

Abstract
Celecoxib is used in the treatment of osteoarthritis, rheumatoid arthritis, acute pain, joint inflammation and sport injuries. Long term administration of the drug results in such complications as gastrointestinaland renal disturbances and cardio-vascular complications. The main objective of the present study was to investigate the feasibility of delivering celecoxib incorporated in gel formulations by iontophoresis. Sodium alginate, sodium carboxymethyl cellulose, hydroxypropyl methylcellulose (HPMC) and carbopol 934P were used to develop topical gel formulations of celecoxib. The gel formulations were evaluated for macroscopic and microscopic properties, pH determination, spreadability, rheological behaviour, and drug release characteristics both in vitro and ex vivo. Drug release was evaluated in the presence of iontophoresis field (0.1 to 0.5 mA/cm2) or without electrical current (passive diffusion) and celecoxib was measured spectrophotometrically at 252 nm. Most gel formulations showed acceptable physicochemical properties. Amongst formulations, gel formulation containing HPMC K4M which indicated greater performance in drug release behaviour was selected for further in vivo studies. The cumulative percent of drug released in vitro at the end of each experiment was 36%, 63%, and 89.7% for passive diffusion, direct electric current (DC) current density of 0.3 mA/cm2, and 0.5 mA/cm2, respectively. The findings of ex vivo drug transport across rat skin also showed a significantly higher release of celecoxib compared to passive flux for both AC and DC currents. A 0.5 mA/cm2 of DC current increased drug flux to 73% compared to 41.5% of passive diffusion. It can be concluded from the results of this study that the application of iontophoresis enhances the flux of celecoxib, as compared to the passive diffusion.
Keywords: Iontophoresis, Celecoxib, Gel formulation, NSAIDs, In vitro, Ex vivo, Rat skin
INTRODUCTION
Drug penetration enhancement across the skin is facilitated by using chemical enhancers and/or physical techniques. Among physical approaches, phonophoresis, electroporation and iontophoresis, i.e. the application of a low voltage electric current (<0.5 mA/cm2), are progressively used to enhance drug transport into and through the skin (1). In iontophoresis, usually a direct electric current (DC) is applied as a driving force, however; alternating current (AC) pulses have also been used (2).
Transdermal iontophoresis appears to be a promising technique for local and systemic drug delivery of charged particles; however, it can also be efficient to a lesser extent for non-ionized molecules (3). It is sometimes referred to as “an injection without the needle” method (4).
Iontophoresis has found favorable application in the relief of primary hyperhidrosis of palms and soles by poldine methane sulphonate (5). The method has also been successfully used in the delivery of lidocaine, pilocarpine (6), prednisolone (7), and small proteins e.g. leuprolide and calcitonin (5). A combination of iontophoresis and electroporation has shown to be effective in the delivery of peptides, proteins, genes and oligonucleotides (8). Clinical effectiveness of iontophoresis has been shown for both steroidal and non-steroidal anti-inflammatory drugs (NSAIDs) (7,9,10).
NSAIDs are one type of the most prescribed medicines usually applied topically for the treatment of soft tissue inflammation and sport injuries such as muscle pulls and tears, ligament sprains and tendonitis. There are some reports describing the relationship between physicochemical properties of drugs and the transdermal enhancement effect of iontophoresis for some NSAIDs such as aspirin, ibuprofen, indomethacin, salicylic acid, ketoprofen, and naproxen (11,12).
Celecoxib, a Cox-2 selective inhibitor, inhibits the transformation of arachidonic acid to prostaglandin precursors and hence has potent anti-inflammatory and analgesic effects. This drug is used in the treatment of osteoarthritis, rheumatoid arthritis, acute pain, joint inflammation and both pre and postsurgical pain (13). Oral celecoxib is also prescribed in sport injuries e.g. shoulder girdle tendon inflammation, ankle pain and rheumatoid arthritis due to heavy exercises such as wrestling, body building, boxing etc (14). Long term oral administration of this drug is associated with upper gastrointestinal (GI) intolerance, gastric mucosal ulcers, bleeding, and cardiovascular complications (15). The side effects might be overcome by using a transdermal formulation with a high degree of drug permeation through skin. However, low aqueous solubility of celecoxib (4 mg/L) and intact barrier function of stratum corneum are two major limiting factors in formulation of a desired topical preparation (16). Several approaches including formulation of micro or nanoemulsion systems, niosomal or liposomal gels (17), nanostructured lipid carrier based gel (18) and application of natural or chemical penetration enhancers (19) have been tried to increase the passage of celecoxib into the skin and provide localized action at the inflamed tendons, ligaments, and joints.
To the best of our knowledge, no study has been reported on the application of iontophoresis for permeation of celecoxib across the skin. Therefore, the aim of the present study was to develop a topical gel formulation of celecoxib suited for iontophoretic delivery and to investigate the effects of some iontophoretic parameters on the transport of drug through excised rat skin. This method of administration has drawn interests from both medical practitioners and physiotherapists due to its certain advantages over oral administration such as bypassing the primary hepatic drug clearance, avoiding GI side effects and depositing a high concentration of the medication into the affected areas like muscles and joints (20).
MATERIALS AND METHODS
Materials
Celecoxib was a kind gift from Amin Pharmaceutical Company (Isfahan, Iran). Carbopol 934P and sodium carboxymethyl cellulose (Na CMC, Nymcel) were obtained from BFG (USA) and Colorcon (England), respectively. Both two grades of hydroxypropyl methylcellulose (HPMC K4M and HPMC E5) as well as sodium alginate were provided by Fluka (Switzerland). All other reagents and chemicals used in the study were of analytical grade and purchased from Merck (Germany).
Animals
All animal experiments were approved by the Ethics Committee of Isfahan University of Medical Science and performed in accordance with National Institute of Health Guide for the Care and Use of Laboratory Animals.
Male Wistar rats (200 ± 25g) were bred in the animal house. The animals were housed individually in wire-bottomed cages under a uniform condition of temperature and humidity and fed with normal rat chow and tap water. Rat full thickness skin was excised post sacrifice from the abdominal region and hairs were removed with an electric hair clipper. The subcutaneous tissue and the fat adhering on the dermis side were removed using a scalpel, cleaned with isopropyl alcohol and washed with distilled water. The skin was then stored in physiologic saline solution (0.9%) at room temperature for half an hour before the use (15,16,20).
Preparation of celecoxib gel formulations
Different gelling agents including sodium alginate, Na CMC, HPMC K4M, HPMC E5, and carbopol 934 were used to prepare celecoxib gel formulations. An appropriate amount of celecoxib together with methylparaben as a preservative which had already been dissolved in propylene glycol, ethanol or PEG 400, were added to one of the gel bases previously described. Some other ingredients were also used (Table 1). For carbopol gel, triethanolamine (TEA) was used as an alkalizing agent (15,21). The prepared gels were carefully checked for their turbidity, color, texture, and presence of air bubbles, and then stored in wide mouth vessels till further physical and chemical tests were made (21).
Table 1.
The compositions used to prepare celecoxib containing gel formulations.
Macroscopic and microscopic evaluation
All prepared gels were macroscopically inspected for the presence of flocculated particles, smoothness, fluidity, color, and transparency 48 h after preparation (15). The formulations also were microscopically examined for homogeneity, texture, and the presence of air bubbles (20).
Determination of pH
The pH of the formulations was measured using an electrical pH meter (Metrohm-6320, Switzerland) immediately after preparation and every two weeks (21).
Spreadability test
The spreadability property of celecoxib gel was determined by measuring the spreading diameter of 1 g gel mass between two horizontal glass slabs (20 × 20 cm) after 1 min. The weight of the upper plate was 125 g (22,23).
Drug assay in gels
A portion of gel containing 100 μg of celecoxib was weighed carefully, dissolved in propylene glycol or ethanol. Drug concentration was determined at 252 nm using a UV-Visible spectrophotometer (Shimadzu, Japan).
Rheological properties of the gel formulations
The selected celecoxib gel formulations were studied for rheological behaviour at 25 °C using a digital viscometer (Rheomat RM180; Mettler, UK). The machine is a rotational viscometer that uses a motor driven bob (or spindle) rotating within a fixed cylinder cup. Each sample was carefully loaded into the gap using a tablespoon to ensure a minimum shearing and allowed to equilibrate for at least 1 h prior to the test. Rheological properties of gels were measured using the loop test, in which a specified amount of shearing stress (0.1 to 40 Pa) was increasingly applied on the sample over a set period of time (60 s) and then returned to the starting point to record the resulting strain (up and down curves). At least three replicate experiments were conducted for each sample (24,25).
In vitro drug release studies
Franz diffusion cells made of two glass components, donor and receptor, separated by a membrane barrier was used for drug release studies. The cellulose acetate membrane was mounted between two chambers which were held in place with a clamp. The receiver chamber (of 35 ml capacity) was filled with phosphate buffer (pH 5.6) and 2 g celecoxib gel was evenly spread on the surface of membrane facing the donor chamber. The content of the receiver was stirred using a magnetic bar at 200 rpm. The assembled cell was placed in a water bath where the temperature was maintained at 25 ± 1 °C.
The open mouth of the donor chamber was covered with Parafilm to prevent evaporation and provide occlusive conditions. Samples of 0.6 ml of the receptor phase were withdrawn via the sampling arm at regular time intervals (15, 30, 45 min, 1, 2 and 5 h) and replaced with freshly-prepared buffer to maintain the release medium at sink condition. The samples were filtered through a 0.45 μm membrane filter and analyzed for drug content by a UV-Visible spectrophotometer (Shimadzu, Japan) at 252 nm. The mean percentage of celecoxib released was plotted as a function of time. All experiments were carried out in triplicate and the results were presented as mean values ± S.D. (15,16,21).
Ex vivo drug permeation studies
Transport of celecoxib across the skin from tested gel formulations was conducted according to the procedure adopted for the release study. In brief, the excised rat skin was mounted between Franz donor and receiver cells where the stratum corneum side was facing the donor cell and the dermal side of the skin was in contact with phosphate buffer (pH 5.6) in the receiver compartment. The cells were fixed using a clamp (16,26) and the gel formulation (2 g) was applied evenly on the epidermal surface of the skin. Aliquots of 1 ml were periodically withdrawn from the receptor compartment up to 5 h, which were quickly replaced with an equal volume of fresh medium to maintain the volume of the release medium constant.
The absorbance of the withdrawn samples was measured spectrophotometrically at 252 nm and the rate of drug transport through the skin was determined.
Iontophoresis studies
Both in vitro drug release and ex vivo drug transport experiments were carried out in the presence of iontophoresis field or without application of any electrical current (passive diffusion). In the iontophoretic experiments, a portable iontophoresis system (NEURADYN 710F, Iran) was applied to generate a weak current (DC or AC) of 0.1 mA/cm2 to 0.5 mA/cm2 using silver/silver chloride electrodes, shown to possess both stability and reversibility (3,12).
This is the most commonly used cathode in iontophoretic systems which prevents electrolysis of water and shift in pH (27). The AC voltage with a low frequency of 12.5 Hz and moderate voltages of 2.5 V were applied between two electrodes. The electrode connected to the positive pole of the power supply (silver wire) was used as the anode and the electrode connected to the negative pole of the generator (silver/silver chloride) was used as the cathode. The cathode surface was placed on the celecoxib gel (donor phase) and the anode was immersed in the receptor solution which was stirred using a Teflon-coated magnetic stirrer at 300 rpm. Sampling and drug content analyses were carried out as described earlier. The effect of various iontophoresis parameters including applied current density, type of electrical current (AC or DC), working cycles of the current were investigated. As the control, similar experiments were accomplished without application of any electrical current (passive flux) (27).
Data analysis
The cumulative percent of celecoxib released from the gels or the amount of drug transported through the rat skin was plotted against time. The cumulative percents of drug released or permeated obtained at the end of experiment (5 h) were compared using one-way analysis of variance (ANOVA) followed by Duncan multiple-comparison test. Mean differences were considered significant at the level of P<0.05. The data analysis was carried out using SPSS software, version 12.
RESULTS
Physicochemical properties of formulations
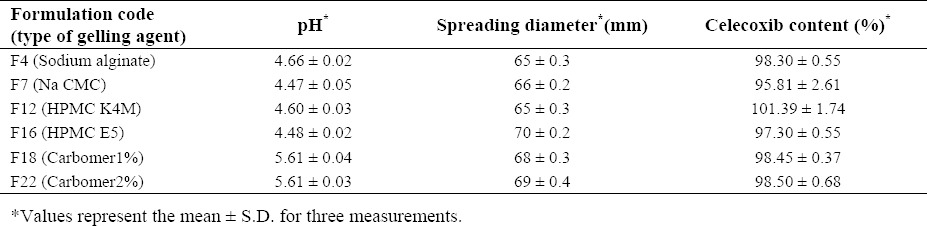
Most of the prepared formulations were acceptable in visual inspection and microscopical examination after 48 h. The gels consisting of carbomer, HPMC and Na CMC were transparent or white (opaque) in nature, while sodium alginate gels were brownish in color. They also showed good homogeneity, smoothness, and spreadability with no phase separation or presence of air bubbles (Table 2). The pH of the gel formulations was in the range of 4.66 ± 0.02 to 5.61 ± 0.04 with no significant changes over time. The drug content of the selected gel formulations also are shown in Table 2.
Table 2.
Physicochemical properties of celecoxib gel formulations.
Rheological properties of celecoxib gel formulations
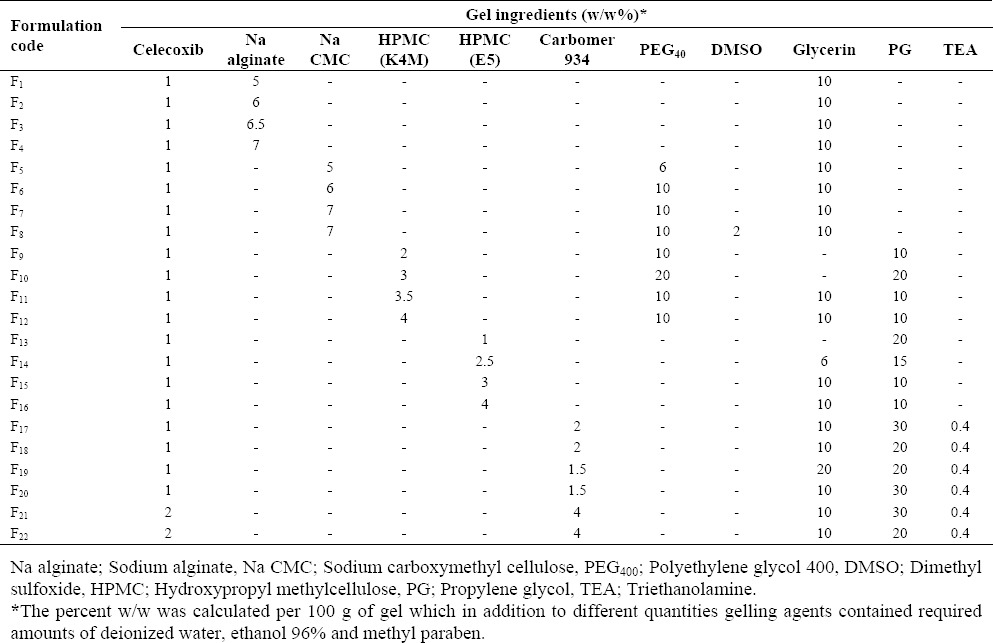
The rheological study of celecoxib gels was tested in a practical shear rate interval ranging from 0.01 s-1 to 1300 s-1. The flow properties of selected gels (formulations F4, F7, F12, F16, F18, and F22) presented in Fig. 1 indicated a non-Newtonian, pseudoplastic behaviour, which is typically seen in hydrophilic gels. Reproducibility of the rheological experiments for all six formulations was in acceptable limit with coefficients of variation (CV%) lower than 6%.
Fig. 1.

The effects of type and concentration of gelling agents on the flow properties of celecoxib gels. A; F4 (Na alginate), B; F7 (Na CMC), C; F12 (HPMC K4M), D; F16 (HPMC E5), E; F18 (Carbomer1%), F; F22 (Carbomer2%)(25 °C, n=3).
In vitro drug release, ex vivo drug transport, and iontophoresis studies
Franz diffusion cells holding cellulose acetate membranes were used to compare the drug release from six formulations prepared with different gelling agents are indicated in Table 2. The mean percent of drug released after five hours was found to be minimum (13.7 %) from sodium alginate base and maximum (41.5%) from HPMC K4M gel (Table 3). HPMC K4M gel formulation (formulation F12) was therefore adopted for both in vitro drug release and ex vivo drug transport studies in the presence of iontophoresis field.
Table 3.
Comparative drug release from selected celecoxib gel formulations using Franz cell and cellulosic membrane (37 °C, n=3).
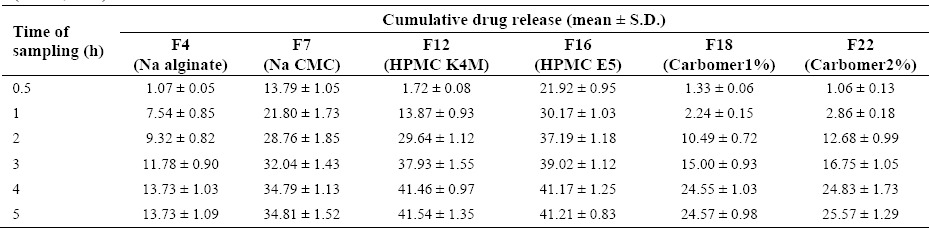
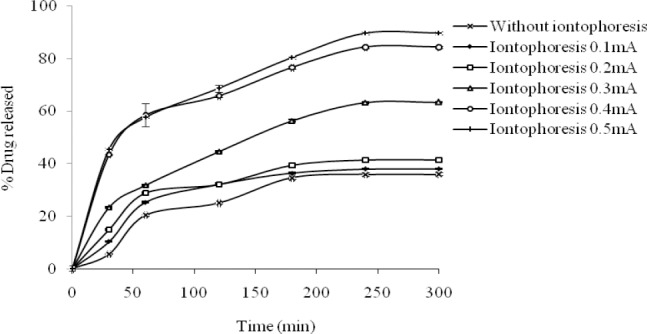
Fig. 2 shows the release profile of celecoxib from HPMC gel formulation, when subjected to DC iontophoresis current. The release of drug was significantly increased when current intensity was increased from 0.3 mA/cm2 to 0.5 mA/cm2. The flux enhancement (Figs. 3 and 4) was also higher with both DC and AC iontophoresis current compared to passive diffusion across the rat skin.
Fig. 2.
The effect of iontophoresis field (DC, 25 °C, n=3) on the in vitro release of celecoxib from HPMC- based gel (F12) using Franz cell and cellulosic membrane.
Fig. 3.
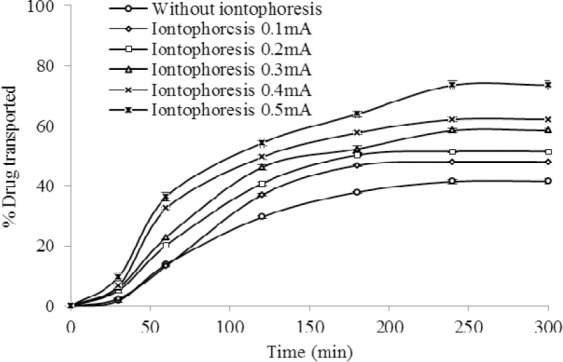
The effect of iontophoresis field (DC, 25 °C, n=3) on the ex vivo transport of celecoxib from HPMC gel (F12) through rat skin.
Fig. 4.
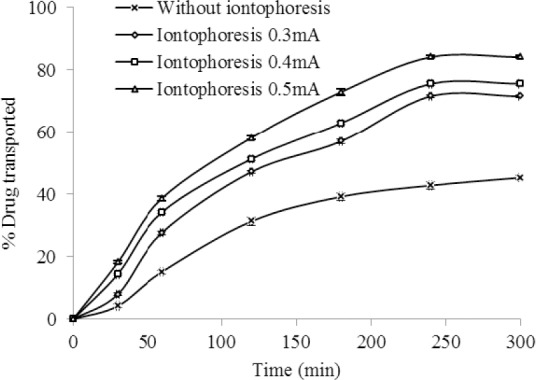
The effect of iontophoresis AC current 30:70 (30% on and 70% off, 25 °C, n=3) on the ex vivo transport of celecoxib from HPMC gel (F12) through rat skin.
DISCUSSION
Iontophoretic devices for delivery of charged drug molecules across or into the skin have been applied since the early 1900's by Galvani and Vota. In this physical approach a negligible amount of low-level electric current (0.5 mA/cm2 or less) are used to actively drive ionic drugs into the body (10). The method appears to be a promising technique for clinical applications (such as dentistry, dermatology, ophthalmology and physiotherapy) as well as the transdermal delivery of a variety of compounds in a controlled manner (8).
Transdermal enhancement effect of iontophoresis for some NSAIDs has previously reported to overcome the barrier of stratum corneum (11,12). Furthermore, iontophoresis has been clinically proven to increase the local anti-inflammatory activity of NSAIDs in soft tissue injuries and to avoid GI and other systemic effects caused by oral administration of these drugs especially in younger athletes who suffer from muscle pulls and tears or tendonitis (28).
In attempts to overcome the upper GI intolerance and cardiovascular side effect of oral celecoxib, some topical formulations of the drug have been prepared and the role of natural or chemical penetration enhancers have been evaluated in drug passage through the skin (17,18,19,21). Karade and colleagues (21) used DMSO 2% (penetration enhancer) in celecoxib gel formulations to improve drug release using diffusion cell. Furthermore, Shamsher and coworkers (19) formulated a carbopol gel of celecoxib and evaluated the effect of natural penetration enhancers (turpentine and tulsi oil) on drug permeation across rat skin. Their findings showed that the presence of natural terpenes of two essential oils could increase the transport of drug by altering either physicochemical properties of the drug or changing the structure of horny layer of the stratum corneum.
In the current study, a physical enhancement method, i.e. iontophoresis, was applied to investigate the effect of low voltage electric field on in vitro drug release from celecoxib gel and ex vivo drug transport through the rat skin.
Gel formulations are commonly acceptable delivery systems for iontophoresis because of their several advantages such as safety, stability and easiness of handling (26). The high percentage of water used in the gel formulations provides an electro-conductive medium for rapid movement of charged molecules. Furthermore, it has been reported that a decrease in the viscosity of gel, results in an increase in the formulation conductivity (27).
In our study, the prepared celecoxib gels presented a pseudoplastic (non-Newtonian) behaviour (Fig. 1), providing improved spreadability of gel on the skin. Among different types of prepared formulations, Na CMC and Na alginate gels showed less pseudoplasticity than HPMC K4M indicating that HPMC gels are potentially easier to spread and retain on the skin. This base (formulation F12) also showed acceptable physicochemical properties along with greater percentage of drug release (Table 3) and hence, it was considered the optimal formulation for subsequent iontophoretic studies.
Different parameters including operational and biological factors have shown to affect iontophoretic delivery of a drug (2). The physicochemical properties of the drug (molecular weight and size, charge and polarity), the formulation characteristics (pH, ionic strength, drug concentration, inactive ingredients, and the type of dosage form) as well as the experimental conditions (e.g. current density and current profile) are the important variables which have been investigated in iontophoretic studies. On the other hand, biological parameters such as pH and condition of the skin should be taken into account in clinical applications of iontophoresis (2).
The amide hydrogen of celecoxib is weakly acidic (pKa, 11.1) and hence the drug is completely ionized at pH 4.6-5.6 (equation 1). During iontophoresis, the positively charged species of the drug (the prominent form) are pushed and repelled under the positive charge of the anode (27).
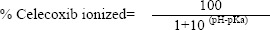
In a parallel line, findings of a study by Kotwal and colleagues (27) revealed that the ionization of diphenhydramine (a very weak acid, pKa 9.1) which was increased at pH 4.2 resulted in greater repulsion of ionized drug molecules and increased permeation during iontophoresis.
On iontophoresis experiments performed in the present study, as the current density of the electric field increased (to at least 0.3 mA/cm2), the percent celecoxib released was consequently augmented (Fig. 2). At the end of experiment, the cumulative percent of drug released was 36%, 63% and 89.7% for passive diffusion, current density of 0.3 mA/cm2, and 0.5 mA/cm2, respectively.
The findings of ex vivo drug transport across rat skin also showed a significantly higher iontophoretic transport of celecoxib compared to the passive flux for both AC and DC currents (P<0.05). A 0.5 mA/cm2 of DC increased drug flux to 73% compared to 41.5% of passive diffusion (Fig. 3). The maximum drug transport of 84.2% at DC current density of 0.5 mA/cm2 was also obtained and found to be significantly higher (P<0.05) than passive flux of 45.3% (Fig. 4).
Both in AC and DC current, a change in the electric density (0.3 mA/cm2-0.5 mA/cm2) has remarkable influence on transport of the drug through the membrane. In other words, the extents of ionized molecules, which may transfer through the skin, are proportional to the applied current density (10,29).
A linear relationship (R2=0.97) between the cumulative percent of drug transported after 5 h and the applied DC current density at the limit of 0.1 mA/cm2 to 0.5 mA/cm2 was observed. Similar linear correlation between the applied current and its iontophoretic flux have also been reported for methylphenidate, TRH, verapamil, GRH, diclofenac sodium and ketorolac (2). The high-end current density of 0.5 mA/cm2 used in this study is often considered to be the maximum permissible limit of iontophoretic current which should be used in clinical applications (29).
The mechanism of iontophoresis for celecoxib may be similar to that recently reported by Zuo and coworkers (12) for a few NSAIDs. They proposed that both strong lipophilicity and the dissociation extent of these drugs are the key factors to determine the transdermal enhancement effect of iontophoresis. On the other hand, iontophoresis not only enlarges the intercellular gaps of stratum corneum but also leads to the unordered arrangement of skin intercellular lipids (12).
CONCLUSION
Iontophoresis is a promising and progressively used physical technique to enhance transdermal drug delivery with safety and high efficiency. The findings of the present study showed that celecoxib transdermal flux through rat skin increases as a function of both DC and AC current density. Because of complete ionization of celecoxib, iontophoretic drug transport was almost twice as much as of the passive flux. Our results suggest that celecoxib may be considered a suitable candidate for transdermal transport through HPMC gel formulation by iontophoresis to cure soft tissue inflammation and sport injuries. However, to support in vitro results, more in vivo investigations and clinical studies are required.
ACKNOWLEDGEMENTS
The content of this paper is extracted from the Pharm.D thesis NO. 388499 submitted by R. Musavinasab which was financially supported by the Research Department of Isfahan University of Medical Sciences, Isfahan, I.R. Iran. The authors gratefully acknowledge Mrs. Moazzen and Mr. Sharifi the laboratory technicians of Isfahan School of Pharmacy for their technical assistance.
REFERENCES
- 1.Jorge LL, Feres CC, Teles V. Topical preparations for pain relief: efficacy and patient adherence. J Pain Res. 2011;4:11–24. doi: 10.2147/JPR.S9492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixit N, Bali V, Baboota S, Ahuja A, Ali J. Iontophoresis-an approach for controlled drug delivery: a review. Curr Drug Deliv. 2007;4:1–10. doi: 10.2174/1567201810704010001. [DOI] [PubMed] [Google Scholar]
- 3.Sebastiani P, Nicoli S, Santi P. Effect of lactic acid and iontophoresis on drug permeation across rabbit ear skin. Int J Pharm. 2005;292:119–126. doi: 10.1016/j.ijpharm.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 4.Minakshi Verma M, Gupta AK, Vinay Kumar Verma VK. An Injection without the Needle: Iontophoresis. J Biomed Pharm Res. 2013;2:65–71. [Google Scholar]
- 5.Tanner T, Marks R. Delivering drugs by the transdermal route: review and comment. Skin Res Technol. 2008;14:249–260. doi: 10.1111/j.1600-0846.2008.00316.x. [DOI] [PubMed] [Google Scholar]
- 6.Rawat S, Vengurlekar S, Rakesh B, Jain S, Srikarti G. Transdermal delivery by iontophoresis. Indian J Pharm Sci. 2008;70:5–10. doi: 10.4103/0250-474X.40324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costello CT, Jeske AH. Iontophoresis: applications in transdermal medication delivery. Phys Ther. 1995;75:554–563. doi: 10.1093/ptj/75.6.554. [DOI] [PubMed] [Google Scholar]
- 8.Kanikkannan N. Iontophoresis based transdermal delivery systems. Bio Drugs. 2002;16:339–347. doi: 10.2165/00063030-200216050-00003. [DOI] [PubMed] [Google Scholar]
- 9.Curdy C, Yogeshvar N, Guy R. Piroxicam delivery into human stratum corneum in vivo: iontophoresis versus passive diffusion. J Control Release. 2001;76:73–79. doi: 10.1016/s0168-3659(01)00418-7. [DOI] [PubMed] [Google Scholar]
- 10.Dhote V, Bhatnagar P, Mishra PK, Mahajan SC, Mishra DK. Iontophoresis: a potential emergence of a transdermal drug-delivery system. Sci Pharm. 2012;80:1–28. doi: 10.3797/scipharm.1108-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tashiro Y, Shichibe S, Kato Y, Hayakawa E, Itoh K. Effect of lipophilicity on in vivo iontophoretic delivery. I. NSAIDs. Biol Pharm Bull. 2001;24:278–283. doi: 10.1248/bpb.24.278. [DOI] [PubMed] [Google Scholar]
- 12.Zuo J, Du L, Li M, Liu B, Zhu W, Jin Y. Transdermal enhancement effect and mechanism of iontophoresis for non-steroidal anti-inflammatory drugs. Int J Pharm. 2014;466:76–82. doi: 10.1016/j.ijpharm.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 13.Katzung BG, Masters SB, Trevor AJ. Basic and clinical pharmacology. 12th ed. New York: The McGraw-Hill Inc; 2012. pp. 635–656. [Google Scholar]
- 14.Petri M, Hufman SL, Waser G, Cui H, Snabs MC, Verburg KM. Celecoxib effectively treats patients with acute shoulder tendinitis/bursitis. J Rheumatol. 2004;31:1614–1620. [PubMed] [Google Scholar]
- 15.Baboota S, Shakeel F, Ahuja A, Ali J, Shafiq S. Design, development and evaluation of novel nanoemulsion formulations for transdermal potential of celecoxib. Acta Pharm. 2007;57:315–332. doi: 10.2478/v10007-007-0025-5. [DOI] [PubMed] [Google Scholar]
- 16.Soliman SM, Abdel Malak NS, El-Gazayerly ON, Abdel Rehim AA. Formulation of microemulsion gel systems for transdermal delivery of celecoxib: In vitro permeation, anti-inflammatory activity and skin irritation tests. Drug Discov Ther. 2010;4:459–471. [PubMed] [Google Scholar]
- 17.Fetih G, Fa thalla D, El-Badry M. Liposomal gels for site-specific, sustained delivery of celecoxib: in vitro and in vivo evaluation. Drug Dev Res. 2014;75:257–266. doi: 10.1002/ddr.21179. [DOI] [PubMed] [Google Scholar]
- 18.Joshi M, Patravale V. Nanostructured lipid carrier (NLC) based gel of celecoxib. Int J Pharm. 2008;346:124–132. doi: 10.1016/j.ijpharm.2007.05.060. [DOI] [PubMed] [Google Scholar]
- 19.Shamsher AA, Charoo NA, Rahman Z, Pillai KK, Kohli K. Tulsioil as a potential penetration enhancer for celecoxib transdermal gel formulations. Pharm Dev Technol. 2014;19:21–30. doi: 10.3109/10837450.2012.751403. [DOI] [PubMed] [Google Scholar]
- 20.Tavakoli N, Minaiyan M, Saghaei E. Preparation of diltiazem topical gel for the treatment of anal fissure and in vitro, ex-vivo drug release evaluations. J Kerman Univ Med Sci. 2007;14:167–175. [Google Scholar]
- 21.Karade Preeti G, Shah Rohit R, Chougule DD, Bhise SB. Formulation and evaluation of celecoxib gel. J Drug Deliv Ther. 2012;2:132–135. [Google Scholar]
- 22.Vannat B, Gross D, Pouget MP, Pourrat A, Pourrat H. Comparison of the physical stability of astringent hydrogels based on cellulose derivatives. Drug Dev Ind Pharm. 1995;21:559–570. [Google Scholar]
- 23.Jelvehgari M, Rashidi MR, Mirzamohammadi SH. Adhesive and spreading properties of pharmaceutical gel composed of cellulose polymer. Jundishapur J Nat Pharm Prod. 2007;2:45–58. [Google Scholar]
- 24.Rudraraju VS, Wyandt CM. Rheological characterization of microcrystalline cellulose/sodium carboxymethyl cellulose hydrogels using a controlled stress rheometer: part I. Int J Pharm. 2005;292:53–61. doi: 10.1016/j.ijpharm.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Jones DS, Muldoon BC, Woolfson AD, Sanderson FD. An examination of the rheological and mucoadhesive properties of poly(acrylicacid) organogels designed as platforms for local drug delivery to the oral cavity. J Pharm Sci. 2007;96:2632–2646. doi: 10.1002/jps.20771. [DOI] [PubMed] [Google Scholar]
- 26.Pillai O, Panchagnula R. Transdermal delivery of insulin from poloxamer gel: ex vivo and in vivo skin permeation studies in rat using iontophoresis and chemical enhancers. J Control Release. 2003;14(89):127–140. doi: 10.1016/s0168-3659(03)00094-4. [DOI] [PubMed] [Google Scholar]
- 27.Kotwal V, Bhise K, Thube R. Enhancement of iontophoretic transport of diphenhydramine hydrochloride thermosensitive gel by optimization of pH, polymer concentration, electrode design, and pulse rate. AAPS Pharm SciTech. 2007;28:E120. doi: 10.1208/pt0804120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heim B. Transdermal administration of anti-inflammatory medications in sports injuries: use of iontophoresis and phonophoresis to enhance delivery. Int J Pharm Compd. 2006;10:14–18. [PubMed] [Google Scholar]
- 29.Patni M, Puranik P, Sonawane A, Panzade P. Transdermal iontophoretic delivery of timolol maleate. Braz J Pharm Sci. 2012;48:819–827. [Google Scholar]