadenylated mRNAs with short 3′ UTRs (Fig. 3F). Binding to the stop codon of all other detectable mRNAs with longer 3′ UTRs in general rose at the stop codon and remained high throughout the 3′ UTR. These data suggest that PABPC1 accumulates in the proximity of the start and stop codons on all mRNAs, although this binding pattern is most marked in genes lacking a poly(A) tail or with short UTRs. The difference in the contour of the CLIP tags mapping to the stop codon between histone and nonhistone mRNAs may reflect, at least in part, the abundant PABPC1 binding at the PAS in polyadenylated mRNAs. Together, these results suggest that PABPC1 interacts with the translation initiation and termination sites in a poly(A) and PAS-independent fashion.
FIGURE 3.

PABPC1 binds to a subset of mRNAs at their translation initiation and termination sites. (A) Distribution of PABPC1 CLIP tags along histone mRNA transcripts. Histone mRNAs were binned into 100 evenly sized regions and the CLIP tag coverage in each bin was used to create a composite profile. (B) Screenshots from the UCSC genome browser for two representative histone genes showing CLIP tags proximal the 5′ and 3′ end of the CDS. Green and Red boxes indicate annotated start and stop codons, respectively. (C) Absolute distribution of CLIP tags proximal to the start codon of histone genes. (D) Absolute distribution of CLIP tags proximal to the stop codon of histone genes. (E) Absolute distribution of CLIP tags proximal to the start codon of all mRNAs split into quartiles by 5′ UTR length. (F) Absolute distribution of CLIP tags proximal to the stop codon of all mRNAs split into quartiles by 3′ UTR length.
PABPC1 binds to A-rich sequences within a subset of 5′ UTRs
Our initial analysis revealed a small subset of PABPC1 CLIP tags localized within 5′ UTRs (Fig. 1E). Interestingly, we found a relatively low correlation between number of CLIP-seq tags in 5′ UTRs and mRNA abundance (mRNA-seq) (Fig. 4A, Spearman correlation coefficient; R = 0.356). This low correlation suggested that the binding of PABPC1 to the 5′ UTR is heterogeneous across the population of mRNAs in MEL cells and occurs at determinants that are specific to subset(s) of transcripts. The number of CIMS sites within 5′ UTRs (Supplemental Fig. S2D; 79 sites across 39 genes) was insufficient to identify a 5′-UTR-specific motif. However, analysis of the 5′ UTR CLIP tags by a low stringency approach (Pyicoclip implementation of the modified false discovery rate [mFDR] approach (Althammer et al. 2011)), identified ∼2800 PABPC1 CLIP-tag clusters located in 5′ UTRs (see Materials and Methods and Supplemental Table S3). Although 5′ UTRs are generally G–C rich (Pesole et al. 1997), MEME analysis of PABPC1 5′ UTR cluster sites revealed an A-rich sequence motif that mapped to ∼300 unique 5′ UTR clusters (Fig. 4B). We examined the correlation of CLIP tags from A-rich motif containing 5′ UTRs with the mRNA-seq data set and found an even weaker correlation (Fig. 4C, Spearman correlation coefficient; R = 0.186) than what was observed for all 5′ UTR CLIP tags (Fig. 4A). This lower correlation coefficient suggests that PABPC1 interactions with A-rich motifs in the 5′ UTR are further uncoupled from mRNA steady-state expression levels and more likely to reflect binding to a small subset of mRNAs. A gene ontology analysis (DAVID; (Huang da et al. 2009)) on this subset of mRNAs revealed enrichment for gene function terms involved in the regulation of transcription, DNA binding, nuclear processes, and cell cycle control (Fig. 4D). Together, these results suggest that PABPC1 may coordinately regulate mRNAs involved in these basic cellular processes through binding to a shared 5′ UTR motif.
FIGURE 4.

Enrichment of PABPC1 binding to A-rich sequences in the 5′ UTRs of a subset of murine transcripts. (A) Correlation analysis of PABPC1 CLIP tags in 5′ UTRs of protein-coding transcripts and mRNA-seq RPKM per gene values (Spearman correlation coefficient, R = 0.356). (B) Motif logo uncovered by MEME analysis of PABPC1 CLIP-tag clusters in the 5′ UTRs of mRNAs. (C) Correlation analysis of PABPC1 CLIP tags in 5′ UTRs of transcripts containing the A-rich motif (B) and RNA-seq RPKM per gene values (Spearman correlation coefficient, R = 0.186). (D) Gene ontology analysis of genes with A-rich motifs within CLIP-tag clusters in the 5′ UTR. (E) Screenshot of PABPC1 CLIP tags from the UCSC genome browser for two transcripts with PABPC1 binding within their 5′ UTRs (Pabpc1 [top] and Ccnd2 [bottom]).
Interestingly, we found that the top A-rich PABPC1 binding site in the 5′ UTR corresponds to Pabpc1 mRNA (Fig. 4E). This is of note, because it has been previously reported that PABPC1 represses the translation of its own mRNA via binding to a 5′ UTR A-rich determinant (Melo et al. 2003a,b). Among other transcripts with significant PABPC1 binding to the A-rich sequence in the 5′ UTR were, cell cycle control protein Cyclin D2 (Ccnd2) (Fig. 4E); scaffold attachment Factor B (Safb), an RNA-binding protein that impacts on both transcription and splicing (Nayler et al. 1998); and adenosylmethionine decarboxylase 1 (Amd1), a protein associated with cell and tumor growth (Paasinen-Sohns et al. 2011), metabolism, and obesity (Tabassum et al. 2012). These observations led us to hypothesize that PABPC1 binds to an A-rich determinant within the 5′ UTR of a subset of mRNAs and this binding may be of particular importance to the regulation of their translation.
PABPC1 auto-regulates its expression by binding to an A-rich element in the 5′ UTR of its mRNA
PABPC1 was previously found to repress its own translation by binding to a 5′ UTR A-rich determinant (de Melo Neto et al. 1995, 2000). Interestingly, we identified two distinct clusters of PABPC1 binding sites in the Pabpc1 mRNA 5′ UTR (Fig. 5A, red and green bars, respectively), a more 5′ cluster, overlapping with the previously identified A-rich element (“5′ cluster”, red bar), and a second, larger cluster of unknown function (“3′ cluster”, green bar). To determine whether these clusters have overlapping or unique roles in regulation of PABPC1 expression we cloned the intact Pabpc1 5′ UTR into a firefly luciferase reporter plasmid and separately inserted derivative 5′ UTRs specifically lacking each of the two individual PABPC1 CLIP-tag clusters (Fig. 5B). Each of these plasmids was transfected into NIH-3T3 cells. NIH-3T3 cells were chosen because they are more effectively transfected than MEL cells. Firefly luciferase protein and RNA expression were quantified 48 h post transfection (Fig. 5C). Luciferase protein was significantly (C1-5′ UTR versus Luc; P < 0.01, two-tailed t-test) repressed in the presence of native Pabpc1 5′ UTR in the absence of an appreciable impact on mRNA accumulation (Fig. 5C). This impact on expression was fully consistent with a mechanism of translation inhibition by PABPC1. Deletion of the more 5′ located interaction site resulted in a significant increase in protein expression as compared with the intact Pabpc1 5′ UTR (Mut1 versus C1-5′UTR; P < 0.001, two-tailed t-test) (red cluster in Fig. 5C), while deletion of the more 3′ located cluster had a statistically significant but marginal impact on protein output (Mut 2 versus C1-5′UTR; P < 0.001, two-tailed t-test). To further extend these observations we deleted both of the clusters together (Fig. 5B, Mut 3) and measured reporter mRNA and protein expression. This dual deletion resulted in a significant increase in protein and mRNA expression compared with the intact Pabpc1 5′ UTR (Mut1 versus 5′ UTR; P < 0.001, two-tailed t-test) (Fig. 5C), similar in magnitude to deletion of the 5′ cluster (red cluster, Mut 2) alone. These luciferase reporter assays suggest that the more 5′ cluster mediates an autoregulatory control over PABPC1 protein expression, while the function of the more 3′ cluster, if any, remains to be defined (see below).
FIGURE 5.

PABPC1 CLIP tag clusters co-localize to a 5′ UTR translational control region. (A) Screenshot from UCSC genome browser of PABPC1 CLIP tags in the 5′ UTR for Pabpc1 mRNA. Red and Green bars highlight two major PABPC1 CLIP tag clusters (5′ and 3′ clusters, respectively). (B) Insertion of the Pabpc1 5′ UTR and three derivatives in an expression vector in frame with the firefly luciferase ORF. Mutants represent deletion of either one or both of the CLIP tag clusters denoted in A. (C) Quantification of firefly luciferase mRNA levels (qRT-PCR; light gray bars) and luciferase enzymatic activity levels as a proxy for protein abundance (luciferase assay; dark gray bars). (D) Agarose gel of genomic DNA PCR showing Pabpc1 5′ UTR region for WT cells (untransfected C2C12 cells), cells transfected with a vector expressing Cas9 without guide RNAs (Cas9), and cells transfected with vectors expressing Cas9 and gRNAs targeting sites flanking both of the PABPC1 CLIP tag clusters (Cas9/gRNA). Upper and lower arrows denote WT and mutant loci, respectively. (E) Quantification of Pabpc1 mRNA levels (qPCR) for mutant clones relative to WT cells. (F) PABPC1 immunoblot of WT C2C12 cells and mutant clones. PABPC1 levels were quantified by densitometry and normalized to β-actin. (**) P < 0.01; (***) P < 0.001, two-tailed t-test.
While PABPC1 has been reported to auto-regulate mRNA translational efficiency by binding to the 5′ UTR A-rich sequence, this was not clearly delineated from the luciferase assays as deletion of the 5′ cluster also resulted in a significant (Mut 1 versus C1-5′ UTR; P < 0.05, two-tailed t-test) increase in luciferase reporter mRNA expression (Fig. 5C). Also of interest, we found that the 3′ cluster, whose deletion did not impact upon the reporter expression (Fig. 5C), overlapped the start of a predicted upstream open reading frame (uORF) and co-localized with initiating ribosomes as determined by ribosome profiling with harringtonine-treated mouse ES cells (Supplemental Fig. S3; Ingolia et al. 2011). Therefore, this region may regulate control mechanisms that are more complex than an isolated impact on translational efficiency.
To further address the mechanism of this translation regulation, we chose to examine the in vivo function of Pabpc1 5′ UTR binding clusters by ablating them in cell lines with CRISPR/Cas9-mediated site-directed deletion. Two guide RNAs (gRNAs) corresponding to sites flanking the Pabpc1 5′ UTR binding clusters were cloned into separate vectors expressing the Cas9 nuclease. These two vectors were cotransfected into the mouse myoblast cell line C2C12 and Puromycin-resistant clones containing a heterozygous deletion of this region (Supplemental Fig. S4) were identified (Fig. 5D). Analysis of multiple Pabpc1 5′ UTR Mut+/− clones revealed that deletion of the Pabpc1 5′ UTR clusters resulted in ∼1.5-fold to twofold (P < 0.01, two-tailed t-test) increase in PABPC1 protein levels in the absence of an alteration in steady-state mRNA levels. PABPC1 protein and corresponding mRNA levels for one such clone shown (Fig. 5E,F). These in vivo results strongly support the model in which PABPC1 binding within the 5′ UTR of its encoding mRNA is critical for the homeostasis of PABPC1 protein expression. Specifically, deletion of the PABPC1 binding site in the 5′ UTR results in increased levels of PABPC1 protein expression in vivo in the absence of an alteration in mRNA levels.
PABPC1 inhibits synthesis of a subset of proteins by binding to 5′ UTR A-rich determinants encoded in their mRNAs
We next sought to determine whether PABPC1 binding to 5′ UTR A-rich elements mediated regulatory control over additional mRNAs. Three mRNAs with prominent PABPC1 binding clusters at A-rich sites within their 5′ UTR were chosen for study; Safb, Amd1, and Ccnd2 (Fig. 6A,C,E). The full 5′ UTR of each of these mRNAs was cloned into the firefly luciferase reporter plasmid and its impact was compared with the corresponding 5′ UTRs lacking the PABPC1 binding site (Fig. 6B,D,F). Deletion of the PABPC1 binding region from both the Safb and Amd1 5′ UTRs significantly enhanced luciferase expression levels (Amd1: P < 0.05 and Safb: P < 0.01, two-tailed t-tests) without altering corresponding mRNA levels (Fig. 6G). A significant (P < 0.01, two-tailed t-test) increase in luciferase activity was also observed upon deletion of the PABPC1 binding site from the Ccnd2 5′ UTR (Fig. 6G) although in this case there was a corresponding increase in mRNA levels (P < 0.05, two-tailed t-test). This increase in mRNA levels was similar to what was observed for deletion of the Pabpc1 5′ cluster region (Fig. 5C). These results demonstrate that PABPC1 binding to A-rich sites within the 5′ UTR of specific mRNAs can repress protein expression by repressing mRNA levels and/or by impeding effective translation. Thus, PABPC1 is involved in autoregulatory and transreguatory post-transcriptional control of gene expression in mammalian cells via an array of mechanistic pathways.
FIGURE 6.

PABPC1 binds in the 5′ UTR of transcripts and negatively regulates translation. (A,C,E) Screenshots from the UCSC genome browser of PABPC1 CLIP tags in the 5′ UTR for Safb (A), Amd1 (D), and Ccnd2 (G) mRNA. (B,D,F) The native Safb (B), Amd1 (D), and Ccnd2 (F) 5′ UTRs (top) or mutant derivatives lacking the A-rich PABPC1 binding site cluster (bottom) were separately inserted in-frame with the firefly luciferase ORF in a standard expression vector. The PABPC1 binding site, corresponding to the CLIP tag cluster in (A, C, and E), is represented by the black rectangle within the 5′ UTR. (G) Quantification of firefly luciferase mRNA levels (qRT-PCR; light gray) and luciferase enzymatic activity levels (luciferase assay; dark gray) for the Safb (B), Amd1 (D), and Ccnd2 (F) 5′ UTR constructs. (*) P < 0.05; (**) P < 0.01, two-tailed t-test.
DISCUSSION
PABPC1 is an abundant cytoplasmic RNA-binding protein that is expressed in all somatic cells. The functions of PABPC1 are best understood in the context of its binding to the homopolymeric poly(A) tails of mRNAs. This PABPC1/poly(A) tail complex has been linked to pathways that control mRNA stability and translation activity and exerts significant impact on multiple cell functions (Mangus et al. 2003; Goss and Kleiman 2013; Smith et al. 2014). In Saccharomyces cerevisiae, deletion of its single poly(A) binding protein, Pab1p, is incompatible with cell viability (Sachs et al. 1987) and in Drosophila melanogaster, homozygosity for P-element disruption of the cytoplasmic PABP gene results in embryonic lethality (Sigrist et al. 2000). The impact of PABPC1 depletion or ablation in mammalian cells remains undefined.
Limited in vitro studies suggest that PABPC1 can bind to mRNAs at sites other than the poly(A) tail (Sladic et al. 2004). In vivo analysis in yeast provided further evidence that Pab1p binds to genomically encoded A- and A/U-rich sequences in mRNAs (Tuck and Tollervey 2013; Baejen et al. 2014). In the present report, we performed CLIP-seq on PABPC1 in MEL cells with the goal of revealing the extent and role of PABPC1 binding to genomically encoded sequences in the mammalian transcriptome. These studies reveal that PABPC1 binds to complex sequences within different regions of annotated mRNAs and that subsets of these interactions have a defined impact on gene expression.
PABPC1 binds to the PAS of mRNAs throughout the mammalian transcriptome
A key observation from our study is that the majority of the PABPC1 CLIP tags cluster within mRNA 3′ UTRs (73.89%) (Fig. 1E). This is not surprising as proteins that bind to the CDS are generally susceptible to displacement by the elongating ribosome and stably assembled RNP complexes are preferentially localized to the 3′ UTR “sanctuary” (de Moor et al. 2005; Gebauer et al. 2012). As PABP's are well characterized for their strong association to mRNA poly(A) tails it was necessary to rigorously demonstrate that the enrichment within 3′ UTRs reflected direct binding rather than “bleed over” from binding to the adjoining poly(A) tails. Mapping of PABPC1 binding at single-nucleotide resolution by a CIMS analysis (Fig. 2; Supplemental Fig. S2) unambiguously identified that PABPC1 binds directly within the mRNA 3′ UTRs (Fig. 2A). Coupled with two orthogonal approaches, we were able to further determine that the majority of these binding interactions are localized to the canonical cleavage and polyadenylation signal sequence (AAUAAA) (Fig. 2C–H). These observations are consistent with the prior Pab1p PAR-CLIP analysis in yeast in which binding was mapped to the AU-rich efficiency element within the 3′ UTR (Baejen et al. 2014). Our finding that PABPC1 CLIP-seq also demonstrates in vivo binding to AU-rich sequences suggests that this previous report in the yeast model did not reflect a technological bias introduced by PAR-CLIP preferentially crosslinking to U residues. These results lead us to conclude that binding of cytoplasmic PABPs to polyadenylation elements has been conserved from yeast to mammalian cells.
While the function(s) of non-poly(A) tail PABP RNP complexes in the cytoplasmic compartment remains unclear, related binding activities have been functionally linked to post-transcriptional pathways of gene regulation. For example, the cytoplasmic polyadenylation element binding (CPEB) protein binds to an A-rich element (CPE) within the 3′ UTR where it recruits the cleavage and specificity factor (CPSF) to the PAS with consequent cytoplasmic extension of the poly(A) tail and translational enhancement in certain cellular settings (Richter 2007). CPEB can also recruit proteins such as Maskin to regulate mRNA translation in Xenopus oocytes (Richter 2007) and mouse hippocampus (Theis et al. 2003). It is plausible that PABPC1, once bound to the PAS, can recruit additional trans-acting factors that modulate mRNA stability and/or translation by impacting on the length and/or function of the poly(A) tail. Alternatively, PABPC1 may also bind to the PAS without any functional significance. Based on our mapping data, these and related models can now be more fully explored.
PABPC1 occupancy is enriched at sites in close proximity to the translation initiation and termination codons
The mapping of CLIP tags within the MEL cell transcriptome revealed robust binding to replication-dependent histone mRNAs. These mRNAs are unique among polymerase II transcribed mRNAs in that they lack PAS elements and poly(A) tails. Analysis of the histone mRNAs thus allowed us to focus on PABPC1 interactions in the absence of the predominant poly(A) tail and PAS binding activities. Intriguingly, the CLIP tags within histone mRNAs localized to the sites of translation initiation and termination (Fig. 3B–D). We observed similar enrichment for CLIP tags at the start codons throughout the transcriptome while the signal at stop codons was somewhat overshadowed in the bulk of mRNAs by PAS binding (Fig. 3E,F). Importantly, the small number of CIMS sites and lack of any enriched sequence motif for PABPC1 binding at sites flanking ORFs, suggests that enrichment of CLIP tags bracketing the open reading frame may reflect indirect association of PABPC1 at these sites. This indirect positioning of PABPC1 is consistent with the model proposed by others that PABPC1 remains associated with the elongating ribosome during translation (Uchida et al. 2002; Peixeiro et al. 2012). Further study will be necessary to understand the role and functional consequences of PABPC1 binding to these regions.
PABPC1 binds to A-rich sites within the 5′ UTR of a restricted subset of mRNAs with resultant post-transcriptional repression of gene expression
The binding of PABPC1 within 5′ UTRs appears to be limited to a highly restricted subset of mRNAs (Fig. 4). This specificity is indicated by the lack of correspondence between mRNAs bound in this region by PABPC1 and overall mRNA representation in the transcriptome (Fig. 4A,C). MEME analysis of 5′ UTR clusters revealed enrichment for a predominantly A-rich motif, consistent with the binding site preference of the PABPs (Fig. 4B). Interestingly, the highest ranked PABPC1 binding target within this mRNA subset was Pabpc1 mRNA. PABPC1 has been previously reported to auto-regulate its own translation by binding to an A-rich domain within the 5′ UTR (Melo et al. 2003a). This translational control domain is coincident with a prominent PABPC1 CLIP-tag cluster identified in the current study (5′ cluster, highlighted in red, Fig. 5A). Remarkably, our analysis also revealed an adjacent and even more prominent cluster of CLIP tags (3′ cluster, highlighted in green, Fig. 5A) that did not impact on translation (C1-5′ UTR versus Mut 2) (Fig. 5C). Interestingly, this second PABPC1 binding region tracks with the positioning of the initiating ribosome in mouse ES cells as mapped by ribosomal profiling and was predicted to encode the start of a uORF (Supplemental Fig. S3; Ingolia et al. 2011). Thus, this PABPC1 binding element within the 5′ UTR may yet play a role in translation regulation not captured by the luciferase assay (Fig. 5C).
To validate the in vivo function of Pabpc1 5′ UTR clusters in translational control, we deleted the region of the Pabpc1 5′ UTR spanning both of the PABPC1 CLIP-tag clusters via Crispr/Cas9 endonuclease targeting. The twofold increase in PABPC1 protein expression in cells heterozygous for the 5′ UTR deletion in the absence of any alteration in mRNA steady-state levels, confirmed that this region acts to auto-regulate Pabpc1 translation (Fig. 5F). Importantly, PABPC1 overexpression has been associated with defective spermiogenesis in mice (Yanagiya et al. 2010), deadenylation, and translation inactivation in Xenopus oocytes (Wormington et al. 1996), and with variations in cell cycle and apoptosis in certain leukemias (Verlaet et al. 2001). Thus, this autoregulatory feature of the Pabpc1 5′ UTR likely mediates a regulatory pathway relevant to critical aspects of cell differentiation and proliferation.
The potential for PABPC1 to control gene expression was further extended by the analysis of additional mRNAs identified with 5′ UTR PABPC1 CLIP-tag clusters. Deletion of these binding site regions enhanced the translation of reporter expressing Safb and Amd1 mRNA 5′ UTRs (Fig. 6G). Protein expression was also enhanced by similar deletion within the 5′ UTR of the Ccnd2 mRNA, although in this case there was a concomitant increase in the steady-state mRNA levels (Fig. 6G). We note that deletion of the 5′ cluster (Mut 1) in the Pabpc1 5′ UTR (Fig. 5C) also enhanced mRNA levels, although to a lesser extent than the corresponding protein expression. These data highlight the potential for the 5′ UTR binding of PABPC1 to impact on a variety of mechanisms that repress gene expression, including both mRNA stability as well as translation regulation. The intimate linkage of mRNA translation with mRNA stability makes a clear delineation of the primary mechanisms difficult using the luciferase assay and will necessitate subsequent studies more clearly focused on the impact of this binding activity on ribosome loading and mRNA turnover. The relative importance of each of these pathways may reflect specifics of the binding site, including interaction with other trans-factors and/or adoption of specific RNA secondary structures. Together, we reveal that PABPC1 regulates a repertoire of gene regulatory pathways and establish a foundation for the exploration of additional targets and cellular functions mediated by PABPC1 binding to genomic mRNA sequences.
MATERIALS AND METHODS
Cell culture and CLIP-seq analysis
MEL and NIH-3T3 cells were grown under standard conditions in minimal essential medium (MEM) and Dulbecco's modified Eagle medium (DMEM), respectively, supplemented with 10% (vol/vol) fetal bovine serum (FBS) and 1× antibiotic–antimycotic (Invitrogen). MEL cells were washed with Hanks’ balanced salt solution (HBSS) and cross-linked with UV (400 mJ/cm2) three times on ice. CLIP was performed according to previously published protocol (Ule et al. 2005; Chi et al. 2009). Briefly UV cross-linked MEL cells were lysed with 1× PMPG in the presence of RNase 1 (2.5 U, Promega), DNAse I (Promega) treated for 15 min. The lysates were ultracentrifuged at 90,000g for 20 min. Immunoprecipitation was performed with protein A Dynabeads coated with PABPC1 antibody (Abcam). Following the wash steps radiolabeled 3′ adaptor was ligated to the complexes on the beads using T4 RNA ligase (Thermo Scientific) for 16 h at 16°C. The beads were then washed, treated with T4 polynucleotide kinase (NEB), and the RNP complexes were eluted off of the beads. Ninety percent of the eluate were used for autoradiography and the remainder was used for immunoblotting. The RNP complexes were resolved on 4%–12% NuPage gels (Invitrogen), transferred to nitrocellulose membrane, and then exposed to X-ray film. Using the X-ray film as a guide, the portion of the nitrocellulose membrane corresponding to PABPC1-RNA complexes was excised, Proteinase K (Roche) treated, and the RNA was Phenol extracted. The purified RNA was ligated to a 5′ adaptor, amplified, and sequencing libraries were constructed. Libraries generated from biological triplicates were individually bar coded, pooled, and sequenced on an Illumina HiSeq 2000 platform at the University of Pennsylvania Next Generation Sequencing Core (NGSC).
CLIP-seq read processing and alignment
Adapter sequences (GTGTCAGTCACTTCCAGCGGTCGTATGCCGTCTTCTGCTTG) were removed from raw reads and only trimmed reads were used for downstream analysis. Trimmed reads from each individual replicate CLIP-seq experiment were collapsed and mapped to the mouse genome (mm10) with Novoalign (Novocraft, Selagnor, MYS) with the parameters –t 85 –l 15 –s 1 –o Native –r None. Replicate experiments were merged and only uniquely mapped reads were used for subsequent analysis.
CIMS and cluster analysis
CIMS analysis was applied to identify single-nucleotide RBP-RNA interaction sites (as described; Moore et al. 2014). Briefly, deletion sites were extracted for each CLIP tag from novoalign output and a negative binomial test was used to assess significance. Sites with FDR < 0.001 were used for downstream analysis. To identify significant CLIP-seq clusters we used Pyicoclip (Althammer et al. 2011) with an mFDR < 0.01. Gencode annotation vM2 was used for all analyses.
mRNA-seq
mRNA-seq was performed as previously described (Elliott et al. 2013). Briefly, total RNA was purified from the MEL cell cultures (miRNeasy; Qiagen). Poly(A)+ RNA was isolated using oligo(dT) beads (Life Technologies). RNA was fragmented for 7 min using Fragmentation Reagent (Life Technologies). mRNA-seq libraries were then generated using the Illumina smRNA-seq kit (Illumina). Reads were trimmed with Cutadapt, mapped with Tophat2, and gene expression was quantified using HTseq (Trapnell et al. 2009; Martin 2011; Anders et al. 2015). Custom python scripts were used to calculate RPKM.
Motif analysis
Motif analysis was carried out by aligning CIMS sites and extracting sequences ±10 nt from each site. A custom script was used to create a position–weight matrix and used the R package SeqLogo to generate motif logos (Bembom et al. 2007). For hexanucleotide enrichment analysis, the equally sized regions in the exonic portion of the mRNA transcriptome were shuffled10 times and the prevalence of each hexanucleotide was calculated and compared with the abundance in ±15-nt CIMS regions. A position weight matrix was created from the top 20 hexanucleotides. For de novo motif discovery, MEME was used with a maximum width of 12 nt (Bailey et al. 2009).
Active polyadenlyation addition site identification
To identify high-confidence polyadenylation additions sites, we used a custom python script to filter raw mRNA-seq reads with at least 20 adenines at the 3′ end. We then removed these poly(A) stretches, mapped the remaining sequence to the mouse genome with Tophat2, and calculated the density of 3′ ends using bedtools genomecov (Quinlan and Hall 2010). Only sites with >10 reads per million were considered bona fide poly(A) sites.
Luciferase assays
5′ UTR or defined variants were cloned into a firefly luciferase vector. These constructs were transfected into NIH-3T3 cells in 12-well plate using Turbofect transfection reagent (Thermo Scientific). After 48-h luciferase activity was measured using Dual Luciferase assay kit (Promega) and the corresponding mRNA levels were quantified by qPCR.
CRISPR targeted deletion of PABPC1 5′ UTR region
gRNA oligos (ATAAATGTGTGTTCCGAGCCCGG) and (TCGGTCTCGGCTGCTTCACCGGG) were designed using the Broad Institute CRISPR design tool (http://www.crispr.mit.edu). After restrictions digest with BbsI they were cloned into px330 vector and then transfected into C2C12 cells using Lipofectamine 3000 (Life Technologies). After 72 h, purmomycin was added at 1 µg/mL to and colonies with targeted 5′ UTR deletions were selected for genomic DNA PCR.
Quantitative Western blotting
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer, and the following primary and secondary antibodies were used: rabbit anti-PABPC1 (Abcam), rabbit anti-actin (Bethyl), and goat-anti-rabbit IgG (Licor). Blots were visualized and scanned with Odyssey scanner and software (Li-Cor Bioscience).
Re-analysis of ribosome profiling data
Ribosome profiling data from harringtonine-treated mouse embryonic stem cells (mESCs) were obtained from GSE30839. We processed the ribosome profiling data as previously described (Ingolia et al. 2012). Briefly, reads were trimmed for adapter sequence (CTGTAGGCACCATCAATTCGTATGCCGTCTTCTGCTTGAA), filtered by mapping to mouse ribosomal RNA sequences. Filtered reads were mapped to the mouse transcriptome and genome using TopHat2 (Trapnell et al. 2009; Kim et al. 2013). Only mapped reads with no mismatches were used for further analysis. Aminoacyl-tRNA sites were identified as previously described (Ingolia et al. 2012).
Data access
We have created an interactive publicly available genome browser to house the data generated in this study at https://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=isilver1&hgS_otherUserSessionName=PABPC1_CLIP_Public.
DATA DEPOSITION
The data sets supporting the results of this article are available in the GEO repository, under accession number GSE69755.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Anastasios Vourekas (Mourelatos Laboratory, UPenn) for assistance with CLIP-seq design and reagents; Dr. Ihab Younis (Dreyfuss Laboratory, UPenn) for the luciferase plasmids; and members of the Gregory and Liebhaber laboratories for helpful discussions. This work was funded by RO1HL065449 to S.A.L.
Author contributions: H.K.K. and S.A.L. conceived of the study and designed the experiments. H.K.K., X.J., and I.M.S. performed the experiments. I.M.S. analyzed the data with input from H.K.K., S.A.L., and B.D.G. H.K.K., I.M.S., X.J., B.D.G., and S.A.L. interpreted the results and wrote the manuscript. All authors have read and approved the manuscript for publication.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.053447.115.
REFERENCES
- Adam SA, Nakagawa T, Swanson MS, Woodruff TK, Dreyfuss G. 1986. mRNA polyadenylate-binding protein: gene isolation and sequencing and identification of a ribonucleoprotein consensus sequence. Mol Cell Biol 6: 2932–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althammer S, González-Vallinas J, Ballaré C, Beato M, Eyras E. 2011. Pyicos: a versatile toolkit for the analysis of high-throughput sequencing data. Bioinformatics 27: 3333–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baejen C, Torkler P, Gressel S, Essig K, Söding J, Cramer P. 2014. Transcriptome maps of mRNP biogenesis factors define pre-mRNA recognition. Mol Cell 55: 745–757. [DOI] [PubMed] [Google Scholar]
- Bag J, Wu J. 1996. Translational control of poly(A)-binding protein expression. Eur J Biochem 237: 143–152. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37(Web Server issue):W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bembom O, Keles S, van der Laan MJ. 2007. Supervised detection of conserved motifs in DNA sequences with cosmo. Stat Appl Genet Mol Biol 6: Article8. [DOI] [PubMed] [Google Scholar]
- Blobel G. 1973. A protein of molecular weight 78,000 bound to the polyadenylate region of eukaryotic messenger RNAs. Proc Natl Acad Sci 70: 924–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CG, Dreyfuss G. 1994. Conserved structures and diversity of functions of RNA-binding proteins. Science 265: 615–621. [DOI] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. 2009. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460: 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melo Neto OP, Standart N, Martins de Sa C. 1995. Autoregulation of poly(A)-binding protein synthesis in vitro. Nucleic Acids Res 23: 2198–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melo Neto OP, Walker JA, Martins de Sa CM, Standart N. 2000. Levels of free PABP are limited by newly polyadenylated mRNA in early Spisula embryogenesis. Nucleic Acids Res 28: 3346–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moor CH, Meijer H, Lissenden S. 2005. Mechanisms of translational control by the 3′ UTR in development and differentiation. Semin Cell Dev Biol 16: 49–58. [DOI] [PubMed] [Google Scholar]
- Elliott R, Li F, Dragomir I, Chua MM, Gregory BD, Weiss SR. 2013. Analysis of the host transcriptome from demyelinating spinal cord of murine coronavirus-infected mice. PLoS One 8: e75346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F, Preiss T, Hentze MW. 2012. From cis-regulatory elements to complex RNPs and back. Cold Spring Harb Perspect Biol 4: a012245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good PJ, Abler L, Herring D, Sheets MD. 2004. Xenopus embryonic poly(A) binding protein 2 (ePABP2) defines a new family of cytoplasmic poly(A) binding proteins expressed during the early stages of vertebrate development. Genesis 38: 166–175. [DOI] [PubMed] [Google Scholar]
- Gorgoni B, Richardson WA, Burgess HM, Anderson RC, Wilkie GS, Gautier P, Martins JP, Brook M, Sheets MD, Gray NK. 2011. Poly(A)-binding proteins are functionally distinct and have essential roles during vertebrate development. Proc Natl Acad Sci 108: 7844–7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss DJ, Kleiman FE. 2013. Poly(A) binding proteins: are they all created equal? Wiley Interdiscip Rev RNA 4: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Sherman F. 1995. 3′-end-forming signals of yeast mRNA. Mol Cell Biol 15: 5983–5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornstein E, Git A, Braunstein I, Avni D, Meyuhas O. 1999. The expression of poly(A)-binding protein gene is translationally regulated in a growth-dependent fashion through a 5′-terminal oligopyrimidine tract motif. J Biol Chem 274: 1708–1714. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. 2012. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7: 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahvejian A, Svitkin YV, Sukarieh R, M'Boutchou MN, Sonenberg N. 2005. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev 19: 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kini HK, Kong J, Liebhaber SA. 2014. Cytoplasmic poly(A) binding protein C4 serves a critical role in erythroid differentiation. Mol Cell Biol 34: 1300–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleene KC, Wang MY, Cutler M, Hall C, Shih D. 1994. Developmental expression of poly(A) binding protein mRNAs during spermatogenesis in the mouse. Mol Reprod Dev 39: 355–364. [DOI] [PubMed] [Google Scholar]
- Mangus DA, Evans MC, Jacobson A. 2003. Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol 4: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17: 10–12. [Google Scholar]
- Melo EO, de Melo Neto OP, Martins de Sá C. 2003a. Adenosine-rich elements present in the 5′-untranslated region of PABP mRNA can selectively reduce the abundance and translation of CAT mRNAs in vivo. FEBS Lett 546: 329–334. [DOI] [PubMed] [Google Scholar]
- Melo EO, Dhalia R, Martins de Sa C, Standart N, de Melo Neto OP. 2003b. Identification of a C-terminal poly(A)-binding protein (PABP)-PABP interaction domain: role in cooperative binding to poly (A) and efficient cap distal translational repression. J Biol Chem 278: 46357–46368. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, Darnell RB. 2014. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat Protoc 9: 263–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayler O, Stratling W, Bourquin JP, Stagljar I, Lindemann L, Jasper H, Hartmann AM, Fackelmayer FO, Ullrich A, Stamm S. 1998. SAF-B protein couples transcription and pre-mRNA splicing to SAR/MAR elements. Nucleic Acids Res 26: 3542–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paasinen-Sohns A, Kaariainen E, Yin M, Järvinen K, Nummela P, Hölttä E. 2011. Chaotic neovascularization induced by aggressive fibrosarcoma cells overexpressing S-adenosylmethionine decarboxylase. Int J Biochem Cell Biol 43: 441–454. [DOI] [PubMed] [Google Scholar]
- Peixeiro I, Inácio Â, Barbosa C, Silva AL, Liebhaber SA, Romão L. 2012. Interaction of PABPC1 with the translation initiation complex is critical to the NMD resistance of AUG-proximal nonsense mutations. Nucleic Acids Res 40: 1160–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesole G, Liuni S, Grillo G, Saccone C. 1997. Structural and compositional features of untranslated regions of eukaryotic mRNAs. Gene 205: 95–102. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD. 2007. CPEB: a life in translation. Trends Biochem Sci 32: 279–285. [DOI] [PubMed] [Google Scholar]
- Sachs AB, Davis RW, Kornberg RD. 1987. A single domain of yeast poly(A)-binding protein is necessary and sufficient for RNA binding and cell viability. Mol Cell Biol 7: 3268–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist SJ, Thiel PR, Reiff DF, Lachance PE, Lasko P, Schuster CM. 2000. Postsynaptic translation affects the efficacy and morphology of neuromuscular junctions. Nature 405: 1062–1065. [DOI] [PubMed] [Google Scholar]
- Sladic RT, Lagnado CA, Bagley CJ, Goodall GJ. 2004. Human PABP binds AU-rich RNA via RNA-binding domains 3 and 4. Eur J Biochem 271: 450–457. [DOI] [PubMed] [Google Scholar]
- Smith RW, Blee TK, Gray NK. 2014. Poly(A)-binding proteins are required for diverse biological processes in metazoans. Biochem Soc Trans 42: 1229–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, König J, Hussain S, Zupan B, Curk T, Frye M, Ule J. 2012. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein–RNA interactions. Genome Biol 13: R67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabassum R, Jaiswal A, Chauhan G, Dwivedi OP, Ghosh S, Marwaha RK, Tandon N, Bharadwaj D. 2012. Genetic variant of AMD1 is associated with obesity in urban Indian children. PLoS One 7: e33162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarun SZ Jr, Sachs AB. 1995. A common function for mRNA 5′ and 3′ ends in translation initiation in yeast. Genes Dev 9: 2997–3007. [DOI] [PubMed] [Google Scholar]
- Theis M, Si K, Kandel ER. 2003. Two previously undescribed members of the mouse CPEB family of genes and their inducible expression in the principal cell layers of the hippocampus. Proc Natl Acad Sci 100: 9602–9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Hu J, Zhang H, Lutz CS. 2005. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res 33: 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck AC, Tollervey D. 2013. A transcriptome-wide atlas of RNP composition reveals diverse classes of mRNAs and lncRNAs. Cell 154: 996–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Hoshino S, Imataka H, Sonenberg N, Katada T. 2002. A novel role of the mammalian GSPT/eRF3 associating with poly(A)-binding protein in cap/poly(A)-dependent translation. J Biol Chem 277: 50286–50292. [DOI] [PubMed] [Google Scholar]
- Ule J, Jensen K, Mele A, Darnell RB. 2005. CLIP: a method for identifying protein–RNA interaction sites in living cells. Methods 37: 376–386. [DOI] [PubMed] [Google Scholar]
- Verlaet M, Deregowski V, Denis G, Humblet C, Stalmans MT, Bours V, Castronovo V, Boniver J, Defresne MP. 2001. Genetic imbalances in preleukemic thymuses. Biochem Biophys Res Commun 283: 12–18. [DOI] [PubMed] [Google Scholar]
- Wahle E. 1991. A novel poly(A)-binding protein acts as a specificity factor in the second phase of messenger RNA polyadenylation. Cell 66: 759–768. [DOI] [PubMed] [Google Scholar]
- Wang Z, Kiledjian M. 2000. The poly(A)-binding protein and an mRNA stability protein jointly regulate an endoribonuclease activity. Mol Cell Biol 20: 6334–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SE, Hillner PE, Vale RD, Sachs AB. 1998. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 2: 135–140. [DOI] [PubMed] [Google Scholar]
- Williams KR, Konigsberg WH. 1991. Identification of amino acid residues at interface of protein-nucleic acid complexes by photochemical cross-linking. Methods Enzymol 208: 516–539. [DOI] [PubMed] [Google Scholar]
- Wilusz CJ, Gao M, Jones CL, Wilusz J, Peltz SW. 2001. Poly(A)-binding proteins regulate both mRNA deadenylation and decapping in yeast cytoplasmic extracts. RNA 7: 1416–1424. [PMC free article] [PubMed] [Google Scholar]
- Wormington M, Searfoss AM, Hurney CA. 1996. Overexpression of poly(A) binding protein prevents maturation-specific deadenylation and translational inactivation in Xenopus oocytes. EMBO J 15: 900–909. [PMC free article] [PubMed] [Google Scholar]
- Yanagiya A, Delbes G, Svitkin YV, Robaire B, Sonenberg N. 2010. The poly(A)-binding protein partner Paip2a controls translation during late spermiogenesis in mice. J Clin Invest 120: 3389–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Darnell RB. 2011. Mapping in vivo protein–RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol 29: 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}