

In this study, Ye et al. show that activation of GCN2, an amino acid-sensing kinase, during amino acid deprivation leads to ATF4-dependent transcription of the stress response protein Sestrin2. These results demonstrate an important link between GCN2 and mTORC1 signaling during amino acid homeostasis.
Keywords: GCN2, mTORC1, Sestrin, amino acid deprivation
Abstract
Mammalian cells possess two amino acid-sensing kinases: general control nonderepressible 2 (GCN2) and mechanistic target of rapamycin complex 1 (mTORC1). Their combined effects orchestrate cellular adaptation to amino acid levels, but how their activities are coordinated remains poorly understood. Here, we demonstrate an important link between GCN2 and mTORC1 signaling. Upon deprivation of various amino acids, activated GCN2 up-regulates ATF4 to induce expression of the stress response protein Sestrin2, which is required to sustain repression of mTORC1 by blocking its lysosomal localization. Moreover, Sestrin2 induction is necessary for cell survival during glutamine deprivation, indicating that Sestrin2 is a critical effector of GCN2 signaling that regulates amino acid homeostasis through mTORC1 suppression.
Proteins are the most abundant macromolecules in living cells. The basic building blocks of proteins, amino acids, are not only required for regular cellular functions but are also key determinants for cell survival. Eukaryotes have evolved two major regulatory mechanisms for amino acid sensing to adapt to fluctuating amino acid levels in the environment: the mechanistic target of rapamycin complex 1 (mTORC1) signaling pathway, which is activated in the presence of amino acids, and the general control nonderepressible 2 (GCN2) signaling pathway, which is activated by the absence of amino acids.
mTORC1 integrates environmental signals such as nutrients, growth factors, oxygen, and stress to regulate protein translation, metabolism, and cell growth (Laplante and Sabatini 2012; Jewell et al. 2013; Shimobayashi and Hall 2014). Although growth factors stimulate mTORC1 activity, amino acids are required for complete activation of mTORC1 (Hara et al. 1998). Activated mTORC1 phosphorylates ribosomal S6 kinase (S6K) and eIF4E-binding proteins (4EBPs) to promote 5′ cap-dependent translation (Hara et al. 1998; Wang et al. 1998). In addition, mTORC1 phosphorylates and inactivates unc-51-like autophagy-activating kinase 1 (ULK1), the initiating kinase of autophagy, to inhibit autophagic protein degradation (Kim et al. 2011; Shang et al. 2011). Through these two mechanisms, mTORC1 enhances protein anabolism under amino acid-replete conditions. However, the mechanisms by which mTORC1 senses amino acids remain elusive. When amino acids are present, the Rag family of GTPases plays an essential role in recruiting mTORC1 to the surface of the lysosome (Kim et al. 2008; Sancak et al. 2008, 2010), where another small GTPase, Rheb, stimulates the kinase activity of mTORC1. Several recent reports demonstrate that a family of stress response proteins, Sestrins, inhibit the Rag complex to disrupt mTORC1 localization to the lysosome (Chantranupong et al. 2014; Parmigiani et al. 2014; Peng et al. 2014; Kim et al. 2015). However, how Sestrins are regulated by amino acid availability and which Sestrin family members are critical for this process remain unknown.
In yeast and mammals, GCN2 is the direct sensor of amino acid deprivation (AAD). Upon starvation, uncharged tRNAs bind to GCN2 and stimulate its dimerization and autophosphorylation (Wek et al. 1989, 1995; Diallinas and Thireos 1994; Dong et al. 2000; Qiu et al. 2002; Narasimhan et al. 2004). Activated GCN2 phosphorylates eIF2α to block translation initiation of most mRNAs (Dever et al. 1992). However, particular mRNAs that contain an upstream ORF (uORF) cluster in their 5′ untranslated region (UTR) are efficiently translated upon eIF2α phosphorylation, including the yeast transcription factor GCN4 (Hinnebusch 1984) and its mammalian ortholog, ATF4 (Harding et al. 2000; Vattem and Wek 2004). Once ATF4 is up-regulated, it induces expression of many genes involved in amino acid metabolism, including amino acid synthetases and transporters, amino acyl tRNA synthetases (Harding et al. 2003; Bunpo et al. 2009; Ye et al. 2010, 2012), and regulators of autophagy (B'chir et al. 2013). Together, these ATF4 targets promote cellular adaptation to the amino acid shortage.
GCN2 activation leads to attenuated global translation, autophagy induction, and growth arrest (Lehman et al. 2015), all of which act in opposition to mTORC1 activity. In this study, we discovered that GCN2 activation can lead to sustained suppression of mTORC1 kinase activity. Upon AAD, GCN2 signaling transcriptionally up-regulated the stress response protein Sestrin2. Sestrin2 induction is required to block lysosomal recruitment and activation of mTORC1. Furthermore, Sesn2 deletion sensitizes cells to glutamine starvation-induced cell death, which is rescued by the mTORC1 inhibitor rapamycin, suggesting that Sestrin2 is not only a critical mediator of the two amino acid-sensing machineries but also a key determinant of cell fate during AAD.
Results and Discussion
GCN2 is required to inhibit mTORC1 kinase activity and lysosomal localization upon prolonged AAD
Genetic experiments have suggested that GCN2 activation can contribute to mTORC1 inhibition following leucine depletion (Anthony et al. 2004), but the molecular mechanism and the dynamics of the cross-talk between GCN2 and mTORC1 have not been identified. When wild-type mouse embryonic fibroblasts (MEFs) were cultured in leucine-free medium, an acute repression of mTORC1 activity, as assessed by decreased S6K phosphorylation (T389), was detected within 30 min, followed by a brief recovery phase. After 8 h, mTORC1 activity started decreasing again, with maximum repression reached by 24 h (Fig. 1A). ATF4 induction was monitored as a measure of GCN2 activity, which inversely correlated with mTORC1 activity during prolonged starvation (Fig. 1A). To investigate whether GCN2 activation is required for sustained mTORC1 suppression upon leucine depletion, wild-type and Gcn2−/− MEFs were starved in leucine-free medium for up to 24 h. Leucine depletion induced ATF4 in wild-type cells but not in Gcn2−/− cells (Fig. 1B). Interestingly, although acute mTORC1 suppression was observed in both wild-type and Gcn2−/− cells upon leucine depletion, mTORC1 activity did not decrease in Gcn2−/− cells during prolonged starvation (8–24 h) (Fig. 1B), suggesting that GCN2 is required for long-term mTORC1 suppression but not short-term suppression. Since ATF4 is a major downstream effector of GCN2 that regulates amino acid homeostasis (Harding et al. 2003; Ye et al. 2010), we next tested whether ATF4 was required for mTORC1 suppression upon leucine deprivation. Similar to what we observed in Gcn2−/− cells, Atf4−/− cells displayed sustained S6K phosphorylation upon leucine withdrawal (Fig. 1B), indicating that GCN2-dependent mTORC1 suppression relies on ATF4. To confirm that the acute mTORC1 regulation by leucine does not depend on GCN2–ATF4 signaling, wild-type, Gcn2−/−, and Atf4−/− MEFs were starved in leucine-free medium for 20 min, and then leucine was added back for another 20 min. All three cell lines were able to reactivate mTORC1 by leucine stimulation, indicating that GCN2–ATF4 signaling is not required for the acute activation of mTORC1 by leucine (Supplemental Fig. S1).
Figure 1.
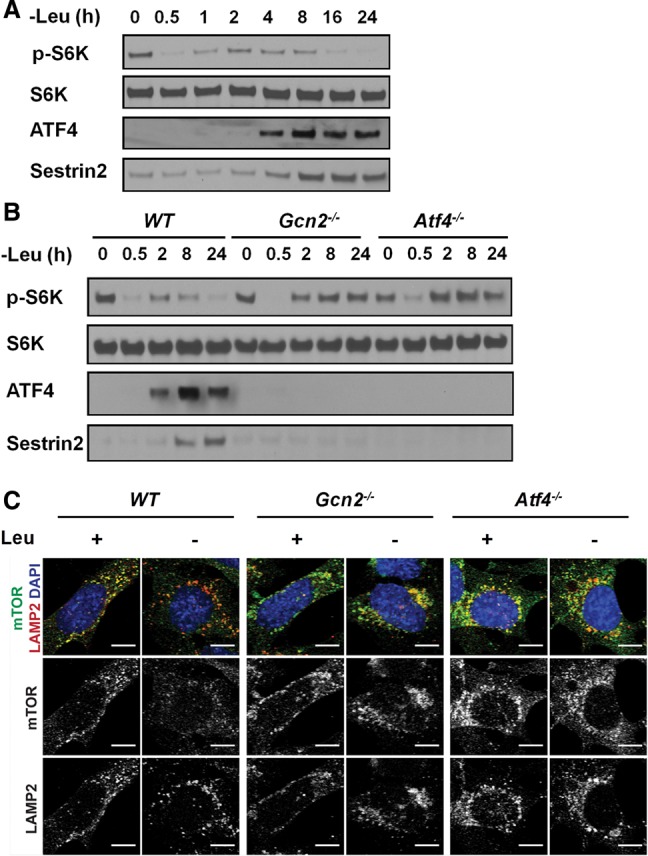
Sustained mTORC1 repression during leucine deprivation requires the GCN2–ATF4 pathway. (A) Immunoblots of lysates from wild-type MEFs that were cultured in leucine-free medium for up to 24 h. (B) Immunoblots of lysates from wild-type, Gcn2−/−, and Atf4−/− MEFs that were cultured in leucine-free medium for up to 24 h. (C) Lysosomal recruitment of mTOR, analyzed by immunofluorescence against mTOR and the lysomal marker LAMP2. MEFs were cultured in ±leucine medium for 24 h. Bars, 10 µm.
Since amino acids regulate mTORC1 activity by inducing its recruitment to lysosomal membranes (Sancak et al. 2008, 2010), we examined whether the GCN2–ATF4 pathway was required to block lysosomal localization of mTORC1 during AAD. In wild-type MEFs cultured in full medium, mTOR was present in punctate structures that colocalized with the lysosomal membrane protein LAMP2. After 24 h of leucine starvation, mTOR was distributed throughout the cytoplasm (Fig. 1C). In contrast, in both Gcn2−/− and Atf4−/− MEFs, mTOR remained in punctate structures upon leucine starvation. Together, these data show that the GCN2–ATF4 pathway represses mTORC1 activity by inhibiting its lysosomal localization.
The GCN2–ATF4 pathway transcriptionally up-regulates Sestrin2 during AAD
The lysosomal localization of mTORC1 requires the Rag GTPases, which localize to the lysosomal surface and are responsible for mTORC1 recruitment in the presence of amino acids (Kim et al. 2008; Sancak et al. 2008, 2010). There is emerging evidence that members of the Sestrin family of stress response proteins are negative regulators of the Rag GTPases (Chantranupong et al. 2014; Parmigiani et al. 2014; Peng et al. 2014; Kim et al. 2015). Given that Sestrins disrupt mTORC1 recruitment to the lysosome and inhibit its kinase activity, we examined whether expression of Sestrins was regulated by the GCN2–ATF4 pathway under AAD. The levels of Sestrin2 increased significantly during leucine starvation and correlated with eIF2α phosphorylation and ATF4 induction (Figs. 1A, 2A; Supplemental Fig. S2A). To determine whether Sestrin2 induction was regulated by GCN2–ATF4 signaling, Sestrin2 levels upon AAD were measured in Gcn2−/− and Atf4−/− MEFs. Strikingly, loss of GCN2–ATF4 signaling abrogated Sestrin2 induction during leucine starvation. Moreover, the basal expression of Sestrin2 was considerably lower in Atf4−/− MEFs as compared with wild-type MEFs (Figs. 1B, 2A; Supplemental Fig. S2A). Since Sestrins function upstream of Rag GTPases, we generated Rag A/B knockout HEK293T cells using the CRISPR/Cas9 system to determine whether Rag signaling is necessary for the sustained mTORC1 suppression upon leucine deprivation. In contrast to control cells, the Rag A/B knockout cells displayed sustained S6K phosphorylation even after 24 h of leucine starvation, indicating that long-term mTORC1 suppression is dependent on the Rag GTPases (Fig. 2B).
Figure 2.
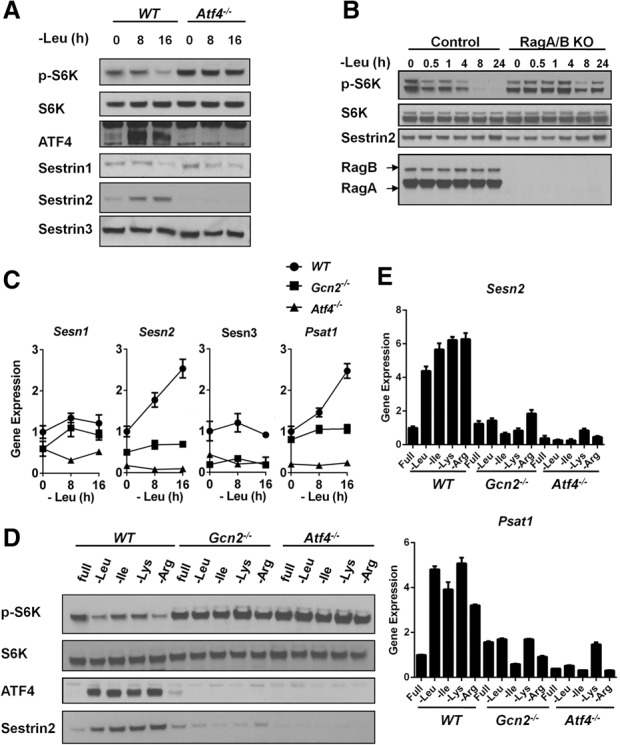
ATF4 is necessary for the transcriptional up-regulation of Sestrin2 under AAD. (A) Immunoblots of lysates from wild-type and Atf4−/− MEFs that were cultured in leucine-free medium for 0, 8, and 16 h. (B) Immunoblots of lysates from control and Rag A/B knockout HEK293T cells that were cultured in leucine-free medium for up to 24 h. (C) Quantitative PCR (qPCR) measuring mRNA expression in wild-type, Gcn2−/−, and Atf4−/− MEFs cultured in leucine-free medium for 0, 8, and 16 h. Data represent mean ± SD of triplicate PCR reactions; a representative of two independent experiments is shown. (D) Immunoblots of lysates from wild-type, Gcn2−/−, and Atf4−/− MEFs that were cultured in media lacking individual amino acids as indicated for 24 h. (E) qPCR for measuring mRNA expression in wild-type, Gcn2−/−, and Atf4−/− MEFs that were cultured in media lacking individual amino acids as indicated for 24 h. Data represent mean ± SD of triplicate PCR reactions; a representative of two independent experiments is shown.
Since ATF4 is a transcription factor, we next examined whether ATF4 up-regulates Sestrin2 transcriptionally. Indeed, Sestrin2 mRNA was significantly induced during leucine deprivation in a time-dependent manner in wild-type MEFs but not in Gcn2−/− or Atf4−/− MEFs (Fig. 2C). The kinetics of Sestrin2 induction resembled those of an established ATF4 target, phosphoserine amino transferase 1 (PSAT1) (Ye et al. 2012). In contrast to Sestrin2, the mRNA and/or protein levels of Sestrin1 and Sestrin3 were not induced in response to AAD. Sestrin1 and Sestrin3 protein levels were similar between wild-type and Atf4−/− MEFs despite lower levels of mRNA (Fig. 2A,C).
To confirm that ATF4 played a direct role in Sestrin2 induction, we examined a publicly available data set of genome-wide sequencing of ATF4 chromatin immunoprecipitation (ChIP) (Han et al. 2013). ATF4 was potently enriched at the Sesn2 promoter at regions that contained multiple ATF4 consensus binding sequences (Supplemental Fig. S2B). We tested whether these regions were bound in response to leucine deprivation by performing ATF4-specific ChIP. We found that ATF4 was highly enriched at a site located 3.6 kb upstream of the Sesn2 transcription start site upon leucine deprivation (Supplemental Fig. S2B). In contrast, ATF4 was not enriched at the Sesn1 promoter.
To determine whether mTORC1 suppression and Sestrin2 induction by the GCN2–ATF4 pathway were specific to leucine deprivation or general consequences of AAD, we next examined mTORC1 activity during isoleucine, lysine, or arginine withdrawal. Similar to leucine deprivation, isoleucine, lysine, and arginine deprivation all reduced mTORC1 activity significantly in wild-type MEFs. In contrast, Gcn2−/− and Atf4−/− MEFs maintained mTORC1 activity under these starvation conditions (Fig. 2D). Withdrawal of each of these three amino acids (isoleucine, lysine, or arginine) increased Sestrin2 protein and mRNA levels in wild-type MEFs but not in Gcn2−/− or Atf4−/− MEFs (Fig. 2D,E). These data suggest that GCN2–ATF4-dependent Sestrin2 induction and mTORC1 suppression are general consequences of AAD and are not caused by the absence of a specific amino acid. In contrast, the endoplasmic reticulum (ER) stress-inducing agent thapsigargin induced Sestrin2 and inhibited mTORC1 in wild-type and Gcn2−/− cells but not in Atf4−/− cells (Supplemental Fig. S2C), indicating that GCN2-dependent Sestrin2 induction specifically responds to AAD but not other stress conditions that induce ATF4. Furthermore, using two human cancer cell lines (HT1080 and DLD1) with stable expression of an ATF4 shRNA (Ye et al. 2010), we demonstrated that the induction of Sestrin2 upon amino acid starvation in these human cell lines also depended on ATF4 (Supplemental Fig. S3A,B).
Sestrin2 is required for mTORC1 suppression upon AAD
To determine whether Sestrin2 was necessary for mTORC1 suppression during leucine withdrawal, wild-type or Sesn2−/− MEFs were cultured in leucine-free medium for up to 24 h. The Sesn2−/− cells displayed an attenuated short-term mTORC1 suppression in response to leucine deprivation and lacked long-term mTORC1 suppression (Fig. 3A). A requirement for Sestrin2 in long-term mTORC1 suppression upon withdrawal of other amino acids was demonstrated by culturing wild-type or Sesn2−/− MEFs in medium lacking either leucine, isoleucine, lysine, or arginine for 24 h. Although ATF4 induction was not affected by Sestrin2 deletion, AAD treatment failed to repress mTORC1 activity in Sesn2−/− MEFs (Fig. 3B), indicating that Sestrin2 is indispensable for mTORC1 suppression during amino acid withdrawal. The phosphorylation of another mTORC1 target, ULK1 (S757), was also reduced in wild-type cells but not in Sesn2−/− cells upon AAD (Fig. 3B). Moreover, similar to what was observed in Gcn2−/− MEFs, mTOR remained in punctate structures in Sesn2−/− MEFs after 24 h of leucine starvation, indicating that Sestrin2 induction is also required for inhibition of mTORC1 lysosomal localization (Fig. 3C). Together, these data indicate that Sestrin2 is essential for GCN2–ATF4-mediated mTORC1 suppression upon AAD.
Figure 3.
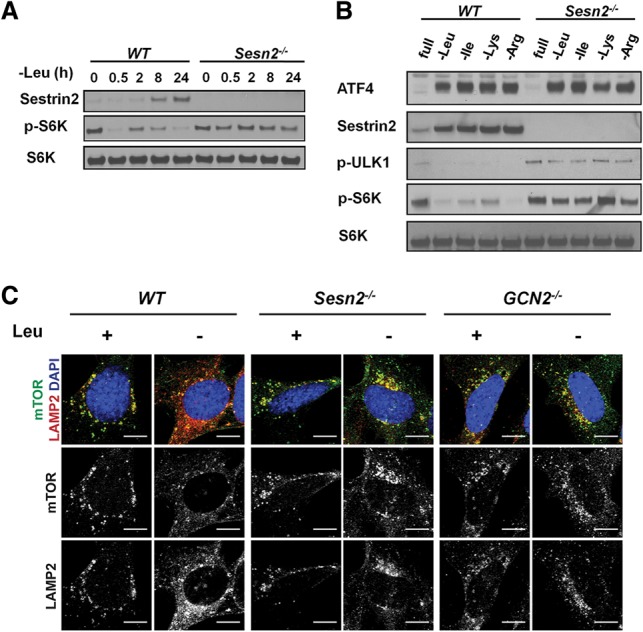
Sestrin2 is necessary for mTORC1 repression during AAD. (A) Immunoblots of lysates from wild-type and Sesn2−/− MEFs cultured in leucine-free medium for up to 24 h. (B) Immunoblots of lysates from wild-type and Sesn2−/− MEFs cultured in amino acid-free media as indicated for 24 h. (C) Immunofluorescence against mTOR and the lysosomal marker LAMP2 in wild-type, Gcn2−/−, and Sesn2−/− MEFs cultured in ±leucine medium for 24 h. Bars, 10 µm.
Sestrin2-dependent mTORC1 suppression is necessary for cell survival during glutamine withdrawal
Glutamine is a nonessential amino acid that can activate mTORC1 through mechanisms that are distinct from mTORC1 activation by essential amino acids (Nicklin et al. 2009; Kim et al. 2013; Jewell et al. 2015). Interestingly, unlike leucine deprivation, glutamine starvation only repressed mTORC1 after long-term treatment (24 h), and no acute repression was observed (Fig. 4A). To determine whether glutamine starvation could repress mTORC1 activity through Sestrin2 induction, wild-type, Gcn2−/−, or Atf4−/− MEFs were cultured in glutamine-free medium for 24 h. Similar to withdrawal of essential amino acids, glutamine deprivation significantly induced Sestrin2 expression in a GCN2/ATF4-dependent manner (Fig. 4B). In addition, mTORC1 suppression in the context of glutamine depletion required GCN2, ATF4, and Sestrin2 (Fig. 4B,D). Since GCN2–ATF4 signaling has been reported to promote cell survival during glutamine starvation (Ye et al. 2010), we investigated whether Sestrin2 also contributes to cell survival during glutamine starvation. Strikingly, Sesn2−/− MEFs were particularly sensitive to glutamine starvation-induced cell death. Approximately 80% of Sesn2−/− cells died after 48 h of glutamine depletion, compared with 30% of wild-type MEFs (Fig. 4C). Immunoblotting for the apoptosis marker cleaved caspase3 confirmed that cell death was due to apoptosis (Fig. 4D). To confirm that repressing mTORC1 is a critical downstream function of Sestrin2, we next examined whether mTORC1 repression could suppress death of Sesn2−/− cells upon glutamine starvation. Indeed, the mTORC1 inhibitor rapamycin significantly reduced caspase3/7 activity and rescued the survival of Sesn2−/− cells upon glutamine starvation (Fig. 5; Supplemental Fig. S4A). Moreover, rapamycin also rescued the survival of Gcn2−/− cells upon glutamine withdrawal (Supplemental Fig. S4B). Together, these data show that mTORC1 suppression by the GCN2–ATF4–Sestrin2 pathway is required for maintaining cellular homeostasis and survival during glutamine starvation.
Figure 4.
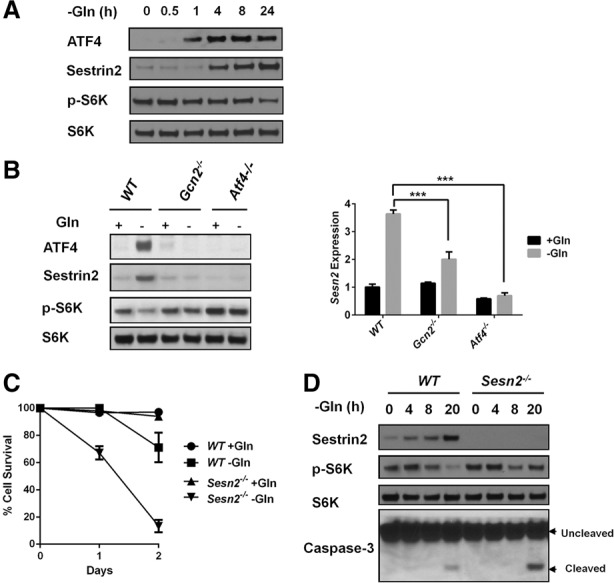
Sestrin2 is necessary for mTORC1 repression and cell survival upon glutamine deprivation. (A) Immunoblots of lysates from wild-type MEFs that were cultured in glutamine-free medium for up to 24 h. (B, left panel) Immunoblots of lysates from wild-type, Gcn2−/−, and Atf4−/− MEFs cultured in ±glutamine medium for 24 h. (Right panel) qPCR for Sestrin2 mRNA expression in wild-type, Gcn2−/−, and Atf4−/− MEFs cultured in ±glutamine medium for 24 h. Data represent mean ± SD of triplicate PCR reactions; a representative of two independent experiments is shown. (***) P < 0.001, determined by Student's two-tailed t-test. (C) Percentage cell survival measured by Trypan Blue staining of wild-type and Sesn2−/− MEFs cultured in ±glutamine medium for 1 or 2 d. (D) Immunoblots of lysates from wild-type and Sesn2−/− MEFs cultured in ±glutamine medium for up to 20 h.
Figure 5.
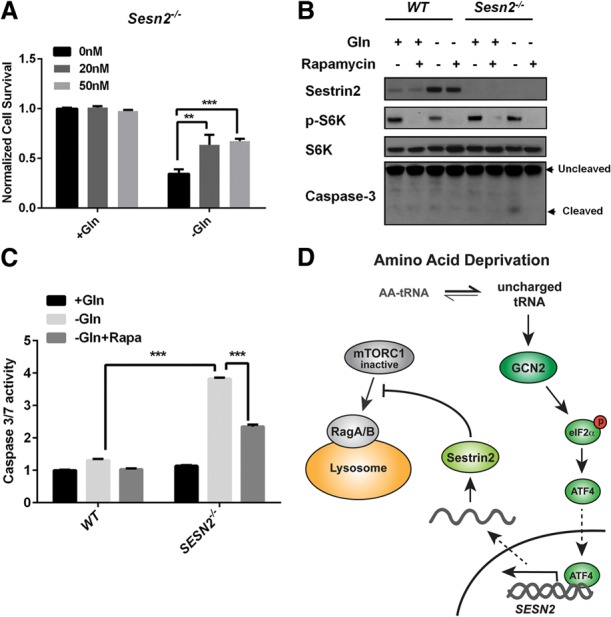
Rapamycin inhibits apoptosis of Sesn2−/− MEFs upon glutamine starvation. (A) Survival of Sesn2−/− MEFs cultured in ±glutamine medium for 48 h with the indicated concentrations of rapamycin, normalized to survival of wild-type cells. Data represent mean ± SD of three biological repeats. (**) P < 0.01; (***) P < 0.001, determined by Student's two-tailed t-test. (B) Immunoblots of lysates from wild-type and Sesn2−/− MEFs cultured in ±glutamine medium with or without 20 nM rapamycin for 24 h. (C) Wild-type and Sesn2−/− MEFs were cultured in ±glutamine medium with or without 50 nM rapamycin for 24 h. Caspase3/7 activity was determined using the Caspase-Glo 3/7 assay kit. Data represent mean ± SD of three biological repeats. (***) P < 0.001, determined by Student's two-tailed t-test. (D) Model of mTORC1 regulation by GCN2. Upon AAD, uncharged tRNAs activate the GCN2/ATF4 pathway, which induces expression of Sestrin 2, which inhibits translocation of mTORC1 to lysosomal membranes. This mechanism allows GCN2 to communicate AAD to the mTORC1 pathway.
GCN2 is an evolutionarily conserved sensor of AAD in eukaryotes (Hinnebusch 1994; Baird and Wek 2012). Upon activation by uncharged tRNAs, GCN2 phosphorylates eIF2α to repress global protein translation, which is one of the most energy-consuming processes in the cell. By conserving amino acids and energy, cells are more likely to survive starvation. In addition, eIF2α phosphorylation causes a selective translational up-regulation of a group of stress-responsive transcripts with a unique 5′ UTR structure. Among them, ATF4 is critical for increasing nonessential amino acid synthesis (Harding et al. 2003). For instance, the GCN2–ATF4 pathway up-regulates all three enzymes in the serine biosynthetic pathway (PHGDH, PSAT1, and PSPH), which are essential for cell proliferation under serine depletion (Ye et al. 2012). Additionally, upon glutamine withdrawal, ATF4 up-regulates asparagine synthetase (ASNS), which increases asparagine synthesis to support cell survival (Ye et al. 2010). Here, we identify a novel mechanism by which GCN2 signaling maintains the viability of cells under AAD through sustained repression of mTORC1 (Fig. 5D). It was reported that leucine, arginine, and glutamine are major amino acids that are required for mTORC1 activation, although via different mechanisms (Nicklin et al. 2009; Kim et al. 2013; Jewell et al. 2015; Jung et al. 2015; Rebsamen et al. 2015; Wang et al. 2015). Our results demonstrate that withdrawal of any of these amino acids also results in GCN2-dependent induction of Sestrin2 to maintain long-term repression of mTORC1. The established ability of GCN2 to become activated as a general response to uncharged tRNAs therefore links single amino acid deficiencies to mTORC1-dependent translation. Thus, GCN2-induced Sestrin2 expression constitutes a unifying mechanism through which mTORC1 activity is suppressed during long-term starvation of individual amino acids.
Materials and methods
Cell culture
All MEFs used in this study were immortalized with SV40 large T antigen. MEF, HEK293T, DLD1, and HT1080 cells were cultured in DMEM/F12 supplemented with penicillin, streptomycin, 10% FBS, 1× nonessential amino acids (Gibco), and 55 µM β-mercaptoethanol (β-ME). The glutamine-free medium was DMEM. To generate the HEK293T Rag A/B knockout cells, the following primers were used for cloning gRNA targeting of RagA/B into LenticripsrV2 (Addgene #52961): sgRRAGA-F (caccgGGATGGCCTCCAGACACGAC), sgRRAGA-R (aaacGTCGTGTCTGGAGGCCATCCc), sgRRAGB-F (caccgTTGTCCTTAGGTGCTGTTGA), and sgRRAGB-R (aaacTCAACAGCACCTAAGGACAAc).
All of the amino acid-free special media were produced by the Media Preparation Core Facility at Memorial Sloan Kettering Cancer Center.
Immunoblots
Antibodies were purchased from Cell Signaling (p-S6K1, #9234; S6K, #2708; p-eIF2α, #9271; eIF2α, #9722; Rag A, #4357; p-Ulk1, #14202; and Caspase-3, #9662), Santa Cruz Biotechnology (ATF4, sc-200), ProteinTech (Sestrin2, 10795-1-AP), and Abgent (Sestrin1, AP7650a; Sestrin3, AP12471c).
Reverse transcription and real-time PCR
Total RNA was extracted following the TRIzol reagent (Invitrogen) protocol. Three micrograms of total RNA was used in reverse transcription following the iScript cDNA synthesis kit (Bio-Rad) protocol. Quantitative PCR (qPCR) was performed on a 7900HT sequence detection system (Applied Biosystems). Expression of the Sestrins was measured using SYBR Green master mix. Primers used were Sesn1-F (GCCATTACCCCTCCATTATCG), Sesn1-R (GCATTCTCTAAACCATTGAGCC), Sesn2-F (CAGCCTCACCTATAACACCATC), Sesn2-R (CTCGCCGTAATCATAGTCATCG), Sesn3-F (TTACTTGAACGGAGCCTGAAG), and Sesn3-R (TCCATCAGAAGCAGATTCACG).
Expression of other genes was measured using TaqMan gene expression assays (Applied Biosystems). Gene expression data were normalized to 18S rRNA. All of the values were normalized to wild-type cell samples in full medium.
Cell death and caspase activity assay
To measure cell death, cells were stained with Trypan Blue. Stained/unstained cells were counted, and cell death percentages were calculated. Caspase3/7 activity was determined using the Caspase-Glo 3/7 assay kit (Promega). The luminescence signal was normalized to the protein concentration of the lysate.
Immunofluorescence
For immunofluorescence staining, cells were rinsed with ice-cold PBS, fixed with 4% formaldehyde in PBS for 15 min, and permeabilized with 0.05% Triton X-100 in PBS for 5 min. After rinsing with PBS, cells were blocked with PBG (0.5% BSA, 0.2% cold water fish gelatin in PBS) for 30 min, incubated with primary antibodies in PBG for 1.5 h, washed twice with PBG + 4% normal goat serum, and then incubated with fluorescent secondary antibodies in PBG + 4% normal goat serum for 45 min. After three washes with PBS, cells were mounted on microscope slides with Prolong anti-fade + DAPI and imaged using a Leica TCS SP5-II. Antibodies were purchased from Abcam (LAMP2, ab13524) and Cell Signaling (mTOR, #2983). Secondary antibodies (Alexa fluor 488 anti-rabbit, Alexa fluor 555 anti-rat) and Prolong anti-fade + DAPI were from Life Technologies.
ChIP assay
Wild-type or Atf4−/− MEFs (2 × 107) were fixed for 10 min in 1% formaldehyde (Tousimis), and this reaction was quenched by incubation for 5 min in 0.125 M glycine. Nuclear pellets were lysed in 10 mM Tris (pH 8.0), 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, and 0.5% N-laurylsarcosine and sheared for 30 min (30 sec on, 30 sec off) in TPX microtubes using a Bioruptor bath sonicator (Diagenode). Complexes bearing ATF4 were captured with 5 µg of antibody (Santa Cruz Biotechnology, sc-200) bound to 50 µL of protein G Dynabeads (Life Technologies), washed five times in RIPA wash buffer (50 mM HEPES-KOH at pH 7.6, 300 mM lithium chloride, 1 mM EDTA, 1% NP-40 substitute, 0.7% sodium deoxycholate) and once in TE wash buffer (10 mM Tris at pH 8.0, 1 mM EDTA, 50 mM NaCl), and eluted in 0.1 M sodium bicarbonate/1% SDS. Cross-links were reversed by incubation overnight at 65°C followed by treatment with RNase A and proteinase K. DNA was isolated using PCR purification columns (Qiagen). Enrichment of ATF4 at target loci relative to input was determined by quantitative real-time PCR using the following primers: Sesn2 -3kb F (AGTGTTTGGTCAGGCAAGGT), Sesn2 -3kb R (TCAGTGGCTTTAACACGGCT), Sesn1 pr F (GATGACTGCTGGATTGCGGG), and Sesn1 pr R (AACCCTGCCGGTTCTTGTC).
Competing interest statement
C.B.T. is a founder of Agios Pharmaceuticals and a member of its scientific advisory board. C.B.T. also serves on the Board of Directors at Merck.
Supplementary Material
Acknowledgments
We thank Dr. David Ron (University of Cambridge) for the Gcn2−/− MEFs, and the Media Preparation and Molecular Cytology Core Facilities of the Memorial Sloan Kettering Cancer Center, which are supported by the cancer center support grant P30 CA008748. This work was supported by National Institutes of Health grants R01 CA105463, P01 CA104838, and K99 CA184239. W.P. is the Genentech Foundation Fellow of the Hope Funds Cancer Research Foundation. B.K. is the Berger Foundation Fellow of the Damon Runyon Cancer Research Foundation.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.269324.115.
References
- Anthony TG, McDaniel BJ, Byerley RL, McGrath BC, Cavener DR, McNurlan MA, Wek RC. 2004. Preservation of liver protein synthesis during dietary leucine deprivation occurs at the expense of skeletal muscle mass in mice deleted for eIF2 kinase GCN2. J Biol Chem 279: 36553–36561. [DOI] [PubMed] [Google Scholar]
- Baird TD, Wek RC. 2012. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr 3: 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- B'chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. 2013. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 41: 7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunpo P, Dudley A, Cundiff JK, Cavener DR, Wek RC, Anthony TG. 2009. GCN2 protein kinase is required to activate amino acid deprivation responses in mice treated with the anti-cancer agent L-asparaginase. J Biol Chem 284: 32742–32749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, Sabatini DM. 2014. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep 9: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, Hinnebusch AG. 1992. Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68: 585–596. [DOI] [PubMed] [Google Scholar]
- Diallinas G, Thireos G. 1994. Genetic and biochemical evidence for yeast GCN2 protein kinase polymerization. Gene 143: 21–27. [DOI] [PubMed] [Google Scholar]
- Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG. 2000. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell 6: 269–279. [DOI] [PubMed] [Google Scholar]
- Han J, Backa SH, Hur J, Lin YH, Gildersleeve R, Shan JX, Yuan CL, Krokowski D, Wang SY, Hatzoglou M, et al. 2013. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15: 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. 1998. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem 273: 14484–14494. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG. 1984. Evidence for translational regulation of the activator of general amino acid control in yeast. Proc Natl Acad Sci 81: 6442–6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. 1994. The eIF-2α kinases: regulators of protein synthesis in starvation and stress. Semin Cell Biol 5: 417–426. [DOI] [PubMed] [Google Scholar]
- Jewell JL, Russell RC, Guan KL. 2013. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol 14: 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, Guan KL. 2015. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347: 194–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Genau HM, Behrends C. 2015. Amino acid dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol Cell Biol 35: 2479–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. 2008. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10: 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG, Hoffman GR, Poulogiannis G, Buel GR, Jang YJ, Lee KW, Kim BY, Erikson RL, Cantley LC, Choo AY, et al. 2013. Metabolic stress controls mTORC1 lysosomal localization and dimerization by regulating the TTT-RUVBL1/2 complex. Mol Cell 49: 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Ro SH, Kim M, Park HW, Semple IA, Park H, Cho US, Wang W, Guan KL, Karin M, et al. 2015. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci Rep 5: 9502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149: 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman SL, Cerniglia GJ, Johannes GJ, Ye J, Ryeom S, Koumenis C. 2015. Translational upregulation of an individual p21Cip1 transcript variant by GCN2 regulates cell proliferation and survival under nutrient stress. PLoS Genet 11: e1005212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan J, Staschke KA, Wek RC. 2004. Dimerization is required for activation of eIF2 kinase Gcn2 in response to diverse environmental stress conditions. J Biol Chem 279: 22820–22832. [DOI] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. 2009. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136: 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, Guan KL, Karin M, Budanov AV. 2014. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep 9: 1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yin N, Li MO. 2014. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 159: 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Hu C, Dong J, Hinnebusch AG. 2002. Mutations that bypass tRNA binding activate the intrinsically defective kinase domain in GCN2. Genes Dev 16: 1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen M, Pochini L, Stasyk T, de Araujo MEG, Galluccio M, Kandasamy RK, Snijder B, Fauster A, Rudashevskaya EL, Bruckner M, et al. 2015. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519: 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. 2010. Ragulator–Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141: 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X. 2011. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci 108: 4788–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M, Hall MN. 2014. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol 15: 155–162. [DOI] [PubMed] [Google Scholar]
- Vattem KM, Wek RC. 2004. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci 101: 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Campbell LE, Miller CM, Proud CG. 1998. Amino acid availability regulates p70 S6 kinase and multiple translation factors. Biochem J 334(Pt 1): 261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W, et al. 2015. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347: 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek RC, Jackson BM, Hinnebusch AG. 1989. Juxtaposition of domains homologous to protein kinases and histidyl-tRNA synthetases in GCN2 protein suggests a mechanism for coupling GCN4 expression to amino acid availability. Proc Natl Acad Sci 86: 4579–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek SA, Zhu S, Wek RC. 1995. The histidyl-tRNA synthetase-related sequence in the eIF-2α protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol 15: 4497–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova-Marjon E, Diehl JA, Ron D, Koumenis C. 2010. The GCN2–ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 29: 2082–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, Thompson CB. 2012. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci 109: 6904–6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.