

Erythropoietin-producing hepatocellular carcinoma A3 (EphA3) can form dimers in the absence of ligand binding, which are stabilized by the sterile α-motif (SAM) domain. This challenges the current understanding of EphA3 activation events and establishes a new role for the EphA3 SAM domain in receptor-receptor interactions.
Keywords: dimerization, EphA3, SAM domain, signaling
Abstract
The erythropoietin-producing hepatocellular carcinoma A3 (EphA3) receptor tyrosine kinase (RTK) regulates morphogenesis during development and is overexpressed and mutated in a variety of cancers. EphA3 activation is believed to follow a ‘seeding mechanism’ model, in which ligand binding to the monomeric receptor acts as a trigger for signal-productive receptor clustering. We study EphA3 lateral interactions on the surface of live cells and we demonstrate that EphA3 forms dimers in the absence of ligand binding. We further show that these dimers are stabilized by interactions involving the EphA3 sterile α-motif (SAM) domain. The discovery of unliganded EphA3 dimers challenges the current understanding of the chain of EphA3 activation events and suggests that EphA3 may follow the ‘pre-formed dimer’ model of activation known to be relevant for other receptor tyrosine kinases. The present work also establishes a new role for the SAM domain in promoting Eph receptor lateral interactions and signalling on the cell surface.
INTRODUCTION
Erythropoietin-producing hepatocellular carcinoma A3 (EphA3) is a receptor tyrosine kinase (RTK) that regulates axon guidance and morphogenesis during development by controlling cell adhesion, motility and contractility [1–4]. EphA3 also plays an important role in tumorigenesis and is often overexpressed and/or mutated in a variety of cancers including colorectal, pancreatic and ovarian cancer, melanoma, hepatocellular carcinoma and glioblastoma [5–7]. In addition, recent data show that EphA3 is up-regulated in the stromal micro-environment of many different tumour types, where it plays an important role in promoting tumour growth [7]. Intriguingly, activation of the overexpressed EphA3 in tumours can inhibit tumour growth [7], suggesting that EphA3 may be an attractive molecular target for cancer therapies [8]. Because EphA3 is scarcely expressed in healthy adult tissues [9], a molecular therapy that is specific for EphA3 could offer significant benefits and low side effects [4,10,11]. Therefore, new basic knowledge about the mechanism of activation of this receptor could pave the way to such new specific therapies.
The EphA3 receptor, like most RTKs, has a large extracellular region, a single transmembrane segment and an intracellular region. The extracellular region is composed of an N-terminal ligand-binding domain (LBD), a Sushi domain, an epidermal growth factor (EGF)-like domain and two fibronectin type III repeats [10]. The intracellular region encompasses a juxtamembrane (JM) segment known to regulate kinase activity, followed by the kinase domain, a sterile α-motif (SAM) domain and a PDZ-binding motif at the C-terminus.
The ligands of the Eph receptors are called ephrins and are anchored to neighbouring cells [10]. Eph receptor activation is believed to occur in response to dimerization/clustering of ephrin-bound Eph receptors [3,12,13]. A ‘seeding mechanism’ for clustering has been proposed to occur in three steps: (i) an Eph receptor binds to an ephrin ligand, (ii) the ligand-bound receptors form dimers and (iii) these dimers assemble into larger clusters. Thus, clustering and activation are believed to be triggered by ligand binding occurring in the first step of this process. Yet, there is evidence that direct receptor–receptor contacts are also important for Eph function. In particular, solved crystal structures of isolated Eph extracellular domains in complex with ephrins reveal that direct receptor–receptor interactions could also contribute to dimerization/clustering [12,14,15]. Furthermore, the SAM domain, located in the intracellular region of the receptors, has also been suggested to mediate dimerization/clustering [15–19]. However, there is no conclusive experimental evidence that the SAM domain plays a critical role in promoting or stabilizing Eph–Eph interactions in the cellular environment. In fact, it has also been proposed that the SAM domain mediates the interaction of the Eph receptors with signalling effectors such as SH2 domain-containing inositol 5-phosphatase 2 (SHIP2) [20,21].
The mechanism of Eph receptor activation is unique within the RTK superfamily, as the rest of the RTKs generally signal as dimers without forming clusters [22,23]. RTK dimerization brings the two kinase domains in close proximity, so that they can phosphorylate and activate each other. In the canonical ‘diffusion-based’ model of RTK activation [24], RTKs are monomers in the absence of ligand, but dimerize and cross-phosphorylate/activate each other upon ligand binding. However, previous work has identified unliganded RTK dimers [25–29] and thus an alternative model was proposed, the so-called ‘pre-formed dimer model’ [30]. In this model, the RTKs form dimers in the absence of ligand and ligand binding induces a structural change in the receptor that re-orients the catalytic domains for efficient activation. Now it is established that for some RTKs (such as epidermal growth factor receptors (EGFRs), fibroblast growth factor receptors (FGFRs) and tropomyosin receptor kinases (Trk)), the unliganded dimer is an important signalling intermediate [29–33]. Some unliganded dimers are tyrosine phosphorylated and their basal phosphorylation has been suggested to ‘prime’ the kinase for rapid activation upon ligand binding [29,30]. Furthermore, the response of the pre-formed dimers to ligand binding is not limited by the diffusion of the receptors within the plasma membrane and is thus faster.
In our exploration of the mechanism of EphA3 activation, in the present study we use a Förster resonance energy transfer (FRET)-based approach to investigate if EphA3 can form dimers on the surface of live cells in the absence of ephrin ligand binding. The results demonstrate that EphA3 has a strong propensity to dimerize in the absence of ligand binding. Furthermore, they show that EphA3 unliganded dimers are stabilized by interactions involving the SAM domain.
MATERIALS AND METHODS
EphA3 cloning and mutagenesis
Monomeric eYFP was obtained from Professor M. Betenbaugh (Johns Hopkins University). Monomeric turquoise (mTurq) was a generous gift from Professor Paul S. Park (Case Western Reserve University, Cleveland, OH, U.S.A.).
The EphA3 receptors were linked to eYFP and mTurq at their C-termini via (GGS)5 unstructured linkers. The cDNA encoding the EphA3 receptor was originally cloned in the pIRES2-EGFP vector as described [6]. We sub-cloned EphA3 in the pcDNA3.1(+) vector (Invitrogen) between the BamHI and EcoRI restriction sites. We used 5′-GCC CCC GGATCC ACC AGC AAC ATG GAT TGT CAG C-3′ as a forward primer and 5′-GGG CCC GAA TTC CAA CAC GGG AAC TGG GCC-3′ as a reverse primer in the PCR. The PCR products for (i) the (GGS)5 linker with mTurq and (ii) the (GGS)5 linker with monomeric eYFP were generated from existing pcDNA 3.1(+) plasmids in the laboratory, with EcoRI and XhoI restriction sites at the ends using 5′-GCC CCC GAATTC GGA GGA AGT GGC GGA AGT GGC-3′ as a forward primer and 5′-GGG CCC CTCGAG TTA CTT GTA CAG CTC GTC CAT GC-3′ as a reverse primer. The PCR products were double digested and ligated into pcDNA 3.1(+) EphA3 between the EcoRI and XhoI sites to obtain the plasmid constructs pcDNA 3.1(+) EphA3-15aa-mTurq and pcDNA3.1(+) EphA3-15aa-eYFP.
To delete the SAM domains from pcDNA 3.1(+) EphA3-15aa-mTurq and pcDNA3.1(+) EphA3-15aa-eYfp, we made use of the naturally occurring EcoRV restriction site right before the SAM domain. First, the sequences after the SAM domains, including the EphA3 C-terminus, the (GGS)5 linker and the fluorescent protein were amplified by PCR using 5′-GCG ATA TCT CAA AGA ATG GCC CAG TTC C-3′ as a forward primer and 5′- ACC CCC CAG AAT AGA ATG ACA C-3′ as a reverse primer. Then, the pcDNA 3.1(+) EphA3-15aa-mTurq plasmid and the PCR products were double digested using the restriction enzymes EcoRV and XhoI. The double digested PCR products were ligated to obtain the plasmid constructs pcDNA3.1(+) EphA3 ΔSAM-15aa-mTurq and pcDNA3.1(+) EphA3 ΔSAM-15aa-eYFP.
Cell culture and transfection
Human embryonic kidney 293T (HEK293T) cells were purchased from the A.T.C.C. The cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% FBS (Hyclone), 3.5 g/l (19.4 mM) D-glucose and 1.5 g/l (17.9 mM) sodium bicarbonate. For FRET experiments, the cells were plated in collagen-coated 35 mm glass bottom dishes (MatTek Corporation). The cells were co-transfected with either pcDNA3.1(+) EphA3-15aa-mTurq and pcDNA3.1(+) EphA3-15aa-eYFP or EphA3 ΔSAM-pcDNA3.1(+) EphA3-15aa-mTurq and EphA3 ΔSAM-pcDNA3.1(+) EphA3-15aa-eYFP. Various amounts of DNA, ranging from 130 to 4 μg, were used for transfection, in order to achieve a broad range of receptor expression in individual cells. Transfections were performed using Lipofectamine 3000 (Invitrogen) following the manufacturer's protocol.
We used HEK293T cells under reversible osmotic stress as a novel model system to characterize EphA3 dimerization (Figure 1). To swell the cells, we followed the protocol described in [34], exposing the cells to hypo-osmotic medium (10% medium+90% distilled water+25 mM HEPES) under conditions ensuring that cell adhesion is preserved so that the cells can be imaged. Since HEK293 cells are known to express ephrins that may interact with overexpressed Eph receptors on neighbouring adherent cells [13], in our experiments we imaged only isolated membranes not near other cells, as shown in Supplementary Figure S1 in the Supplementary Data. All experiments were further performed under starvation conditions, to ensure that no soluble ephrin-A ligand is present and that unliganded EphA3 dimerization is studied.
Figure 1. Images of HEK293T cells expressing full-length EphA3 (top) and EphA3 ΔSAM (bottom) before, during and after the application of reversible osmotic stress [34].

The results are in agreement with the complete reversibility of the treatment reported previously [34].
Two-photon microscopy
A spectrally resolved two-photon microscope with line-scanning capabilities was used to acquire spectral images as described previously [35,36]. A MaiTai femtosecond mode locked laser (Spectra- Physics) was used as the excitation energy source.
Measurement of dimerization propensity
In the two-photon microscope, we measure the donor intensity in the presence of the acceptor, FDA, as well as the acceptor intensity FA, as described in [35,37,38]. This is accomplished by performing two scans. In the first scan, we excite the donor with minimal acceptor excitation (800 nm) and in the second scan we excite the acceptor only (960 nm). As described in [35], we then calculate the FRET efficiency E and the intensity of the donor, FD, as if no FRET occurred, using the following equation:
| 1 |
Next, the FRET efficiency E is corrected for the so-called ‘proximity FRET’, which occurs when donors and acceptors approach each other by chance within distances of 100 Å (1 Å=0.1 nm) or so without engaging in specific interactions. This correction, which is required for membrane proteins, has been discussed in detail and experimentally verified [39].
To calculate donor and acceptor concentrations, [D] and [A] from FD and FA, we image solutions of purified fluorescent proteins of known concentrations in order to calibrate the fluorescence intensity of the EphA3 receptors as discussed in previous publications [40,41]. We determine the acceptor fraction as:
| 2 |
The dimeric fraction is determined from the corrected FRET efficiency according to:
| 3 |
The constant Ẽ in eqn (4) is the ‘intrinsic FRET’, a structural parameter that depends only on the separation and the orientation of the two fluorescent proteins in the dimer, not on the dimerization propensity. The dependence of the intrinsic FRET, Ẽ, on the distance between the fluorescent proteins in the dimer, d, is given by:
| 4 |
where R0 is the Forster radius for the mTurq–YFP FRET pair, 54.5 Å. Based on the law of mass action, the dimeric fraction can be written as a function of the total receptor concentration, T and the 2D dissociation constant Kdiss according to eqn (5):
| 5 |
We use eqns (3) and (5) to fit the measured dimeric fractions while optimizing for the two adjustable parameters: the dissociation constant Kdiss and the intrinsic FRET Ẽ.
The Gibbs free energy of dimerization is calculated according to:
| 6 |
with the standard state defined as 1nm2/receptor [41].
Western blots
For Western blots, 4 × 105 HEK293T cells per dish were transfected with 0.05–2 μg of pcDNA3.1(+) EphA3-15aa-eYFP or EphA3 ΔSAM-pcDNA3.1(+) EphA3-15aa-eYFP. Transfection was performed using Lipofectamine 3000 following the manufacturer's protocol. Twenty four hours post transfection, cells were lysed in lysis buffer (25 mM Tris/Cl, 0.5% Triton X-100, 20 mM NaCl, 2 mM EDTA, phosphatase inhibitors and protease inhibitors; Roche Applied Science). The lysed samples were subjected to centrifugation at 15000 g for 15 min at 4°C. The lysates were collected and stored at −20°C. The total protein concentrations in the lysates were measured using the BCA Protein Assay Kit (BioRad). The same total protein amounts were loaded on 3%–8% NuPAGEHNovexHTris-Acetate mini gels (Invitrogen) and the proteins were separated using SDS/PAGE. The proteins were transferred on to a nitrocellulose membrane and blocked using 5% non-fat milk in 1× TBST. Total EphA3 expression was probed with anti-N-EphA3 antibodies (Santa Cruz Biotechnology). The membranes were then stripped and re-probed with anti-phospho-Y779-EphA3 antibodies (Cell Signaling) to measure EphA3 phosphorylation. The secondary antibodies were anti-rabbit horseradish peroxidase-conjugated antibodies (Promega). The membranes were incubated with Amersham ECL Plus™ Western Blotting Detection Reagent (GE HealthCare Life Sciences) for 2 min and the bands were visualized in a ChemiDoc Molecular Imager (Bio-Rad). The intensities of the bands were quantified with the ChemiDoc Quantity One software.
RESULTS
EphA3 forms dimers in the absence of ligand binding
We have previously shown that dimerization curves for membrane receptors can be measured in plasma membrane vesicles, derived from mammalian cells, using FRET [42,43]. This model system offers a critical advantage over live cells by allowing precise measurements of the 2D concentration of the receptors, which is known to govern the dimerization behaviour according to the law of mass action. On the other hand, the plasma membrane of live cells is highly ‘wrinkled’, as cells possess 2–3 times the membrane surface needed to sustain their shape [44,45]. The complex membrane topology prevents the conversion of effective 3D receptor concentrations, determined by comparing the fluorescence intensities with standard solutions of known concentrations, into 2D receptor concentrations within the plasma membrane. It has been previously shown, however that cells ‘un-wrinkle’ their membranes in a reversible manner [34] when subjected to controlled osmotic stress. This controlled osmotic stress is not lethal and leads to disassembly of the caveolae, which are 60–80 nm cup-shaped invaginations of the plasma membrane.
In the present study, we use HEK293T cells under reversible osmotic stress to study whether the EphA3 receptor forms dimers in the plasma membrane of live cells in the absence of ligand binding to the ephrin-binding pocket. The two-photon images in Figure 1 demonstrate the reversibility of the osmotic stress effect on cells expressing EphA3. The pre-swelling images were captured after cell starvation for 12 h. The dish was then removed from the microscope stage and the starvation medium was replaced with swelling medium (10% medium+90% distilled water +25 mM HEPES). After 5 min, the swollen cells were imaged again. After ∼2 h (a typical length of an imaging session), the swelling medium was replaced with starvation medium and the cells were placed in the incubator. After 2 h more, the post-swelling images of the cells were recorded. They are indistinguishable from the pre-swelling images (Figure 1), consistent with prior reports of complete reversibility [34].
The cells under reversible osmotic stress exhibit large areas of stretched membrane with homogeneously distributed EphA3 fluorescence (Supplementary Figure S1 in the Supplementary Data), such that EphA3 concentration in the plasma membrane can be calculated, along with the FRET efficiencies. This allows us to calculate two parameters that describe EphA3 dimerization [41,46]. The first parameter is the dissociation constant, Kdiss, which is used to calculate the dimer stability or dimerization free energy ΔG. The second parameter is the structural parameter ‘intrinsic FRET’, Ẽ [41,47,48], which depends on the dimer structure and in particular on the distance and orientation of the fluorescent proteins in the dimer.
We used full-length EphA3 tagged at the C-terminus with the fluorescent proteins mTurq or eYFP, which are a FRET pair, via a (GGS)5 flexible linker. This linker has been previously shown to not perturb dimerization [47] and to lack secondary structure [49] and was used to ensure free rotation of the fluorescent proteins. HEK293T cells were co-transfected with plasmids encoding EphA3-eYFP and EphA3-mTurq at a 1:3 ratio, while varying the total amount of plasmids. After receptor expression, reversible osmotic stress was applied and images of the swollen cells were captured in the two-photon microscope. For data analysis, regions of homogenous, diffraction-limited membrane fluorescence of approximately 3 μm in length were chosen and analysed (Supplementary Figure S1). FRET efficiency and donor and acceptor concentrations were calculated for each region. Because EphA3 expression levels after transient transfection vary from cell to cell, a wide range of receptor concentrations were sampled. Two hundred and seven individual cells expressing various levels of EphA3 were imaged in five independent experiments and the images were analysed to construct a dimerization curve.
The FRET efficiencies measured for full-length EphA3 are shown in Figure 2(A) (black symbols), with each data point representing a single membrane region. Figure 2(B) shows the donor concentration versus the acceptor concentration in each membrane region. From the FRET efficiencies and the donor to acceptor ratios in Figures 2(A) and 2(B), we calculated the EphA3 dimeric fraction at each receptor concentration following the step-by step protocol described in [41]. A model describing the equilibrium between EphA3 monomers and dimers was fitted to these data according to eqn 5, yielding the dimerization curves in Figure 2(C), as described in published work [50–53]. The fit also yielded the optimal dissociation constant Kdiss and the intrinsic FRET constant Ẽ and their 95% confidence intervals (Table 1). In Figure 2(C), the dimeric fractions are binned and the averages and standard errors are shown together with the best-fit dimerization curve. The data closely follow the dimer prediction over the entire receptor concentration range, suggesting that EphA3 forms dimers in the absence of ligand binding.
Figure 2. FRET measurements of EphA3 dimerization propensities.
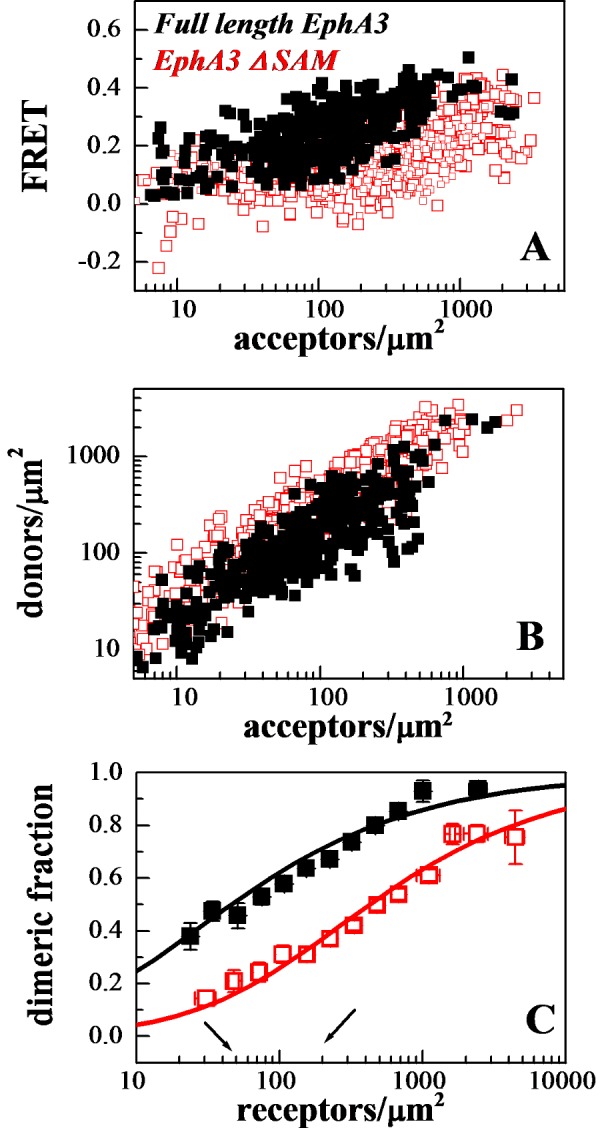
(A) FRET as a function of acceptor concentration for full-length EphA3 (solid black symbols) and EphA3 lacking the SAM domain (open red symbols). Every data point represents a single membrane region. (B) EphA3 donor concentration plotted as a function of EphA3 acceptor concentration in each membrane region analysed. (C) Dimeric fraction as a function of receptor concentrations. The dimeric fractions are binned and averages and standard errors are shown for each bin. The solid line, given by 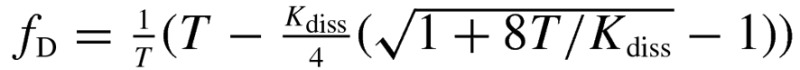 , is the theoretical curve for the best-fit dimerization model. The full-length EphA3 receptor forms dimers in the absence of ligand binding (black). Deletion of the SAM domain decreases the dimerization propensity of the EphA3 receptor (red).
, is the theoretical curve for the best-fit dimerization model. The full-length EphA3 receptor forms dimers in the absence of ligand binding (black). Deletion of the SAM domain decreases the dimerization propensity of the EphA3 receptor (red).
Table 1. Parameters describing the stability and structure of EphA3 unliganded dimers.
Kdiss is the dissociation constant (receptors/μm2), ΔG is the dimerization free energy, Ẽ is the intrinsic FRET efficiency and d is the calculated distance between the fluorescent proteins in the EphA3 dimers. Kdiss and Ẽ are determined from a fit of the dimerization model to the FRET data and the uncertainties are the 95% confidence intervals from the fit. ΔG and d are calculated using eqns (6) and (4) respectively. Deletion of the EphA3 SAM domain increases the dissociation constant by an order of magnitude, corresponding to a dimerization free energy change of −1.3±0.3 kcal/mole.
| Kdiss (rec/μm2) | ΔG (kcal/mol) | Ẽ | d (Å) | |
|---|---|---|---|---|
| EphA3 | 55±16 | −5.8±0.2 | 0.60±0.03 | 50±1 |
| EphA3 ΔSAM | 471±130 | −4.5±0.2 | 0.49±0.04 | 54±1 |
To further confirm that EphA3 forms dimers, we plotted the FRET efficiency as a function of acceptor fraction, xA, under conditions where dimerization does not strongly depend on concentration (Figure 3). In particular, we selected data points corresponding to membrane regions in which EphA3 concentration exceeded 600 receptors/μm2, such that the dimeric fractions in these regions are fairly constant (between ∼80% and ∼90%, as shown in Figure 2C). Under these conditions, the dependence of FRET efficiency on the acceptor fraction is known to be linear for a dimer and non-linear for higher order oligomers [54–56]. We see that the data in Figure 3 are well described by a linear function (P<0.001), a finding that lends further support to the conclusion that EphA3 forms dimers in the absence of ligand binding.
Figure 3. FRET as a function of receptor acceptor fraction, for total EphA3 concentrations that exceed 600 receptors/μm2.

Under these conditions, the EphA3 receptors are ∼80%–90% dimeric and the FRET signal depends primarily on the acceptor fraction. The linear dependence is indicative of a dimer [54–56].
The 2D dissociation constant, Kdiss, calculated for EphA3 is 55 receptors/μm2 (Table 1). This value and the dimerization curves in Figure 2(C) allow us to directly evaluate the physiological relevance of EphA3 dimerization using reported values of EphA3 expression in cancer lines. For example, the human acute lymphoblastic pre-B (LK63) pre-B leukaemia cell line has been reported to express 20000–80000 EphA3 receptors per cell [11,57]. Using an estimated cell area of ∼400 μm2 [58,59], we calculate receptor densities in the range of 50–200 receptors per μm2. This concentration range, bracketed by the two arrows in Figure 2(C), matches or exceeds the measured 2D dissociation constant of 55 receptors/μm2 (Table 1). Therefore, a substantial fraction (50%–70%) of the EphA3 receptor is dimeric in this concentration range (Figure 2C). Thus, EphA3 dimerization can be very significant at physiological receptor expression levels.
A question may arise as to whether the interactions between EphA3 receptors in the membrane are modulated by ephrin-A ligands co-expressed on the cell surface, through ligand–receptor interactions in cis [60]. There are reports that ephrin-A3 is expressed in HEK293T cells [61], although at low levels that are not detectable by Western blotting. In addition, in our experiments the dimerization curves are measured over a broad range of EphA3 concentrations, including very high EphA3 concentrations. As a consequence, there probably is a large excess of EphA3 over ephrin-A molecules in the plasma membrane and thus ephrin-A effects are expected to be negligible. Nevertheless, to experimentally address this issue we measured EphA3 dimerization curves in cells treated with phosphatidylinositol-specific phospholipase C (PI-PLC), which is known to remove glycosylphosphatidylinositol (GPI)-anchored proteins such as ephrin-As from the cell surface. We have previously shown by Western blotting that a 4 h PI-PLC treatment of cells expressing high ephrin-A levels efficiently removes these GPI-linked ligands [60]. In addition, a 4 h PI-PLC treatment also completely eliminated the fluorescence of GPI-anchored YFP transiently transfected in HEK293T cells, demonstrating the effectiveness of GPI-linked protein removal from the cell surface (Figure 4A). We therefore performed the FRET experiments after treating the cells with PI-PLC for 4 h and then inducing swelling (Figure 4B). These dimerization curves are essentially identical with those previously obtained without PI-PLC treatment, demonstrating that EphA3 dimerization in our experiments is not affected by the presence of ephrin-A ligands (Supplementary Table S1 in the Supplementary Data).
Figure 4. PI-PLC treatment does not alter the dimerization propensity of EphA3 and EphA3 ΔSAM.

(A) In control experiments, PI-PLC treatment completely cleaves YFP-GL-GPI [76] from the surface of HEK293T cells. (B) Dimerization curves with and without PI-PLC treatment.
We further reasoned that if EphA3 dimers exist and are phosphorylated, the phosphorylation per receptor should increase with the increase in receptor expression, similarly to the increase in dimeric fraction. We therefore used Western blotting to assess EphA3 phosphorylation in the absence of ligand, as a function of receptor expression, using antibodies recognizing the phosphorylated Tyr779 motif in the EphA3 activation loop. One typical Western blot is shown in Figure 5(A) and a quantification based on four independent experiments is shown in Figure 5(B). These results demonstrate higher levels of EphA3 phosphorylation when receptor expression is higher, consistent with the FRET results.
Figure 5. The average EphA3 receptor phosphorylation increases with expression levels, supporting the idea of EphA3 dimerization in accord with the law of mass action.

(A) Western blots showing the expression and activation of EphA3-eYFP as a function of DNA amounts used for transfection. (B) Quantification of EphA3 phosphorylation as a function of DNA amounts used for transfection, from four independent experiments. Shown is the ratio of anti-p-Tyr779 antibody staining and anti-EphA3 antibody staining, as a measure of the average receptor phosphorylation.
Deletion of the EphA3 SAM domain decreases EphA3 dimerization
Given the fact that SAM domains are known to mediate protein–protein interactions, we investigated if the EphA3 SAM domain contributes to EphA3 dimerization in the absence of ligand binding. We therefore characterized the dimerization propensity of EphA3 lacking the SAM domain (EphA3 ΔSAM). We analysed 291 individual cells with various levels of EphA3 ΔSAM expression, in five independent experiments. The comparison of the raw FRET measurements for EphA3 ΔSAM (red open symbols in Figure 2A) and full-length EphA3 (black solid symbols) suggests that EphA3 dimerization is substantially decreased in the absence of the SAM domain. The dimerization curves in Figure 2(C) demonstrate a highly significant (P<0.001) decrease in dimerization upon the deletion of the SAM domain, with the dissociation constant increasing by an order of magnitude (Table 1). Over the physiologically relevant EphA3 concentration range (50–200 receptors per μm2), the dimeric fraction is reduced from 50%–70% down to 15%–40% upon the deletion of the SAM domain. These results demonstrate that EphA3 unliganded dimers are stabilized by receptor–receptor interactions that involve the SAM domain.
Deletion of the EphA3 SAM domain decreases EphA3 tyrosine phosphorylation
We probed lysates expressing full-length EphA3-eYFP and EphA3 lacking the SAM domain (EphA3 ΔSAM-eYFP) with antibodies recognizing the phosphorylated Tyr779 motif in the EphA3 activation loop. The cells were placed in fresh medium without serum to ensure that there was no soluble ligand present. The results in Figure 6 show that the tyrosine phosphorylation of eYFP-labelled EphA3 ΔSAM is ∼40% lower than full-length EphA3 phosphorylation, whereas Supplementary Figure S2 shows a similar decrease in phosphorylation for mTurq-labelled EphA3 ΔSAM. Thus, deletion of the SAM domain decreases tyrosine phosphorylation of the EphA3 receptor. This is consistent with a role of the SAM domain in promoting EphA3 dimerization and tyrosine phosphorylation.
Figure 6. Phosphorylation of full-length EphA3-eYFP and EphA3 ΔSAM-eYFP.

(A) A typical Western blot experiment. Lane 1: empty vector. Lane 2: full-length EphA3. Lane 3: EphA3 ΔSAM. (B) Quantification from three independent experiments. Tyr779 phosphorylation decreases by ∼40% when EphA3 lacks the SAM domain.
DISCUSSION
Whereas most RTKs have been long believed to dimerize only in response to ligand binding, new knowledge is emerging that some RTKs can form dimers in the absence of ligand [25–29]. Whereas unliganded RTK dimerization is sometimes overlooked, its importance can be profound. First, various cancers have been linked to dysregulation of unliganded dimerization due to RTK overexpression [62–70]. Second, some pathogenic RTK mutations have been shown to cause aberrant signalling by increasing RTK unliganded dimerization [69,71,72]. Third, previous work has demonstrated that unliganded dimerization potentiates the response of RTKs to their ligands [29,30].
In the present study, we present the first study of unliganded dimerization of the EphA3 receptor in cells. Dimerization of unliganded Eph receptors has been previously surmised based on information under the high Eph receptor concentrations present in crystals [15] and we now show that EphA3 forms dimers in the absence of ligand at physiologically relevant EphA3 concentrations. Therefore, the ‘pre-formed dimer’ model, previously shown to be relevant for many RTKs, such as EGFR, fibroblast growth factor (FGF) and Trk receptors [29–33], is also relevant for EphA3. This implies that the Eph receptors exhibit a mode of activation that shares some similarities with other RTKs.
Our results suggest that EphA3 unliganded dimers are an intermediate in the EphA3 activation process. The data further suggest that in response to ligand binding, EphA3 clusters are assembled from pre-formed EphA3 dimers, not just EphA3 monomers. In fact, EphA3 is predominantly dimeric for typical EphA3 concentrations that are physiologically relevant and thus clustering in some cases may occur predominantly via ligand-induced lateral association of pre-formed EpA3 dimers. Therefore, the ‘seeding mechanism’ of Eph receptor cluster formation is probably incomplete, as it does not take into account the formation of unliganded dimers.
Our work further uncovers a novel role of the SAM domain in Eph receptor interactions. In particular, we find that the dimerization of the EphA3 receptor is significantly inhibited when the SAM domain is deleted, demonstrating that EphA3 dimers are stabilized by contacts that involve the SAM domain. Furthermore, the deletion of the SAM domain leads to a decrease in EphA3 phosphorylation in the absence of ligand. Interestingly, two EphA3 non-sense somatic mutations identified in lung cancer result in a truncated receptor lacking part or most of the SAM domain [73,74]. These mutations and possibly other EphA3 cancer mis-sense mutations affecting residues in the SAM domain (www.cbioportal.org), could decrease the tumour suppressing effects of EphA3 signalling by inhibiting dimerization and phosphorylation of the unliganded EphA3 receptor [6,75]. Thus, the discovery that the EphA3 receptor can be predominantly dimeric in live cells in the absence of ligand binding offers a new mechanistic hypothesis for how these mutations contribute to cancer progression.
Acknowledgments
We thank Kunal Saluja and Manasee Gedam for technical support and Dr Anne Kenworthy for the YFP-GL-GPI plasmid.
Abbreviations
- EphA3
erythropoietin-producing hepatocellular carcinoma A3
- FRET
Förster resonance energy transfer
- GPI
glycosylphosphatidylinositol
- HEK293T
human embryonic kidney 293T
- mTurq
monomeric turquoise
- PI-PLC
phosphatidylinositol-specific phospholipase C
- RTK
receptor tyrosine kinase
- SAM
sterile α-motif
AUTHOR CONTRIBUTION
Deo Singh, Elena Pasquale and Kalina Hristova designed the experiments and wrote the paper. Deo Singh, QingQing Cao and Xiang Zhou generated the plasmids. Deo Singh, QingQing Cao and Matt Salotto performed the FRET experiments. Christopher King developed the FRET imaging and analysis methodologies and wrote the requisite software. Deo Singh and Fozia Ahmed performed the Western blot experiments.
FUNDING
This work was supported by the National Science Foundation [grant number 1157687]; the National Institute of Health [grant numbers GM068619, GM095930 (to K.H.) and CA138390 (to E.B.P.)] and a NSF Graduate Research Fellowship [grant number DGE-1232825 (to C.K.)].
References
- 1.Lawrenson I.D., Wimmer-Kleikamp S.H., Lock P., Schoenwaelder S.M., Down M., Boyd A.W., Alewood P.F., Lackmann M. Ephrin-A5 induces rounding, blebbing and deadhesion of EphA3-expressing 293T and melanoma cells by CrkII and Rho-mediated signalling. J. Cell Sci. 2002;115:1059–1072. doi: 10.1242/jcs.115.5.1059. [DOI] [PubMed] [Google Scholar]
- 2.Smith F.M., Vearing C., Lackmann M., Treutlein H., Himanen J., Chen K., Saul A., Nikolov D., Boyd A.W. Dissecting the EphA3/ephrin-A5 interactions using a novel functional mutagenesis screen. J. Biol. Chem. 2004;279:9522–9531. doi: 10.1074/jbc.M309326200. [DOI] [PubMed] [Google Scholar]
- 3.Janes P.W., Nievergall E., Lackmann M. Concepts and consequences of Eph receptor clustering. Semin. Cell Dev. Biol. 2012;23:43–50. doi: 10.1016/j.semcdb.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Janes P.W., Slape C.I., Farnsworth R.H., Atapattu L., Scott A.M., Vail M.E. EphA3 biology and cancer. Growth Factors. 2014;32:176–189. doi: 10.3109/08977194.2014.982276. [DOI] [PubMed] [Google Scholar]
- 5.Day B.W., Stringer B.W., Al Ejeh F., Ting M.J., Wilson J., Ensbey K.S., Jamieson P. R., Bruce Z.C., Lim Y.C., Offenhaser C., et al. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell. 2013;23:238–248. doi: 10.1016/j.ccr.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Lisabeth E.M., Fernandez C., Pasquale E.B. Cancer somatic mutations disrupt functions of the epha3 receptor tyrosine kinase through multiple mechanisms. Biochemistry. 2012;51:1464–1475. doi: 10.1021/bi2014079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vail M.E., Murone C., Tan A., Hii L., Abebe D., Janes P.W., Lee F.T., Baer M., Palath V., Bebbington C., et al. Targeting EphA3 inhibits cancer growth by disrupting the tumor stromal microenvironment. Cancer Res. 2014;74:4470–4481. doi: 10.1158/0008-5472.CAN-14-0218. [DOI] [PubMed] [Google Scholar]
- 8.Barquilla A., Pasquale E.B. Eph receptors and ephrins: therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 2015;55:465–487. doi: 10.1146/annurev-pharmtox-011112-140226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiari R., Hames G., Stroobant V., Texier C., Maillere B., Boon T., Coulie P.G. Identification of a tumor-specific shared antigen derived from an eph receptor and presented to CD4 T cells on HLA class II molecules. Cancer Res. 2000;60:4855–4863. [PubMed] [Google Scholar]
- 10.Boyd A.W., Bartlett P.F., Lackmann M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov. 2014;13:39–62. doi: 10.1038/nrd4175. [DOI] [PubMed] [Google Scholar]
- 11.Keane N., Freeman C., Swords R., Giles F.J. EPHA3 as a novel therapeutic target in the hematological malignancies. Expert Rev. Hematol. 2012;5:325–340. doi: 10.1586/ehm.12.19. [DOI] [PubMed] [Google Scholar]
- 12.Himanen J.P., Yermekbayeva L., Janes P.W., Walker J.R., Xu K., Atapattu L., Rajashankar K.R., Mensinga A., Lackmann M., Nikolov D.B., Dhe-Paganon S. Architecture of Eph receptor clusters. Proc. Natl. Acad. Sci. U.S.A. 2010;107:10860–10865. doi: 10.1073/pnas.1004148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seiradake E., Harlos K., Sutton G., Aricescu A.R., Jones E.Y. An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nat. Struct. Mol. Biol. 2010;17:398–U27. doi: 10.1038/nsmb.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu K., Tzvetkova-Robev D., Xu Y., Goldgur Y., Chan Y.P., Himanen J.P., Nikolov D.B. Insights into Eph receptor tyrosine kinase activation from crystal structures of the EphA4 ectodomain and its complex with ephrin-A5. Proc. Natl. Acad. Sci. U.S.A. 2013;110:14634–14639. doi: 10.1073/pnas.1311000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikolov D.B., Xu K., Himanen J.P. Homotypic receptor-receptor interactions regulating Eph signaling. Cell Adh. Migr. 2014;8:360–365. doi: 10.4161/19336918.2014.971684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thanos C.D., Goodwill K.E., Bowie J.U. Oligomeric structure of the human EphB2 receptor SAM domain. Science. 1999;283:833–836. doi: 10.1126/science.283.5403.833. [DOI] [PubMed] [Google Scholar]
- 17.Smalla M., Schmieder P., Kelly M., Ter Laak A., Krause G., Ball L., Wahl M., Bork P., Oschkinat H. Solution structure of the receptor tyrosine kinase EphB2 SAM domain and identification of two distinct homotypic interaction sites. Protein Sci. 1999;8:1954–1961. doi: 10.1110/ps.8.10.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Behlke J., Labudde D., Ristau O. Self-association studies on the EphB2 receptor SAM domain using analytical ultracentrifugation. Eur. Biophys. J. 2001;30:411–415. doi: 10.1007/s002490100164. [DOI] [PubMed] [Google Scholar]
- 19.Stapleton D., Balan I., Pawson T., Sicheri F. The crystal structure of an Eph receptor SAM domain reveals a mechanism for modular dimerization. Nat. Struct. Biol. 1999;6:44–49. doi: 10.1038/4917. [DOI] [PubMed] [Google Scholar]
- 20.Zhuang G.L., Hunter S., Hwang Y., Chen J. Regulation of EphA2 receptor endocytosis by SHIP2 lipid phosphatase via phosphatidylinositol 3-kinase-dependent Rac1 activation. J. Biol. Chem. 2007;282:2683–2694. doi: 10.1074/jbc.M608509200. [DOI] [PubMed] [Google Scholar]
- 21.Lee H.J., Hota P.K., Chugha P., Guo H., Miao H., Zhang L.Q., Kim S.J., Stetzik L., Wang B.C., Buck M. NMR Structure of a Heterodimeric SAM:SAM Complex: Characterization and Manipulation of EphA2 Binding Reveal New Cellular Functions of SHIP2. Structure. 2012;20:41–55. doi: 10.1016/j.str.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemmon M.A., Ferguson K.M. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem. J. 2000;350:1–18. doi: 10.1042/bj3500001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/S0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 24.Fantl W.J., Johnson D.E., Williams L.T. Signaling by Receptor Tyrosine Kinases. Annu. Rev. Biochem. 1993;62:453–481. doi: 10.1146/annurev.bi.62.070193.002321. [DOI] [PubMed] [Google Scholar]
- 25.Chung I., Akita R., Vandlen R., Toomre D., Schlessinger J., Mellman I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature. 2010;464:783–707. doi: 10.1038/nature08827. [DOI] [PubMed] [Google Scholar]
- 26.Low-Nam S.T., Lidke K.A., Cutler P.J., Roovers R.C., van Bergen en Henegouwen P.M., Wilson B.S., Lidke D.S. ErbB1 dimerization is promoted by domain co-confinement and stabilized by ligand binding. Nat. Struct. Mol. Biol. 2011;18:1244–1249. doi: 10.1038/nsmb.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maruyama I., Shen J.Y. Brain-derived neurotrophic factor receptor TrkB exists as a preformed dimer in living cells. FASEB J. 2012:26. doi: 10.1186/1750-2187-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mischel P.S., Umbach J.A., Eskandari S., Smith S.G., Gundersen C.B., Zampighi G.A. Nerve growth factor signals via preexisting TrkA receptor oligomers. Biophys. J. 2002;83:968–976. doi: 10.1016/S0006-3495(02)75222-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin C.C., Melo F.A., Ghosh R., Suen K.M., Stagg L.J., Kirkpatrick J., Arold S.T., Ahmed Z., Ladbury J.E. Inhibition of basal FGF receptor signaling by dimeric Grb2. Cell. 2012;149:1514–1524. doi: 10.1016/j.cell.2012.04.033. [DOI] [PubMed] [Google Scholar]
- 30.Belov A.A., Mohammadi M. Grb2, a double-edged sword of receptor tyrosine kinase signaling. Sci. Signal. 2012;5:pe49. doi: 10.1126/scisignal.2003576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burgess A.W., Cho H.S., Eigenbrot C., Ferguson K.M., Garrett T.P.J., Leahy D.J., Lemmon M.A., Sliwkowski M.X., Ward C.W., Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell. 2003;12:541–552. doi: 10.1016/S1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 32.Brewer M.R., Choi S.H., Alvarado D., Moravcevic K., Pozzi A., Lemmon M.A., Carpenter G. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol. Cell. 2009;34:641–651. doi: 10.1016/j.molcel.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tao R.H., Maruyama I.N. All EGF(ErbB) receptors have preformed homo- and heterodimeric structures in living cells. J. Cell Sci. 2008;121:3207–3217. doi: 10.1242/jcs.033399. [DOI] [PubMed] [Google Scholar]
- 34.Sinha B., Koster D., Ruez R., Gonnord P., Bastiani M., Abankwa D., Stan R.V., Butler-Browne G., Vedie B., Johannes L., et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. 2011;144:402–413. doi: 10.1016/j.cell.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raicu V. In: FRET-based determination of protein complex structure at nanometer length scale in living cells. Diaspro A., editor. Boca Raton: CRC press; 2010. Nanoscopy. Multidimensional Optical Fluorescence Microscopy, 13.1–13.18. [Google Scholar]
- 36.Biener G., Stoneman M.R., Acbas G., Holz J.D., Orlova M., Komarova L., Kuchin S., Raicu V. Development and experimental testing of an optical micro-spectroscopic technique incorporating true line-scan excitation. Int. J. Mol. Sci. 2014;15:261–276. doi: 10.3390/ijms15010261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh D.R., Mohammad M.M., Patowary S., Stoneman M.R., Oliver J.A., Movileanu L., Raicu V. Determination of the quaternary structure of a bacterial ATP-binding cassette (ABC) transporter in living cells. Integr. Biol. 2013;5:312–323. doi: 10.1039/C2IB20218B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patowary S., Alvarez-Curto E., Xu T.R., Holz J.D., Oliver J.A., Milligan G., Raicu V. The muscarinic M-3 acetylcholine receptor exists as two differently Sized complexes at the plasma membrane. Biochem. J. 2013;452:303–312. doi: 10.1042/BJ20121902. [DOI] [PubMed] [Google Scholar]
- 39.King C., Sarabipour S., Byrne P., Leahy D.J., Hristova K. The FRET signatures of non-interacting proteins in membranes: simulations and experiments. Biophys. J. 2014;106:1309–1317. doi: 10.1016/j.bpj.2014.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li E., Placone J., Merzlyakov M., Hristova K. Quantitative measurements of protein interactions in a crowded cellular environment. Anal. Chem. 2008;80:5976–5985. doi: 10.1021/ac800616u. [DOI] [PubMed] [Google Scholar]
- 41.Chen L., Novicky L., Merzlyakov M., Hristov T., Hristova K. Measuring the energetics of membrane protein dimerization in mammalian membranes. J. Am. Chem. Soc. 2010;132:3628–3635. doi: 10.1021/ja910692u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Del Piccolo N., Placone J., He L., Agudelo S.C., Hristova K. Production of plasma membrane vesicles with chloride salts and their utility as a cell membrane mimetic for biophysical characterization of membrane protein interactions. Anal. Chem. 2012;84:8650–8655. doi: 10.1021/ac301776j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarabipour S., Chan R.B., Zhou B., Di Paolo G., Hristova K. Analytical characterization of plasma membrane-derived vesicles produced via osmotic and chemical vesiculation. Biochim. Biophys. Acta. 2015;1848:1591–1598. doi: 10.1016/j.bbamem.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adler J., Shevchuk A.I., Novak P., Korchev Y.E., Parmryd I. Plasma membrane topography and interpretation of single-particle tracks. Nat. Methods. 2010;7:170–171. doi: 10.1038/nmeth0310-170. [DOI] [PubMed] [Google Scholar]
- 45.Parmryd I., Onfelt B. Consequences of membrane topography. FEBS. J. 2013;280:2775–2784. doi: 10.1111/febs.12209. [DOI] [PubMed] [Google Scholar]
- 46.Chen L., Placone J., Novicky L., Hristova K. The extracellular domain of fibroblast growth factor receptor 3 inhibits ligand-independent dimerization. Sci. Signal. 2010;3:ra86. doi: 10.1126/scisignal.2001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarabipour S., Hristova K. FGFR3 unliganded dimer stabilization by the juxtamembrane domain. J. Mol. Biol. 2015;427:1705–1714. doi: 10.1016/j.jmb.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Del Piccolo N., Placone J., Hristova K. Effect of thanatophoric dysplasia type i mutations on fgfr3 dimerization. Biophys. J. 2015;108:272–278. doi: 10.1016/j.bpj.2014.11.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Evers T.H., E.M. van Dongen W.M., Faesen A.C., Meijer E.W., Merkx M. Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry. 2006;45:13183–13192. doi: 10.1021/bi061288t. [DOI] [PubMed] [Google Scholar]
- 50.Sarabipour S., Hristova K. Glycophorin A transmembrane domain dimerization in plasma membrane vesicles derived from CHO, HEK 293T, and A431 cells. Biochim. Biophys. Acta. 2013;1828:1829–1833. doi: 10.1016/j.bbamem.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sarabipour S., Hristova K. FGFR3 transmembrane domain interactions persist in the presence of its extracellular domain. Biophys. J. 2013;105:165–171. doi: 10.1016/j.bpj.2013.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Placone J., Hristova K. Direct assessment of the effect of the Gly380Arg achondroplasia mutation on fgfr3 dimerization using quantitative imaging FRET. PLoS One. 2012;7:e46678. doi: 10.1371/journal.pone.0046678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Placone J., He L., Del Piccolo N., Hristova K. Strong dimerization of wild-type ErbB2/Neu transmembrane domain and the oncogenic Val664Glu mutant in mammalian plasma membranes. Biochim. Biophys. Acta. 2014;1838:2326–2330. doi: 10.1016/j.bbamem.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adair B.D., Engelman D.M. Glycophorin a helical transmembrane domains dimerize in phospholipid bilayers - a resonance energy transfer study. Biochemistry. 1994;33:5539–5544. doi: 10.1021/bi00184a024. [DOI] [PubMed] [Google Scholar]
- 55.Li M., Reddy L.G., Bennett R., Silva N.D., Jr, Jones L.R., Thomas D.D. A fluorescence energy transfer method for analyzing protein oligomeric structure: application to phospholamban. Biophys. J. 1999;76:2587–2599. doi: 10.1016/S0006-3495(99)77411-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schick S., Chen L.R., Li E., Lin J., Koper I., Hristova K. Assembly of the M2 tetramer is strongly modulated by lipid chain length. Biophys. J. 2010;99:1810–1817. doi: 10.1016/j.bpj.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tomasevic N., Luehrsen K., Baer M., Palath V., Martinez D., Williams J., Yi C., Sujatha-Bhaskar S., Lanke R., Leung J., et al. A high affinity recombinant antibody to the human EphA3 receptor with enhanced ADCC activity. Growth Factors. 2014;32:223–235. doi: 10.3109/08977194.2014.984808. [DOI] [PubMed] [Google Scholar]
- 58.Wimmer-Kleikamp S. H., Nievergall E., Gegenbauer K., Adikari S., Mansour M., Yeadon T., Boyd A.W., Patani N.R., Lackmann M. Elevated protein tyrosine phosphatase activity provokes Eph/ephrin-facilitated adhesion of pre-B leukemia cells. Blood. 2008;112:721–732. doi: 10.1182/blood-2007-11-121681. [DOI] [PubMed] [Google Scholar]
- 59.Tingbeall H.P., Needham D., Hochmuth R.M. Volume and osmotic properties of human neutrophils. Blood. 1993;81:2774–2780. [PubMed] [Google Scholar]
- 60.Falivelli G., Lisabeth E.M., de la Torre E.R., Perez-Tenorio G., Tosato G., Salvucci O., Pasquale E.B. Attenuation of Eph receptor kinase activation in cancer cells by coexpressed ephrin ligands. PLoS One. 2013;8:e81445. doi: 10.1371/journal.pone.0081445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shaw G., Morse S., Ararat M., Graham F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002;16:869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- 62.Harari D., Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 2000;19:6102–6114. doi: 10.1038/sj.onc.1203973. [DOI] [PubMed] [Google Scholar]
- 63.Browne B.C., O'Brien N., Duffy M.J., Crown J., O'Donovan N. HER-2 Signaling and Inhibition in Breast Cancer. Curr. Cancer Drug Targets. 2009;9:419–438. doi: 10.2174/156800909788166484. [DOI] [PubMed] [Google Scholar]
- 64.Ross J.S., Slodkowska E.A., Symmans W.F., Pusztai L., Ravdin P.M., Hortobagyi G.N. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. 2009;14:320–368. doi: 10.1634/theoncologist.2008-0230. [DOI] [PubMed] [Google Scholar]
- 65.Sakai K., Yokote H., Murakami-Murofushi K., Tamura T., Saijo N., Nishio K. Pertuzumab, a novel HER dimerization inhibitor, inhibits the growth of human lung cancer cells mediated by the HER3 signaling pathway. Cancer Sci. 2007;98:1498–1503. doi: 10.1111/j.1349-7006.2007.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cho H.S., Mason K., Ramyar K.X., Stanley A.M., Gabelli S.B., Denney D.W., Leahy D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 67.Webster M.K., Donoghue D.J. FGFR activation in skeletal disorders: too much of a good thing. Trends Genet. 1997;13:178–182. doi: 10.1016/S0168-9525(97)01131-1. [DOI] [PubMed] [Google Scholar]
- 68.Adar R., Monsonego-Ornan E., David P., Yayon A. Differential activation of cysteine-substitution mutants of fibroblast growth factor receptor 3 is determined by cysteine localization. J. Bone Miner. Res. 2002;17:860–868. doi: 10.1359/jbmr.2002.17.5.860. [DOI] [PubMed] [Google Scholar]
- 69.Robertson S.C., Tynan J.A., Donoghue D.J. RTK mutations and human syndromes - when good receptors turn bad. Trends Genet. 2000;16:265–271. doi: 10.1016/S0168-9525(00)02021-7. [DOI] [PubMed] [Google Scholar]
- 70.Galvin B.D., Hart K.C., Meyer A.N., Webster M.K., Donoghue D.J. Constitutive receptor activation by Crouzon syndrome mutations in fibroblast growth factor receptor (FGFR) 2 and FGFR2/Neu chimeras. Proc. Natl. Acad. Sci. U.S. A. 1996;93:7894–7899. doi: 10.1073/pnas.93.15.7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen L.I., Webster M.K., Meyer A.N., Donoghue D.J. Transmembrane domain sequence requirements for activation of the p185(c-neu) receptor tyrosine kinase. J. Cell Biol. 1997;137:619–631. doi: 10.1083/jcb.137.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Webster M.K., Donoghue D.J. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J. 1996;15:520–527. [PMC free article] [PubMed] [Google Scholar]
- 73.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhuang G.L., Song W.Q., Amato K., Hwang Y., Lee K., Boothby M., Ye F., Guo Y., Shyr Y., Lin L.P., et al. Effects of cancer-associated epha3 mutations on lung cancer. J. Natl. Cancer Inst. 2012;104:1182–1197. doi: 10.1093/jnci/djs297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Keller P., Toomre D., Diaz E., White J., Simons K. Multicolour imaging of post-Golgi sorting and trafficking in live cells. Nat. Cell Biol. 2001;3:140–149. doi: 10.1038/35055042. [DOI] [PubMed] [Google Scholar]