

Abstract
Drivers of population genetic structure are still poorly understood in marine micro-organisms. We exploited the North Sea–Baltic Sea transition for investigating the seascape genetics of a marine diatom, Skeletonema marinoi. Eight polymorphic microsatellite loci were analysed in 354 individuals from ten locations to analyse population structure of the species along a 1500-km-long salinity gradient ranging from 3 to 30 psu. To test for salinity adaptation, salinity reaction norms were determined for sets of strains originating from three different salinity regimes of the gradient. Modelled oceanographic connectivity was compared to directional relative migration by correlation analyses to examine oceanographic drivers. Population genetic analyses showed distinct genetic divergence of a low-salinity Baltic Sea population and a high-salinity North Sea population, coinciding with the most evident physical dispersal barrier in the area, the Danish Straits. Baltic Sea populations displayed reduced genetic diversity compared to North Sea populations. Growth optima of low salinity isolates were significantly lower than those of strains from higher native salinities, indicating local salinity adaptation. Although the North Sea–Baltic Sea transition was identified as a barrier to gene flow, migration between Baltic Sea and North Sea populations occurred. However, the presence of differentiated neutral markers on each side of the transition zone suggests that migrants are maladapted. It is concluded that local salinity adaptation, supported by oceanographic connectivity patterns creating an asymmetric migration pattern between the Baltic Sea and the North Sea, determines genetic differentiation patterns in the transition zone.
Keywords: local adaptation, marine phytoplankton, oceanographic connectivity, population genetics
Introduction
Marine microbial organisms often have large population sizes, vast dispersal capabilities and fast growth rates and display mainly asexual reproduction. Therefore, marine phytoplankton species have typically been considered to have continuous distribution and weak biogeographic population structure (Finlay 2002). Intraspecific genetic diversity of a phytoplankton bloom was initially documented by Gallagher (1980). Since then, the paradigm of panmixia has been challenged by several studies showing apparent genetic structure among phytoplankton populations (Rynearson & Armbrust 2000, 2004; Nagai et al. 2007; Casteleyn et al. 2010; Tahvanainen et al. 2012). The biology of whatever organism in focus may also contribute to its population genetic structure. Studies on marine gastropods and fish show that differential selection along environmental gradients and variable life history traits may lead to differentiated populations (Kyle & Boulding 2000; Nanninga et al. 2014). With respect to phytoplankton species, Rynearson & Armbrust (2004) proposed that genetic differentiation may be maintained by a combination of differential selection from the environment and by restricted water flow between two intermixing estuaries. However, a comprehensive study of population genetic structure in phytoplankton species along an environmental gradient combined with experimental data is lacking. In a spatially heterogeneous environment with various selective pressures, populations may develop locally adapted physiological features provided that the balance between gene flow and local selection is favourable (Blanquart et al. 2013). It is also becoming increasingly apparent that physical features such as seascape topography or oceanographic currents may contribute to the structuring of phytoplankton populations (Casabianca et al. 2012). Currents driving the oceanographic connectivity may lead to asymmetric migration patterns and spatial differentiation of populations (Godhe et al. 2013), which may promote adaptation of populations to local environmental conditions (Kawecki & Holt 2002).
Population structure may also in part be a consequence of isolation by distance (IBD). Isolation by distance often corresponds to the intraspecific population structure distributed over large spatial scales (Wright 1943). The geographic distance is, however, a poor predictor of pairwise genetic differentiation on smaller scales. Connectivity patterns may be a better predictor of genetic structure on a regional level (White et al. 2010). Regional oceanographic circulation patterns in the Baltic Sea–North Sea transition have been shown to correlate with the genetic structure of, for example, fish species (Teacher et al. 2013). Johannesson & Andre (2006) reviewed the available data on genetic diversity of macro-organisms in the Baltic Sea and compared it to sister populations in the North Sea. That study revealed a clear pattern of comparably low genetic diversity in the Baltic Sea.
The Baltic Sea is a geologically young regional sea which was colonized by marine species from the adjacent North Sea during the onset of the Littorina Sea phase (7500 years BP) (Björck 1995). Today, the Baltic Sea is the world's second largest brackish sea connected to the North Sea by several narrow straits representing a transition zone. A horizontal salinity gradient ranging from 3 to 30 practical salinity units (psu) traverses the Baltic Sea and parts of the North Sea in a NE–SW direction. This gradient is maintained by restricted water exchange through the narrow straits and by the high influence of freshwater run-off to the inner basins (Leppèaranta & Myrberg 2009). The low salinity and the subarctic conditions in the distant basins reduce species richness (Zettler et al. 2007), and populations living in the Baltic Sea ecosystem are considered genetically impoverished compared to the populations of the same species in the NE Atlantic (Johannesson & Andre 2006). The natural stressors in terms of low salinity and temperature together with increasing anthropogenic pressures may impose strong selection and a reduction in genetic diversity in the Baltic Sea. Also, bottlenecks during the recent colonization and genetic drift through ‘allelic surfing’ (Excoffier & Ray 2008) may have contributed to the present low diversity. Added up, the Baltic Sea is characterized by relatively low diversity with evidence for local adaptation and endemic speciation (Pereyra et al. 2009).
While genetic diversity patterns and the underlying factors of Baltic macro-organisms, particularly fish, are more frequently studied and better understood, genetic diversity of Baltic Sea micro-organisms has received little attention so far. One study on the toxic dinoflagellate Alexandrium ostenfeldii revealed high genotypic diversity and strong genetic differentiation among local bloom populations (Tahvanainen et al. 2012). However, this study did not include a comparison to populations outside the Baltic Sea and provided merely a conceptual explanation of differentiation patterns. A seascape approach, including the role of physical and hydrographic boundaries and their influence on gene flow, is lacking from the Baltic Sea. Knowledge about seascape diversity patterns and connectivity dynamics is a prerequisite to understand the response of phytoplankton to environmental change, for example local adaptation. The genetic variability, selection pressures and the adaptative potential of phytoplankton populations across the Baltic Sea and parts of the North Sea may be different along the salinity gradient. The theory of phenotype–environment mismatch (Marshall et al. 2010) predicts that total adaptation may be reinforced in such gradients through selection against migrants (Hendry 2004) leading to reduced realized connectivity through gene flow producing a pattern of isolation by adaptation (Nosil et al. 2009).
The diverse diatom genus Skeletonema is found from temperate to tropical ecosystems, but in Scandinavian waters, S. marinoi is the exclusively dominant species. It occurs across the Baltic Sea and the North Sea coasts of Scandinavia, where it often is a prominent component of the spring phytoplankton community and an important primary producer contributing up to 60% of the total primary production (Jochem 1989). As several other diatom species in temperate areas, the planktonic cells of S. marinoi transform into resting stages when conditions turn unfavourable, sink to the ocean floor and may remain viable in the sediments for several decades (Härnström et al. 2011). For diatoms, asexual reproduction mediates progressive diminution and, at a critical size, cells undergo sexual reproduction. Sexual reproduction has been shown to occur in Baltic Sea isolates of S. marinoi within a determined size window and triggered by shifts in environmental parameters (Godhe et al. 2014). Auxosporulation seems to be possible when cells reach about 50% of the maximum valve diameter. Even though sexual reproduction may be infrequent, it is expected to support genetic diversity within Baltic Sea populations. Population structure of S. marinoi has previously been studied in the coastal waters of Kattegat and Skagerrak, and findings indicate that populations are genetically diverse and stable over time (Härnström et al. 2011). Investigations from the same area show a significant correlation between regional gene flow and oceanographic connectivity (Godhe et al. 2013), emphasizing the role of seascape dynamics when assessing drivers behind population structure of phytoplankton species.
We hypothesized that the salinity gradient of the Baltic Sea–North Sea transition creates genetic divergence and local adaptation in S. marinoi populations in conjunction with physical barriers. Additionally, we wanted to test whether genetic diversity increased towards the high-saline part of the studied gradient as has been shown for multicellular organisms (Johannesson & Andre 2006). We combine a population genetic approach with experimental data to assess the phenotypic reaction norm along the salinity gradient. We attempt to increase the robustness of our results by modelling physical dispersal mechanisms that may be involved in the potential gene flow among the studied populations. With this overarching approach, we aim towards revealing the drivers shaping genetic diversity patterns and populations structure of common marine planktonic protists in the Baltic Sea and the NE North Sea.
Material and methods
Study area
We follow the definition of ICES on ecoregions (ICES 2004) dividing the study area into the Baltic Sea and the North Sea (Fig.1). According to this definition, Kattegat and Skagerrak are parts of the North Sea ecoregion. The transition zone, the Danish Straits, is part of the Baltic Sea ecoregion. The hydrographic front separating Baltic Sea water from North Sea water is close to the northern entrance of the straits but may shift its positions in the transition zone depending on prevailing wind conditions (Ehlin 1981). On a timescale of months to years, a baroclinic, wind-independent principal circulation occurs in the Baltic Sea due to a large horizontal gradient of salinity. There is a concurrent outflow of low-saline water through the Danish Straits at the surface layer and inflow of more saline water in deep layers. The long-term mean surface circulations are also affected by winds. These are, however, highly variable, and the observed outcome is a nonlinear combination of baroclinic mean circulation and mean wind-driven circulation (Leppèaranta & Myrberg 2009). Sea surface salinity data were retrieved from HELCOM (Axe 2010) (Fig.1, Table 1).
Fig. 1.
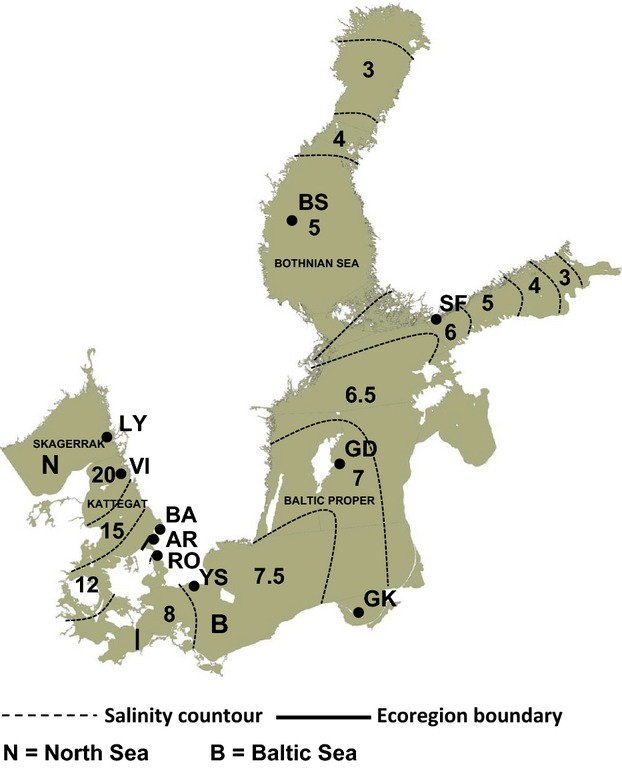
Baltic Sea ecoregion as defined by the ICES, which divides the study area into the Baltic Sea and the North Sea with a boundary (thick line) at the NE and SE parts of the Danish Belts. The salinity gradient is visualized by sea surface salinity at respective areas divided by dashed lines. Black circles indicating locations for sediment samples. Five of them were located in the North Sea (LY = Lysekil, VI = Vinga, BA = Båstad, AR = Arild and RO = Öresund) and five in the Baltic Sea (YS = Ystad, GK = Gdansk, GD = Gotland, SF = Storfjärden and BS = Bothnian Sea).
Table 1.
Summary of sampled populations and characteristics at sampling stations. The sample size (No. of genotyped) varied between 17 and 58 individuals per local population, which accounted for approximately 65–95% of the originally established strains (No. of strains). The column showing month/year refers to the time point when sediment samples were taken. We recorded the coordinates (decimal values), surface salinity (psu) and depth (m) of the ten sampling stations along the salinity gradient
| Population | No. of strains | Month/year | No. of genotyped | Longitude | Latitude | Salinity, psu | Depth, m |
|---|---|---|---|---|---|---|---|
| Bothnian Sea (BS) | 42 | 06/2011 | 40 | 18.55E | 62.12N | 3–6 | 83 |
| Gulf of Finland (SF) | 35 | 04/2013 | 32 | 23.25E | 59.88N | 4–6 | 20 |
| Gotland (GD) | 37 | 09/2011 | 33 | 19.05E | 57.41N | 6–8 | 96 |
| Gdansk (GK) | 39 | 04/2012 | 33 | 18.95E | 54.56N | 7–10 | 50 |
| Ystad (YS) | 25 | 05/2011 | 17 | 13.85E | 55.42N | 7–10 | 1 |
| Öresund (RO) | 61 | 03/2009 | 58 | 12.73E | 55.98N | 10–20 | 14 |
| Arild (AR) | 27 | 05/2011 | 17 | 12.34E | 56.16N | 11–23 | 1 |
| Båstad (BA) | 36 | 05/2011 | 33 | 12.85E | 56.43N | 11–23 | 1 |
| Vinga (VI) | 57 | 03/2009 | 45 | 11.52E | 57.55N | 12–35 | 78 |
| Lysekil (LY) | 61 | 03/2009 | 46 | 11.05E | 58.25N | 12–35 | 101 |
Sample collection and establishment of clonal cultures
We collected surface sediment samples (1–5 cm) with a gravity corer (LIMNOS) from ten locations along the salinity gradient (Fig.1, Table 1). We use the term ‘local population’ when referring to these geographically separated populations. Sampling stations Bothnian Sea (BS), Storfjärden (SF), Gotland (GD), Bay of Gdansk (GK) and Ystad (YS) are in the Baltic Sea ecoregion and stations Arild (AR), Båstad (BA), Vinga (VI) and Lysekil (LY) are in the North Sea ecoregion. Station Öresund (RO), located closest to the ecoregion border, was grouped together with other North Sea stations in subsequent statistical analyses because of similar salinity conditions.
Mixed subsamples of sediment were added to algal growth medium f/2+Si (Guillard 1975) of respective salinity in 24-well NUNC plates and incubated at 4 °C and 40 μmol photons m−2 s−1 (12:12 light:dark cycle). After 7 days, chains of S. marinoi were isolated (one from each well) by transferring it through several drops of f/2 media under an inverted microscope (Leica DMI3000 B) with a micropipette. Single chains were transferred into separate wells containing medium of respective salinities. After 7 days, clonal cultures were transferred to 50-mL NUNC culturing flasks and kept in the same conditions as described above. We isolated 30–60 clonal strains per location.
DNA extraction and microsatellite genotyping
Clonal cultures in exponential growth phase were filtered onto membrane filters (Versapor 3000, pore size 3.0 μm) and stored in −80 °C. DNA was extracted from frozen cells using a CTAB-based protocol previously described by Kooistra et al. (2003). We used eight microsatellite loci (Almany et al. 2009), and the PCR amplification was carried out according to the protocol described in Godhe & Härnström (2010). The PCR products were analysed in an ABI 3730 (Applied Biosystems) using an internal standard (GS600LIZ). genemapper (ABI Prism GeneMapper Software v.3.0) was used to determine the allele sizes for the individual loci.
Data analyses
Microsatellite Tools for Excel (Park 2001) was used for detecting identical eight-locus genotypes. Estimated deviations from Hardy–Weinberg equilibrium (HWE) and genotypic linkage disequilibrium (LD) were calculated using the software genepop v. 4.2 (Raymond & Rousset 1995) (10 000 Markov chain dememorizations, 20 batches and 5000 iterations per batch) including each locus in each local population following Bonferroni correction to adjust the level of statistical significance (Rice 1989). Technical artefacts, such as potential large allele dropout, stuttering or presence of null alleles, were computed using microchecker v. 2.2 (Van Oosterhout et al. 2004; 95% confidence interval and 1000 randomizations). Null-allele frequencies of non-HWE loci were calculated as in the study by Brookfield (1996).
Observed heterozygosity (Ho), gene diversity (Hs) and the inbreeding coefficient (FIS) were calculated using fstat v. 2.9.3 (Goudet 1995) per locus and for each local population separately. Confidence limits (95%) were obtained by 2000 bootstrapping replicates (using fstat). Significant differences of Ho, Hs, FIS and allelic richness were estimated with two-tailed pairwise sampled t-test performed in spss version 16.0. The level of intrapopulation diversity was estimated by performing rarefraction on measures of allelic diversity using the software hp-rare version 1.1 (Kalinowski 2005), which enabled the usage of allelic richness as a valid measure of diversity despite differences in sample size.
FST values between each pair of local populations were computed using the genepop software including all loci. The statistical significances of FST values were estimated following Fisher's method where probabilities of exact tests are combined. Pairwise Jost D values (Dest) (Jost 2008) were calculated using smogd (Crawford 2010) version 1.2.5 (CI 95% were obtained by 1000 bootstrapping replicates). Genetic population structure was analysed by Bayesian probabilistic population assignment in structure 2.3.3 (Pritchard et al. 2000; Falush et al. 2003) using the admixture model and correlated allele frequencies. The log-likelihoods of the generated data were used to infer the most likely ΔK. Pairwise IBD analyses were performed by Mantel tests in the isolde (Rousset 2000) subprogram embedded within genepop. Three separate IBD analyses were performed using (i) the entire data set, (ii) the Baltic Sea population genotype frequencies and (iii) the North Sea data. Geographic distances (shortest waterway in nautical miles) were estimated using qgis 2.4.0 (QGIS 2011).
Local adaptation experiment
We tested population-specific fitness along the salinity gradient by estimating maximum growth rates of three local populations in two discrete salinity regimes. We included two local populations from within the Baltic Sea (BS and GD) and one from the North Sea (BA). Strains from GD were included to control for the geographic distance between stations. Each population was represented by 10 strains to account for potential variability among genotypes (or individuals). For every population, maximum growth rates were determined at eleven different salinities (0, 3, 5, 7, 10, 12, 15, 20, 25, 30 and 35 psu). Populations were tested through the entire salinity gradient starting from their native salinity, that is 5 psu (BS), 7 psu (GD) and 15 psu (BA), in a stepwise manner towards the extremes (0 and 35 psu). Before growth rates were measured, strains were allowed to acclimate in culture medium set to the next salinity (≤ five practical salinity units at a time) for 7 days. From acclimated cultures, subsamples (equivalent to 10 000 cells/mL) were inoculated into 50-mL NUNC culturing flasks containing fresh medium and growth was monitored daily for 5–10 days until stationary phase was reached. The growth conditions were 75 μmol photons m−2 s−1 (12:12 light:dark cycle) at 10 °C. Growth was estimated by daily fluorescence measurements on a Varian Cary Eclipse Fluorescence Spectrophotometer (excitation 430 nm, emission 680 nm) equipped with a well plate reader. Significant differences in maximum growth rates were analysed by one-way anova in spss (Bonferroni correction). Potential changes in cell size as a response to transfers into new salinities, which would affect the fluorescence, was accounted for by calibrating cell numbers vs. fluorescence after each acclimatization period. Growth rates (r, intrinsic rate of increase) were calculated based on the longest period of exponential growth as in the study by Wood et al. (2005).
Directional relative migration
A new approach was used to calculate directional relative migration. The approach is a directional extension to measures of genetic differentiation and is based on defining a pool of migrants for each combination of two samples in pairwise comparison. The pool of migrants is calculated as the geometric means of the frequencies of the respective alleles in the two populations. A measure of genetic differentiation is then calculated for both the first and the second population compared to the pool, generating two directional measures of genetic differentiation. These directional measures are then used to calculate directional relative migration. The method is explained in detail by Sundqvist et al. (2013). Directional relative migration is here calculated using jost's d (Jost 2008) as a measure of genetic differentiation. Equation 22 in Jost (2008) is used to calculate the relative migration rates. To test whether migration is significantly higher in one direction than the other (i.e. asymmetric), 95% confidence intervals are calculated from 1000 bootstrap iterations. Directional relative migration calculations are performed using the function divmigrate from the r-package diversity (Keenan et al. 2013).
Simulation of dispersal and oceanographic connectivity
Dispersal and connectivity of S. marinoi were simulated with a biophysical model combining velocity fields from an ocean circulation model with a particle-tracking routine to generate dispersal trajectories. Velocity fields were first calculated with the 3-dimensional ocean circulation model baltix [for details of the model, see Hordoir et al. (2013)]. baltix is a regional model for the Baltic Sea, Kattegat, Skagerrak and the North Sea [configured from the NEMO ocean engine (Madec 2010)]. Velocity fields were modelled in hindcast mode for 8 years (1995–2002) with a horizontal resolution of 3.7 km (2 nm), a vertical resolution of 3–22 m and a temporal baroclinic resolution of 6 min.
Dispersal was simulated as particle trajectories calculated with the Lagrangian trajectory model tracmass, based on the study by Döös (1995). Trajectories were calculated in offline mode using the velocity fields generated by the baltix model with a 3-h update. In the first analysis, dispersal from the sampling stations was calculated for all 12 months and for diatoms drifting in the depth intervals 0–2 m and 10–12 m. All trajectories were simulated for 10 and 20 days of drifting based on the typical longevity of a Skeletonema bloom in the area (Wasmund et al. 1998). All dispersal calculations were repeated for the 8 years between 1995 and 2002. From each sampling station, 490 particles were released each month every year for each combination of depth interval and drift time, making a total of 1 881 600 particles. We calculated the dispersal probability and the multigenerational connectivity between all pairs of sampling stations. The dispersal probability was estimated by calculating the proportion of particles released from site i that ended up in site j. This will generate a connectivity matrix for each combination of month, drift depth and drift time estimating the probability of dispersal between all stations. Multigenerational connectivity was calculated by allowing stepping-stone dispersal over many single dispersal events and summed over all possible dispersal routes (White et al. 2010). Calculation of multigenerational connectivity is motivated by the life history of S. marinoi with the successive transformation between dispersing, vegetative cells and the benthic resting stage and may predict long-term connectivity between locations and barriers to gene flow (Nilsson Jacobi et al. 2012). This is achieved by multiplying the single-event connectivity matrix with itself n times, in this case, 8, 16 or 32 times. All calculations of connectivity matrices were performed using matlab r2013a (MathWorks Inc).
Correlation tests of oceanographic connectivity and directional relative migration
Multiple Mantel tests performed using R were used to explore possible correlation (R2) between directional relative migrations based on allele frequencies and modelled oceanographic trajectories. Significance levels were obtained by 5000 permutations. Mantel tests are widely applied in studies investigating the relationship between genetic and geographic distance (IBD) (Rousset 2000). The test is also appropriate in our case as oceanographic distance based on dispersal probability between locations is analogous to geographic distance. Mantel tests have previously been applied to correlation testing between genetic and oceanographic distances (White et al. 2010). The single generational matrix was compared against directional relative migration for each month separately and at 0–2 m for 10 and 20 days and at 10–12 m for 10 and 20 days. Skeletonema marinoi produces resting stages that may be resuspended and transported into a neighbouring area to seed a bloom during the next season. Therefore, it was appropriate to test the correlation between directional relative migration and oceanographic trajectories over multiple generations. The multigenerational matrices (8, 16, 32 generations) were compared against migration at the same depths and using the same drifting periods as above. The statistical approach was identical to the tests of single generational matrices, except that the multigenerational matrices included average values of connectivity for the whole year.
Results
The percentage of established clones that yielded genetic data was 60–95% per local population. All eight microsatellite loci were polymorphic. The most variable locus was Smar5 with 12–26 alleles per local population. Smar3 was the least variable locus displaying 1–5 alleles per local population. Significant departures from HWE were observed for all loci in a varying number of populations (P < 0.05, Table S1). Loci Smar1 and Smar5 showed heterozygote deficiency in all local populations. Locus Smar2 showed heterozygote deficiency in one population out of ten. No signs of large allele dropout or stuttering were found using microchecker. Frequencies of Brookfield null-allele estimates were low or moderate in loci Smar2, Smar3, Smar4, Smar6 and Smar7. Higher frequencies of null-allele estimates were found in Smar1, Smar5 and Smar8 (Table S1). Heterozygote deficiency is commonly observed in clonal or partially clonal diploid organisms (Balloux et al. 2003) and leads to elevated estimates of null-allele frequencies. In S. marinoi, estimates may therefore be falsely overestimated. The correlation between loci with high null-allele frequency and high heterozygote deficiency was significant (Spearman correlation, n = 80, P < 0.01). We did not observe any correlation between Brookfield null-allele estimates and any particular geographic location (two-tailed paired samples t-test, P > 0.05, Bonferroni correction). Therefore, a systematic error of null-allele frequencies could be refuted and all loci were included in further analyses of genetic differentiation. Analyses of the eight microsatellite loci across all populations showed no signs of LD. None of the genotyped individuals displayed identical eight microsatellite loci genotypes.
Ho ranged from 0.20 to 0.37, Hs from 0.62 to 0.74, and allelic richness from 3.88 to 4.28 in local populations from the Baltic Sea. The respective indices from local populations in the North Sea were 0.38–0.44, 0.73–0.77 and 4.42–4.63. Genetic diversity was significantly lower in populations in the Baltic Sea compared to populations from the North Sea (two-tailed paired samples t-test, HO, P = 0.019, HS, P = 0.007, allelic richness, P = 0.0004 Table 2). The inbreeding coefficients (FIS) were positive and on average higher in the Baltic Sea (0.54) compared to the North Sea (0.44). However, no statistically significant differences were found (two-tailed paired samples t-test, P = 0.090).
Table 2.
Comparison of genetic diversity by testing pooled Baltic Sea populations vs. North Sea populations. The average value of Ho, Hs and allelic richness (corrected for sample size) in the Baltic Sea were significantly lower compared to the North Sea (two-tailed paired samples t-test, Ho, P = 0.019, Hs, P = 0.007, allelic richness, P = 0.0004). The inbreeding coefficient (FIS) was not significantly different (two-tailed paired samples t-test, P = 0.090)
| Population | Sample size | Loci | Ho | Hs | FIS | Allelic richness |
|---|---|---|---|---|---|---|
| BS | 40 | 8 | 0.25 | 0.68 | 0.64 | 3.96 |
| SF | 32 | 8 | 0.37 | 0.65 | 0.42 | 4.00 |
| GD | 33 | 8 | 0.20 | 0.64 | 0.68 | 3.75 |
| GK | 33 | 8 | 0.36 | 0.74 | 0.51 | 4.28 |
| YS | 17 | 8 | 0.35 | 0.62 | 0.44 | 3.88 |
| BALTIC SEA, mean | 0.31* | 0.66** | 0.54 | 3.97*** | ||
| RO | 58 | 8 | 0.38 | 0.74 | 0.44 | 4.52 |
| AR | 17 | 8 | 0.44 | 0.77 | 0.41 | 4.63 |
| BA | 33 | 8 | 0.44 | 0.74 | 0.41 | 4.48 |
| VI | 45 | 8 | 0.43 | 0.75 | 0.43 | 4.59 |
| LY | 46 | 8 | 0.38 | 0.73 | 0.48 | 4.42 |
| NORTH SEA, mean | 0.41 | 0.74 | 0.44 | 4.53 |
Value significant at the P = 0.05 level.
Value significant at the P = 0.01 level.
Value significant at the P = 0.001 level.
Bayesian probability assignment conducted in structure revealed two genetically divergent populations (K = 2, evaluated as in the study by Evanno et al. (2005)): one in the Baltic Sea and another in the North Sea (Fig.2). Pairwise FST ranged from 0.004 to 0.200 (Table 3). All 45 pairs were significantly differentiated after Bonferroni correction (P < 0.05). Jost d (Dest) ranged from 0.017 to 0.459 (95% CI, 1000 bootstraps, Table 3). We found a significant isolation-by-distance pattern between FST and geographical distance (nautical miles) (R2 = 0.38, P = 0.002, Fig.3a) across the salinity gradient. No significant IBD was detected among local Baltic Sea populations (R2 = 0.12, P = 0.21, Fig.3b) nor among the local North Sea populations (R2 = 0.01, P = 0.57, Fig.3c).
Fig. 2.
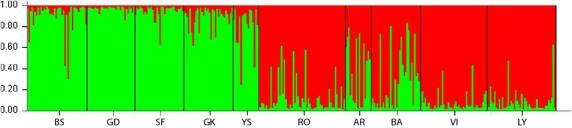
Bayesian probability assignment (structure) displaying genetic differentiation in the data set. The green colour represents individuals that were assigned to the Baltic Sea, and the red colour represents individuals that were assigned to North Sea populations. The structure analysis included admixture; therefore, most of the individuals are a mix of red and green. A split in genetic resemblance was observed at the entrance of the Baltic Sea.
Table 3.
Genetic differentiation measures of pairwise comparisons. Pairwise Jost D (Dest) values (95% CI) are shown above the diagonal. Pairwise FST values (below the diagonal) showed significant differentiation between all pairs of samples rejecting a hypothesis of panmixia
| population | BS | SF | GD | GK | YS | RO | AR | BA | VI | LY |
|---|---|---|---|---|---|---|---|---|---|---|
| BS | — | 0.086 | 0.024 | 0.189 | 0.119 | 0.165 | 0.113 | 0.243 | 0.255 | 0.196 |
| SF | 0.077 | — | 0.076 | 0.136 | 0.356 | 0.397 | 0.234 | 0.329 | 0.453 | 0.459 |
| GD | 0.033 | 0.042 | — | 0.234 | 0.233 | 0.275 | 0.174 | 0.123 | 0.294 | 0.304 |
| GK | 0.105 | 0.093 | 0.093 | — | 0.056 | 0.214 | 0.046 | 0.163 | 0.225 | 0.251 |
| YS | 0.087 | 0.200 | 0.137 | 0.087 | — | 0.133 | 0.075 | 0.170 | 0.184 | 0.156 |
| RO | 0.075 | 0.147 | 0.120 | 0.076 | 0.047 | — | 0.068 | 0.058 | 0.049 | 0.017 |
| AR | 0.040 | 0.130 | 0.080 | 0.050 | 0.050 | 0.020 | — | 0.072 | 0.052 | 0.083 |
| BA | 0.091 | 0.131 | 0.077 | 0.045 | 0.067 | 0.040 | 0.030 | — | 0.080 | 0.036 |
| VI | 0.096 | 0.155 | 0.128 | 0.074 | 0.078 | 0.013 | 0.020 | 0.039 | — | 0.045 |
| LY | 0.077 | 0.159 | 0.129 | 0.084 | 0.048 | 0.004 | 0.030 | 0.041 | 0.017 | — |
Fig. 3.
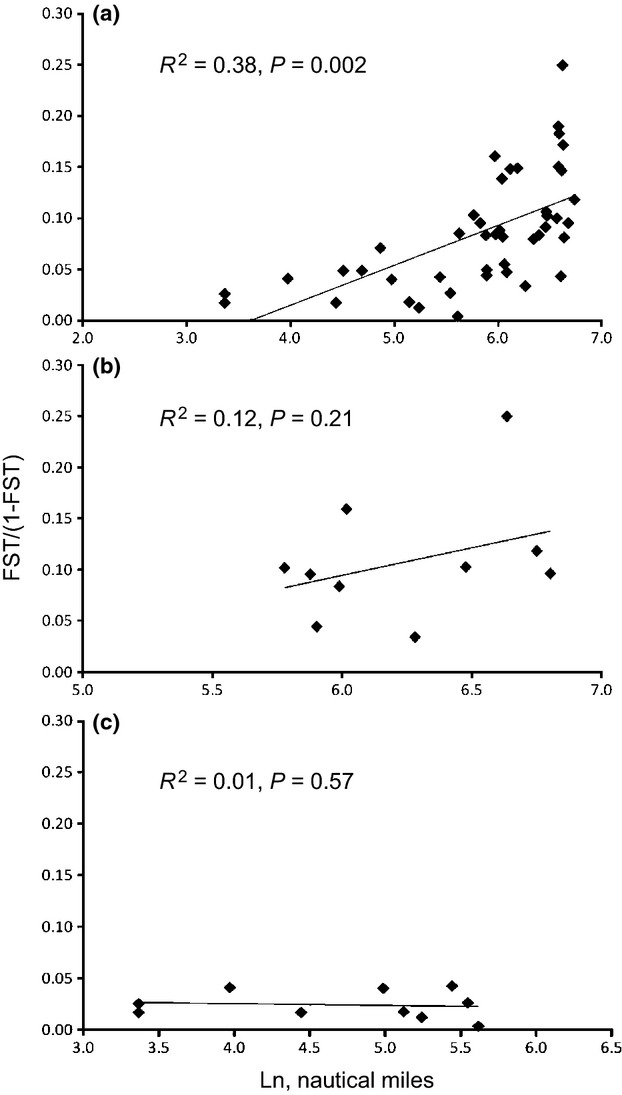
(a) IBD analyses showed significant correlation when all samples were included. (b) Genetic versus geographic distance of the Baltic Sea. FST of Baltic Sea populations were not isolated by geographic distance. (c) The North Sea stations were not significantly isolated by distance. Note the different scale on x-axes.
Maximum growth rates were recorded at different salinities in each local population. The Bothnian Sea (BS) population displayed maximum growth rate at 7 psu, Gotland (GD) population at 10 psu and Båstad (BA) population at 35 psu. BA had a significantly higher growth rate in 30 and 35 psu compared to BS and GD (one-way anova, BA vs. BS 30 psu, P < 0.0001, BA vs. BS 35 psu, P < 0.0001, BA vs. GD 30 psu, P < 0.0001, BA vs. GD 35 psu, P < 0.0001, Bonferroni correction). BS had a significantly higher growth rate in 5, 7 and 12 psu compared to BA (one-way anova, BS vs. BA 5 psu, P = 0.0003, BS vs. BA 7 psu, P = 0.034, BS vs. BA 12 psu, P = 0.013, Bonferroni correction). GD had significantly higher growth rates at 5 and 10 psu compared to BA (one-way anova, GD vs. BA 5 psu, P < 0.0001, GD vs. BA 10 psu, P < 0.0001) (Fig.4). There were also some other significant differences; for example, GD had significantly higher growth rate in 25 psu compared to BS (one-way anova, GD vs. BS 25 psu, P < 0.0001). All growth rate data may be obtained from the Data accessibility section (doi:10.5061/dryad.j5cf0).
Fig. 4.

Experimental assessments of the reaction norm of three local populations along the salinity gradient. (a) The Bothnian Sea (BS) population had its maximum growth rate at the native salinity of 5 psu. Growth was significantly reduced in salinities exceeding 10 psu. (b) The Gotland (GD) population had its maximum growth rate close to the native salinity of 7 psu. Growth rates were reduced with increasing salinity. (c) The Båstad (BA) population had reduced growth rates at lower salinities (<12 psu). The highest growth rate was observed at 35 psu. Dashed line represents the reaction norm and grey-shaded box the observed salinity range at each location. Vertical lines represent standard error (SE) of ten replicates.
The relative migration network (Fig.5a) shows all relative migration rates. As in the structure plot, this result shows that gene flow was strongest between populations within the two subareas. In Figure5b, directional relative migration rates below 0.50 were filtered out to emphasize the major geneflow barrier, the Danish straits. Gene flow among local populations in the northern Baltic Sea (BS, SF, GD) had higher rates of relative migration compared to gene flow among southern Baltic populations (GK, YS). Of all 45 population pairs, the migration is only found to be significantly asymmetric (95% CI, 1000 bootstrap iterations) in two pairs (YS-RO and YS-VI). Interestingly, these pairs are on each side of the transition zone. The strongest link between the Baltic Sea population and the North Sea population is displayed from YS to RO, contributing with an asymmetric directional relative migration rate of 0.58 over the transition zone in the south to north direction. GK was the most isolated local population with all relative migration rates below 0.50.
Fig. 5.
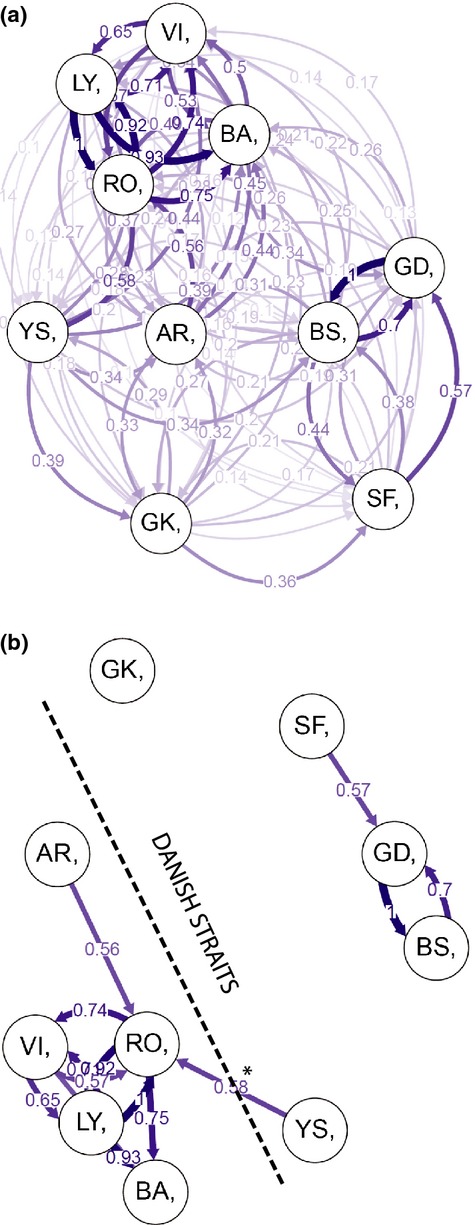
(a) The directional relative migration network including all relative migrations values indicates stronger gene flow within the subareas than between. (b) Directional relative migration network displaying relative migrations above 0.5. The direction of the relative migration between RO and YS was significantly asymmetric (*CI 95%).
Oceanographic connectivity between all ten local populations was assessed by modelling trajectories of dispersal at the two depths separately for each month. Virtual particles were allowed to disperse for 10 or 20 days. Multigenerational models were performed to find potential patterns by a stepping-stone approach, simulating a scenario where resting stages initiate the bloom after transport to a neighbouring area. The oceanographic connectivity is visualized in one example (Fig. S1) showing connectivity to Lysekil from the rest of the stations. Connectivity did not change significantly among stations within the North Sea. However, oceanographic connectivity dropped with approximately ten orders of magnitude when entering the Baltic Sea.
Mantel tests were used to investigate potential correlation (R2) between directional relative migrations and modelled oceanographic trajectories. Significant correlations (obtained by 5000 permutations) were found throughout the year at 0–2 m and at 10–12 m depth at drifting periods of 10 and 20 days (Table 4). Strongest correlations between directional relative migrations and oceanographic trajectories were observed at surface water with dispersal time of 10 or 20 days. During summer, that is in June, July and August, the least number of significant correlations was found. Significant correlations were additionally observed using 8, 16 and 32 generations. Mantel tests with data on 8 and 16 generations resulted in significant correlations for the surface water with a drifting period of 10 days. The 32-generational scenario was best supported by the statistical analyses as significant correlation between directional relative migrations and oceanographic trajectories was found at each depth and for each drifting period (Table S2).
Table 4.
Correlation coefficients of multiple Mantel tests between directional relative migration and oceanographic connectivity matrices. Results are presented for each month separately for 10 or days and at two different depths. All of the analyses, except two, showed significant correlation at 0–2 m. Correlation at 10–12 m was less frequent. Correlation coefficients given as R2. Significance levels obtained by 5000 permutations
| 0–2 m |
10–12 m |
|||
|---|---|---|---|---|
| Month | 10 days | 20 days | 10 days | 20 days |
| Jan | 0.34* | 0.47* | 0.38* | 0.25 |
| Feb | 0.33* | 0.33* | 0.30 | 0.35* |
| Mar | 0.37* | 0.63** | 0.21 | 0.49** |
| Apr | 0.35* | 0.41* | 0.25 | 0.25 |
| May | 0.35* | 0.45* | 0.21 | 0.32* |
| Jun | 0.31* | 0.33* | 0.18 | 0.18 |
| Jul | 0.23 | 0.40* | 0.18 | 0.21 |
| Aug | 0.33* | 0.36* | 0.18 | 0.13 |
| Sep | 0.49** | 0.32* | 0.18 | 0.35* |
| Oct | 0.31* | 0.27 | 0.33* | 0.35* |
| Nov | 0.40* | 0.58** | 0.42* | 0.29 |
| Dec | 0.47** | 0.61** | 0.25 | 0.49** |
Statistically significant values in italics,
P < 0.05,
P < 0.001.
Discussion
We compared the genetic diversity of a planktonic micro-organism on both sides of the Danish Straits, representing a transition zone between the Baltic Sea and the North Sea. Diversity estimates of S. marinoi populations in the area decreased from the high-saline basins in the North Sea towards the low-salinity areas east of the Danish Straits, which coincide with the general pattern of decreased genetic diversity of marine macro-organisms (e.g. fish, macro-algae and bivalves) observed in the Baltic Sea (reviewed by Johannesson & Andre (2006)). As in macro-organisms, our analyses also revealed distinct genetic divergence of a low-salinity Baltic Sea population coinciding with the most evident physical dispersal barrier in the area, the Danish Straits. Skeletonema marinoi populations showed strong differentiation patterns, driven by adaptation to the local salinity regimes and oceanographic connectivity patterns.
Genetic diversity of Baltic populations
Recent results from studies on, for example, fish and mussels have revealed taxon-specific diversity patterns across the salinity gradient, complicating a generalization of diversity clines in the area (Wennerström et al. 2013). To our knowledge, there is no similar comparison conducted with plankton organisms in the area, except for a study conducted by Holmborn et al. (2011) using mtDNA that showed a reduced level of genetic diversity in a calanoid copepod in the Baltic Sea. A significant reduction in genetic diversity and allelic richness of S. marinoi populations over the Danish Straits complies with the pattern of lower genetic diversity in marginal habitats (Kawecki 2008). The reduced neutral genetic diversity in the Baltic Sea may be due to historical events such as bottlenecks and founder events. Calculation of bottlenecks is dependent on estimates of the effective population sizes, and given the lack of proper estimates, such exercise would require too many assumptions. Inbreeding, estimated by the FIS, may be defined as mating among relatives, which may increase homozygosity. Inbreeding may be a result of purely nonrandom mating and life history traits or because of population structure, leading to increased FIS by restricted interpopulation gene flow (Wright 1968). Skeletonema marinoi isolates from the Baltic Sea are homothallic and recombine by outbreeding and inbreeding. Sexual reproduction and the formation of sexual auxospores can be induced by altered salinity conditions (Godhe et al. 2014). However, the frequency of sexual recombination in situ is largely unknown, complicating the interpretation of the observed FIS. The relatively high FIS (range 0.41–0.68) may suggest that local populations are inbred and isolated with low outside recruitment. Low genetic diversity of populations inhabiting geographically isolated and marginal ecosystems, such as the Baltic Sea, is commonly regarded as specifically adapted genetic lineages with a high degree of asexual reproduction (Tatarenkov et al. 2005; Kawecki 2008). Asexual reproduction reduces recombination events prolonging the prevalent pattern of LD (Alpermann et al. 2009), and mixtures of non-interbreeding populations increase heterozygote deficiency (Wahlund effect) (Sinnock 1975). Significant LD was, however, not detected in any pairs of loci when the data set was analysed across all populations. In relation to this, our findings suggest that sexual events occur in Baltic populations. Positive FIS further support the idea that all studied populations reproduce sexually, but the extent of genetic inbreeding was on average higher within the Baltic Sea, suggesting a higher degree of asexual reproduction. With the current data, it is difficult to determine a modelled frequency of sexual events as the signature of sex in genomes may only require sporadic recombination events (Halkett et al. 2005).
The future consequences of reduced genetic diversity within the Baltic Sea are hard to predict but may involve several ecological risks. For example, the adaptation potential in Baltic Sea populations may be reduced. Low genetic diversity may also cause a more homogenized functional diversity (Olden et al. 2004), implying potential disruptions in, for example, primary productivity or fluxes of energy and nutrients (Hughes et al. 2008). Phytoplankton species diversity promotes stability of the pelagic ecosystem and efficient use of resources (Ptacnik et al. 2008). The role of genetic diversity may be of comparable significance, especially in systems with low species diversity (Crutsinger et al. 2008) such as the Baltic Sea. Taking into account the predicted climate change scenarios (Meier et al. 2012), lower genetic diversity of phytoplankton populations in the Baltic Sea may lead to distinctive ecological effects compared to the NE Atlantic. However, despite the evidence of, for example, reduced fitness and productivity as a consequence of low genetic diversity in many other systems and organisms (Bell 1991; Gamfeldt et al. 2005), the overall reduction in diversity in S. marinoi in the Baltic Sea might not lead to ecological risks. Skeletonema marinoi populations in the Baltic Sea actually display genetic diversity comparable to other marine diatom species (Rynearson & Armbrust 2000; Evans et al. 2005). Direct comparisons of the level of genetic diversity between species, based on, for example, Hs in microsatellite loci are not possible without a certain degree of ambiguity as individual loci have their own evolutionary history. Nevertheless, the reason for the reduction in genetic diversity of S. marinoi in the Baltic Sea probably stems from their mode of reproduction, featuring possibly lower frequency of obligate sexual reproduction (Chepurnov et al. 2004).
Genetic differentiation
Based on the 354 individuals sampled across the entire salinity gradient covering 1500 km, we found evidence for a strong population genetic structure with the most pronounced genetic differentiation across the Danish Straits. Bayesian probalistic population assignment combined with a ΔK evaluation supported two genetically differentiated populations on each side of the Danish Straits. This particular geneflow barrier was expected as several other studies on macro-organisms have shown similar results (Väinölä & Hvilsom 1991). Apart from physical impediment, the Danish Straits may also function as an environmental barrier where salinity increases steeply along a short geographic distance. Our results indicate that this transition zone is a barrier for eukaryotic micro-organisms.
The role of benthic resting stages, which may anchor local populations to specific habitats, may be an important factor for maintaining population structures. This is supported by a strong local link between benthic and pelagic assemblages of S. marinoi populations at coastal sites on the Swedish west coast (Godhe & Härnström 2010) where one gram of sediment can contain up to 50 000 propagules (McQuoid 2002). The genetic structure of the benthic seed bank is stable over time and serves as a local reservoir for pelagic populations, reinforcing the regional genetic population structure. The locally well-adapted populations continuously shift between pelagic and benthic life stages.
Species may also be genetically structured because of the geographic distance between subpopulations. A greater distance between two populations leads to reduced gene flow and thus comparatively larger genetic differences (Wright 1943). FST of the 10 local populations displayed significant differentiation and were correlated with geographic distance. Geographic distance could, however, not explain genetic differentiation when the Baltic Sea and the North Sea were analysed separately, indicating that IBD is only a crude explanation of the observed population structure. Geographic distance may not adequately infer gene flow in the ocean where organisms often disperse by asymmetric circulation patterns. Stochasticity in modes of dispersal may lead to seemingly chaotic genetic patchiness that may be more satisfactorily inferred by ecological and oceanographic drivers (Selkoe et al. 2010). As the geographic distance and salinity is confounded in this study, the effect of geographic distance on genetic differentiation cannot be entirely disentangled from the effect of salinity. However, experiments conducted within this study support the conclusion that salinity may be an important driver of genetic differentiation. Genetic population structure of eukaryotic phytoplankton in the Baltic Sea has previously been studied in Alexandrium ostenfeldii (Tahvanainen et al. 2012). This dinoflagellate has genetically differentiated populations from south to north, indicating impeded gene flow across the Baltic Proper. A similar pattern of reduced gene flow between northern and southern local populations in the Baltic Sea was also found in our study (Fig. S2) and has likewise been shown for, for example, Baltic herring (Jørgensen et al. 2005), suggesting that it reflects a general pattern.
Local salinity adaptation
Recently, Dupont et al. (2014) showed that salinity shifts in the Baltic Sea area create diverging taxonomic units of bacteria and that community composition is mainly determined by subtle differences in metabolic pathways, enabling microbes to inhabit different salinity regimes. Salinity and temperature are critical aspects in the marginal habitat of the Baltic Sea, setting the limits for the distribution of various species (Bonsdorff & Pearson 1999). The phenotypic fitness of S. marinoi strains has been shown to vary with different salinity regimes (Saravanan & Godhe 2010). In accordance with earlier observations, where Baltic Sea strains of S. marinoi grew between 2.5 and 35 psu (Balzano et al. 2011), we expected a wide salinity tolerance range. We also expected subtle local adaptation across the salinity gradient, according to the results of Dupont et al. (2014).
All the tested individuals grew in 3–35 psu, but maximum growth rates were displayed in different salinity regimes. These results conform well to earlier comparisons of, for example, species of red algae in the same area. Populations of Delesseria sanguinea and Membranoptera alata from the North Sea and the Baltic Sea have a wide salinity tolerance, but display ecotypic differences in salinity optima (Rietema 1993). We found support for local adaptation as populations originated from a low-salinity area exhibited a peak shaped reaction norm and the population originating outside the Baltic Sea showed an inverse sigmoid reaction norm with increasing salinity. Our results suggest that the exchange of genetic material between the Baltic Sea and the North Sea is restricted enough to create locally adapted populations. Within the next century, salinity is predicted to decrease significantly in the Baltic basin (Meier et al. 2012), further enhancing the salinity difference across the Danish Straits. This may lead to an even stronger genetic cline across the Danish Straits. The success of the locally adapted populations in neighbouring areas is likely to be hampered in concurrence with the theory of ‘selection against immigrants’ (Hendry 2004). This phenomenon would be intensified by the predicted increased salinity difference across the Danish Straits.
Oceanographic connectivity and migration
Our results conform to the findings by Godhe et al. (2013) who showed that genetic differentiation of S. marinoi populations is in part driven by oceanographic connectivity. Specifically, our study suggests that relative migration was higher within the subareas than between, indicating that the dramatic reduction in oceanographic connectivity at the entrance of the Baltic Sea had an effect on the dispersal between the two areas. Interestingly, the only cases in which the relative migration was significantly asymmetric were between RO-YS and VI-YS. In both cases, the sites are located at each side of the Danish Straits. The relative migration was significantly higher from the Baltic Sea out to the North Sea than that in the other direction following the direction of surface currents (Leppèaranta & Myrberg 2009). The dramatic reduction in oceanographic connectivity at the entrance of the Baltic Sea emphasizes its effect on potential dispersal.
The asymmetric pattern of gene flow also suggests that the Baltic Sea S. marinoi population is not an unequivocal demographic sink, which by definition marginal populations are. Typically, marginal populations are geographically situated at the outskirts of a central population and exhibit lower genetic diversity because of nonoptimal environmental conditions, endurable by only a few genotypes (Kawecki 2008). Marginal populations function as demographic sinks where the direction of gene flow mainly radiates from the central (source) population. The central population is ideally adapted to its environment, expressing a high level of genetic diversity and feeds the marginal populations with poorly adapted migrants (Kawecki & Holt 2002). Our demonstrated directional relative migration network does not conform to the classical source–sink model with a poorly adapted and genetically impoverished marginal population because we observe a strong gene flow out of the Baltic Sea. This demonstrates an example where asymmetric dispersal patterns imposed by environmental conditions may reverse the source–sink structure and promote niche evolution in marginal habitats favouring adaptation to the less suitable environment. This has been shown in theory by Kawecki & Holt (2002) who demonstrated that adaptation to a new habitat is favoured when the extent of gene flow is higher towards the presupposed central population. Our results suggest that oceanographic connectivity reinforces a reversed source–sink structure, promoting adaptation of the Baltic population to low salinity. The probability of successful adaptation increases with increasing levels of genetic diversity in the adapting population (Kawecki 2008). Therefore, despite the moderate level of genetic diversity within the local Baltic Sea populations, our results indicate that S. marinoi has adapted to the low-salinity conditions.
Conclusions
Genetic diversity of S. marinoi was reduced in the low-salinity habitat within the Baltic Sea compared to high-salinity areas in the NE Atlantic. Population structure coincided with the most evident dispersal barrier in the area, the steep salinity shift and respective local salinity adaptation. The pattern of differentiation was supported by the extent and direction of migration in our data set, which further correlated partly with oceanographic connectivity. The aspect of potential local salinity adaptation in our populations is therefore supported by both genetic and experimental data. Our study also exemplifies possible local adaptation in a marginal habitat facilitated by asymmetric gene flow with the central population. Finally, our results increase the understanding of phytoplankton metapopulation dynamics in an area faced by a strong environmental gradient and rapid environmental change. Indications of local salinity adaptation may be an important aspect in understanding the observed population structure. From an evolutionary perspective, preference for distinctive salinity regimes among genetically differentiated populations indicates the capacity of adaptation in one of the most important primary producers in the area.
Acknowledgments
This study was funded by the Academy of Finland Grant # 256074 to CS and AK and by a Linnaeus grant from the Swedish Research Councils, VR and Formas (2010-751) (AG, PJ). Financial support was also received from Walter and Andrée de Nottbeck Foundation (CS) and NordForsk – 44881 (CS, AK). The fragment analysis was performed at the Genomics Core Facility, University of Gothenburg, by Dr Elham Rekabdar. We would also like to thank Matti Ollikainen for support with laboratory analyses and Anna Palmbo, Alf Norkko, Kenneth Sjöqvist and Justyna Kobos for assistance with sediment collection.
Data accessibility
Microsatellite sequences: GenBank Accession nos EU855763, EU855769-EU855771, EU855775, EU855777, GQ250935, GQ250937. The Skeletonema marinoi strains are available from Gothenburg University's Marine Algal Culture Collection (GUMACC) and assessed through http://assemblemarine.org/the-sven-lov-n-centre-for-marinesciences-tj-rn/ and from the Algal Culture collection at the Finnish Environment Institute. Sampling locations, growth rate data, oceanographic connectivity matrices, microsatellite genotypes and the directional relative migration matrix are publicly available at Dryad (doi:10.5061/dryad.j5cf0).
Supporting information
Additional supporting information may be found in the online version of this article.
Table S1 Summary of N (sample size), Na (number of alleles per population per loci), expected (He) and observed (Ho) heterozygosity and Brookfield null allele frequencies (a) in all populations across eight genotyped loci. Heterozygote deficiency after Bonferroni correction, P < 0.05*.
Table S2 Correlation between directional relative migration and oceanographic connectivity was also tested over multiple generations based on stepping stone dispersal.
Fig. S1 Oceanographic connectivity measured as the minimum connectivity over 16 generations and visualized as the connectivity between Lysekil (North Sea) and to all other stations.
Fig. S2 structure plot for the Baltic Sea only (K = 3) suggesting southern populations (GK, YS) are genetically differentiated from northern populations.
References
- Almany GR, De Arruda MP, Arthofer W, et al. Permanent genetic resources added to molecular ecology resources database 1 May 2009–31 July 2009. Molecular Ecology Resources. 2009;9:1460–1466. doi: 10.1111/j.1755-0998.2009.02759.x. [DOI] [PubMed] [Google Scholar]
- Alpermann TJ, Beszteri B, John U, Tillmann U, Cembella AD. Implications of life-history transitions on the population genetic structure of the toxigenic marine dinoflagellate Alexandrium tamarense. Molecular Ecology. 2009;18:2122–2133. doi: 10.1111/j.1365-294X.2009.04165.x. [DOI] [PubMed] [Google Scholar]
- Axe P. 2010. Baltic Sea environment fact sheet. HELCOM Baltic Sea Environment Fact Sheets. Online. 17.9.2014, http://www.helcom.fi/baltic-sea-trends/environment-fact-sheets/
- Balloux F, Lehmann L, de Meeus T. The population genetics of clonal and partially clonal diploids. Genetics. 2003;164:1635–1644. doi: 10.1093/genetics/164.4.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzano S, Sarno D, Kooistra WH. Effects of salinity on the growth rate and morphology of ten Skeletonema strains. Journal of Plankton Research. 2011;33:937–945. [Google Scholar]
- Bell G. The ecology and genetics of fitness in Chlamydomonas. IV. The properties of mixtures of genotypes of the same species. Evolution. 1991;45:1036–1046. doi: 10.1111/j.1558-5646.1991.tb04368.x. [DOI] [PubMed] [Google Scholar]
- Björck S. A review of the history of the Baltic Sea, 13.0–8.0 ka BP. Quaternary International. 1995;27:19–40. [Google Scholar]
- Blanquart F, Kaltz O, Nuismer SL, Gandon S. A practical guide to measuring local adaptation. Ecology Letters. 2013;16:1195–1205. doi: 10.1111/ele.12150. [DOI] [PubMed] [Google Scholar]
- Bonsdorff E, Pearson TH. Variation in the sublittoral macrozoobenthos of the Baltic Sea along environmental gradients: a functional-group approach. Australian Journal of Ecology. 1999;24:312–326. [Google Scholar]
- Brookfield J. A simple new method for estimating null allele frequency from heterozygote deficiency. Molecular Ecology. 1996;5:453–455. doi: 10.1111/j.1365-294x.1996.tb00336.x. [DOI] [PubMed] [Google Scholar]
- Casabianca S, Penna A, Pecchioli E, Jordi A, Basterretxea G, Vernesi C. Population genetic structure and connectivity of the harmful dinoflagellate Alexandrium minutum in the Mediterranean Sea. Proceedings of the Royal Society B: Biological Sciences. 2012;279:129–138. doi: 10.1098/rspb.2011.0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteleyn G, Leliaert F, Backeljau T, et al. Limits to gene flow in a cosmopolitan marine planktonic diatom. Proceedings of the National Academy of Sciences. 2010;107:12952–12957. doi: 10.1073/pnas.1001380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chepurnov VA, Mann DG, Sabbe K, Vyverman W. Experimental studies on sexual reproduction in diatoms. International Review of Cytology. 2004;237:91–154. doi: 10.1016/S0074-7696(04)37003-8. [DOI] [PubMed] [Google Scholar]
- Crawford NG. SMOGD: software for the measurement of genetic diversity. Molecular Ecology Resources. 2010;10:556–557. doi: 10.1111/j.1755-0998.2009.02801.x. [DOI] [PubMed] [Google Scholar]
- Crutsinger GM, Souza L, Sanders NJ. Intraspecific diversity and dominant genotypes resist plant invasions. Ecology Letters. 2008;11:16–23. doi: 10.1111/j.1461-0248.2007.01118.x. [DOI] [PubMed] [Google Scholar]
- Döös K. Interocean exchange of water masses. Journal of Geophysical Research: Oceans (1978–2012) 1995;100:13499–13514. [Google Scholar]
- Dupont CL, Larsson J, Yooseph S, et al. Functional tradeoffs underpin salinity-driven divergence in microbial community composition. PLoS ONE. 2014;9:e89549. doi: 10.1371/journal.pone.0089549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlin U. Hydrology of the baltic Sea. The Baltic Sea. 1981:123–134. (ed. Voipio A). Elsevier, Amsterdam. [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software Structure: a simulation study. Molecular Ecology. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Evans KM, Kühn SF, Hayes PK. High levels of genetic diversity and low levels of genetic differentiation in North Sea Pseudo-Nitzschia pungens (BACILLARIOPHYCEAE) populations. Journal of Phycology. 2005;41:506–514. [Google Scholar]
- Excoffier L, Ray N. Surfing during population expansions promotes genetic revolutions and structuration. Trends in Ecology & Evolution. 2008;23:347–351. doi: 10.1016/j.tree.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay BJ. Global dispersal of free-living microbial eukaryote species. Science. 2002;296:1061–1063. doi: 10.1126/science.1070710. [DOI] [PubMed] [Google Scholar]
- Gallagher J. Population genetics of Skeletonema costatum (BACILLARIOPHYEAE) in Narragansett Bay. Journal of Phycology. 1980;16:464–474. [Google Scholar]
- Gamfeldt L, Wallén J, Jonsson PR, Berntsson KM, Havenhand JN. Increasing intraspecific diversity enhances settling success in a marine invertebrate. Ecology. 2005;86:3219–3224. [Google Scholar]
- Godhe A, Härnström K. Linking the planktonic and benthic habitat: genetic structure of the marine diatom Skeletonema marinoi. Molecular Ecology. 2010;19:4478–4490. doi: 10.1111/j.1365-294X.2010.04841.x. [DOI] [PubMed] [Google Scholar]
- Godhe A, Kremp A, Montresor M. Genetic and microscopic evidence for sexual reproduction in the centric diatom Skeletonema marinoi. Protist. 2014;165:401–416. doi: 10.1016/j.protis.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Godhe A, Egardt J, Kleinhans D, Sundqvist L, Hordoir R, Jonsson PR. Seascape analysis reveals regional gene flow patterns among populations of a marine planktonic diatom. Proceedings. Biological sciences/The Royal Society. 2013;280:20131599. doi: 10.1098/rspb.2013.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet J. FSTAT (version 1.2): a computer program to calculate F-statistics. Journal of Heredity. 1995;86:485–486. [Google Scholar]
- Guillard RR. Culture of phytoplankton for feeding marine invertebrates. In: Smith W, Chanley M, editors. Culture of Marine Invertebrate Animals. New York: Plenum Press; 1975. pp. 29–60. [Google Scholar]
- Halkett F, Simon J, Balloux F. Tackling the population genetics of clonal and partially clonal organisms. Trends in Ecology & Evolution. 2005;20:194–201. doi: 10.1016/j.tree.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Hendry AP. Selection against migrants contributes to the rapid evolution of ecologically dependent reproductive isolation. Evolutionary Ecology Research. 2004;6:1219–1236. [Google Scholar]
- Holmborn T, Goetze E, Põllupüü M, Põllumäe A. Genetic species identification and low genetic diversity in Pseudocalanus acuspes of the Baltic Sea. Journal of Plankton Research. 2011;33:507–515. [Google Scholar]
- Hordoir R, Dieterich C, Basu C, Dietze H, Meier H. Freshwater outflow of the Baltic Sea and transport in the Norwegian current: a statistical correlation analysis based on a numerical experiment. Continental Shelf Research. 2013;64:1–9. [Google Scholar]
- Hughes AR, Inouye BD, Johnson MT, Underwood N, Vellend M. Ecological consequences of genetic diversity. Ecology Letters. 2008;11:609–623. doi: 10.1111/j.1461-0248.2008.01179.x. [DOI] [PubMed] [Google Scholar]
- Härnström K, Ellegaard M, Andersen TJ, Godhe A. Hundred years of genetic structure in a sediment revived diatom population. Proceedings of the National Academy of Sciences. 2011;108:4252–4257. doi: 10.1073/pnas.1013528108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICES. 2004. Report of the Regional Ecosystem Study Group for the North Sea, 5–7 April 2004. ICES CM 2004/ACE:06.
- Jochem F. Distribution and importance of autotrophic ultraplankton in a boreal inshore area(Kiel Bight, Western Baltic) Marine Ecology Progress Series. Oldendorf. 1989;53:153–168. [Google Scholar]
- Johannesson K, Andre C. INVITED REVIEW: life on the margin: genetic isolation and diversity loss in a peripheral marine ecosystem, the Baltic Sea. Molecular Ecology. 2006;15:2013–2029. doi: 10.1111/j.1365-294X.2006.02919.x. [DOI] [PubMed] [Google Scholar]
- Jørgensen HB, Hansen MM, Bekkevold D, Ruzzante DE, Loeschcke V. Marine landscapes and population genetic structure of herring (Clupea harengus L.) in the Baltic Sea. Molecular Ecology. 2005;14:3219–3234. doi: 10.1111/j.1365-294X.2005.02658.x. [DOI] [PubMed] [Google Scholar]
- Jost L. GST and its relatives do not measure differentiation. Molecular Ecology. 2008;17:4015–4026. doi: 10.1111/j.1365-294x.2008.03887.x. [DOI] [PubMed] [Google Scholar]
- Kalinowski ST. hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Molecular Ecology Notes. 2005;5:187–189. [Google Scholar]
- Kawecki TJ. Adaptation to marginal habitats. Annual Review of Ecology, Evolution, and Systematics. 2008;39:321–342. [Google Scholar]
- Kawecki TJ, Holt RD. Evolutionary consequences of asymmetric dispersal rates. The American Naturalist. 2002;160:333–347. doi: 10.1086/341519. [DOI] [PubMed] [Google Scholar]
- Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA. diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods in Ecology and Evolution. 2013;4:782–788. [Google Scholar]
- Kooistra WH, De Stefano M, Mann DG, Salma N, Medlin LK. Phylogenetic position of Toxarium, a pennate-like lineage within centric diatoms (BACILLARIOPHYCEAE) Journal of Phycology. 2003;39:185–197. [Google Scholar]
- Kyle C, Boulding E. Comparative population genetic structure of marine gastropods (Littorina spp.) with and without pelagic larval dispersal. Marine Biology. 2000;137:835–845. [Google Scholar]
- Leppèaranta M, Myrberg K. Physical oceanography of the Baltic Sea. Chichester: Springer; 2009. [Google Scholar]
- Madec G. 2010. Nemo ocean engine, version 3.3, Tech. rep http://www.nemo-ocean.eu/
- Marshall D, Monro K, Bode M, Keough M, Swearer S. Phenotype–environment mismatches reduce connectivity in the sea. Ecology Letters. 2010;13:128–140. doi: 10.1111/j.1461-0248.2009.01408.x. [DOI] [PubMed] [Google Scholar]
- McQuoid MR. Pelagic and benthic environmental controls on the spatial distribution of a viable diatom propagule bank on the Swedish west coast. Journal of Phycology. 2002;38:881–893. [Google Scholar]
- Meier HM, Andersson HC, Arheimer B, et al. Comparing reconstructed past variations and future projections of the Baltic Sea ecosystem—first results from multi-model ensemble simulations. Environmental Research Letters. 2012;7:034005. [Google Scholar]
- Nagai S, Lian C, Yamaguchi S, et al. Microsatellite markers reveal population genetic structure of the dinoflagellate Alexandrium tamarense (DINOPHYCEAE) in Japanese coastal waters. Journal of Phycology. 2007;43:43–54. [Google Scholar]
- Nanninga GB, Saenz-Agudelo P, Manica A, Berumen ML. Environmental gradients predict the genetic population structure of a coral reef fish in the Red Sea. Molecular Ecology. 2014;23:591–602. doi: 10.1111/mec.12623. [DOI] [PubMed] [Google Scholar]
- Nilsson Jacobi M, André C, Döös K, Jonsson PR. Identification of subpopulations from connectivity matrices. Ecography. 2012;35:1004–1016. [Google Scholar]
- Nosil P, Funk DJ, Ortiz-Barrientos D. Divergent selection and heterogeneous genomic divergence. Molecular Ecology. 2009;18:375–402. doi: 10.1111/j.1365-294X.2008.03946.x. [DOI] [PubMed] [Google Scholar]
- Olden JD, LeRoy Poff N, Douglas MR, Douglas ME, Fausch KD. Ecological and evolutionary consequences of biotic homogenization. Trends in Ecology & Evolution. 2004;19:18–24. doi: 10.1016/j.tree.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Park SDE. 2001. Trypanotolerance in West African cattle and the population genetic effects of selection. Ph.D. thesis, University of Dublin.
- Pereyra RT, Bergström L, Kautsky L, Johannesson K. Rapid speciation in a newly opened postglacial marine environment, the Baltic Sea. BMC Evolutionary Biology. 2009;9:70. doi: 10.1186/1471-2148-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptacnik R, Solimini AG, Andersen T, et al. Diversity predicts stability and resource use efficiency in natural phytoplankton communities. Proceedings of the National Academy of Sciences. 2008;105:5134–5138. doi: 10.1073/pnas.0708328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QGIS. Quantum GIS Version 2.4.0. Open Source Geospatial Foundation Project. 2011. http://www.qgis.org/
- Raymond M, Rousset F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. Journal of Heredity. 1995;86:248–249. [Google Scholar]
- Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x. [DOI] [PubMed] [Google Scholar]
- Rietema H. Ecotypic differences in Baltic and North Sea populations of Delesseria sanguinea and Membranoptera alata. Botanica Marina. 1993;36:15–22. [Google Scholar]
- Rousset F. Genetic differentiation between individuals. Journal of Evolutionary Biology. 2000;13:58–62. [Google Scholar]
- Rynearson TA, Armbrust E. DNA fingerprinting reveals extensive genetic diversity in a field population of the centric diatom Ditylum brightwellii. Limnology and Oceanography. 2000;45:1329–1340. [Google Scholar]
- Rynearson TA, Armbrust E. Genetic differentiation among populations of the planktonic marine diatom ditylum brightwellii (BACILLARIOPHYCEAE) Journal of Phycology. 2004;40:34–43. [Google Scholar]
- Saravanan V, Godhe A. Genetic heterogeneity and physiological variation among seasonally separated clones of Skeletonema marinoi (BACILLARIOPHYCEAE) in the Gullmar Fjord, Sweden. European Journal of Phycology. 2010;45:177–190. [Google Scholar]
- Selkoe KA, Watson JR, White C, et al. Taking the chaos out of genetic patchiness: seascape genetics reveals ecological and oceanographic drivers of genetic patterns in three temperate reef species. Molecular Ecology. 2010;19:3708–3726. doi: 10.1111/j.1365-294X.2010.04658.x. [DOI] [PubMed] [Google Scholar]
- Sinnock P. The Wahlund effect for the two-locus model. American Naturalist. 1975;109:565–570. [Google Scholar]
- Sundqvist L, Zackrisson M, Kleinhaus D. Directional genetic differentiation and asymmetric migration. Journal of Theoretical Biology. 2013 http://arxiv.org/abs/1304.0118. [Google Scholar]
- Tahvanainen P, Alpermann TJ, Figueroa RI, et al. Patterns of post-glacial genetic differentiation in marginal populations of a marine microalga. PLoS ONE. 2012;7:e53602. doi: 10.1371/journal.pone.0053602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatarenkov A, Bergström L, Jönsson RB, Serrão EA, Kautsky L, Johannesson K. Intriguing asexual life in marginal populations of the brown seaweed Fucus vesiculosus. Molecular Ecology. 2005;14:647–651. doi: 10.1111/j.1365-294X.2005.02425.x. [DOI] [PubMed] [Google Scholar]
- Teacher AG, André C, Jonsson PR, Merilä J. Oceanographic connectivity and environmental correlates of genetic structuring in Atlantic herring in the Baltic Sea. Evolutionary Applications. 2013;6:549–567. doi: 10.1111/eva.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väinölä R, Hvilsom M. Genetic divergence and a hybrid zone between Baltic and North Sea Mytilus populations (Mytilidae: Mollusca) Biological Journal of the Linnean Society. 1991;43:127–148. [Google Scholar]
- Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes. 2004;4:535–538. [Google Scholar]
- Wasmund N, Nausch G, Matthäus W. Phytoplankton spring blooms in the southern Baltic Sea—spatio-temporal development and long-term trends. Journal of Plankton Research. 1998;20:1099–1117. [Google Scholar]
- Wennerström L, Laikre L, Ryman N, et al. Genetic biodiversity in the Baltic Sea: species-specific patterns challenge management. Biodiversity and Conservation. 2013;22:3045–3065. [Google Scholar]
- White C, Selkoe KA, Watson J, Siegel DA, Zacherl DC, Toonen RJ. Ocean currents help explain population genetic structure. Proceedings of the Royal Society B: Biological Sciences. 2010;277:1685–1694. doi: 10.1098/rspb.2009.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AM, Everroad RC, Wingard LM. Measuring growth rates in microalgal cultures. In: Anderson RA, editor. Algal culturing techniques. Burlington, Massachusetts: Elsevier Academic Press; 2005. pp. 269–286. [Google Scholar]
- Wright S. 1968. Evolution and the genetics of populations. Vol. 1. Genetic and biometrie foundations.
- Wright S. Isolation by distance. Genetics. 1943;28:114. doi: 10.1093/genetics/28.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zettler ML, Schiedek D, Bobertz B. Benthic biodiversity indices versus salinity gradient in the southern Baltic Sea. Marine Pollution Bulletin. 2007;55:258–270. doi: 10.1016/j.marpolbul.2006.08.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Summary of N (sample size), Na (number of alleles per population per loci), expected (He) and observed (Ho) heterozygosity and Brookfield null allele frequencies (a) in all populations across eight genotyped loci. Heterozygote deficiency after Bonferroni correction, P < 0.05*.
Table S2 Correlation between directional relative migration and oceanographic connectivity was also tested over multiple generations based on stepping stone dispersal.
Fig. S1 Oceanographic connectivity measured as the minimum connectivity over 16 generations and visualized as the connectivity between Lysekil (North Sea) and to all other stations.
Fig. S2 structure plot for the Baltic Sea only (K = 3) suggesting southern populations (GK, YS) are genetically differentiated from northern populations.
Data Availability Statement
Microsatellite sequences: GenBank Accession nos EU855763, EU855769-EU855771, EU855775, EU855777, GQ250935, GQ250937. The Skeletonema marinoi strains are available from Gothenburg University's Marine Algal Culture Collection (GUMACC) and assessed through http://assemblemarine.org/the-sven-lov-n-centre-for-marinesciences-tj-rn/ and from the Algal Culture collection at the Finnish Environment Institute. Sampling locations, growth rate data, oceanographic connectivity matrices, microsatellite genotypes and the directional relative migration matrix are publicly available at Dryad (doi:10.5061/dryad.j5cf0).