

Whole-genome duplication (WGD) doubles the DNA content in the nucleus and leads to polyploidy. In this review, Yant and Bomblies discuss both the adaptive potential and problems associated with WGD, focusing primarily on cellular effects.
Keywords: DNA content, evolution, whole-genome duplication
Abstract
Whole-genome duplication (WGD) doubles the DNA content in the nucleus and leads to polyploidy. In whole-organism polyploids, WGD has been implicated in adaptability and the evolution of increased genome complexity, but polyploidy can also arise in somatic cells of otherwise diploid plants and animals, where it plays important roles in development and likely environmental responses. As with whole organisms, WGD can also promote adaptability and diversity in proliferating cell lineages, although whether WGD is beneficial is clearly context-dependent. WGD is also sometimes associated with aging and disease and may be a facilitator of dangerous genetic and karyotypic diversity in tumorigenesis. Scaling changes can affect cell physiology, but problems associated with WGD in large part seem to arise from problems with chromosome segregation in polyploid cells. Here we discuss both the adaptive potential and problems associated with WGD, focusing primarily on cellular effects. We see value in recognizing polyploidy as a key player in generating diversity in development and cell lineage evolution, with intriguing parallels across kingdoms.
Whole-genome duplication (WGD) has played a role in the evolution of all major eukaryotic lineages and can involve single somatic cells or entire organisms. At the whole-organism level, WGD has been linked to phenotypic novelty, speciation, and adaptation as well as the evolution of genomic complexity (e.g., see Levin 1983; Ramsey and Schemske 1998, 2002; Otto and Whitton 2000; Soltis et al. 2003; Doyle et al. 2008; Hegarty et al. 2013). However, WGD also occurs in somatic cells in otherwise diploid organisms, where it plays important roles in normal development and likely also in inducible wound repair and stress responses. Polyploid cells can also be a hallmark of aging and disease and may be intermediates in the progression of many tumors, where they increase genetic and karyotypic diversity (e.g., see Storchova and Pellman 2004; Storchova et al. 2006; Thorpe et al. 2007; Storchova 2014; Coward and Harding 2014). A role in disease, while detrimental to the organism, nevertheless highlights the adaptive potential of genome duplication at the level of cell lineages.
Despite their adaptive potential, proliferating polyploid cell lineages or organisms face challenges, particularly to chromosome segregation in both mitosis and meiosis, which we discuss below. In most of the cases in which WGD is associated with pathological outcomes, this seems to arise from the propensity for chromosome missegregation, thus emphasizing that understanding both the nature of the problem and how evolution might confer adaptation to it in some cases is important.
In this review, we discuss how the adaptive potential and cellular novelty provided by genome duplication can contribute to normal development, environmental responses, and disease states. We discuss challenges that polyploid cells or organisms face, especially with chromosome segregation, and how this might relate to some of the risks associated with unplanned polyploidy. We focus exclusively on within-species polyploidy (autopolyploidy), as this is relevant in a wide range of situations from whole-organism polyploidy to cellular WGD in normal development and disease. We do not discuss allopolyploids, which arise from hybridization coupled with WGD; interested readers are referred instead to thorough reviews that cover this topic (e.g., see Ramsey and Schemske 1998, 2002; Otto and Whitton 2000; Soltis et al. 2003; Doyle et al. 2008; Gaeta and Pires 2009; Hegarty et al. 2013). While the underlying biologies of different systems are, of course, distinct, we support the idea that considering potentially informative parallels across systems can provide new testable hypotheses in a range of fields.
Somatic WGD
Polyploid cells are a normal part of development in otherwise diploid plants and animals and play beneficial and often essential roles arising from the phenotypic novelty of these cells compared with their diploid counterparts. Somatic polyploid cells can arise either by cell fusion or when cell division aborts before cytokinesis (for review, see Nagl 1982; Edgar and Orr-Weaver 2001; Edgar et al. 2014). Two primary paths by which somatic polyploid cells are generated—endocycling and endomitosis—differ in timing of cell cycle exit (Fig. 1A), which has important consequences for a cell's capacity for future division. Endocycling cells entirely skip mitosis (M phase) and have only S and G phases (Fig. 1A, a,b). Sometimes S phase is truncated, in which case chromosome replication may be incomplete (Fig. 1A, a), resulting in chromosomes that cannot properly separate and segregate in mitotic division (Fig. 1B; Nagl 1982; Edgar and Orr-Weaver 2001; Edgar et al. 2014). Endomitosis is distinct from endocycling in that the process aborts later in the cell cycle (Fig. 1A, c,d) and thus has at least some mitotic features, including completion of chromosome replication, chromosome condensation, nuclear envelope breakdown, and sometimes even spindle formation (for review, see Nagl 1982; Edgar and Orr-Weaver 2001; Edgar et al. 2014). Because partial mitotic progression results in complete chromosome replication and separation, endomitotic cells can better retain the ability to re-enter mitosis than endocycled cells (Nagl 1982).
Figure 1.
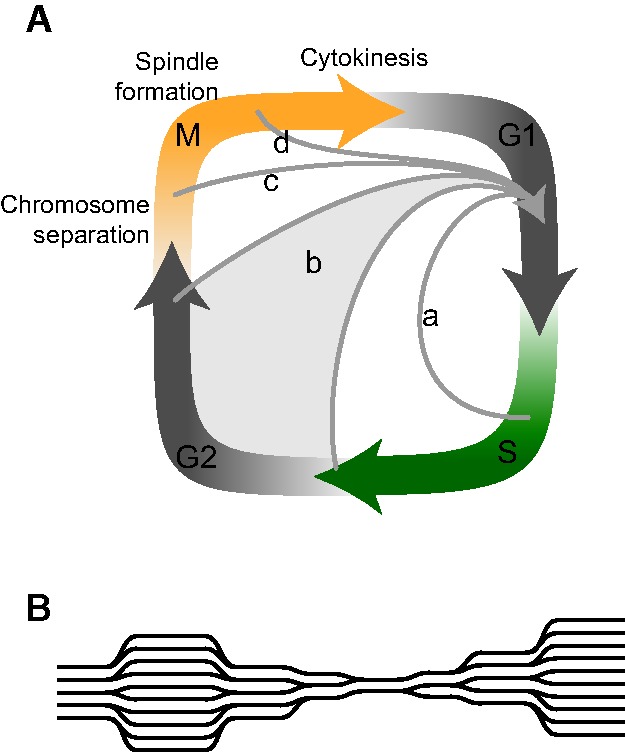
Cell cycle truncations leading to endopolyploidy. (A) Genome duplication can result from cell cycle truncations at any point after DNA replication has commenced but before cytokinesis fully divides cells. Different exits have distinct effects on cell biology and replicative potential. Endocycles (a and b) exit the cell cycle prior to mitosis; early exit prior to completion of S phase (a) leads to incomplete chromosome replication focused mostly on euchromatic regions. Cells that remain capable of mitosis have full-length S phases (b) (e.g., see Fox et al. 2010). Exit after M phase has begun (c) allows chromosome separation, while exit after spindle formation (d) likely contributes to nuclear shape changes (Castellano and Sablowski 2008). Adapted with permission from Macmillan Publishers Ltd.: Nature Reviews Molecular Cell Biology (Edgar et al. 2014), © 2014. (B) Possible architecture of chromosomes with partial DNA replication, showing amplified euchromatic regions and underreplicated heterochromatic regions (see Nagl 1982; Edgar et al. 2014).
Endopolyploid cells have provided lessons in how WGD can alter the biology of cells, highlighting the important roles ploidy variation can play in development, stress resilience, and disease. The ability to generate endopolyploid cells seems to have re-evolved many times and is likely an important adaptation in those tissues or cell types where mitotic division would be deleterious for structural reasons, when rapid growth or large cell size are required, or to allow cell survival when DNA damage makes mitotic division untenable (for review, see Vinogradov et al. 2001; Edgar et al. 2014; Orr-Weaver 2015). Although there is clearly variation in the biology of endopolyploid cell types in different tissues or species, they do share several important consistent features, such as increased cell size and perhaps altered growth potential and physiology, which we discuss below (e.g., see Levin 1983; Butterfass 1987; Galitski et al. 1999; Sugimoto-Shirasu and Roberts 2003; Barow 2006; Orr-Weaver 2015; Scholes and Paige 2015).
Big cells and rapid growth—developmental roles of somatic polyploidy
Polyploid cells often arise in diploids as a normal and regulated part of development. Examples include cells in the blood–brain barrier in insects, where tissue expansion is necessary but mitotic division would disrupt critical septate junctions, and cardiomyocytes, where mitotic division can destroy important intracellular structures (for review, see Orr-Weaver 2015). In placental trophoblast cells, endopolyploidy is important for invasive and nutritive functions (Parisi et al. 2003), and in megakaryocytes and glial cells, it is important for achieving their very large cell sizes (Orr-Weaver 2015). In moths and butterflies, large color-carrying wing scale cells are also endopolyploid (Cho and Nijhout 2013). In plants, there are also numerous examples of specialized large endopolyploid cells such as cotton fibers, cells in the pericarp of tomato fruits, giant cells in Arabidopsis sepals, leaf hairs, and cells essential for the formation of nitrogen-fixing nodules of legumes (for review, see De Veylder et al. 2011; Roeder et al. 2012). In all of these cases, endopolyploidy is a tightly regulated aspect of development and generally a terminally differentiated state. Its wide taxonomic distribution indicates that organisms have evolved to modulate ploidy to fit their needs, suggesting that variation in DNA content and/or cell size via endopolyploidy is adaptive in most multicellular eukaryotes. Conversely, symbionts and parasites can also induce endopolyploidy in their hosts; for example, at “nutrient exchange sites” in plants (for review, see Wildermuth 2010).
Endopolyploidy can also be inducible by variable conditions; e.g., when rapid growth is required or under stress conditions. Again, in most instances, these responses are tightly regulated normal processes. For example, a likely important conditional role for somatic polyploidy is in wound repair. Here endopolyploidy likely becomes important because polyploid cells can grow rapidly into vacant space without time-consuming mitotic divisions (for review, see Edgar et al. 2014). In humans, well-healing wounds have abundant tetraploid cells, while chronic wounds lack them (Ermis et al. 1998; Oberringer et al. 1999). Endopolyploidy is also involved in wound healing in Drosophila melanogaster, where polyploidy and cell fusion were shown to be directly important in repairing damaged epithelium at wound sites (Losick et al. 2013). This effect may also translate to whole-organism polyploids: A tetraploid morph of a New Zealand snail has faster wound repair than its diploid counterpart, although whether this arises directly from its polyploid state rather than an associated effect remains to be seen (Krois et al. 2013).
In plants, somatic genome duplication is linked to another kind of wounding response called overcompensation. Plants with this trait respond to herbivory by regrowing larger than before, often producing greater seed yield than undamaged controls (Scholes and Paige 2014). Recent work in Arabidopsis thaliana shows that strains with higher proportions of endopolyploid cells can overcompensate more effectively than those with fewer, and increasing the ability of strains to endocycle increases their ability to overcompensate (Scholes and Paige 2014, 2015). Although functionally distinct from localized wound repair in animals, the systems share a need for generating rapid tissue growth, and this may explain the shared reliance on endopolyploidy as a mechanism.
Another context in which inducible endopolyploidy seems to be important is stress response and resilience, where increased levels of endopolyploidy have been hypothesized to confer direct benefits (for review, see De Veylder et al. 2011; Schoenfelder and Fox 2015; Scholes and Paige 2015). However, in yeast cultures, isogenic diploid and tetraploid cells do not differ in stress tolerance, showing that stress tolerance is not a universal feature of polyploid cells (Andalis et al. 2004). Nevertheless, a number of interesting correlations have been found that hint that somatic WGD may contribute to stress resilience in at least some multicellular organisms. For example, in plant species Medicago and sorghum, root endopolyploidy is associated with salt tolerance (Ceccarelli et al. 2006; Elmaghrabi et al. 2013) and can be induced by salt in tolerant, but not sensitive, strains of sorghum (Ceccarelli et al. 2006). This suggests that the ability to induce endopolyploidy may be directly responsible for the resistance to salt, likely due to cell size changes in the roots that could alter ion uptake. Higher proportions of endopolyploid cells also contribute to greater drought tolerance in plants (Cookson and Granier 2006). Levin (1983) pointed out that biochemical and physiological changes that follow from WGD may also play important roles in adapting polyploid organisms to novel habitats. Indeed, there are hints that at least some of what has been demonstrated for endopolyploidy in plants might translate to whole-organism effects: For example, autotetraploid rice has greater resilience to drought and lower superoxide levels than diploid rice (Yang et al. 2014), and A. thaliana autotetraploids have greater salt tolerance than isogenic diploids (Chao et al. 2013). Just as with endopolyploidy, these effects may arise at least in part from larger cell size.
In animals, endopolyploidy in the liver was reported to increase after injury or toxic exposure, and WGD in this case was proposed to be a direct response to stress, since treatments that attenuate oxidative stress also reduce endopolyploidy (Gentric and Desdouets 2014). It has been argued that these polyploid cells can sometimes continue to divide and that aneuploidy resulting from chromosome segregation problems might be adaptive for the selectable diversity that it provides in toxic liver environments (Duncan et al. 2010). However, aneuploid cells also have the potential to give rise to pathologic cell lineages, suggesting that this would be a risky strategy at best (Gupta 2000). Furthermore, a recent single-cell resequencing study found evidence that aneuploidy is actually very rare across normal mammalian tissues, including the liver, suggesting that aneuploidy is not an adaptive feature of organ function and remains characteristic only of disease states (Knouse et al. 2014).
In both animals and plants, endopolyploidy is also implicated in resilience to DNA damage. DNA double-strand breaks trigger a signaling cascade that can switch off mitosis and trigger the initiation of endocycling (Ciccia and Elledge 2010; Adachi et al. 2011; Davoli and de Lange 2011). Endocyling after DNA damage is likely important for preventing cell death while halting mitotic proliferation of DNA-damaged cells (Ciccia and Elledge 2010; Adachi et al. 2011; Davoli and de Lange 2011). It could also provide a protective buffer against complete gene loss or haploinsufficiency due to the presence of additional DNA copies. An effect on resilience to DNA damage is also evident in A. thaliana, where increasing the proportion of endopolyploid cells in diploids as well as whole-organism tetraploidy both increase UV tolerance (Hase et al. 2006; Gegas et al. 2014). However, resistance to DNA damage of polyploid cells is certainly not universal: Isogenic ploidy series in yeast have shown that higher-ploidy cells in many cases do not have higher DNA damage resistance than diploids, and polyploids are actually more sensitive to some DNA-damaging agents (for review, see Storchova and Pellman 2004). This raises the possibility of differences among species or among types of DNA damage as to whether polyploidy is beneficial.
It has been proposed that endopolyploidy might directly affect the physiology and metabolic state of cells (Lee et al. 2009), perhaps due to the altered scaling ratios that accompany WGD-associated cell size increases (Weiss et al. 1975; Cavalier-Smith 1978; Levin 1983; Galitski et al. 1999; Storchova and Pellman 2004). Metabolic changes are evident and particularly well studied in cancers, and while, of course, cancers are highly complex and heterogeneous, in some cases, these shifts may be directly linked to polyploidy. Cancer cells commonly show increased reliance on glycolysis and resilience to hypoxic conditions, a set of traits called the Warburg effect (see e.g., Kim and Dang 2006; Vander Heiden et al. 2009). It has been shown that increased glycolysis precedes tumor hypoxia and thus seems to be a facilitator of tumor growth rather than a consequence of it (Vander Heiden et al. 2009). Recently, it was suggested that polyploid, but not diploid, tumor cells show the Warburg traits of resistance to hypoxia and reliance on glycolysis (Zhang et al. 2013). Why this is and whether it arises as a direct or indirect consequence of WGD are unclear, but similar patterns are seen elsewhere: Inhibition of Aurora kinases in acute myeloid leukemia cells also induces polyploidy, again coupled with increased glycolysis (Liu et al. 2011), and, in glioblastoma, polyploid cells similarly show increased glycolysis and are hypersensitive to glycolysis inhibitors relative to their diploid counterparts (Donovan et al. 2014). Other metabolic effects may also be linked to WGD: Aspirin and resveratrol selectively target polyploid cells by activating AMP kinase, a core sensor of cellular energy whose hyperactivation tetraploid cells are more sensitive to than diploid cells, for unknown reasons (Lissa et al. 2014). Whether similar metabolic shifts occur in endopolyploid cells in other systems will be interesting to explore. In this light, it is intriguing that, in Arabidopsis arenosa, a screen for selection after organismal WGD identified an AMP kinase-like protein of unknown function as having been under strong selection (Yant et al. 2013), perhaps reflecting a need for metabolic retuning after WGD, although the function of the selected alleles remains to be tested.
Are there costs of endopolyploidy?
While regulated endopolyploidy can clearly be beneficial, in some circumstances, there may also be costs. In particular, induced or spontaneous polyploidy can have risks associated with the unscheduled resumption of mitosis, as emphasized by the presence in animals of checkpoints that have evolved specifically to limit the proliferation of polyploid cells (Ganem and Pellman 2007). While endopolyploid cells are often terminally differentiated, some do remain capable of mitosis. For example, megakaryocytes, which give rise to platelets, are sometimes mitotically competent and can give rise to still-polyploid daughter cells (Leysi-Derilou et al. 2014). A big potential cost of allowing endopolyploid cells to return to mitosis is the risk of chromosome missegregation. In Drosophila and Culex, for example, polyploid cells in the digestive tract and rectum remain capable of mitosis, but these divisions often produce aneuploid progeny cells (Fox et al. 2010). Whether these cause trouble for the organism is not known, but they nevertheless highlight that mitosis from polyploid cells can be problematic. In the mammalian liver, polyploid cells can sometimes also continue to proliferate, but these also experience challenges with chromosome segregation and spindle geometry (Gentric and Desdouets 2014). The danger of mitotic divisions in endopolyploid cells is further highlighted by the observation that tumorigenesis can be promoted by re-entry to mitosis of DNA-damaged polyploid cells, which produces proliferative aneuploid populations (Davoli and de Lange 2011; Edgar et al. 2014). We discuss challenges associated with polyploid chromosome segregation in more detail below.
In normal development, there may also be costs associated with cell size increases and the usual irreversibility of polyploidy (Orr-Weaver 2015; Scholes and Paige 2015). For example, it has been proposed that high proportions of polyploid cells in an organ may decrease “metabolic scope” (the ratio of maximum to basal metabolic rate), suggesting that tissues with more polyploid cells have a poorer “safety margin” when operating at high capacity (Vinogradov et al. 2001). Furthermore, because endopolyploid cells cannot generally resume mitotic division (although they can continue to endocycle), the proliferative capacity of tissues with high proportions of endopolyploid cells can be limited. For example, endopolyploidy that occurs in response to toxicity or injury in the liver is thought to limit long-term regenerative capacity and may be a major factor in aging (Gupta 2000). The problem of limited mitotic potential could be particularly prominent in fluctuating environments, where one set of conditions may favor endopolyploidy, but, as conditions change, endopolyploidy could become disfavored, at which point it cannot generally be undone (Scholes and Paige 2015).
Another potential problem associated with endopolyploidy is that surface to volume ratio decreases with WGD-driven cell size increases. This could result in a problem of communication—a decrease in the efficiency of nuclear–cytoplasmic transport (Orr-Weaver 2015; Scholes and Paige 2015). That cell size matters for polyploid cells is supported by direct comparisons of isogenic diploid and tetraploid yeast cells, where expression differences attributable purely to a ploidy shift included mostly genes whose expression is responsive to cell size independent of DNA content (Galitski et al. 1999; Wu et al. 2010). In some cells, the shift in surface to volume ratio may be compensated to some extent by alterations in nuclear shape that increase surface area, such as deep indentations in the nuclear membrane and/or flattening of the nucleus (Nagl 1982; Buntrock et al. 2012; Pirrello et al. 2013; Orr-Weaver 2015). Indeed, in tomato fruits, complexity of nuclear shape correlates positively with cell ploidy (Nagl 1982; Pirrello et al. 2013), and highly endopolyploid cells in a moth have bizarre giant nuclei that are flat and elaborately branched (Buntrock et al. 2012). These shape changes have been proposed to increase nuclear–cytoplasmic communication, but whether they are actually important for the cells in which they occur is not clear. It could be that nuclear shape modification is an adaptive and regulated response to polyploidy, but it may also be incidental to the endoreduplication process: In A. thaliana mutants with partially disrupted mitotic spindles, endopolyploid cells form with complexly branched nuclei. This distortion is thought to arise because the nuclei in these cells have been subjected to repeated but ultimately unsuccessful application of spindle forces (Castellano and Sablowski 2008). These findings suggest that complex nuclear shapes could arise sporadically but, by lessening the surface area to volume challenge, could nevertheless be advantageous in cells with high ploidy.
WGD and adaptation in cell lineages
Some proliferating polyploid cell lineages (in tissue culture or tumors, for example) persist, thrive, and diversify, highlighting the adaptive potential of WGD. What is the nature of that potential? Experimental evolution studies have probed the adaptive potential of WGD in cell lineages, and there are good examples of immediate as well as longer-term fitness effects resulting from ploidy shifts (e.g., see Gerstein and Otto 2009). Here we discuss examples from fungi, plants, and animals.
Fungi
Natural variation for ploidy exists in Saccharomyces cerevisiae, with haploids, diploids, and tetraploids endemic to the same microsite, suggesting that ploidy variation could play an adaptive role (Ezov et al. 2006). The ploidy state flexibility of yeast allows the study of strains that are genetically identical except for ploidy, a useful tool for directly plumbing the effects of WGD in evolving populations (Galitski et al. 1999). In stationary culture (nutrient-limited conditions) in yeast, tetraploidy was detrimental to cell survival. Although tetraploid cells could sense nutrient deprivation just as diploid cells could, they did not respond by aborting mitosis and instead continued to proliferate, which, in these conditions, was lethal (Andalis et al. 2004). Leveraging a similar approach, Selmecki et al. (2015) compared the longer-term evolvability of otherwise genetically identical haploids, diploids, and tetraploids and found that evolutionary adaptation to a poor carbon source medium was significantly faster in tetraploids than in diploids. Population genomic resequencing, modeling, and phenotyping indicated that more beneficial mutations arose in autotetraploids than in diploids and that these also have stronger fitness effects (Selmecki et al. 2015). This study provides some of the clearest evidence to date of the greater adaptability of polyploid lineages. However, these results, taken together, also highlight that polyploidy is advantageous in some, but not all, situations.
Plants
Most studies of ploidy dynamics in plant cell lineages focus on either callus (three-dimensional growths of “dedifferentiated” cells on solid medium) or liquid cell culture. Results from these studies also highlight potential adaptive roles for ploidy shifts as well as the potential influence of environment on whether ploidy is beneficial. Long-term callus growth of a broad range of plant species indicates a common progression over time from diploid to tetraploid cells followed by devolution into aneuploid swarms (e.g., see Murashige and Nakano 1967; Torrey 1967; Singh and Harvey 1975). Importantly, although highly repeatable in callus, a trend to higher ploidy is not universal in plant tissue culture: Liquid-grown suspended cultures in Haplopappus gracilis and Haplopappus ravenii are exclusively made up of diploid cells, while callus growths of the same species are quickly dominated by polyploid cells (Singh and Harvey 1975). This underscores that whether polyploidy is adaptive for cells depends on growth conditions. It has been noted that the progression from diploid to tetraploid and aneuploid cells seen in callus cultures is at least superficially similar to events in neoplastic progression in carcinogenesis (e.g., see Gaspar et al. 1991; Häsler et al. 2012). To what degree these similar paths are (or are not) driven by similar selection pressures in tumors and callus culture remains to be seen. One shared feature may be a selection for tolerance to hypoxia in crowded tumor or callus conditions, as there is some evidence that tolerance to hypoxia may be higher in polyploid cells than diploid cells, at least in humans (Zhang et al. 2013).
Whether aneuploidy that follows from tetraploidy in callus growth is selectively advantageous to cell lineages in this context or merely an unavoidable consequence of mitotic divisions in tetraploid calli is not clear. In any case, attempts to generate embryos from callus are usually successful in both the diploid and tetraploid stages but progressively fail as calli become aneuploid, suggesting that, while tolerated in culture, aneuploidy is detrimental to multicellular development, while both diploidy and tetraploidy are tolerated (Torrey 1967; Gaspar et al. 1991). In those cases where regeneration does succeed from aneuploid calli, the plants that result are nevertheless euploid (fully diploid or tetraploid), suggesting that there is strong selection for euploid lineages in multicellular development (Feher et al. 1989; Raja et al. 1992). Understanding the dynamics of this process and the nature and strength of selection against aneuploidy in multicellular plant growth will be very interesting, especially in light of the fact that aneuploidy in plant leaves is not incompatible with cell survival (e.g., see Wright et al. 2009).
Mammals
In mammals, the anarchic proliferation that characterizes within-host cancer evolution commonly includes a high diversity of aneuploid cell lineages associated with disease progression, and at least some of these are thought to arise via chromosome missegregation from tetraploid intermediates (for review, see Ganem et al. 2007; Davoli and de Lange 2011; Burrell and Swanton 2014; Gerlinger et al. 2014b). Tetraploid intermediates may be quite common and have been suggested to occur in >50% of liver adenocarcinomas and ∼30% of pancreas and lung adenocarcinomas, cervical carcinomas, neuroblastomas, and Hodgkin's lymphomas (see references in Davoli and de Lange 2011). While polyploid intermediates can arise spontaneously, viruses linked to cancer can also trigger this progression. For example, viral-induced cell fusion can trigger the proliferation of autotetraploid human cells if oncogenes or a mutated version of p53 are expressed (Duelli et al. 2005, 2007). Human papilloma virus (HPV) infection promotes cell fusion in humans (Hu et al. 2009) and mice (Gao and Zheng 2010) and contributes to the etiology of cervical cancer.
A limitation to understanding the role that polyploid cells might play in tumorigenesis has been that it is rarely possible to observe very early events in human cancer etiology. However, Barrett's esophagus provides such a glimpse. This condition predisposes to esophageal adenocarcinoma but is recognizable before neoplastic progression (Galipeau et al. 1996). Biopsies containing elevated quantities of tetraploid cells portend inactivation of the p53 tumor suppressor, disease progression, and the onset of gross aneuploidy. This provides direct evidence that even though some tissue abnormalities were already present, tetraploidy preceded aneuploidy and disease progression (Galipeau et al. 1996). Increased frequency of polyploid cells also occurs during the progression of cervical (Olaharski et al. 2006), breast (Dutrillaux et al. 1991), and other cancers (for review, see Davoli and de Lange 2011). This raises the hypothesis that unstable tetraploid intermediates, even if not primarily causal, can facilitate the generation of highly aneuploid malignancies. Beyond aneuploidy, cancer cells have a large array of additional problems with the maintenance of genome stability (Box 1). That polyploidy may serve as an intermediate promoter of tumorigenesis has empirical support from the observation that tetraploid, but not diploid, p53-null mouse mammary epithelial cells promote tumorigenesis when transplanted into immune-compromised mice (Fujiwara et al. 2005).
Box 1. Engines of genome diversification
Aggressive aneuploid cancers, some of which may arise via tetraploid intermediates, exhibit striking genomic modifications, some of which are recognized in many systems, while others are specific to the cancer literature. Recent work has demonstrated that these diversifying processes engender the establishment of genetically rich cell communities upon which natural selection acts (Stephens et al. 2011; Nik-Zainal et al. 2012a,b; Baca et al. 2013). The recognition of most of these processes challenges the view that cancer evolution is simply a process of gradual serial mutation accumulation, arguing instead that punctuated or saltational evolutionary trajectories can also be important (Baca et al. 2013; Lazebnik 2014). Several dramatic examples of the types of genome modification that have been reported in cancer evolution include the following:
Chromoplexy (from the Greek pleco, to weave or braid): A phenomenon of complex genome restructuring in which DNA translocations and deletions emerge in a highly interdependent manner; observed first in prostate cancers, where it frequently accounts for dysregulation of important cancer loci (Baca et al. 2013). It appears to disrupt multiple cancer genes in a coordinated fashion, and the level of chromoplexy is correlated with tumor histological grade (Baca et al. 2013)
Chromothripsis (from the Greek thripsis, shattering): A cataclysmic burst of genome rearrangement during a single cell cycle, in which hundreds of genomic rearrangements occur in a one-generation crisis usually focusing on one or a small number of chromosomes (Stephens et al. 2011).
Kataegis (Greek for shower or thunderstorm): A localized storm of hypermutation that normally colocalizes with somatic rearrangements; common in breast cancer cells (Nik-Zainal et al. 2012a). It is sometimes associated with arrangements that have features of chromothripsis.
Aneuploidy: The state of harboring a chromosome complement that differs from simple multiples of haploid chromosome sets. This can be greater or less than the diploid quantity. Aneuploidy can provide a strong selective advantage, e.g., in response to multiple environmental stressors in yeast (Rancati et al. 2008). Tetraploid cells commonly missegregate chromosomes on account of their supernumerary centrosomes (Ganem et al. 2009), readily generating subclones with aneuploid chromosome complements. Aneuploidy is a hallmark of cancers found in ∼90% of solid tumors and 50% of blood cancers (Beroukhim et al. 2010; Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer [http://cgap.nci.nih.gov/Chromosomes/Mitelman]).
Chromosomal instability (CIN): A persistently elevated rate of chromosome gain/loss common in many cancers that leads to aneuploidy (Lengauer et al. 1997).
Expression of meiosis genes: Meiosis genes, normally expressed in the mammalian germline, are misexpressed in many cancers. Genome instability, seen commonly as part of the neoplasmic phenotype, could be caused by an admixture of mitotic and meiotic complexes (Simpson et al. 2005).
Masking: Increased allelic redundancy in polyploid genomes covers the effect of deleterious mutations (as they are less likely to be homozygous and are often a smaller proportion of the allelic complement). It has been suggested that this aspect of tetraploidy would be especially beneficial in the face of a mutator phenotype, as encountered in many cancers (Davoli and de Lange 2011). In addition to buffering potentially deleterious single-nucleotide polymorphisms (SNPs), polyploidy may also buffer the effect of rampant aneuploidy found in cancers (Varetti et al. 2014). This is potentially an opportunity, as retained alleles may provide low-frequency variants upon which selection can act as a sublineage encounters novel tumor microenvironmental challenges.
How exactly WGD might facilitate tumor progression is an open question. This is no surprise given the range of evolutionary trajectories that characterize diverse cancer types and the difficulty of obtaining a truly representative picture of the evolutionary paths that cell lineages follow within a single tumor (Gerlinger et al. 2014b; Walther et al. 2015). Beyond the unresolved population genetic considerations of the effects of variation in a polyploid context (e.g., see Otto and Whitton 2000; Gerstein and Otto 2009), on the phenotypic side, recent work suggested that polyploidy may indeed promote rampant aneuploidy and genetic and phenotypic diversity (Lagadec et al. 2012; Erenpreisa and Cragg 2013; Zhang et al. 2013). Furthermore, polyploid giant cells, which are often found in human solid tumors, are highly resistant to low oxygen conditions, cycle slowly (Zhang et al. 2013), and have been proposed to also contribute to lineage expansion and heterogeneity upon induction by chemotherapy or radiation treatment (Lagadec et al. 2012; Zhang et al. 2013). The data available to date thus suggest that polyploidy could substantially contribute to cell lineage diversity and adaptability in diverse situations.
Polyploid cell lineages as evolving populations
Genome duplication increases the number of available alleles for mutations to accumulate and upon which selection can then act (Otto 2007). The doubling of segregating alleles at each locus means that recessive alleles can be better masked in autotetraploids, allowing for the retention of greater allelic diversity and likely also greater deleterious genetic load than in diploids (Otto 2007). Because nascent polyploid lineages instantly differ in at least some respects (e.g., cell size) from their diploid progenitors, alleles suddenly find themselves “a stranger in a strange land,” and thus even pre-existing variants experience novel selection pressures. This alone may increase phenotypic diversity. Adding to this are the genomic instabilities that arise when mitotic progression resumes in neo-autopolyploids (whether individual cells or entire organisms), which may generate additional genetic diversity via chromosome rearrangements, insertions, and deletions. Aneuploidy not only creates novel variation in its own right but can also expose retained recessive mutations on the remaining chromosomes as additional sheltering copies are lost.
Genetic diversity in tumors can predict progression to disease (Maley et al. 2006), likely because selection for phenotypic novelty can arise rapidly in a tumor environment, where heterogeneity develops as distinct lineages encounter variable microenvironmental selection pressures (Marusyk et al. 2012). An emerging view of tumors is one of heterogeneous “communities” that consist of a diversity of highly branched evolutionary trajectories (Gerlinger et al. 2014a). Recent whole-genome resequencing of cell subpopulations from a single tumor sorted by DNA content emphasized tumor complexity and that both sequence and structural variation are common (Malhotra et al. 2015). The potential dangers associated with diversity and polyploidy are highlighted by a study of multiple tumor regions in a single patient with kidney cancer. The cell sublineage in the primary tumor that was most similar to those metastatic sites with the greatest chromosomal instability consisted of tetraploid cells, while remaining regions were diploid, raising the possibility that polyploidy may have contributed to metastatic potential in this case (Gerlinger et al. 2012). Another recent study gave additional evidence that WGD in human colon tumor cell lines promotes tolerance of chromosome abnormalities relative to isogenic diploids. Independent autotetraploid lines showed convergent changes, including repeated losses of a region of chromosome 4q that is commonly absent in colorectal cancers in vivo (Dewhurst et al. 2014). Taken together, there is now considerable evidence to support the idea that tumor transformation involves aneuploidy, with tetraploidy sometimes serving as an intermediate “gateway state” that both promotes diversity and aneuploidy and provides greater tolerance of them (Storchova and Pellman 2004; Storchova et al. 2006; Thorpe et al. 2007; Storchova 2014; Coward and Harding 2014). There will likely be much to gain from considering cancer, at least in some respects, as a potentially predictable evolutionary process of heterogeneous cell lineages and considering polyploidy and the aneuploidy that can follow from it as central engines of diversity in that process (Coward and Harding 2014).
Polyploid chromosome segregation
One of the most repeatable costs of WGD is the instability of chromosome segregation. In whole-organism polyploidy, chromosome segregation problems are strikingly evident in meiosis, where failures in the sorting of additional homologous chromosomes can lead to infertility or developmental abnormalities in progeny. Similarly, mitosis in polyploid cells also often leads to aneuploidy. Below we discuss chromosome segregation problems that polyploids face in meiosis and mitosis in turn.
Polyploid meiosis
Segregating additional chromosomes in meiosis is a vexing challenge for newly formed autopolyploids. When more than two homologs are present in a meiotic cell, they can form aberrant associations called multivalents among more than two of the available homologs, which can cause segregation problems (e.g., for review, see Ramsey and Schemske 2002; Comai 2005; Bomblies and Madlung 2014). Both the nature of the problem and the solutions that can evolve are most extensively studied in plants, where whole-organism polyploids are especially common. Many newly formed autopolyploids exhibit extensive multivalent formation, often coupled with reduced fertility, whereas most established autopolyploids form primarily or exclusively diploid-like bivalents (e.g., see Shaver 1962; Charpentier et al. 1986; Wolf et al. 1989; Srivastava et al. 1992; Ramsey and Schemske 2002; Santos et al. 2003; Yant et al. 2013). Even in shorter-term selection experiments for fertility in newly generated autopolyploids, quadrivalent frequency declined and bivalent frequency increased in several generations in, e.g., Hyoscyamus albus (Srivastava and Lavania 1990), Pennisetum typhoides (Jauhar 1970), Zea mays (Gilles and Randolph 1951), Secale cereale (Bremer and Bremer-Reinders 1954; Hilpert 1957), and A. thaliana (Santos et al. 2003). There are a handful of exceptions to the general trend of reduced crossover formation in autotetraploids. In several related newly autopolyploid grasses, fertility is positively correlated with chiasma number and quadrivalent formation (Myers 1945; Müntzing 1951; McCollum 1958; Hazarika and Rees 1967; Simonsen 1975). These species are unusual in two important ways: (1) They form quadrivalents with only terminal chiasmata that disjoin regularly, and (2) unlike in other species, a decline in crossover frequency in these species leads to increased univalent frequency, which is strongly linked to infertility. Thus, the main selective force in these species seems to be on univalent prevention rather than multivalent suppression. Taking the above together, the evolution of meiotic stability after WGD in autopolyploids seems to involve either (1) multivalent prevention via reductions in genome-wide crossover rates or, (2) more rarely, univalent prevention via increases in recombination coupled with modifications of crossover placement to facilitate segregation.
Previous observations are consistent with the existence of genetic multivalent suppression systems in autopolyploids that (in most cases) reduce crossover frequency, often to one per chromosome pair, and/or alter the localization of crossovers (e.g., see Shaver 1962; Hazarika and Rees 1967; Watanabe 1983; Srivastava and Lavania 1990). Crossover reduction may be a major route of autopolyploid meiotic stabilization because it reduces the likelihood of multivalent formation. This is particularly clear in the extreme example when each chromosome can form only a single crossover, in which case only bivalents can persist to metaphase. This is further supported by the observation that lower crossover rates in diploids correlate with increased meiotic stability of the neopolyploids derived from them, where diploids with low crossover rates are effectively preadapted for polyploid success (e.g., see Murray et al. 1984; Srivastava et al. 1992; Jenczewski et al. 2002). In autotetraploid Arabidopsis arenosa, selection after WGD acted on genes encoding structural proteins important for the formation of chromosome axes, crossover designation, and synapsis, suggesting that this reflects a coordinated multigenic shift in meiosis that reduces crossing over (Hollister et al. 2012; Yant et al. 2013). A reduction in crossover rates could risk the formation of unpaired univalents if there is not also assurance that at least one crossover forms per bivalent, which is needed for regular chromosome segregation (Jones and Franklin 2006). One mechanism of lowering crossovers without risking univalents and thus aneuploidy is to increase the strength or distance of crossover interference. Crossover interference prevents formation of new crossovers near previously designated ones and has been suggested as a mechanism for crossover reduction in autopolyploids (Shaver 1962; Lavania 1991). By this mechanism, a single crossover forms on each chromosome uninhibited, but additional crossovers would be suppressed. Although the molecular nature of crossover interference is not yet known, a leading theory proposes that the signal is a physical force transmitted along the chromosome axes (Zhang et al. 2014), making it especially interesting that axis components and interacting proteins are under selection in tetraploid A. arenosa (Yant et al. 2013); how these genes might contribute to multivalent suppression is an important as yet unanswered question.
Polyploid mitosis
Unlike with meiosis, it is not immediately obvious, when considering only genome duplication, why mitosis should be problematic for autopolyploids; it is, as discussed above, nevertheless consistently linked with aneuploidy, showing that polyploid cells clearly do face problems in mitosis. Important insights into possible underlying mechanisms came from an elegant study in yeast in which the investigators screened for mutations that were lethal to tetraploids but not diploids (Storchova et al. 2006). Among thousands tested, the investigators identified 39 tetraploid-specific lethal mutants, which collectively indicate that genes important for spindle geometry, sister chromatid cohesion, and homologous recombination are essential specifically in tetraploids (Storchova et al. 2006). The importance of spindle geometry for polyploids highlighted in the yeast study likely arises from a scaling problem. Cell and nuclear volume increase with ploidy, but spindle size does not. This mismatch leads to increased spindle attachment abnormalities that threaten the regularity of chromosome segregation in polyploid cells (Storchova et al. 2006; Storchova and Kuffer 2008). The importance of cohesion and homologous recombination for polyploids may be linked to a saturation of DNA repair due to the presence of additional DNA (Storchova et al. 2006; Storchova and Kuffer 2008). The work in yeast highlights that mitotic problems faced by polyploid cells can arise as direct by-products of both the altered geometry and increased DNA content of polyploid cells.
There are also indications that whole-organism polyploids can suffer mitotic instabilities. For example, in leaf tissues of polyploid plants, aneuploidy has been reported (e.g., see Greilhuber and Weber 1975; Wright et al. 2009). However, more direct comparisons of diploids and tetraploids are needed to conclude that this is a general trend and that it is specific to polyploidy rather than other aspects of the biology of these species. However, if the somatic aneuploidy is truly due to problems with polyploid mitosis in these plants, we expect that the challenges that cause it are likely similar to those noted above for yeast. Importantly, however, mitotic segregation problems may be excluded from plant stem cell tissues. For example, even though mitotic instability has been reported in leaves of an autotetraploid A. arenosa strain (Wright et al. 2009), wild-collected accessions consistently exhibit euploid chromosome complements (Comai et al. 2000; Hollister et al. 2012; Schmickl et al. 2012; Arnold et al. 2015). Thus, either stem cells that give rise to gametes do not become aneuploid, or any aneuploid lineages that do arise are strongly selected against during development such that aneuploid lineages do not persist to contribute to the gamete pool. This is consistent with results from plants regenerated from tissue culture, which suggest selection for euploid cells in multicellular development (Feher et al. 1989; Raja et al. 1992). Thus, it seems that the aneuploidy seen in leaves of polyploids is sporadic. An important question that remains is how such failures are excluded from tissues that ultimately form gametes. Is this purely a selective process, or is there some important difference in the way mitosis is controlled in distinct tissues that makes polyploid cells in some contexts more prone to subsequent aneuploidy than others?
Reduction divisions in somatic cells
A mysterious process that highlights the sometimes fluid boundaries between mitosis and meiosis is somatic reduction, which refers to the observation that some somatic polyploid cells undergo meiosis-like reduction divisions (e.g., see Huskins 1948; Wilson and Cheng 1949; Rajaraman et al. 2007). While not explicitly tested in most species, in Trillium plants, somatic reduction divisions that occur in polyploids were shown to indeed segregate homologous chromosomes from one another as in meiosis (Wilson and Cheng 1949). Likely the same is true in other species in which the progeny cells survive, suggesting that somatic reduction divisions are, at least in this fundamental way, meiosis-like. Cytological studies have provided further hints that somatic reduction divisions often show meiosis-like features: In the onion Allium cepa, chemically induced polyploid somatic cells undergo reduction divisions that exhibit meiosis-like chiasma formation (Huskins 1948). Polyploid p53-null HeLa cells also undergo reduction divisions in which structures form between homologous chromosomes that are similar to meiotic synaptonemal complexes (Ianzini et al. 2009). Just how meiosis-like somatic reduction divisions are or need to be is unclear, but support for the hypothesis that a full meiotic program is not required for reduction division comes from Candida albicans. This species lacks conventional meiosis as well as components of the crossover formation pathway that are strictly required for meiosis in close relatives (e.g., Saccharomyces cerevisiae) (Tzung et al. 2001). Nevertheless, polyploid C. albicans can diploidize via a meiosis-like program that requires the meiotic proteins SPO11, DMC1, and HOP1 (Bennett and Johnson 2003).
Results to date highlight that the “bleed-over” of meiosis into mitosis can be hazardous to genome stability. For example, in A. cepa, reduction divisions are generally not well organized and frequently show chromosome missegregation (Huskins 1948). In mammals, polyploid hepatocytes undergo reduction divisions with multipolar spindles and lagging chromosomes that yield a range of aneuploid progeny cells (Duncan et al. 2010; Gentric and Desdouets 2014). In response to DNA-damaging agents or radiation, polyploid cells arising in cancers can express meiosis genes and undergo a pseudomeiotic “depolyploidization,” which results in high rates of aneuploidy (Old 2001; Erenpreisa et al. 2005a,b, 2011; Rajaraman et al. 2005, 2007; Kalejs et al. 2006; Puig et al. 2008; Ianzini et al. 2009; Salmina et al. 2010).
Another situation that may or may not be relevant to polyploidy specifically is a phenomenon called “meiomitosis.” Although this process is not a true reduction division, it merits discussion here because it highlights why somatic reduction divisions might commonly be unstable. The concept of “meiomitosis” comes from observations that aggressive cancers with high levels of aneuploidy often express one or a few meiosis genes (for review, see Old 2001; Simpson et al. 2005; Kalejs et al. 2006; Lindsey et al. 2013). Examples of several of these genes as well as their intriguing overlap with some of the genes implicated in adaption to polyploidy in A. arenosa are listed in Box 2. The problem with chromosome segregation in meiomitosis seems to arise from the mixing of systems. Because meiosis evolved from mitosis (Hurst and Nurse 1991), many proteins are shared between the two types of division, but protein complexes generally contain at least some homologs that are either mitosis- or meiosis-specific. Why expressing mixtures of mitotic and meiotic proteins may be problematic was recently laid out in detail for the cohesin complex (Strunnikov 2013); we expect that similar stories apply to other meiosis protein heterocomplexes. Cohesins mediate sister chromatid cohesion in both meiosis and mitosis. In mitosis, the cohesins SMC1 and SMC3 associate with the kleisin Rad21, while, in meiosis, they associate with a related kleisin, Rec8. The Rec8-containing complex remains more strongly associated with sister chromatids, particularly at the centromeres. In meiosis I, this “stickiness” is crucial for retaining sister chromatid cohesion to meiosis II and preventing premature segregation of sisters, but, if aberrantly expressed in mitosis, Rec8 could prevent the timely release of cohesin from sister chromatids and thereby disrupt chromosome segregation (Strunnikov 2013). These problems would apply to both diploid and polyploid cells. Would expression of more meiosis genes in a mitotic cell better ensure regular chromosome segregation? Perhaps certain combinations would indeed help alleviate problems. For polyploid cells, however, the story may be more complex: Work in plants suggests that a diploid meiotic program is ill-suited for polyploid chromosome segregation, and stabilization likely requires a coordinated evolutionary shift in multiple interacting genes (Hollister et al. 2012; Yant et al. 2013).
Box 2. Other meiosis proteins expressed in cancer cells
REC8: A kleisin important for preventing premature sister chromatid separation in meiosis I. REC8 tethers the cohesin complexes to the centromeres of sister chromatids, effectively gluing them together until meiosis II. Its expression in mitosis could cause chromosome missegregation by preventing proper sister chromatid separation (Ishiguro et al. 2010; Lindsey et al. 2013; Strunnikov 2013). REC8 may also drive depolyploidization in polyploid cancer cells by promoting reductional divisions (Kalejs et al. 2006). A homolog of REC8 was also under selection in a tetraploid plant lineage, suggesting a role in stabilizing post-WGD meiotic chromosome segregation (Yant et al. 2013).
DMC1: Together with a related protein, Rad51, DMC1 helps coordinate early events in homologous recombination. DMC1 is overexpressed in several cancers, and targeting its expression in cell culture has been effective in reducing the proliferation and aneuploidy of glioblastoma cells, while it has no effect on nonneoplastic cells (Rivera et al. 2015). Intracranial implantation of glioblastoma cells with knocked down DMC1 levels into immunocompromised mice produced smaller tumors than control cells that express DMC1 (Rivera et al. 2015). In humans, there may be a similar effect that is more dependent on the levels of associated proteins. Reducing the expression of a partner of DMC1, RAD51, sensitizes glioblastoma cells to radiation (Short et al. 2011). Interestingly, while increased DMC1 levels do not lead to negative prognosis in glioblastoma, increased HOP2 and MND1, which are necessary for DMC1–RAD51 to bind to DNA, is correlated with poor survival in The Cancer Genome Atlas (Rivera et al. 2015).
SCP1: During meiosis, the axes of homologous chromosomes are bridged and brought closer by the formation of a zipper-like proteinaceous structure called the synaptonemal complex (SC). The central elements of the SC are SCP1 in humans (Meuwissen et al. 1997), Zip1 in yeast (Sym et al. 1993), C[3]G in Drosophila (Page and Hawley 2001), and ZYP1 in Arabidopsis (Higgins et al. 2005). SCP1 expression in fibroblast cells yields SC-like structures, suggesting that, despite the absence of axial element proteins, SCP1 can sometimes form a SC-like structure on its own when expressed in somatic cells (Öllinger et al. 2005). SCP1 in humans was the first of the cancer-expressed meiosis genes identified (Türeci et al. 1998). In Drosophila, up-regulation of germline genes, including the SC central element C[3]G, is important for brain tumor development (Janic et al. 2010). SC formation could cause inappropriate associations of homologs in mitosis that may not be properly resolved if other meiotic proteins are lacking. Interestingly, the SC central element ZYP1 in A. arenosa also shows strong evidence of having been under selection in the tetraploid lineage, suggesting a role for the SC in stabilizing polyploid chromosome segregation (Yant et al. 2013).
HORMAD1 and HORMAD2, an axis of evil? Meiotic HORMA proteins form linear structures along unsynapsed sets of sister chromatids, help mediate crossing over and the synapsis of homologs (Hollingsworth et al. 1990; Armstrong et al. 2002; Niu et al. 2005; Fukuda et al. 2010; Shin et al. 2010; Daniel et al. 2011), and promote use of the homolog rather than the sister chromatid for double-strand break repair (Schwacha and Kleckner 1994; Niu et al. 2005). In humans, there are two paralogous meiotic HORMA proteins, HORMAD1 and HORMAD2, both of which have been reported to be expressed in aggressive tumors, although not together (Aung et al. 2005; Chen et al. 2005; Liu et al. 2012; Shahzad et al. 2013). At least for HORMAD1, there is evidence that this expression is dangerous: In vitro siRNA silencing of HORMAD1 in ovarian cancer cells decreases their aggressiveness and metastatic potential (Shahzad et al. 2013), and its expression directly contributes to genome instability and aneuploidy in breast cancer cells (Watkins et al. 2015). The latter is apparently due to its usual meiotic role in promoting the use of the homolog as a double-strand break repair template rather than the sister chromatid by suppressing Rad51-mediated double-strand break repair. In the absence of the meiotic recombination machinery, when the Rad51 pathway is blocked, double-strand break repair is instead shuttled to the error-prone nonhomologous end-joining (NHEJ) pathway, resulting in genome instability (Watkins et al. 2015).
Recent work with the yeast homolog Hop1 suggests that purified Hop1 protein can self-associate to form rigid rod-like structures that tightly unite DNA molecules independent of homology (Khan et al. 2012). This finding supports the idea that when these proteins are aberrantly expressed in cells that lack the proteins necessary to subsequently remove them (e.g., Chen et al. 2014; Lambing et al. 2015), they may make chromosomes “sticky,” driving aberrant interactions and missegregation. Recently, we found that a unique allele of ASY1, the A. thaliana homolog of HORMAD1/HORMAD2 and Hop1 (Armstrong et al. 2002), underwent a dramatic selective sweep following WGD in A. arenosa (Hollister et al. 2012; Yant et al. 2013). Whether the chromosome “stickiness” induced by diploid versions of these proteins affects chromosome segregation in cancer cell lineages and polyploid meiosis in similar ways remains to be tested.
Much remains to be learned about somatic reduction. For example, what meiotic genes minimally suffice to drive somatic reduction? Does stability of these divisions correlate with the number of meiosis genes expressed? Are somatic reduction divisions aberrations, or can they be important in normal development? Does somatic reduction ever provide a reliable remedy for the normally irreversible fate of somatic endopolyploidy?
Conclusions
Results from a wide range of eukaryotes clearly show that WGD often provides adaptive opportunities. However, in those cases where polyploid cells continue to divide, they face substantial challenges, especially for the regular segregation of chromosomes. This can lead to chromosome instability and aneuploidy, which can sometimes be adaptive at the level of cell lineages but appears in most cases to be deleterious (or, at best, neutral) for the organism at large. Thus, the regulated management of cellular genome content plays important and beneficial roles in development, tissue repair, and stress responses, while its mismanagement can lead to genome instability and contribute to tissue aging and pathologic states, including cancer progression. Viewing proliferating polyploid cell lineages from an evolutionary and comparative perspective may yield novel insights into the role that the double-edged sword of polyploidy plays in the biology of organisms and their evolution. Many open questions remain, such as understanding the mysterious process and developmental role (if any) of somatic reduction divisions, the role that aneuploidy may play in normal development or stress resilience (if any), and the causes and consequences of expressing partial meiotic programs in somatic cells. Furthermore, many potentially interesting effects are currently only correlated with polyploidy, and more work is required to test causality. Learning which effects are direct outcomes of polyploidization itself and their mechanistic basis has the potential to provide important insights. Where similar correlates are observed across kingdoms, deeper investigation of the underlying causes for the apparent similarities may yield novel insights into the most fundamental effects that polyploidy has on the biology of cells, both individually and in the context of the multicellular organisms in which they are found.
Acknowledgments
We thank Nancy Kleckner and members of the Bomblies and Yant groups for helpful discussions.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.271072.115.
References
- Adachi S, Minamisawa K, Okushima Y, Inagaki S, Yoshiyama K, Kondou Y, Kaminuma E, Kawashima M, Toyoda T, Matsui M, et al. 2011. Programmed induction of endoreduplication by DNA double-strand breaks in Arabidopsis. Proc Natl Acad Sci 108: 10004–10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andalis AA, Storchova Z, Styles C, Galitski T, Pellman D, Fink GR. 2004. Defects arising from whole-genome duplications in Saccharomyces cerevisiae. Genetics 167: 1109–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong SJ, Caryl AP, Jones GH, Franklin FCH. 2002. Asy1, a protein required for meiotic chromosome synapsis, localizes to axis-associated chromatin in Arabidopsis and Brassica. J Cell Sci 115: 3645–3655. [DOI] [PubMed] [Google Scholar]
- Arnold B, Kim ST, Bomblies K. 2015. Single geographic origin of a widespread autotetraploid Arabidopsis arenosa lineage followed by interploidy admixture. Mol Biol Evol 32: 1382–1395. [DOI] [PubMed] [Google Scholar]
- Aung PP, Oue N, Mitani Y, Nakayama H, Yoshida K, Noguchi T, Bosserhoff AK, Yasui W. 2005. Systematic search for gastric cancer-specific genes based on SAGE data: melanoma inhibitory activity and matrix metalloproteinase-10 are novel prognostic factors in patients with gastric cancer. Oncogene 25: 2546–2557. [DOI] [PubMed] [Google Scholar]
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, et al. 2013. Punctuated evolution of prostate cancer genomes. Cell 153: 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barow M. 2006. Endopolyploidy in seed plants. Bioessays 28: 271–281. [DOI] [PubMed] [Google Scholar]
- Bennett RJ, Johnson AD. 2003. Completion of a parasexual cycle in Candida albicans by induced chromosome loss in tetraploid strains. EMBO J 22: 2505–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. 2010. The landscape of somatic copy-number alteration across human cancers. Nature 463: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomblies K, Madlung A. 2014. Polyploidy in the Arabidopsis genus. Chromosome Res 22: 117–134. [DOI] [PubMed] [Google Scholar]
- Bremer G, Bremer-Reinders DE. 1954. Breeding of tetraploid rye in the Netherlands. I. Methods and cytological observations. Euphytica 3: 49–63. [Google Scholar]
- Buntrock L, Marec F, Krueger S, Traut W, Jenkins G. 2012. Organ growth without cell division: somatic polyploidy in a moth, Ephestia kuehniella. Genome 55: 755–763. [DOI] [PubMed] [Google Scholar]
- Burrell RA, Swanton C. 2014. The evolution of the unstable cancer genome. Curr Opin Genet Dev 24: 61–67. [DOI] [PubMed] [Google Scholar]
- Butterfass T. 1987. Cell volume ratios of natural and of induced tetraploid and diploid flowering plants. Cytologia 52: 309–316. [Google Scholar]
- Castellano MM, Sablowski R. 2008. Phosducin-like protein 3 is required for microtubule-dependent steps of cell division but not for meristem growth in Arabidopsis. Plant Cell 20: 969–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T. 1978. Nuclear volume control by nucleoskeletal DNA, selection for cell volume and cell growth rate, and the solution of the DNA C-value paradox. J Cell Sci 34: 247–278. [DOI] [PubMed] [Google Scholar]
- Ceccarelli M, Santantonio E, Marmottini F, Amzallag GN, Cionini PG. 2006. Chromosome endoreduplication as a factor of salt adaptation in Sorghum bicolor. Protoplasma 227: 113–118. [DOI] [PubMed] [Google Scholar]
- Chao D-Y, Dilkes B, Luo H, Douglas A, Yakubova E, Lahner B, Salt DE. 2013. Polyploids exhibit higher potassium uptake and salinity tolerance in Arabidopsis. Science 341: 658–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier AM, Feldman M, Cauderon Y. 1986. Genetic control of meiotic chromosome pairing in tetraploid Agropyron elongatum. I. Pattern of pairing in natural and induced tetraploids and in F1 triploid hybrids. Can J Genet Cytol 28: 783–788. [Google Scholar]
- Chen Y-T, Venditti CA, Theiler G, Stevenson BJ, Iseli C, Gure AO, Jongeneel CV, Old LJ, Simpson AJG. 2005. Identification of CT46/HORMAD1, an immunogenic cancer/testis antigen encoding a putative meiosis-related protein. Cancer Immun 5: 9. [PubMed] [Google Scholar]
- Chen C, Jomaa A, Ortega J, Alani EE. 2014. Pch2 is a hexameric ring ATPase that remodels the chromosome axis protein Hop1. Proc Natl Acad Sci 111: E44–E53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EH, Nijhout HF. 2013. Development of polyploidy of scale-building cells in the wings of Manduca sexta. Arthropod Struct Dev 42: 37–46. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L. 2005. The advantages and disadvantages of being polyploid. Nat Rev Genet 6: 836–846. [DOI] [PubMed] [Google Scholar]
- Comai L, Tyagi AP, Winter K, Holmes-Davis R, Reynolds SH, Stevens Y, Byers B. 2000. Phenotypic instability and rapid gene silencing in newly formed Arabidopsis allotetraploids. Plant Cell 12: 1551–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson SJ, Granier C. 2006. A dynamic analysis of the shade-induced plasticity in Arabidopsis thaliana rosette leaf development reveals new components of the shade-adaptative response. Ann Bot 97: 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coward J, Harding A. 2014. Size does matter: why polyploid tumor cells are critical drug targets in the war on cancer. Front Oncol 4: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel K, Lange J, Hached K, Fu J, Anastassiadis K, Roig I, Cooke HJ, Stewart AF, Wassmann K, Jasin M, et al. 2011. Meiotic homologue alignment and its quality surveillance are controlled by mouse HORMAD1. Nat Cell Biol 13: 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, de Lange T. 2011. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol 27: 585–610. [DOI] [PubMed] [Google Scholar]
- De Veylder L, Larkin JC, Schnittger A. 2011. Molecular control and function of endoreplication in development and physiology. Trends Plant Sci 16: 624–634. [DOI] [PubMed] [Google Scholar]
- Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, et al. 2014. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov 4: 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan P, Cato K, Legaie R, Jayalath R, Olsson G, Hall B, Olson S, Boros S, Reynolds BA, Harding A. 2014. Hyperdiploid tumor cells increase phenotypic heterogeneity within glioblastoma tumors. Mol Biosys 10: 741. [DOI] [PubMed] [Google Scholar]
- Doyle JJ, Flagel LE, Paterson AH, Rapp RA, Soltis DE, Soltis PS, Wendel JF. 2008. Evolutionary genetics of genome merger and doubling in plants. Annu Rev Genet 42: 443–461. [DOI] [PubMed] [Google Scholar]
- Duelli DM, Hearn S, Myers MP, Lazebnik Y. 2005. A primate virus generates transformed human cells by fusion. J Cell Biol 171: 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duelli DM, Padilla-Nash HM, Berman D, Murphy KM, Ried T, Lazebnik Y. 2007. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr Biol 17: 431–437. [DOI] [PubMed] [Google Scholar]
- Duncan AW, Taylor MH, Hickey RD, Newell AEH, Lenzi ML, Olson SB, Finegold MJ, Grompe M. 2010. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 467: 707–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutrillaux B, Gerbault-Seureau M, Remvikos Y, Zafrani B, Prieur M. 1991. Breast cancer genetic evolution: I. Data from cytogenetics and DNA content. Breast Cancer Res Treat 19: 245–255. [DOI] [PubMed] [Google Scholar]
- Edgar BA, Orr-Weaver TL. 2001. Endoreplication cell cycles: more for less. Cell 105: 297–306. [DOI] [PubMed] [Google Scholar]
- Edgar BA, Zielke N, Gutierrez C. 2014. Endocycles: a recurrent evolutionary innovation for post-mitotic cell growth. Nat Rev Mol Cell Biol 15: 294–294. [DOI] [PubMed] [Google Scholar]
- Elmaghrabi AM, Ochatt S, Rogers HJ, Francis D. 2013. Enhanced tolerance to salinity following cellular acclimation to increasing NaCl levels in Medicago truncatula. Plant Cell Tiss Organ Cult 114: 61–70. [Google Scholar]
- Erenpreisa J, Cragg MS. 2013. Three steps to the immortality of cancer cells: senescence, polyploidy and self-renewal. Cancer Cell Int 13: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erenpreisa J, Kalejs M, Cragg M. 2005a. Mitotic catastrophe and endomitosis in tumour cells: an evolutionary key to a molecular solution. Cell Biol Int 29: 1012–1018. [DOI] [PubMed] [Google Scholar]
- Erenpreisa J, Kalejs M, Ianzini F, Kosmacek E, Mackey M, Emzinsh D, Cragg M, Ivanov A, Illidge T. 2005b. Segregation of genomes in polyploid tumour cells following mitotic catastrophe. Cell Biol Int 29: 1005–1011. [DOI] [PubMed] [Google Scholar]
- Erenpreisa J, Salmina K, Huna A, Kosmacek EA, Cragg MS, Ianzini F, Anisimov AP. 2011. Polyploid tumour cells elicit paradiploid progeny through depolyploidizing divisions and regulated autophagic degradation. Cell Biol Int 35: 687–695. [DOI] [PubMed] [Google Scholar]
- Ermis A, Oberringer M, Wirbel R, Koschnick M, Mutschler W, Hanselmann RG. 1998. Tetraploidization is a physiological enhancer of wound healing. Eur Surg Res 30: 385–392. [DOI] [PubMed] [Google Scholar]
- Ezov TK, Boger-Nadjar E, Frenkel Z, Katsperovski I, Kemeny S, Nevo E, Korol A, Kashi Y. 2006. Molecular-genetic biodiversity in a natural population of the yeast Saccharomyces cerevisiae from ‘Evolution Canyon’: microsatellite polymorphism, ploidy and controversial sexual status. Genetics 174: 1455–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feher F, Hangyel Tarczy M, Bocsa I, Dudits D. 1989. Somaclonal chromosome variation in tetraploid alfalfa. Plant Sci 60: 91–99. [Google Scholar]
- Fox DT, Gall JG, Spradling AC. 2010. Error-prone polyploid mitosis during normal Drosophila development. Genes Dev 24: 2294–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. 2005. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437: 1043–1047. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Daniel K, Wojtasz L, Tóth A, Höög C. 2010. A novel mammalian HORMA domain-containing protein, HORMAD1, preferentially associates with unsynapsed meiotic chromosomes. Exp Cell Res 316: 158–171. [DOI] [PubMed] [Google Scholar]
- Gaeta RT, Pires JC. 2009. Homoeologous recombination in allopolyploids: the polyploid ratchet. New Phytol 186: 18–28. [DOI] [PubMed] [Google Scholar]
- Galipeau PC, Cowan DS, Sanchez CA, Barrett MT, Emond MJ, Levine DS, Rabinovitch PS, Reid BJ. 1996. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc Natl Acad Sci 93: 7081–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galitski T, Saldanha AJ, Styles CA, Lander ES, Fink GR. 1999. Ploidy regulation of gene expression. Science 285: 251–254. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Pellman D. 2007. Limiting the proliferation of polyploid cells. Cell 131: 437–440. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Storchova Z, Pellman D. 2007. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev 17: 157–162. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. 2009. A mechanism linking extra centrosomes to chromosomal instability. Nature 460: 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Zheng J. 2010. High-risk HPV E5-induced cell fusion: a critical initiating event in the early stage of HPV-associated cervical cancer. Virol J 7: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar T, Hagege D, Kevers C, Penel C, Crèvecoeur M, Engelmann I, Greppin H, Foidart J-M. 1991. When plant teratomas turn into cancers in the absence of pathogens. Physiol Plant 83: 696–701. [Google Scholar]
- Gegas VC, Wargent JJ, Pesquet E, Granqvist E, Paul ND, Doonan JH. 2014. Endopolyploidy as a potential alternative adaptive strategy for Arabidopsis leaf size variation in response to UV-B. J Exp Bot 65: 2757–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentric G, Desdouets C. 2014. Polyploidization in liver tissue. Am J Pathol 184: 322–331. [DOI] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al. 2014a. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 46: 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, McGranahan N, Dewhurst SM, Burrell RA, Tomlinson I, Swanton C. 2014b. Cancer: evolution within a lifetime. Annu Rev Genet 48: 215–236. [DOI] [PubMed] [Google Scholar]
- Gerstein AC, Otto SP. 2009. Ploidy and the causes of genomic evolution. J Hered 100: 571–581. [DOI] [PubMed] [Google Scholar]
- Gilles A, Randolph LF. 1951. Reduction of quadrivalent frequency in autotetraploid maize during a period of ten years. Am J Bot 38: 12–17. [Google Scholar]
- Greilhuber J, Weber A. 1975. Aneustomaty in Orobanche gracilis. Plant Syst Evol 124: 67–77. [Google Scholar]
- Gupta S. 2000. Hepatic polyploidy and liver growth control. Semin Cancer Biol 10: 161–171. [DOI] [PubMed] [Google Scholar]
- Hase Y, Trung KH, Matsunaga T, Tanaka A. 2006. A mutation in the uvi4 gene promotes progression of endo-reduplication and confers increased tolerance towards ultraviolet B light. Plant J 46: 317–326. [DOI] [PubMed] [Google Scholar]
- Häsler J, Wüest J, Gaspar T, Crèvecoeur M. 2012. Long term in vitro-cultured plant cells show typical neoplastic features at the cytological level. Biol Cell 95: 357–364. [DOI] [PubMed] [Google Scholar]
- Hazarika MH, Rees H. 1967. Genotypic control of chromosome behaviour in rye. X. Chromosome pairing and fertility in autotetraploids. Heredity 22: 317–322. [Google Scholar]
- Hegarty M, Coate J, Sherman-Broyles S, Abbott R, Hiscock S, Doyle J. 2013. Lessons from natural and artificial polyploids in higher plants. Cytogenet Genome Res 140: 204–225. [DOI] [PubMed] [Google Scholar]
- Higgins JD, Sanchez-Moran E, Armstrong SJ, Jones GH, Franklin FCH. 2005. The Arabidopsis synaptonemal complex protein ZYP1 is required for chromosome synapsis and normal fidelity of crossing over. Genes Dev 19: 2488–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilpert G. 1957. Effect of selection for meiotic behaviour in autotetraploid rye. Hereditas 43: 318–322. [Google Scholar]
- Hollingsworth NM, Goetsch L, Byers B. 1990. The HOP1 gene encodes a meiosis-specific component of yeast chromosomes. Cell 61: 73–84. [DOI] [PubMed] [Google Scholar]
- Hollister JD, Arnold BJ, Svedin E, Xue KS, Dilkes BP, Bomblies K. 2012. Genetic adaptation associated with genome-doubling in autotetraploid Arabidopsis arenosa. PLoS Genet 8: e1003093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Plafker K, Vorozhko V, Zuna RE, Hanigan MH, Gorbsky GJ, Plafker SM, Angeletti PC, Ceresa BP. 2009. Human papillomavirus 16 E5 induces bi-nucleated cell formation by cell–cell fusion. Virology 384: 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Nurse P. 1991. A note on the evolution of meiosis. J Theor Biol 150: 561–563. [DOI] [PubMed] [Google Scholar]
- Huskins CL. 1948. Segregation and reduction in somatic tissues. I. Initial observations on Allium cepa. J Hered 39: 311–325. [Google Scholar]
- Ianzini F, Kosmacek EA, Nelson ES, Napoli E, Erenpreisa J, Kalejs M, Mackey MA. 2009. Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Res 69: 2296–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro T, Tanaka K, Sakuno T, Watanabe Y. 2010. Shugoshin–PP2A counteracts casein-kinase-1-dependent cleavage of Rec8 by separase. Nat Cell Biol 12: 500–506. [DOI] [PubMed] [Google Scholar]
- Janic A, Mendizabal L, Llamazares S, Rossell D, Gonzalez C. 2010. Ectopic expression of germline genes drives malignant brain tumor growth in Drosophila. Science 330: 1824–1827. [DOI] [PubMed] [Google Scholar]
- Jauhar PP. 1970. Genotypic control of chromosome behaviour in rye. X. Chromosome pairing and fertility in autotetraploids. Genetica 41: 407–424. [Google Scholar]
- Jenczewski E, Eber F, Manzanares-Dauleux MJ, Chevre AM. 2002. A strict diploid-like pairing regime is associated with tetrasomic segregation in induced autotetraploids of kale. Plant Breed 121: 177–179. [Google Scholar]
- Jones GH, Franklin FCH. 2006. Meiotic crossing-over: obligation and interference. Cell 126: 246–248. [DOI] [PubMed] [Google Scholar]
- Kalejs M, Ivanov A, Plakhins G, Cragg M, Emzinsh D, Illidge T, Erenpreisa J. 2006. Upregulation of meiosis-specific genes in lymphoma cell lines following genotoxic insult and induction of mitotic catastrophe. BMC Cancer 6: 6–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan K, Karthikeyan U, Li Y, Yan J, Muniyappa K. 2012. Single-molecule DNA analysis reveals that yeast Hop1 protein promotes DNA folding and synapsis: implications for condensation of meiotic chromosomes. ACS Nano 12: 10658–10666. [DOI] [PubMed] [Google Scholar]
- Kim JW, Dang CV. 2006. Cancer's molecular sweet tooth and the Warburg Effect. Cancer Res 66: 8927–8930. [DOI] [PubMed] [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, Amon A. 2014. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci USA 111: 13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krois NR, Cherukuri A, Puttagunta N, Neiman M. 2013. Higher rate of tissue regeneration in polyploid asexual versus diploid sexual freshwater snails. Biol Lett 9: 20130422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagadec C, Vlashi E, Donna Della L, Dekmezian C, Pajonk F. 2012. Radiation-induced reprogramming of breast cancer cells. Stem Cells 30: 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambing C, Osman K, Nuntasoontorn K, West A, Higgins JD, Copenhaver GP, Yang J, Armstrong SJ, Mechtler K, Roitinger E, et al. 2015. Arabidopsis PCH2 mediates meiotic chromosome remodeling and maturation of crossovers. PLoS Genet 11: e1005372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavania UC. 1991. Polyploid breeding: meiosis in the diploid progenitor and its predictive value for fertility in the autotetraploid. Proc Indian Nat Sci Acad B 57: 17–24. [Google Scholar]
- Lazebnik Y. 2014. The shock of being united and symphiliosis. Cell Cycle 13: 2323–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HO, Davidson JM, Durunio RJ. 2009. Endoreplication: ploidy with purpose. Genes Dev 23: 2461–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B. 1997. Genetic instability in colorectal cancers. Nature 386: 623–627. [DOI] [PubMed] [Google Scholar]
- Levin DA. 1983. Polyploidy and novelty in flowering plants. Am Natural 122: 1–25. [Google Scholar]
- Leysi-Derilou Y, Robert A, Duchesne C, Garnier A, Boyer L, Pineault N. 2014. Polyploid megakaryocytes can complete cytokinesis. Cell Cycle 9: 2589–2599. [DOI] [PubMed] [Google Scholar]
- Lindsey SF, Byrnes DM, Eller MS, Rosa AM, Dabas N, Escandon J, Grichnik JM. 2013. Potential role of meiosis proteins in melanoma chromosomal instability. J Skin Cancer 2013: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissa D, Senovilla L, Rello-Varona S, Vitale I, Michaud M, Pietrocola F, Boileve A, Obrist F, Bordenave C, Garcia P, et al. 2014. Resveratrol and aspirin eliminate tetraploid cells for anticancer chemoprevention. Proc Natl Acad Sci 111: 3020–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SCS, Dhar SU, Kołodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. 2011. Chromosome Catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146: 889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Chen J, Hu L, Shi X, Zhou Z, Hu Z, Sha J. 2012. HORMAD2/CT46.2, a novel cancer/testis gene, is ectopically expressed in lung cancer tissues. Mol Hum Reprod 18: 599–604. [DOI] [PubMed] [Google Scholar]
- Losick VP, Fox DT, Spradling AC. 2013. Polyploidization and cell fusion contribute to wound healing in the adult Drosophila epithelium. Curr Biol 23: 2224–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques R-A, Rabinovitch PS, et al. 2006. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet 38: 468–473. [DOI] [PubMed] [Google Scholar]
- Malhotra A, Wang Y, Waters J, Chen K, Meric-Bernstam F, Hall IM, Navin NE. 2015. Ploidy-seq: inferring mutational chronology by sequencing polyploid tumor subpopulations. Genome Med 7: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Almendro V, Polyak K. 2012. Intra-tumour heterogeneity: a looking glass for cancer? Nat Genet 12: 323–334. [DOI] [PubMed] [Google Scholar]
- McCollum CD. 1958. Comparative studies of chromosome pairing in natural and induced tetraploid Dactylis. Chromosoma 9: 571–605. [DOI] [PubMed] [Google Scholar]
- Meuwissen RLJ, Meerts I, Hoovers JMN, Leschot NJ, Heyting C. 1997. Human synaptonemal complex protein 1 (SCP1): isolation and characterization of the cDNA and chromosomal localization of the gene. Genomics 39: 377–384. [DOI] [PubMed] [Google Scholar]
- Murashige T, Nakano R. 1967. Chromosome Complement as a determinant of the morphogenic potential of tobacco cells. Am J Bot 54: 963–970. [Google Scholar]
- Murray BG, Sieber VK, Jackson RC. 1984. Further evidence for the presence of meiotic pairing control genes in Alopecurus L. (Gramineae). Genetica 63: 13–20. [Google Scholar]
- Müntzing A. 1951. Cyto-genetic properties and practical value of tetraploid rye. Hereditas 37: 17–84. [Google Scholar]
- Myers WM. 1945. Meiosis in autotetraploid Lolium perenne in relation to chromosomal behavior in autopolyploids. Bot Gaz 106: 304–316. [Google Scholar]
- Nagl W. 1982. Polytene chromosomes of plants. Int Rev Cytol 73: 21–53. [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. 2012a. Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, et al. 2012b. The life history of 21 breast cancers. Cell 149: 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu H, Wan L, Baumgartner B, Schaefer D, Loidl J, Hollingsworth NM. 2005. Partner choice during meiosis is regulated by Hop1-promoted dimerization of Mek1. Mol Biol Cell 16: 5804–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberringer M, Lothschütz D, Jennewein M, Koschnick M, Mutschler W, Hanselmann RG. 1999. Centrosome multiplication accompanies a transient clustering of polyploid cells during tissue repair. Mol Cell Biol Res Commun 2: 190–196. [DOI] [PubMed] [Google Scholar]
- Olaharski AJ, Sotelo R, Solorza-Luna G, Gonsebatt ME, Guzman P, Mohar A, Eastmond DA. 2006. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis 27: 337–343. [DOI] [PubMed] [Google Scholar]
- Old LJ. 2001. Cancer/testis (CT) antigens—a new link between gametogenesis and cancer. Cancer Immun 1: 1. [PubMed] [Google Scholar]
- Öllinger R, Alsheimer M, Benavente R. 2005. Mammalian protein SCP1 forms synaptonemal complex-like structures in the absence of meiotic chromosomes. Mol Biol Cell 16: 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL. 2015. When bigger is better: the role of polyploidy in organogenesis. Trends Genet 31: 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP. 2007. The evolutionary consequences of polyploidy. Cell 131: 452–462. [DOI] [PubMed] [Google Scholar]
- Otto SP, Whitton J. 2000. Polyploid incidence and evolution. Annu Rev Genet 34: 401–437. [DOI] [PubMed] [Google Scholar]
- Page SL, Hawley RS. 2001. c(3)G encodes a Drosophila synaptonemal complex protein. Genes Dev 15: 3130–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi T, Beck AR, Rougier N, McNeil T, Lucian L, Werb Z, Amati B. 2003. Cyclins E1 and E2 are required for endoreplication in placental trophoblast giant cells. EMBO J 22: 4794–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrello J, Bourdon M, Cheniclet C, Coriton O, Bourge MEL, Brown S, Renaudin J-P, Frangne N, Chevalier C. 2013. The structural and molecular analysis of endoreduplicated nuclei in tomato (Solanum lycopersicum) fruit provides evidence for a ploidy-dependent increase in transcriptional activity. Plant Biotechnol 30: 301–307. [Google Scholar]
- Puig P-E, Guilly M-N, Bouchot A, Droin N, Cathelin D, Bouyer F, Favier L, Ghiringhelli F, Kroemer G, Solary E. 2008. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol Int 32: 1031–1043. [DOI] [PubMed] [Google Scholar]
- Raja V, Koul KK, Raina SN, Parida A. 1992. Ploidy-dependent genomic stability in the tissue cultures of ornamental Phlox drummondii Hook. Plant Cell Rep 12: 12–17. [DOI] [PubMed] [Google Scholar]
- Rajaraman R, Rajaraman MM, Rajaraman SR, Guernsey DL. 2005. Neosis—a paradigm of self-renewal in cancer. Cell Biol Int 29: 1084–1097. [DOI] [PubMed] [Google Scholar]
- Rajaraman R, Guernsey DL, Rajaraman MM, Rajaraman SR. 2007. Neosis—a parasexual somatic reduction division in cancer. Int J Hum Genet 7: 29–48. [Google Scholar]
- Ramsey J, Schemske D. 1998. Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu Rev Ecol Syst 29: 467–501. [Google Scholar]
- Ramsey J, Schemske D. 2002. Neopolyploidy in flowering plants. Annu Rev Ecol Syst 33: 589–639. [Google Scholar]
- Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, Perera A, Staehling-Hampton K, Seidel CW, Li R. 2008. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135: 879–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera M, Wu Q, Hamerlik P, Hjelmeland AB, Bao S, Rich JN. 2015. Acquisition of meiotic DNA repair regulators maintain genome stability in glioblastoma. Cell Death Dis 6: e1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder AK, Cunha A, Ohno CK, Meyerowitz EM. 2012. Cell cycle regulates cell type in the Arabidopsis sepal. Development 139: 4416–4427. [DOI] [PubMed] [Google Scholar]
- Salmina K, Jankevics E, Huna A, Perminov D, Radovica I, Klymenko T, Ivanov A, Jascenko E, Scherthan H, Cragg M, et al. 2010. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp Cell Res 316: 2099–2112. [DOI] [PubMed] [Google Scholar]
- Santos JL, Alfaro D, Sanchez-Moran E, Armstrong SJ, Franklin FCH, Jones GH. 2003. Partial diploidization of meiosis in autotetraploid Arabidopsis thaliana. Genetics 165: 1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmickl R, Paule J, Klein J, Marhold K, Koch MA. 2012. The evolutionary history of the Arabidopsis arenosa complex: diverse tetraploids mask the Western Carpathian center of species and genetic diversity. PLoS One 7: e42691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder KP, Fox DT. 2015. The expanding implications of polyploidy. J Cell Biol 209: 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholes DR, Paige KN. 2014. Plasticity in ploidy underlies plant fitness compensation to herbivore damage. Mol Ecol 23: 4862–4870. [DOI] [PubMed] [Google Scholar]
- Scholes DR, Paige KN. 2015. Plasticity in ploidy: a generalized response to stress. Trends Plant Sci 20: 165–175. [DOI] [PubMed] [Google Scholar]
- Schwacha A, Kleckner N. 1994. Identification of joint molecules that form frequently between homologs but rarely between sister chromatids. Cell 76: 51–63. [DOI] [PubMed] [Google Scholar]
- Selmecki AM, Maruvka YE, Richmond PA, Guillet M, Shoresh N, Sorenson AL, De S, Kishony R, Michor F, Dowell R, et al. 2015. Polyploidy can drive rapid adaptation in yeast. Nature 519: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahzad MMK, Shin Y-H, Matsuo K, Lu C, Nishimura M, Shen D-Y, Kang Y, Hu W, Mora EM, Rodriguez-Aguayo C, et al. 2013. Biological significance of HORMA domain containing protein 1 (HORMAD1) in epithelial ovarian carcinoma. Cancer Lett 330: 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaver DL. 1962. A study of meiosis in perennial teosinte, in tetraploid maize and in their tetraploid hybrid. Caryologia 15: 43–57. [Google Scholar]
- Shin YH, Choi Y, Erdin SU, Yatsenko SA, Kloc M, Yang F, Wang PJ, Meistrich ML, Rajkovic A. 2010. Hormad1 mutation disrupts synaptonemal complex formation, recombination, and chromosome segregation in mammalian meiosis. PLoS Genet 6: e1001190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short SC, Giampieri S, Worku M, Alcaide-German M, Sioftanos G, Bourne S, Lio KI, Shaked-Rabi M, Martindale C. 2011. Rad51 inhibition is an effective means of targeting DNA repair in glioma models and CD133+ tumor-derived cells. Neuro Oncol 13: 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen O. 1975. Cytogenetic investigations in diploid and autotetraploid populations of Festuca pratensis. Hereditas 79: 73–108. [DOI] [PubMed] [Google Scholar]
- Simpson AJG, Caballero OL, Jungbluth A, Chen Y-T, Old LJ. 2005. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 5: 615–625. [DOI] [PubMed] [Google Scholar]
- Singh BD, Harvey BL. 1975. Selection for diploid cells in suspension cultures of Haplopappus gracilis. Nature 253: 453.1110794 [Google Scholar]
- Soltis DE, Soltis PS, Tate JA. 2003. Advances in the study of polyploidy since plant speciation. New Phytol 161: 173–191. [Google Scholar]
- Srivastava S, Lavania UC. 1990. Meiotic regularization, restoration of seed fertility and alkaloid content in the induced autotetraploids of Hyoscyamus albus L. Plant Breed 104: 160–166. [Google Scholar]
- Srivastava S, Lavania UC, Sybenga J. 1992. Genetic variation in meiotic behaviour and fertility in tetraploid Hyoscyamus muticus: correlation with diploid meiosis1. Heredity 68: 231–239. [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova Z. 2014. Ploidy changes and genome stability in yeast. Yeast 31: 421–430. [DOI] [PubMed] [Google Scholar]
- Storchova Z, Kuffer C. 2008. The consequences of tetraploidy and aneuploidy. J Cell Sci 121: 3859–3866. [DOI] [PubMed] [Google Scholar]
- Storchova Z, Pellman D. 2004. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 5: 45–54. [DOI] [PubMed] [Google Scholar]
- Storchova Z, Breneman A, Cande J, Dunn J, Burbank K, O'Toole E, Pellman D. 2006. Genome-wide genetic analysis of polyploidy in yeast. Nature 443: 541–547. [DOI] [PubMed] [Google Scholar]
- Strunnikov A. 2013. Cohesin complexes with a potential to link mammalian meiosis to cancer. Cell Regen (Lond) 2: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto-Shirasu K, Roberts K. 2003. ‘Big it up’: endoreduplication and cell-size control in plants. Curr Opin Plant Biol 6: 544–553. [DOI] [PubMed] [Google Scholar]
- Sym M, Engebrecht J, Roeder GS. 1993. ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell 72: 365–378. [DOI] [PubMed] [Google Scholar]
- Thorpe PH, González-Barrera S, Rothstein R. 2007. More is not always better: the genetic constraints of polyploidy. Trends Genet 23: 263–266. [DOI] [PubMed] [Google Scholar]
- Torrey JG. 1967. Morphogenesis in relation to chromosomal constitution in long-term plant tissue cultures. Physiol Plant 20: 265–275. [Google Scholar]
- Türeci Ö, Sahin U, Zwick C, Koslowski M, Seitz G, Pfreundschuh M. 1998. Identification of a meiosis-specific protein as a member of the class of cancer/testis antigens. Proc Natl Acad Sci 95: 5211–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzung KW, Williams RM, Scherer S, Federspiel N, Jones T, Hansen N, Bivolarevic V, Huizar L, Komp C, Surzycki R, et al. 2001. Genomic evidence for a complete sexual cycle in Candida albicans. Proc Natl Acad Sci 98: 3249–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varetti G, Pellman D, Gordon DJ. 2014. Aurea mediocritas: the importance of a balanced genome. Cold Spring Harb Perspect Biol 6: a015842–a015842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov AE, Anatskaya OV, Kudryavtsev BN. 2001. Relationship of hepatocyte ploidy levels with body size and growth rate in mammals. Genome 44: 350–360. [DOI] [PubMed] [Google Scholar]
- Walther V, Hiley CT, Shibata D, Swanton C, Turner PE, Maley CC. 2015. Can oncology recapitulate paleontology? Lessons from species extinctions. Nat Genet 12: 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K. 1983. Studies on the control of diploid-like meiosis in polyploid taxa of Chrysanthemum. Theor Appl Genet 66: 9–14. [DOI] [PubMed] [Google Scholar]
- Watkins J, Weekes D, Shah V, Gazinska P, Joshi S, Sidhu B, Gillett C, Pinder S, Vanoli F, Jasin M, et al. 2015. Genomic complexity profiling reveals that HORMAD1 overexpression contributes to homologous recombination deficiency in triple-negative breast cancers. Cancer Discov 5: 488–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RL, Kukora JR, Adams J. 1975. The relationship between enzyme activity, cell geometry, and fitness in Saccharomyces cerevisiae. Proc Natl Acad Sci 72: 794–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GB, Cheng KC. 1949. Segregation and reduction in somatic tissues. J Hered 39: 3–6. [DOI] [PubMed] [Google Scholar]
- Wildermuth MC. 2010. Modulation of host nuclear ploidy: a common plant biotroph mechanism. Curr Opin Plant Biol 13: 449–458. [DOI] [PubMed] [Google Scholar]
- Wolf PG, Soltis PS, Soltis DE. 1989. Tetrasomic inheritance and chromosome pairing behaviour in the naturally occurring autotetraploid Heuchera grossulariifolia (Saxifragaceae). Genome 32: 655–659. [Google Scholar]
- Wright KM, Pires JC, Madlung A. 2009. Mitotic instability in resynthesized and natural polyploids of the genus Arabidopsis (Brassicaceae). Am J Bot 96: 1656–1664. [DOI] [PubMed] [Google Scholar]
- Wu C-Y, Rolfe PA, Gifford DK, Fink GR. 2010. Control of transcription by cell size. PLoS Biol 8: e1000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PM, Huang QC, Qin GY, Zhao SP, Zhou JG. 2014. Different drought-stress responses in photosynthesis and reactive oxygen metabolism between autotetraploid and diploid rice. Photosynthetica 52: 193–202. [Google Scholar]
- Yant L, Hollister JD, Wright KM, Arnold BJ, Higgins JD, Franklin FCH, Bomblies K. 2013. Meiotic adaptation to genome duplication in Arabidopsis arenosa. Curr Biol 23: 2151–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, Liu J. 2013. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 33: 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Liang Z, Hutchinson J, Kleckner N. 2014. Crossover patterning by the beam-film model: analysis and implications. PLoS Genet 10: e1004042. [DOI] [PMC free article] [PubMed] [Google Scholar]