

In this study, Furth et al. identify a novel link between p53 and the LATS1 and LATS2 kinases, core components of the Hippo tumor suppressor pathway. They show that silencing of LATS1 and LATS2 in nontransformed mammary epithelial cells changes p53 function, resulting in a partially altered p53 conformation and transcriptional output, providing novel insight into the regulation of p53 in tumor cells.
Keywords: TP53, COX-2, cancer, cell migration, NFKB2, homeostasis
Abstract
p53 is a pivotal tumor suppressor and a major barrier against cancer. We now report that silencing of the Hippo pathway tumor suppressors LATS1 and LATS2 in nontransformed mammary epithelial cells reduces p53 phosphorylation and increases its association with the p52 NF-κB subunit. Moreover, it partly shifts p53's conformation and transcriptional output toward a state resembling cancer-associated p53 mutants and endows p53 with the ability to promote cell migration. Notably, LATS1 and LATS2 are frequently down-regulated in breast cancer; we propose that such down-regulation might benefit cancer by converting p53 from a tumor suppressor into a tumor facilitator.
The Hippo pathway, originally found to regulate organ size in Drosophila, inhibits tumorigenesis in mammals by regulating numerous processes (Oren and Aylon 2013). At the core of the pathway are the MST1/2 and LATS1/2 kinases, which inhibit the downstream effectors YAP and TAZ (Zhao et al. 2010) that promote proliferation, oncogenic transformation, and epithelial–mesenchymal transition (EMT) (Moroishi et al. 2015). Some functions of YAP and TAZ are independent of LATS1/2 (jointly referred to here as LATS) (Dupont et al. 2011; Sorrentino et al. 2014). Similarly, LATS kinases may exert functions unrelated to YAP/TAZ. LATS kinases engage in a positive cross-talk with the tumor suppressor p53 under stress and in stem cells (Iida et al. 2004; Aylon et al. 2006, 2010, 2014). Thus, beyond inhibiting YAP/TAZ, LATS possess additional tumor suppressive functions.
p53 is a transcription factor orchestrating numerous processes impinging on cell fate; e.g., growth arrest, senescence, and apoptosis (Bieging et al. 2014). The network governed by p53 operates primarily through differential regulation of target gene expression. Recently, p53 has emerged as a regulator of homeostasis (Vousden and Prives 2009; Bieging et al. 2014). Intriguingly, cancer cells may benefit from p53-mediated adaptive prosurvival processes (Jänicke et al. 2008; Scherz-Shouval et al. 2010).
Point mutations, ablating wild-type p53 function, are frequent in human cancer. Such mutations may disrupt p53–DNA interactions by altering amino acids in the p53 DNA-binding domain that contact DNA directly or may drive conformational changes in p53 (Muller and Vousden 2013). These mutants not only lose tumor suppressor capability but also acquire oncogenic gain of function (GOF), augmenting invasion and tumorigenesis. It is commonly believed that mutant p53 GOF entails new activities not shared with wild-type p53. However, some GOF effects may be due to fixation of pre-existing wild-type p53 activities normally manifested only under particular biological conditions. Genetic and epigenetic aberrations occurring in cancer may affect the delicate regulatory wiring governing p53, shifting it toward a mutant-like state.
We now report that compromised LATS expression, seen in many tumors, alters wild-type p53 to induce migration partly through up-regulation of PTGS2 (prostaglandin-endoperoxide synthase 2; also known as COX-2). LATS knockdown reduces p53 phosphorylation and changes p53's protein interactome, increasing its binding to the NF-κB p52 subunit. In addition, it partially alters p53's conformation and favors a p53 transcriptional program reminiscent of cancer-associated p53 mutants. Hence, by reducing LATS expression, tumors that retain wild-type p53 may convert it from a tumor suppressor to a tumor facilitator.
Results and Discussion
LATS down-regulation reduces p53 phosphorylation
Human breast tumors display significant down-regulation of LATS expression relative to matched normal tissue (The Cancer Genome Atlas [TCGA] breast invasive carcinoma data set) (Supplemental Fig. S1A). Given the positive cross-talk between LATS kinases and p53 (Iida et al. 2004; Aylon et al. 2006, 2010, 2014), we asked whether LATS impacts p53 activity in mammary epithelium. siRNA-mediated knockdown of LATS1 and LATS2 (siLATS1/2) (Supplemental Fig. S1B) did not significantly alter p53 levels in nontransformed MCF10A mammary epithelial cells (Fig. 1A, left panel). p53 is regulated by post-translational modifications (PTMs), including multiple phosphorylations (Meek and Anderson 2009). To assess p53 phosphorylation, we used Phos-tag gels to decrease the mobility of phosphorylated p53. Notably, LATS down-regulation augmented the faster-migrating p53 band (Fig. 1A [right panel], B), confirmed by phosphatase treatment to be hypophosphorylated (Supplemental Fig. S1C). Silencing either LATS1 or LATS2 alone also reduced p53 phosphorylation (Supplemental Fig. S1D). Of note, acute p53 activation by the radiomimetic agent neocarzinostatin (NCS) markedly increased the portion of phosphorylated p53 in both control and LATS-depleted cells, although a mild impact of LATS depletion was retained (Supplemental Fig. S1E). Similar effects were seen also in immortalized human bronchial epithelial cells (HBEC3-KT) and human breast adenocarcinoma MCF7 cells (Supplemental Fig. S1F). Thus, LATS down-regulation compromises p53 phosphorylation.
Figure 1.
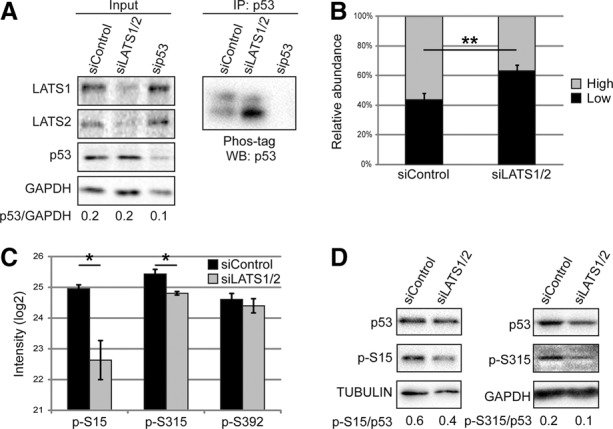
Silencing of LATS1/2 reduces p53 phosphorylation. (A) MCF10A cells were transfected with the indicated siRNAs and subjected to immunoprecipitation (IP) with p53 antibodies (PAb421). (Left panel) Five percent of each extract was taken as input and subjected to standard SDS-PAGE and Western blot (WB). (Right panel) Immunoprecipitation samples were separated by 30 µM Phos-tag SDS-PAGE followed by Western blot analysis with p53-HRP antibody. (B) Quantification of the relative abundance (percentage of total p53) of the upper (high) and lower (low) p53 bands observed with Phos-tag SDS-PAGE. Mean ± SEM from seven experiments. (**) P-value < 0.01. (C) p53 immunoprecipitated with a mix of p53-specific antibodies (PAb1801, DO-1, and PAb421) from MCF10A cells transfected as in A was subjected to mass spectrometry analysis. Mean intensity of phosphorylated peptides ± SEM from three experiments. (*) P-value < 0.05. (D) Lysates of MCF10A cells transfected with the indicated siRNAs were subjected to Western blot analysis with antibodies specific for p53 phosphorylated on either Ser15 (p-S15) or Ser315 (p-S315).
Mass spectrometry (MS) analysis of MCF10A p53 revealed that LATS knockdown caused a significant decrease in Ser15 and Ser315 phosphorylation (Fig. 1C), confirmed by analysis with phospho-specific antibodies (Fig. 1D). Notably, YAP/TAZ knockdown did not rescue these changes (Supplemental Fig. S1G).
LATS down-regulation affects the p53 interactome
PTMs may dictate interaction partners. Indeed, MS analysis revealed increased binding of several proteins to p53 upon LATS knockdown (Fig. 2A). These included promyelocytic leukemia (PML) protein, known to interact and colocalize with p53 (Fogal et al. 2000), as well as products of the NFKB2 gene (Fig. 2A) encoding p52, a member of the NF-κB transcription factor family produced by proteolytic cleavage of its precursor, p100. The increase was specific to p52 (Fig. 2B) and was not observed for the p100-unique portion of the precursor (Supplemental Fig. S2). To test whether this interaction is affected by p53 phosphorylation, we expressed wild-type p53 or p53 mutants S15A and S315A in p53-null H1299 cells followed by immunoprecipitation with anti-p52 antibodies. Notably, although the portion of p53 immunoprecipitated with p52 was relatively small, p53 S315A was selectively, albeit modestly, enriched in the immunoprecipitation (Fig. 2C), suggesting that it bound endogenous p52 more strongly than wild-type p53. Hence, decreased p53 phosphorylation upon LATS down-regulation may increase p53 binding to p52 and to additional partners.
Figure 2.
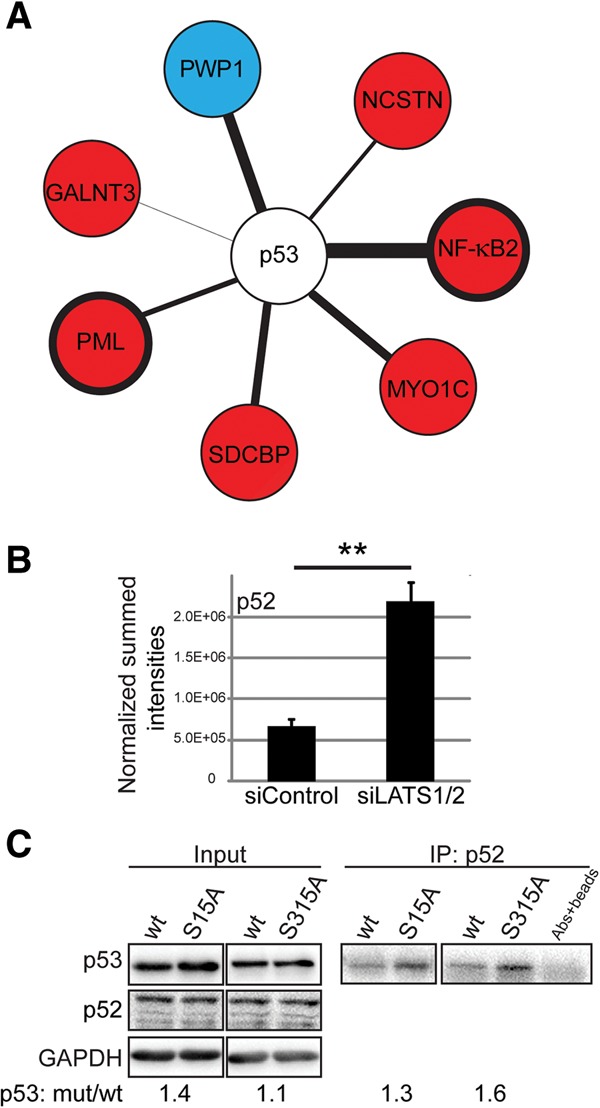
LATS1/2 depletion changes the p53 interactome. (A) Lysates of MCF10A cells transfected with either siControl or siLATS1/2 were subjected to p53 immunoprecipitation. MS analysis of three experiments (same as in Fig. 1C) identified putative p53 interactors whose association with p53 was either up-regulated (red) or down-regulated (blue) significantly upon LATS knockdown. The thickness of the connecting line corresponds to test difference, with a thicker line representing a more robust difference. Welch's t-test, false discovery rate (FDR) ≤ 7%, S0 = 0.1. All expression differences are P-value < 0.02. (B) The sum of intensities of peptides mapping specifically to the NF-κB subunit p52, obtained from the MS analysis in A, was normalized to molecular weight. Mean ± SEM. (**) P-value < 0.01. (C) H1299 cells were transfected with the indicated p53 expression plasmids. Five percent of each extract was retained as input (left panel), and the rest was subjected to immunoprecipitation with anti-p52 antibody (right panel). Coimmunoprecipitation of p53 and p52 was visualized using p53-HRP antibody.
LATS down-regulation favors a mutant p53-like functional state
To test whether LATS down-regulation affects p53's transcriptional activity, we conducted RNA sequencing (RNA-seq) analysis in MCF10A cells transfected with siRNA against LATS1/2 alone, p53 alone, or LATS1/2 and p53 together. Two independent MCF10A batches provided biological replicates. Seven-hundred-thirty-eight genes were differentially expressed between control and siLATS cells (Supplemental Table S1); in 320 of those, the effect of LATS depletion on their differential expression became less pronounced when p53 was simultaneously silenced (Fig. 3A, left panel for siLATS decreased genes, right panel for siLATS increased genes, cf. columns v + vi and vii + viii; see also Supplemental Table S2). Comparing the mean expression of these genes in siLATS cells (Fig. 3A, columns v + vi and vii + viii) and control cells (Fig. 3A, columns i + ii and iii + iv) revealed that LATS depletion rendered them more responsive to p53 (Fig. 3A, top panel). Interestingly, these 320 genes were not significantly enriched for p53-related pathways (Kyoto Encyclopedia of Genes and Genomes [KEGG] and BioCarta), whereas genes differentially expressed upon p53 silencing but not affected by siLATS were significantly enriched for p53 and apoptosis pathways (Supplemental Fig. S3A,B; Supplemental Table S3). Thus, while canonical wild-type p53 target genes were unaffected by LATS status, LATS down-regulation expanded the p53 transcriptional repertoire. Remarkably, the genes in Figure 3A overlapped significantly (P-value < 0.05) with mutant p53-regulated genes in breast cancer cells (Fig. 3A, black bars at the right of each matrix; Supplemental Table S2; Adorno et al. 2009), suggesting that LATS depletion may drive wild-type p53 toward a mutant-like functional state. Furthermore, the 320 genes were enriched for gene ontology (GO) terms associated with adhesion, response to wounding, and cell migration (Fig. 3B). The expression changes of some of those genes and their increased p53 responsiveness upon LATS down-regulation were validated by quantitative RT–PCR (qRT–PCR) analysis (Supplemental Fig. S3C). Notably, the genes rendered responsive to p53 by LATS silencing (Fig. 3A, right matrix) were enriched for NF-κB motifs (Subramanian et al. 2005) near their transcriptional start sites (TSSs; P-value < 0.001) (Supplemental Table S2, gray bars at the right side).
Figure 3.
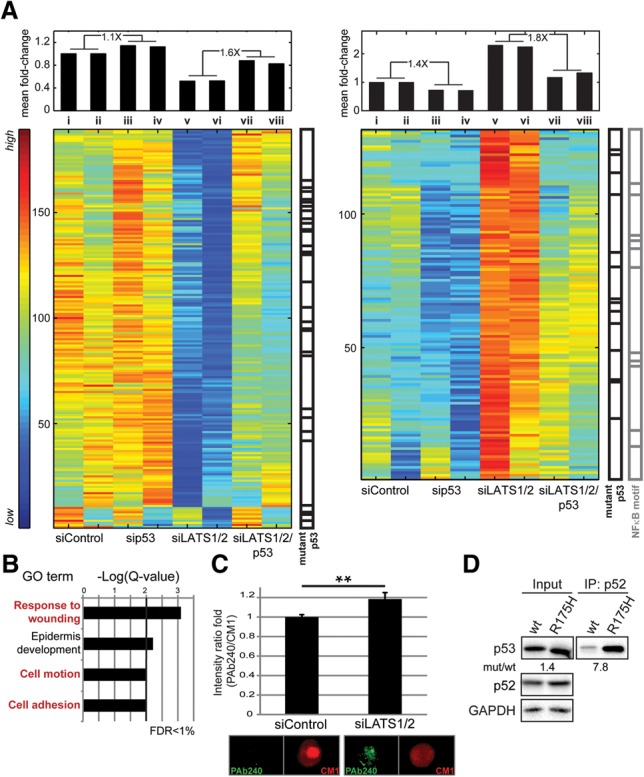
LATS1/2 down-regulation enforces mutant p53-like characteristics. (A) SPIN-ordered expression matrix of genes positively regulated by LATS1/2 and up-regulated upon additional knockdown of p53 (left matrix) or repressed by LATS1/2 and down-regulated upon additional knockdown of p53 (right matrix). Colors indicate expression levels after centering and normalizing each gene (row). The mean fold change relative to control of all genes presented in each column is shown above the matrix. The ratio between pertinent averaged fold change values is indicated at the top. Overlap with mutant p53 target genes (black bars) and genes with NF-κB motifs adjacent to their TSSs (gray bars) is also indicated. Hypergeometric test, P-value < 0.05. (B) GO term enrichment analysis for the genes in A. FDR ≤ 1%. (C) MCF10A cells transfected with the indicated siRNAs were stained with PAb240 and CM1 antibodies and subjected to imaging flow cytometry (ImageStreamX). For each cell, the ratio between the intensity of the PAb240 signal and the CM1 signal was calculated. Fold change of the mean ratio ± SEM. (**) P-value < 0.01. Representative PAb240-positive and PAb240-negative cells are also shown. (D) H1299 cells were transfected with the indicated p53 expression plasmids. Five percent of each extract was retained as input (left panel), and the rest was subjected to immunoprecipitation with anti-p52 antibodies (right panel). Coimmunoprecipitation of p53 and p52 was visualized using p53-HRP antibody.
Our analysis suggested that LATS down-regulation might promote a noncanonical p53 transcriptional program reminiscent of mutant p53. We therefore employed imaging flow cytometry (ImageStreamX) in conjunction with PAb240, a monoclonal antibody recognizing only conformational p53 mutants (Gannon et al. 1990), and CM1, a polyclonal antibody recognizing all p53 molecules. Remarkably, siLATS elicited a mild yet significant increase in PAb240 reactivity (Fig. 3C), implying that some wild-type p53 molecules had undergone a conformational shift. Moreover, the percentage of PAb240+ cells was also increased mildly, although not as much as in MCF10A cells stably expressing the p53R175H mutant (Supplemental Fig. S3D).
NFKB2 is a mutant p53 target gene (Scian et al. 2005) contributing to mutant p53 GOF (Yeudall et al. 2012). Notably, analysis of H1299 cells transiently overexpressing either wild-type p53 or p53R175H revealed preferential p52 binding to mutant p53 (Fig. 3D), further suggesting that LATS silencing renders wild-type p53 more similar to mutant p53.
LATS down-regulation promotes p53-dependent cell migration
As the altered p53 transcriptional program enforced by siLATS is enriched for terms related to cell migration (Fig. 3B), we studied the impact of LATS and p53 depletion on the migration of MCF10A cells, employing a real-time cell analyzer (RTCA). Consistent with earlier studies (Zhang et al. 2008; Aylon et al. 2010), siLATS augmented migration (Fig. 4A,B). Remarkably, simultaneous p53 depletion (Supplemental Fig. S4A) reduced migration nearly to control levels (Fig. 4), implying that the promigratory effect of LATS down-regulation is p53 dependent, as confirmed with an additional p53 siRNA (Supplemental Fig. S4B). Moreover, whereas ectopic p53R175H expression increased migration (Supplemental Fig. S4C), depletion of wild-type p53 alone had no significant effect (Fig. 4). Thus, a mutant-like state of p53, elicited by LATS down-regulation, promotes migration.
Figure 4.
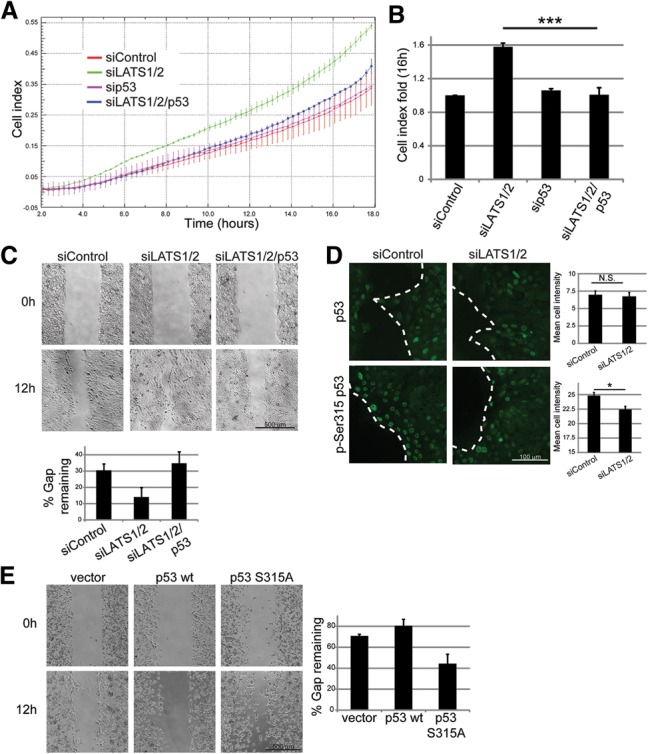
p53 promotes cell migration in LATS-depleted cells. (A) MCF10A cells were transfected with the indicated siRNAs, and migration toward EGF-containing medium was measured. Measurements were performed automatically every 15 min. Each point represents mean ± SD of two technical replicates. (B) Migration cell index fold (relative to control cells) 16 h after start of the migration assay. Mean ± SEM from three to five experiments performed as in A. (***) P-value < 0.001. (C) MCF10A cells were transfected with the indicated siRNAs and transferred to 12-well plates containing ibidi culture inserts. The next day, inserts were removed, and cells were allowed to migrate. (Top panel) Images covering the total gap area of two to three replicates were taken over a period of 12 h. (Bottom panel) Gap width within each field was quantified, and the remaining gap (percentage of initial width at time 0; mean ± SEM) was calculated. (D) Cells treated as in C were seeded on coverslips attached to ibidi culture inserts and subjected to immunostaining for total p53 (top; mix of PAb1801 and DO-1) and for p-Ser315 p53 (bottom) 12 h into the migration assay. The dashed line indicates the border between the leading edge of migrating cells and the gap. Mean staining intensity of particles within 100 µm from the edge was quantified, and mean intensity ± SEM for all cells in each field is shown in the right panel. (E) H1299 cells were transfected with the indicated p53 expression plasmids, and migration assay was performed as in C.
Similar effects were observed in a “wound healing” assay (Fig. 4C; Supplemental Fig. S4D). Overall p53 staining was similar in control and siLATS cells (Fig. 4D, top). However, while control cells showed robust uniform multilayered pSer315 staining near the gap, LATS-depleted cells displayed overall weaker and more heterogeneous staining (Fig. 4D, bottom). Moreover, while wild-type p53 overexpression in H1299 slightly inhibited gap closure, p53S315A overexpression augmented migration (Fig. 4E). Thus, LATS-dependent p53 Ser315 phosphorylation might restrict cell migration. Together, our findings support a novel role for wild-type p53 in inducing cell migration upon LATS down-regulation.
p53-dependent migration involves noncanonical p53 target genes, including PTGS2
PTGS2 is a key enzyme in prostaglandin biosynthesis, overexpressed and linked to tumor aggressiveness in various solid tumors (Dannenberg and Subbaramaiah 2003). Of note, PTGS2 is one of the genes comprising the right panel of Figure 3A, implying that p53 is required for its optimal induction in LATS-deficient cells, as confirmed by qRT–PCR analysis of PTGS2 mRNA and pre-mRNA (Fig. 5A; Supplemental Fig. S5A). Interestingly, although acute activation by NCS induced robust p53 accumulation (Supplemental Fig. S1E) and up-regulation of canonical p53 target genes such as p21 and BTG2 (Supplemental Fig. S5B), it only mildly affected PTGS2 expression in control and LATS-depleted cells (Fig. 5B), indicating that up-regulation of PTGS2 by p53 upon LATS depletion is distinct from p53 activation by genotoxic stress.
Figure 5.
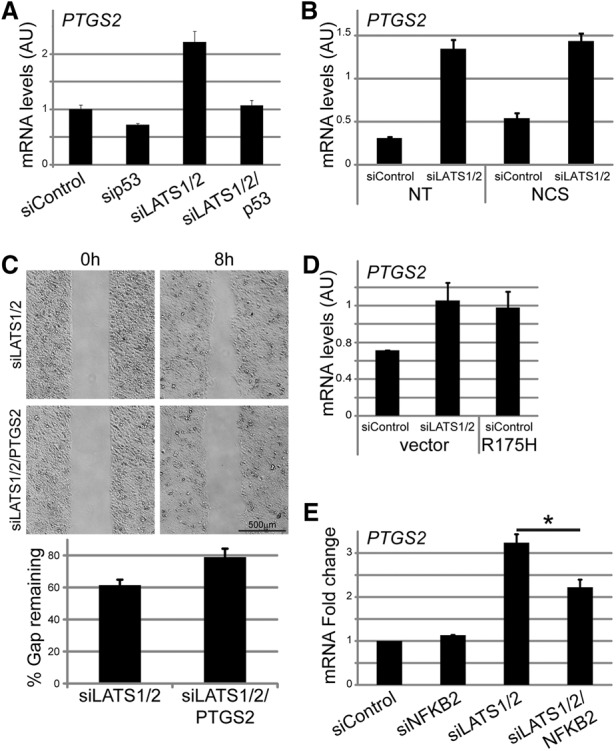
A p53 and p52-dependent increase in PTGS2 expression upon LATS silencing contributes to cell migration. (A) RNA from MCF10A cells transfected with the indicated siRNAs was subjected to PTGS2 mRNA quantification by qRT–PCR. Mean ± SD from two technical replicates. (B) RNA from MCF10A cells transfected with the indicated siRNAs and treated with 200 ng/mL NCS for 2 h was subjected to PTGS2 mRNA quantification by qRT–PCR. Mean ± SD from two technical replicates. (C) MCF10A cells transfected with the indicated siRNAs were subjected to migration analysis as in Figure 4C. Images covering the total gap area were taken over a period of 8 h (top panel) and subjected to gap width quantification (bottom panel). (D) MCF10A cells infected with either control retrovirus or retrovirus encoding p53R175H were transfected with the indicated siRNAs. RNA was extracted, and PTGS2 mRNA was quantified by qRT–PCR. Mean ± SD of two technical replicates. (E) MCF10A cells were transfected with the indicated siRNAs, and PTGS2 mRNA was quantified by qRT–PCR. Mean fold change ± SEM from three experiments.
PTGS2 has been implicated in cell migration and invasion (Singh et al. 2005). Indeed, cells transfected with siLATS together with siPTGS2 (Supplemental Fig. S5C) migrated more slowly than cells transfected with just siLATS (Fig. 5C; Supplemental Fig. S5D), suggesting that the increased migration upon LATS depletion relies partly on PTGS2.
In agreement with our in silico analysis of putative mutant p53 target genes (Fig. 3A; Supplemental Table S2), ectopic expression of p53R175H in MCF10A cells up-regulated PTGS2 mRNA (Fig. 5D). Furthermore, silencing of endogenous mutant p53 in the ductal mammary carcinoma cell line HCC1143 reduced PTGS2 expression (Supplemental Fig. S5E). Interestingly, PTGS2 is also a transcriptional target of NF-κB (Nakao et al. 2000), suggesting that the increased p53–p52 interaction might contribute to PTGS2 induction in LATS-depleted cells. Indeed, NFKB2 knockdown (Supplemental Fig. S5F) reduced PTGS2 mRNA in siLATS but not siControl cells (Fig. 5E). Thus, upon LATS down-regulation, p52 and wild-type p53 might cooperate to up-regulate mutant p53 target genes such as PTGS2, promoting migration.
p53 mutations drive tumor development (Muller and Vousden 2013). However, many tumors retain wild-type p53. We show that LATS down-regulation, observed in breast cancer and other tumors, propels wild-type p53 into noncanonical activities, which might contribute to cancer. TCGA analysis did not reveal a significant correlation between LATS expression and p53 mutations in breast cancer (data not shown), suggesting that mutant p53 may provide the tumors with benefits extending beyond those offered by “altered” wild-type p53. However, it is noteworthy that cancers with low TP53 mutation rates, including breast cancer as a group, tend to down-regulate LATS (Supplemental Fig. S5G).
Our findings reinforce the idea that wild-type p53, a highly flexible protein (Milner 1995), can explore a wide landscape of functional states in a context-dependent manner (Milner 1995; Muller and Vousden 2013). We propose that the state of wild-type p53 can be toggled by other tumor suppressor pathways, exemplified by LATS. The “pseudomutant” wild-type p53 emerging in LATS-compromised cells has distinctive features, including reduced phosphorylation on Ser15 and Ser315 and altered protein–protein interactions. Notably, the altered wild-type p53 drives the preferential expression of noncanonical promigratory genes, regulated also by cancer-associated p53 mutants.
Recently, the CCT/TRiC chaperonin complex was shown to be required for maintaining the wild-type p53 conformation (Trinidad et al. 2013); mutants incapable of CCT binding promote cancer cell migration and invasion. Likewise, embryonic stem cells harboring mutant p53 were found to maintain genomic integrity by forcing the mutant p53 to adopt a wild-type conformation (Rivlin et al. 2014).
Although LATS1 and LATS2 are bona fide Ser/Thr protein kinases, we could not detect direct phosphorylation of p53 by them in vitro (data not shown). Furthermore, p53 does not harbor a LATS consensus site. Hence, the effect on p53 phosphorylation is probably indirect. One candidate is Aurora kinase A (AURKA), which phosphorylates LATS2 and physically interacts with both LATS kinases (Toji et al. 2004; Yabuta et al. 2011); AURKA phosphorylates p53 on S315 (Katayama et al. 2004), and its binding to p53 has been suggested to play a role in maintaining mutant p53 in a wild-type-like conformation (Rivlin et al. 2014). However, other kinases that target p53 Ser315, including various cyclin-dependent kinases and GSK3β, as well as stress kinases such as ATM and ATR that target Ser15 (Bode and Dong 2004) cannot be excluded. LATS depletion may also alter the activity-pertinent phosphatases.
We propose that, under conditions of tissue integrity, when LATS kinases are constitutively active, this might maintain p53 in its canonical tumor-suppressive state. In contrast, during processes involving cell migration, p53 might transiently assume an altered state that favors such processes. As part of p53's emerging role as mediator of cell and tissue homeostasis, its involvement in processes such as EMT and wound healing has been described (Schoppy et al. 2010; Rinon et al. 2011). However, whereas some studies suggest that p53 promotes tissue renewal and wound healing (Schoppy et al. 2010), others indicate that it antagonizes these processes (Nakade et al. 2004). Conceivably, temporary suppression of LATS kinases (e.g., following disruption of tissue architecture) might enable p53 to assume an altered state compatible with tissue regeneration. Such a reversible state might become constitutive in tumors that down-regulate LATS; e.g., through promoter hypermethylation (Takahashi et al. 2005; Jiang et al. 2006).
The impact of LATS on p53 may represent a more general paradigm in which p53 is tuned to a broad network of incoming signals that toggle its functional state. Notably, p53 cooperates with TGF-β-activated Smads (in a manner involving altered p53 phosphorylation) to promote specific cell fates (Cordenonsi et al. 2007). Furthermore, wild-type p53 can acquire a mutant-like conformation in cells exposed to growth factors (Zhang and Deisseroth 1994) or hypoxia (Gogna et al. 2012), which reduces many p53 PTMs (Gogna et al. 2012). The signals that modulate the functional state of p53 may become constitutive as a consequence of pertinent cancer-associated genetic and epigenetic alterations, as suggested by the ability of wild-type p53 to promote migration and invasion of cells from mouse ovarian tumors induced by Pten deletion and mutant Kras (Mullany et al. 2012). It will be of interest to identify additional p53-modulating mechanisms and study their impact on p53 activity in cancers that retain genetically intact TP53.
Materials and methods
MS
For MS analysis of p53, a mixture of PAb1801, DO-1, and PAb421 antibodies was used, and proteins were released from beads using on-beads trypsin digestion. Further details are in the Supplemental Material.
The data have been deposited to the ProteomeXchange Consortium via the Proteomics Identifications Database (PRIDE) partner repository with the data set identifier PXD003120.
RNA expression analysis
RNA from two independent batches of MCF10A cells was subjected to high-throughput RNA-seq. Library preparation, high-throughput sequencing, and analysis are detailed in the Supplemental Material.
The data have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are available through GEO series accession number GSE74493.
Gene expression matrices
A gene was defined as differentially expressed between two conditions if the fold change exceeded 1.5 in both cell batches. The matrices in Figure 3A contain genes differentially expressed in siLATS1/2 samples compared with the control (738 genes), whose expression in siLATS1/2 samples was higher (for the up-regulated genes) or lower (for the down-regulated genes) by at least 1.2-fold relative to siLATS1/2/p53 samples (320 genes) in both cell batches.
Supplementary Material
Acknowledgments
We are grateful to S. Gilad, D. Zalcenstein, S. Saban, and M. Chemla from the Nancy and Stephen Grand Israel National Center for Personalized Medicine for help with RNA-seq, and V. Krizhanovsky for RTCA. We thank C. Prives for p53 expression plasmids, and V. Gorgoulis for HBEC3-KT cells. This work was supported in part by a Center of Excellence grant (1779/11) from the Israel Science Foundation, and research grants from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the Robert Bosch Stiftung, the Estate of John Hunter, the M.D. Moross Institute for Cancer Research at the Weizmann Institute (to M.O.), the Leir Charitable Foundation, the German Research Foundation (Deutsch-Israelische Projektkooperation; to E.D.), and the Israel Cancer Research Fund (to T.G.). M.O. is incumbent of the Andre Lwoff chair in molecular biology. E.D. is incumbent of the Henry J. Leir Professorial Chair.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.268185.115.
References
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, et al. 2009. A mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137: 87–98. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. 2006. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev 20: 2687–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Ofir-Rosenfeld Y, Yabuta N, Lapi E, Nojima H, Lu X, Oren M. 2010. The Lats2 tumor suppressor augments p53-mediated apoptosis by promoting the nuclear proapoptotic function of ASPP1. Genes Dev 24: 2420–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Sarver A, Tovy A, Ainbinder E, Oren M. 2014. Lats2 is critical for the pluripotency and proper differentiation of stem cells. Cell Death Differ 21: 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging KT, Mello SS, Attardi LD. 2014. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 14: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode AM, Dong Z. 2004. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 4: 793–805. [DOI] [PubMed] [Google Scholar]
- Cordenonsi M, Montagner M, Adorno M, Zacchigna L, Martello G, Mamidi A, Soligo S, Dupont S, Piccolo S. 2007. Integration of TGF-β and Ras/MAPK signaling through p53 phosphorylation. Science 315: 840–843. [DOI] [PubMed] [Google Scholar]
- Dannenberg AJ, Subbaramaiah K. 2003. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell 4: 431–436. [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183. [DOI] [PubMed] [Google Scholar]
- Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C, Del Sal G. 2000. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J 19: 6185–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon JV, Greaves R, Iggo R, Lane DP. 1990. Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J 9: 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogna R, Madan E, Kuppusamy P, Pati U. 2012. Re-oxygenation causes hypoxic tumor regression through restoration of p53 wild-type conformation and post-translational modifications. Cell Death Dis 3: e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida S-i, Hirota T, Morisaki T, Marumoto T, Hara T, Kuninaka S, Honda S, Kosai K-i, Kawasuji M, Pallas DC, et al. 2004. Tumor suppressor WARTS ensures genomic integrity by regulating both mitotic progression and G1 tetraploidy checkpoint function. Oncogene 23: 5266–5274. [DOI] [PubMed] [Google Scholar]
- Jänicke RU, Sohn D, Schulze-Osthoff K. 2008. The dark side of a tumor suppressor: anti-apoptotic p53. Cell Death Differ 15: 959–976. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Li X, Hu J, Zhou W, Jiang Y, Li G, Lu D. 2006. Promoter hypermethylation-mediated down-regulation of LATS1 and LATS2 in human astrocytoma. Neurosci Res 56: 450–458. [DOI] [PubMed] [Google Scholar]
- Katayama H, Sasai K, Kawai H, Yuan Z-M, Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA, Sen S. 2004. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet 36: 55–62. [DOI] [PubMed] [Google Scholar]
- Meek DW, Anderson CW. 2009. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol 1: a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner J. 1995. Flexibility: the key to p53 function? Trends Biochem Sci 20: 49–51. [DOI] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, Guan K-L. 2015. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullany LK, Liu Z, King ER, Wong K-K, Richards JS. 2012. Wild-type tumor repressor protein 53 (Trp53) promotes ovarian cancer cell survival. Endocrinology 153: 1638–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PAJ, Vousden KH. 2013. p53 mutations in cancer. Nat Cell Biol 15: 2–8. [DOI] [PubMed] [Google Scholar]
- Nakade K, Zheng H, Ganguli G, Buchwalter G, Gross C, Wasylyk B. 2004. The tumor suppressor p53 inhibits Net, an effector of Ras/extracellular signal-regulated kinase signaling. Mol Cell Biol 24: 1132–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao S, Ogata Y, Shimizu-Sasaki E, Yamazaki M, Furuyama S, Sugiya H. 2000. Activation of NFκB is necessary for IL-1β-induced cyclooxygenase-2 (COX-2) expression in human gingival fibroblasts. Mol Cell Biochem 209: 113–118. [DOI] [PubMed] [Google Scholar]
- Oren M, Aylon Y. 2013. The Hippo signaling pathway and cancer. Springer Science & Business Media, New York. [Google Scholar]
- Rinon A, Molchadsky A, Nathan E, Yovel G, Rotter V, Sarig R, Tzahor E. 2011. p53 coordinates cranial neural crest cell growth and epithelial–mesenchymal transition/delamination processes. Development 138: 1827–1838. [DOI] [PubMed] [Google Scholar]
- Rivlin N, Katz S, Doody M, Sheffer M, Horesh S, Molchadsky A, Koifman G, Shetzer Y, Goldfinger N, Rotter V, et al. 2014. Rescue of embryonic stem cells from cellular transformation by proteomic stabilization of mutant p53 and conversion into WT conformation. Proc Natl Acad Sci 111: 7006–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. 2010. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci 107: 18511–18516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppy DW, Ruzankina Y, Brown EJ. 2010. Removing all obstacles: a critical role for p53 in promoting tissue renewal. Cell Cycle 9: 1313–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scian MJ, Stagliano KER, Anderson MAE, Hassan S, Bowman M, Miles MF, Deb SP, Deb S. 2005. Tumor-derived p53 mutants induce NF-κB2 gene expression. Mol Cell Biol 25: 10097–10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Berry JA, Shoher A, Ramakrishnan V, Lucci A. 2005. COX-2 overexpression increases motility and invasion of breast cancer cells. Int J Oncol 26: 1393–1399. [PubMed] [Google Scholar]
- Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S, et al. 2014. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol 16: 357–366. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Miyoshi Y, Takahata C, Irahara N, Taguchi T, Tamaki Y, Noguchi S. 2005. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin Cancer Res 11: 1380–1385. [DOI] [PubMed] [Google Scholar]
- Toji S, Yabuta N, Hosomi T, Nishihara S, Kobayashi T, Suzuki S, Tamai K, Nojima H. 2004. The centrosomal protein Lats2 is a phosphorylation target of Aurora-A kinase. Genes Cells 9: 383–397. [DOI] [PubMed] [Google Scholar]
- Trinidad AG, Muller PAJ, Cuellar J, Klejnot M, Nobis M, Valpuesta JM, Vousden KH. 2013. Interaction of p53 with the CCT complex promotes protein folding and wild-type p53 activity. Mol Cell 50: 805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137: 413–431. [DOI] [PubMed] [Google Scholar]
- Yabuta N, Mukai S, Okada N, Aylon Y, Nojima H. 2011. The tumor suppressor Lats2 is pivotal in Aurora A and Aurora B signaling during mitosis. Cell Cycle 10: 2724–2736. [DOI] [PubMed] [Google Scholar]
- Yeudall WA, Vaughan CA, Miyazaki H, Ramamoorthy M, Choi M-Y, Chapman CG, Wang H, Black E, Bulysheva AA, Deb SP, et al. 2012. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 33: 442–451. [DOI] [PubMed] [Google Scholar]
- Zhang W, Deisseroth AB. 1994. Conformational change of p53 protein in growth factor-stimulated human myelogenous leukemia cells. Leuk Lymphoma 14: 251–255. [DOI] [PubMed] [Google Scholar]
- Zhang J, Smolen GA, Haber DA. 2008. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res 68: 2789–2794. [DOI] [PubMed] [Google Scholar]
- Zhao B, Li L, Lei Q, Guan K-L. 2010. The Hippo–YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 24: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.