

Abstract
Stroke affects millions of people worldwide every year. Despite this prevalence, mechanisms of long-term injury and repair within the ischemic brain are still understudied. Sterile inflammation occurs in the injured brain after stroke, with damaged tissue exposing central nervous system (CNS)-derived antigen that could initiate potential autoimmune responses. We used a standard immunology-based recall response assay for murine immune cells, isolated from the cervical lymph nodes and spleen after transient stroke, to determine if stroke induces autoreactivity to CNS target antigens. Our assays included novel neuronal peptides, in addition to myelin-, nuclear-, glial-, and endothelial-derived peptides. Autoimmune responses to an antigen were considered positive based on proliferation and activation over non-stimulated conditions. Stroke induced a significant increase in autoreactive CD4+ and CD8+ T cells, as well as autoreactive CD19+ B cells, as early as 4 days after stroke onset. Mice with large infarct volumes exhibited early T and B cell autoreactivity to NR2A, an NMDA receptor subunit, in cells isolated from lymph nodes but not spleen. Mice with small infarct volumes exhibited high autoreactivity to MAP2, a dendritic cytoskeletal protein, as well as myelin-derived peptides. This autoimmunity was maintained through 10 days post-stroke in both lymph nodes and spleen for all lymphocyte subsets. Sham surgery also induced early autoreactive B cell responses to MAP2 and myelin. Based on these observations, we hypothesize that stroke induces a secondary, complex, and dynamic autoimmune response to neuronal antigens with the potential to potentiate, or perhaps even ameliorate, long-term neuroinflammation.
Introduction
Stroke is caused by a loss of blood flow to the brain, which can result in permanent neurological damage. Worldwide, approximately 15 million individuals will suffer a stroke each year, with 800,000 in the U.S. alone (Go et al., 2013). At present, there is only one pharmacological agent, tissueplasminogenactivator, approved for treating ischemicstroke in the acute 4-hour window after onset (Wardlaw et al., 2012). While advances in experimental animal models have identified a substantial number of other promising neurotherapeutics, none have proved efficacious in clinical trials (Kidd, 2009). This suggests the need for a fundamental paradigm shift in stroke research to focus on long-term mechanisms of injury and repair in order to extend potential interventions beyond the acute window. To this end, both preclinical and clinical evidence shows that there is a complex and dynamic interplay between the post-stroke CNS and the immune system that potentially influences stroke pathology for weeks to months after injury onset (Chamorro et al., 2012; Doyle et al., 2015; Planas et al., 2012b; Sardi et al., 2011).
Animal stroke models reveal that circulating leukocytes diapedese into the brain and accumulate at the site of injury within hours after stroke, with various subsets of leukocytes either exacerbating or ameliorating injury (Chu et al., 2014; Gelderblom et al., 2009; Kipnis et al., 2002). B cells, T cells, and natural killer T cells (NK-T) cells comprise the adaptive arm of the immune system, with their interactions initiated by recognition of antigen (see reviews Iadecola and Anrather, 2011; Jin et al., 2010; Sardi et al., 2011). In response to activation, these lymphocytes secrete cytokines, chemokines, and antibodies, in addition to directly binding target cells, to promote an active adaptive immune response. CD4 T cells (i.e., T helper cells) are activated when peptide antigens are presented on the surface of major histocompatibility class (MHC) II antigen presenting cells, such as macrophages, dendritic cells, or B cells. CD4 T cells are robustly activated after stroke, leading to an induction of an effector population that contributes to post-stroke pathology (Chamorro et al., 2012; Kleinschnitz et al., 2010; Lazar-Molnar and Tebo, 2015; Yilmaz et al., 2006). CD8 T cells (i.e., cytotoxic T cells) respond to antigen peptides on MHC I molecules, which are present on most cell types, and are also actively involved in post-stroke inflammation (Gelderblom et al., 2009). Membrane-bound immunoglobulin allows B cells to bind antigens with great specificity and, if activated by cytokines from CD4 T cells, to secrete antibodies specific to a particular antigen. It is a common view that unlike T cells, B cells do not contribute to stroke pathology (Chamorro et al., 2012; Kleinschnitz et al., 2010; Yilmaz et al., 2006), though they do have the potential to contribute to neuroprotection and post-stroke repair (Bodhankar et al., 2013; Monson et al., 2014b).
Most animal studies of CNS-antigenic specificity after stroke have focused on myelin antigens, common targets during demyelinating diseases like multiplesclerosis (Hussain et al., 2014). In our study, using a common form of ischemic stroke injury, we sought to determine if there was an early adaptive autoimmune response to myelin-derived antigens (e.g., myelinbasicprotein; MBP), but also novel neuronally-derived antigens [e.g., microtubule-associated protein 2 (MAP2) and N-methyl-D-aspartate (NMDA) receptor subunit NR2A]. In total, we investigated autoreactive responses to myelin, neuronal, glial, and endothelial peptides, as well as responses to nuclear receptor subfamily 2, group F, member 2 (NR2F), a protein that is critical for neuronal and vascular development (Kim et al., 2009; Lin et al., 2011) and found in the adult mouse brain (Fuentealba et al., 2010). We also determined if the level of braininjury influenced the breadth and scope of antigen-specific responses. Our detailed study of the adaptive immune response following stroke, and in particular the novel rapid response we found even in mice exhibiting small infarct volumes, should be foundational for future studies focusing on neuronal autoimmunity in the injured CNS. Understanding the progression of neuronally-derived autoimmune responses — and whether these responses may contribute to long-term mechanisms of recovery — will aid the development of more effective and clinically translatable immunotherapeutic strategies for the extended treatment of stroke.
Experimental Methods and Design
Transient middle cerebral artery occlusion (tMCAo)
All experiments were approved by the University of Texas Southwestern Medical Institutional Animal Care and Use Committee. Outbred Swiss Webster male mice, 6-8 weeks, were maintained on a 12-hour light cycle, and allowed food and water ad libitum. All experimental time points were replicated on a minimum of two separate days. Mice were initially randomized to either sham or tMCAo groups and researcher-blinded for the remainder of the study. As previously described (Monson et al., 2014b), mice were anesthetized (2% isoflurane/70% NO2/30% O2) at 37°C and the common carotid artery (CCA) permanently ligated. Transcranial laser doppler flowmetry assessed baseline cerebral blood flow (CBF) in the middle cerebral artery (MCA) prior to insertion of an intraluminal filament though the CCA to the MCA origin. Upon successful occlusion (>80% reduction CBF), mice were placed in an incubator. After 60 minutes, mice were re-anesthetized and the suture removed. Reperfusion (>50% baseline CBF) was confirmed, and mice not meeting the occlusion/reperfusion criteria were removed from the study (n=8 from 38 mice). An additional 4 mice died post-operatively. Sham mice were anesthetized for CCA ligation but without filament insertion (n=19). Mice were monitored during recovery from anesthesia and returned to their home cages. At 4, 8, or 10 days after tMCAo, animals were sacrificed by isoflurane overdose and spleens and cervical lymph nodes harvested for autoreactivity assays. Whole brains were dissected, placed into an acrylic brain matrix, and sectioned at 1 mm intervals. Brain slices were placed into 2% 2,3,5-triphenyl tetrazolium chloride (TTC) solution for approximately 5 minutes. TTC solution was replaced with 4% paraformaldehyde and tissues allowed to fix overnight. Infarct volumes were quantified by a blinded observer using Image J analysis software and corrected for edema on corresponding right hemispheric areas as control (Stowe et al., 2011). Five mice did not undergo TTC analysis during the first experiment, as it was not part of the original experimental design.
Determination of autoreactive responses
Individual cervical lymph nodes and spleens were processed through 70 μm mesh filters and splenic cells purified using Lympholyte-M treatment, as per manufacturer’s instructions (Ortega et al., 2013). Antigen-specific responses were determined using the carboxyfluorescein succinimidyl ester (CFSE; Invitrogen)-based dilution assay, adapted for mice, using bulk splenocytes and cervical lymphnode cells. Cells were resuspended (1×106/mL in 0.1 M PBS) and incubated for 7 minutes (0.18 μM CFSE; 37°C). Cells were washed twice with serum-containing media, resuspended in culturemedia (1×106/mL in RPMI 1640, supplemented with 10% fetal calf serum, L-glutamine, penicillin, streptomycin, HEPES buffer, non-essential amino acids, sodium pyruvate, and β-mercaptoethanol), and cultured with indicated antigens at 37°C and 5% CO2. We determined responses to well characterized myelin peptides, such as myelin oligodendrocyte protein (MOG)35-55, proteolipid protein (PLP)178-191, and MBP(Ac1-11), synthesized by the UT Southwestern Protein Chemistry core (Miller et al., 2001; Stromnes and Goverman, 2006). We also generated novel CNS specific antigens NR2A (UniProt Q12879), MAP2 (P11137), and NR2F (Entrezgene proteins) with overlapping 15-mers peptides synthesized by BioSynthesis and purity qualified by RP-HPLC, with specific peptide sequences shown in Table 1. In order to use cells more efficiently, we used peptides pools. On day 6 of culture, cells were washed with fluorescence activated cell sorting (FACS) buffer (PBS with 1% bovine serum albumin and 0.1% Na-Azide) and stained with fluorescently tagged antibodies against TCRβ-BV510 (Biolegend), CD4-v450, CD19-PE, CD8-APC, and CD25-PCY7 (all Tonbo) for 30 minutes at 4°C in the dark. Cells were washed and fixed (1% paraformaldehyde). Flow cytometric data (100,000 lymphocyte-gated events) were acquired on a BD FACS Canto flow cytometer (UT Southwestern Children’s Research Institute FlowCytometry Facility) using BD FACS Diva software v8.0. A response was considered positive when the delta proliferation fraction (ΔPF, test condition − non-stimulated condition) exceeded 1% and the stimulation index (SI, test condition/non-stimulated condition) was greater than 2 (Crawford et al., 2004; Ortega et al., 2013).
Table 1.
Peptides Used in CFSE Proliferation Study.
| Name | Peptide Pool ID |
Peptide ID & Sequences | CD4 CLN |
CD4 SPL |
CDS CLN |
CDS SPL |
CD19 CLN |
CD19 SPL |
|
|---|---|---|---|---|---|---|---|---|---|
| MAP | MAP-1 | MADERKDEAKAPHWT TEASAHSHPPEIKDQ |
APHWDTSAPLTEASA EIKDQGGAGEGLVRS |
+ | + | + | + | + | + |
| MAP-2 | GLVRSANGFPYREDE EHGSQGTYSNTKENG |
YREDEEGAFGEHGSQ TKENGINGELTSADR |
+ | + | + | + | + | + | |
| MAP-3 | TSADRETAEEVLKGE QHKDQTAALPLAAEE |
VLKGEQEKEAQHKDQ LAAEETANLPPSPPP |
− | − | − | + | + | + | |
| MAP-4 | PSPPPSPASEQTVTV GESALAPSVFKQAKD |
QTVTVEEAAGGESAL KQAKDKVSDGVTKSP |
+ | − | + | + | + | + | |
| MAP-5 | VTKSPEKRSSLPRPS RRGVSGDRDENSFSL |
LPRPSSILPPRRGVS NSFSLNSSISSSARR |
− | + | − | + | − | + | |
| MAP-6 | SSARRTTRSEPIRRA STPTTPGSTAITPGT |
PIRRAGKSGTSTPTT ITPGTPPSYSSRTPG |
− | − | − | + | + | + | |
| MAP-7 | SRTPGTPGTPSYPRT TPKSAILVPSEKKVA |
SYPRTPHTPGTPKSA EKKVAIIRTPPKSPA |
− | + | − | + | − | + | |
| MAP-8 | PKSPATPKQLRLINQ KNVKSKIGSTDNIKY |
RLINQPLPDLKNVKS DNIKYQPKGGQVQIV |
− | + | − | − | − | + | |
| MAP-9 | QVQIVTKKIDLSHVT LKNIRHRPGGGRVKI |
LSHVTSKCGSLKNIR GRVKIESVKLDFKEK |
− | − | − | − | − | + | |
| MAP-10 | DFKEKAQAKVGSLDN GGGNVKIDSQKLNFR |
GSLDNAHHVPGGGNV KLNFREHAKARVDHG |
− | − | − | + | − | + | |
| MAP-11 | RVDHGAEIITQSPGR PRRLSNVSSSGSINL |
QSPGRSSVASPRRLS GSINLLESPQLATLA |
− | + | − | + | − | + | |
| MAP-12 | LATLAEDVTAALAKQGL | − | + | − | − | − | + | ||
| NR2F | NR2F-1 | RMPPTQPGQFALT | − | − | − | − | − | + | |
| NR2F-2 | QFALTWGDPLNCKSY | − | + | − | + | + | + | ||
| NR2F-3 | NCHSYLSGYISLLLR | + | − | + | − | + | + | ||
| NR2F-4 | SLLLRAEPYPTSRF | − | − | + | + | + | − | ||
| NR2F-5 | PTSRFGSQCMQPNNI | − | + | + | + | − | + | ||
| NR2F-6 | QPNNIMGIEWICELA | − | + | − | + | − | + | ||
| NR2F-7 | ICELAARMLFSAVEW | − | + | − | + | + | + | ||
| NR2F-8 | SAVEWARNIPFFPDL | + | − | − | − | + | + | ||
| NR2F-9 | ARHIPFFPDLQITD | − | − | − | + | − | + | ||
| NMDA receptor subtype NR2A | NR2A-1 | NRTDPKSLITHVCDL CDLMSGARIHGLVFG |
− | + | − | − | − | + | |
| NR2F-2 | VFGDDTDQEAVAQML QMLDFTSSQTFIPIL |
+ | − | + | − | − | + | ||
| NR2A-3 | PIIGIHGGASMIMAD MADKDPTSTFFQFGA |
+ | − | − | − | + | − | ||
| NR2A-4 | GASIQQQATVMLKIM KIMQDYDWHVFSLVT |
+ | − | + | − | + | − | ||
| NR2A-5 | LVTTIFPGYRDFISF ISFIKTTVDNSFVGW |
+ | − | + | − | + | − | ||
| NR2A-6 | VGWDMQNVITLDTSF TSFEDAKTQVQLKKI |
+ | − | + | − | + | − | ||
| NR2A-7 | KKIHSSVILLYCSKD SKDEAVLILSEARSL |
+ | − | + | − | + | − | ||
| NR2A-8 | RSLGLTGYDFFWIVP IVPSLVSGNTELIPK |
+ | − | + | − | + | − | ||
| NR2A-9 | IPKEFPSGLISVSYD SYDDWDYSLEARVRD |
+ | − | + | − | − | − | ||
| NR2A-10 | VRDGLGILTTAASSM SSMLEKFSYIPEAKA |
− | − | + | − | + | − | ||
| NR2A-11 | AKASCYGQTE | − | − | − | − | + | − | ||
| Myelin oligodendrocyte glycoprotein | MOG35-55 | MEVGWYRSPFSRWHLYRNGK | + | + | + | + | + | + | |
| Proteolipid lipoprotein | PLP178-191 | NTKTTCQSIAFPSK | + | + | − | + | + | + | |
| Myelin basic protein | MBP Ac(l-11) | ASQKRPSQRSK | + | + | + | + | + | + | |
| von Willebrand factor | vWF | Not disclosed by vendor | − | + | + | + | + | + | |
| Cilia fibrillaiy acidic protein | GFAP | GEVIKESKQEHKDVM | + | + | − | + | + | + | |
Note: + indicates a positive response, − indicates a negative lesponse.
Statistics
All studies were performed by researchers blinded to experimental conditions. Statistical analyses between groups were performed using Graph Pad Prism 6.0. Between-group differences in ΔPF by day were evaluated using an unpaired two-tailed Student t test. Within group differences across days were evaluated for each group by one-way ANOVA with Tukey post-hoc analysis. All values are shown as mean ± standard deviation (SD) and p<0.05 was considered statistically significant.
Results
An early splenic CD4 T cell autoimmune response to neuronal and myelin antigen is associated with better recovery
The magnitude of stroke-induced, CNS-antigen specific autoimmunity was determined at 4, 8, and 10 days following tMCAo in immune cells isolated from the cervical lymph nodes and spleen (Figure 1). Four days after stroke, we observed a robust global, CNS-specific autoimmune response in CD4 T cells from both the cervical lymph nodes (p<0.0001; Figure 1C) and spleen (p<0.0001; Figure 1D). This early autoimmune response coincided with elevated infarct volumes (38±36 mm3; n=11; p=0.06) compared to sham (10±7 mm3; n=7; data not graphed). It should be noted that the sham CCA ligation still resulted in areas of injury, as identified by TTC. The largest magnitude of autoreactivity for CD4 T cells isolated from the cervical lymph nodes was found 10 days post stroke (Figure 1C). When considering the between-day progression of autoreactivity by one-way ANOVA, day 10 was elevated over both day 4 (p<0.01) and day 8 (p<0.001) levels (F(2,316)=8.19; p<0.001). CD4 T cells from the cervical lymph nodes were minimally activated at day 8 post-stroke. In contrast, autoreactivity peaked at 8 days after stroke in splenic CD4 T cells (p<0.001) with a subsequent decline at day 10 (p<0.01, day 8 vs. day 10; F(2,467)=6.35; p<0.01).
Figure 1.
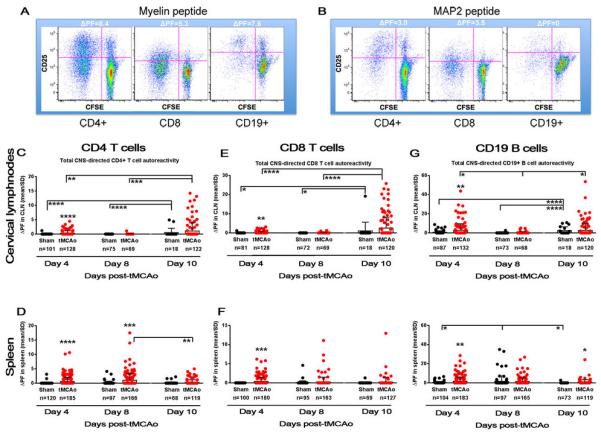
CNS-derived antigen induces autoimmune responses in both B and T cells by four days after stroke. (A-B) Autoreactivity is measured by the change in proliferation fraction (ΔPF) of peptide over non-antigen conditions. Dot plots of individual immune cells following myelin (MOG, left) and MAP2 (right) exposure are shown. CFSE gradient along the x axis (dilution of CFSE intensity from right to left designates proliferation) and CD25 expression on the y axis (increase in CD25 expression from bottom to top designates activation) create the ΔPF. (C-H) The timeframe for autoreactivity (ΔPF; y axis) in both sham populations (black circles) and tMCAo populations (red circles) at 4, 8, and 10 days after tMCAo (x axis) are shown for (C-D) CD4 T cells, (E-F) CD8 T cells, and (G-H) CD19 B cells. Responses from lymphocytes isolated from the cervical lymph nodes (CLN) are shown in the top row, and splenic lymphocyte responses shown in the bottom row of graphs. Values for the number of wells within each group with a measurable response are shown below in parenthesis. Within-day significance is shown as asterisks over each tMCAo group (student's t-test). Between-day significance is shown by bars (one-way ANOVA). Values are mean ± standard deviation (SD). * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Next, we sought to both identify the peptides eliciting the activation, and correlate the responses to the magnitude of infarct volume (Figure 2, Table 1) at 4 days post-stroke. The consistent autoimmune response from CD4 T cells isolated from the cervical lymph nodes included responses to myelin, NR2A (p<0.01), MAP2, and NR2F peptides. Splenic CD4 T cells also responded to myelin (p<0.05), MAP2 (p<0.01), and NR2F, with no response to the NMDAreceptor subunit NR2A. The two animals with the largest infarct volumes exhibited autoimmune responses to NR2A in the cervical lymph nodes, in the complete absence of splenic NR2A autoimmune responses. This suggests a potential activation of T cells by antigen presenting cells migrating from the injured CNS, instead of a blood-borne antigen response subsequent to blood-brain barrier breakdown that could activate splenic populations (Sardi et al., 2011). The specificity of our CNS-derived antigen activation assay was confirmed by the lack of splenic NR2A, splenic, and lymphnode vWF autoimmune responses from these mice. In direct contrast, the most robust myelin (Figure 2B, red circle) and MAP2-derived (Figure 2B, orange circle) autoreactivity occurred in splenic CD4 T cells isolated from animals with the small (2-18 mm3) to moderate (45 mm3) infarct volumes. Surprisingly, these animals, exhibiting the greatest resistance to stroke-induced injury, also showed the highest MAP2 autoreactivity in CD4 T cells from the cervical lymph nodes.
Figure 2.
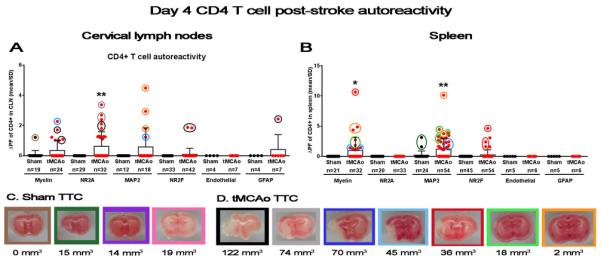
Early splenic CD4 Tcell autoimmune response to neuronal and myelin antigen is associated with smaller infarcts. (A-B) Graphs depict autoreactivity (ΔPF; y axis) for individual peptides in both sham populations (black circles) and tMCAo populations (red circles) at 4 days after tMCAo (x axis). (A) Left graph shows responses from CD4 T cells isolated from the cervical lymph nodes (CLN), and (B) right graph shows splenic CD4 T cell responses. Values for the number of wells within each group with a measurable response are shown below in parenthesis. (C-D) Representative TTC-stained section from each animal in both (C) sham and (D) tMCAo-treated groups. Areas of mitochondrialdysfunction (i.e., infarct volumes) appear white, while healthy tissue stains red. The total infarct volume for each animal is shown below the corresponding pictures (mm3). The border around each infarct is color-coded with the circles around individual data points in the graphs shown in (A, B). Within-day significance is shown as asterisks over each tMCAo group (student's t-test). Values are mean ± standard deviation (SD). * p<0.05, ** p<0.01.
Table 1 shows peptide sequences for MAP2, NR2A, and NR2F that elicited CD4 T cell autoimmune responses. MAP2 is a very large protein, yet both our first and second pool of peptide sequences elicited immunogenic responses from CD4 T cells of the spleen and cervical lymph nodes. Several NR2A sequences also exhibited consistent immunogenicity between compartments. Future studies are needed to verify the location of specific sequence regions on both of these proteins, confirm immunogenicity, and determine if autoreactive responses to MAP2 and NR2A occur in other CNS disease states. Take together, these data show that CD4 T cells specific to CNS antigens are quickly activated, and although brain injury is resolving over the course of 10 days post-tMCAo, there is still an active CNS-specific autoimmune-eliciting signal in the periphery. Furthermore, the majority of the significant response is focused to neuronal and myelin peptides which could be highly oligoclonal in nature, as stroke induces clonal T cell expansion (Liesz et al., 2013), a possibility that needs to be addressed in future experiments.
Transient splenic CD8 T cell responses to CNS antigens precede activation in the cervical lymph nodes
We also quantified CD8 T cell responses to CNS antigen within the same cultures, and again found an early autoimmune response in both the cervical lymph nodes (p<0.01; Figure 1E) and the spleen (p<0.001; Figure 1F). Like the CD4 T cell population, the magnitude of autoreactivity in the cervical lymph node peaked at day 10 (F(2,313)=16.27; p<0.0001) compared to the early elevation at day 4 (p<0.0001) and the complete loss of autoreactivity at day 8 (p<0.0001). CNS-directed autoreactivity of CD8 T cells isolated from the spleen was not significantly sustained beyond the early day 4 time point after stroke. There was no CD8 T cell-originating autoimmune response for the mouse with the largest infarct volume (122 mm3; Figure 3) despite CD4 T cell activation in the cervical lymph node (Figure 2A, black circle). However, the significant response for NR2A in the cervical lymph nodes (p<0.05) was solely reliant upon the mouse with the second largest infarct volume (74 mm3; Figure 3A, grey circles). The only CD8 responses to NR2F and vWF were also from this mouse, with no concomitant autoreactivity in splenocytes isolated from this animal. This again suggests an NR2A-derived antigen signal that activates CD8 T cells, but only for moderately injured mice. Future studies should determine if very large infarcts somehow suppress or delay autoimmune responses in the draining lymph nodes. Similarly in the spleen, a moderately-injured mouse (36 mm3; red circles) dominated the autoimmune responses to myelin and NR2F (p<0.05). Unfortunately, the remaining responders to NR2F do not have corresponding infarct volume data. MAP2 autoreactivity in the spleen occurs only in mice with small and medium-sized infarct volumes, though unlike additional CD4 T cell-mediated MAP2 autoreactivity in the cervical lymph node, the mouse with the smallest infarct volume did not show a CD8 T cell response.
Figure 3.
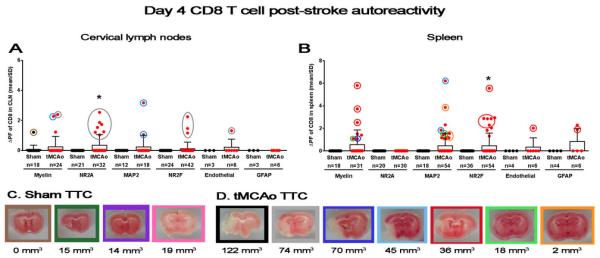
Transient splenic CD8 T cell responses to CNS antigens precede activation in the cervical lymph nodes. (A-B) Graphs depict autoreactivity (ΔPF; y axis) for individual peptides in both sham populations (black circles) and tMCAo populations (red circles) at 4 days after tMCAo (x axis). (A) Left graph shows responses from CD8 T cells isolated from the cervical lymph nodes (CLN), and (B) right graph shows splenic CD8 T cell responses. Values for the number of wells within each group with a measurable response are shown below in parenthesis. (C-D) Representative TTC-stained section from each animal in both (C) sham and (D) tMCAo-treated groups. Areas of mitochondrial dysfunction (i.e., infarct volumes) appear white, while healthy tissue stains red. The total infarct volume for each animal is shown below the corresponding pictures (mm3). The border around each infarct is color-coded with the circles around individual data points in the graphs shown in (A, B). Within-day significance is shown as asterisks over each tMCAo group (student's t-test). Values are mean ± standard deviation (SD). * p<0.05.
The first and second MAP2 peptide sequence pools that elicited an autoreactive CD4 T cell response also induced CD8 T cell activation (Table 1). Though as with CD4 T cell responses, many MAP2 peptide sequence pools that did not activate an autoimmune response in the cervical lymph nodes did trigger CD8 T cell responses in the spleen, again indicating antigen specific activation. There is a broad range of NR2A peptide responses for CD8 T cell activation also found in CD4 T cells, but not for either population isolated from the spleen. Taken together, these data show that CD8 T cells are robustly activated following stroke induction, but this activation may be dependent upon the size of CNSinjury. CD8 T cell responses are lost by one-week post-stroke and this suggests a trafficking of the effector population from the peripheral immune compartment to quite possibly the site of tissue pathology.
Stroke induces a rapid and sustained B cell response to both neuronal and myelin antigen
To our knowledge an autoimmune response following stroke has not been associated with CD19+ cells (Sardi et al., 2011). Interestingly, we found an early day 4 activation of B cells to CNS-specific antigens in both the cervical lymph nodes (p<0.01) and the spleen (p<0.01), with at least one positive response from every sham and tMCAo-induced mouse, with the exception of the mouse with the largest infarct volume (Figure 1G-H, Figure 4). Unlike either T cell population, however, the magnitude of response at day 4 in the cervical lymph nodes was equivalent to the magnitude of response at day 10, suggesting an early and robust B cell activation by CNS antigen that is maintained during the first weeks after stroke. Day 10 autoreactivity remained elevated (p<0.05) in the spleen, which did not occur with either of the T cell subsets. B cells from the cervical lymph nodes exhibited a similar biphasic autoreactivity, with lower levels on day 8 compared to day 4 (p<0.05) and day 10 (p<0.05; F(2,318)=4.02; p<0.05).
Figure 4.
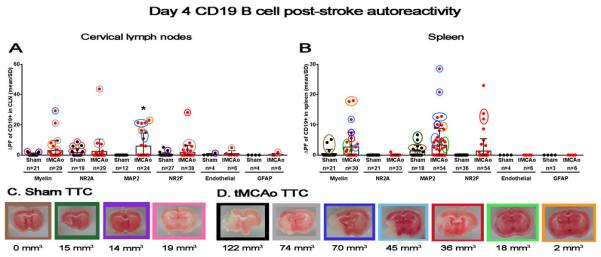
Stroke induces a rapid and sustained B cell response to both neuronal and myelin antigen. (A-B) Graphs depict autoreactivity (ΔPF; y axis) for individual peptides in both sham populations (black circles) and tMCAo populations (red circles) at 4 days after tMCAo (x axis). (A) Left graph shows responses from B cells isolated from the cervical lymph nodes (CLN), and (B) right graph shows splenic B cell responses. Values for the number of wells within each group with a measurable response are shown below in parenthesis. (C-D) Representative TTC-stained section from each animal in both (C) sham and (D) tMCAo-treated groups. Areas of mitochondrial dysfunction (i.e., infarct volumes) appear white, while healthy tissue stains red. The total infarct volume for each animal is shown below the corresponding pictures (mm3). The border around each infarct is color-coded with the circles around individual data points in the graphs shown in (A, B). Within-day significance is shown as asterisks over each tMCAo group (student's t-test). Values are mean ± standard deviation (SD). * p<0.05.
Sham surgery also activated B cell autoimmune responses. In fact, elevations on day 8 in splenic B cells (F(2,271)=5.24; p<0.01) preceded elevations in B cell autoreactivity on day 10 in the cervical lymph nodes (F(2,174)=17.23; p<0.0001). This level of response, across days and in both the sham and stroke populations, may reflect a sustained B cell autoimmune activation after even very mild CNS injury, as may occur after CCA ligation and anesthesia exposure. However, CD8 T cell autoreactivity to myelin peptides has been found in healthy adults in the absence of autoimmunedisease (Crawford et al., 2004). Therefore, the generalized B cell autoimmune response could also reflect a previously undiscovered basal fluctuation in neuronal autoimmunity independent of CNS injury.
While almost every mouse exhibited B cell-mediated autoreactivity at 4 days after tMCAo or sham surgery (Figure 4), only MAP2 autoreactivity in the cervical lymph nodes showed significance (p<0.05). As with T cells, B cell autoreactivity to myelin, NR2A, and NR2F in the cervical lymph nodes was predominated by animals with moderate to large infarct volumes. The mouse with the smallest infarct volume, however, had the highest ΔPF for MAP2 in the cervical lymph node, which seems counterintuitive to CNS antigen exposure being secondary to ischemic tissue damage. Several sham mice also showed responses to myelin, NR2A, and NR2F, a surprising response that again seems independent of injury and warrants further investigation. The heterogeneous response of B cells to CNS antigen was recapitulated in the splenic B cell populations and included trends for both post-stroke myelin (p=0.06) and NR2F autoreactivity (p=0.06). As with T cells, NR2A autoreactivity is predominantly restricted to the cervical lymph nodes, though the second peptide sequence did elicit one weak response in splenic B cells (Table 1). Splenic B cells show the broadest range of MAP2 peptide responses from any of the lymphocyte populations, though future studies should attempt to determine which of these peptides is truly immunogenic. Taken together, these data show that B cells activated following stroke exhibit a global, complex, and sustained autoimmune response to CNS antigen that may indicate a very active role for T and B cells in stroke damage and/or repair.
Discussion
Understanding the role of post-stroke inflammation, including autoimmunity, is an evolving high priority for NIH research (Grotta et al., 2008). Early CNS-directed autoimmunity was studied predominantly in animal models of multiple sclerosis (Hong et al., 2009; York et al., 2010). Now, however, CNS-directed autoimmunity is increasingly observed in other neurodegenerative diseases, including stroke (Planas et al., 2012a), traumaticbraininjury (Schwartz and Moalem, 2001; Weissman et al., 2011), and Alzheimer’s disease (Monson et al., 2014a; Sardi et al., 2011). The role of autoreactive adaptiveimmunity is not only now applicable to many CNS diseases, but is also undergoing a paradigm shift from the concept that all autoimmunity results in detrimental effects. While CD8 and CD4 T cells can target neurons to impede electrical signal, induce excitotoxicity, and promote cell death (Bien et al., 2002; Laguna Goya et al., 2011; Maehlen et al., 1988), MBP-specific autoreactive T cells also function during health to maintain basal neurogenesis and spatial learning (Radjavi et al., 2014). In fact, the adoptive transfer of MBP-specific T cells into neonatal mice does not induce autoimmune disease during adulthood (Kawakami et al., 2005). The mice in our study that exhibited minimal CNS injury, despite an identical duration of stroke, demonstrated high autoreactivity to myelin and MAP2 in splenic lymphocytes. While this could be the result of a sequestration of dangerous autoreactive B and T cells in the spleen, which could reduce brain injury, high MAP2 autoreactivity also is found in the draining lymph nodes. Perhaps, instead, autoreactivity can also be used to home beneficial cell populations, including regulatory T cells and B cells (Bodhankar et al., 2013; Brea et al., 2014; Liesz et al., 2009), to neurons at risk of injury or death, though to what mechanistic end remains to be elucidated. Either way, the adoptive transfer of CD4 T cells can improve neuronal plasticity after CNS trauma (Ishii et al., 2012), which suggests some immune populations may be necessary for post-stroke neuronal anatomical and functional changes that contribute to functional recovery.
After stroke, lymphocytes encounter CNS-derived antigens and acute autoimmune responses against the brain develop (see reviews Becker, 2009; Iadecola and Anrather, 2011; Sardi et al., 2011). One of the few studies of post-stroke neuronal autoimmunity found that MHC II receptors colocalized with brain-derived antigens in the cervical lymph nodes and palatine tonsils of stroke patients (Planas et al., 2012a). Specifically, colocalization with myelin-derived epitopes (e.g., MBP) appeared to be detrimental at 90 days after stroke, as interactions between MBP and lymphocytes correlated with larger infarct volumes. In the same study, MHC II expression of MAP2 and NR2A both correlated with smaller infarct volumes and better long-term recovery at 90 days. Our study, however, revealed that larger infarct volumes 4 days after tMCAo corresponded with increased T and B cell autoreactivity in the cervical lymph node to NR2A, and to a lesser extent, myelin. Autoimmunity to the NMDA receptor is involved in encephalitis, with clinical presentations including memory deficits, seizures and psychosis (Lazar-Molnar and Tebo, 2015). Although this is believed to be mediated by NMDA autoantibodies from B cells, it is possible that CD4 T cell responses to NR2A, which is concomitant with B cell activation in our data, may predispose a patient to this disease. Multiple strokes in patients increase serum levels of NR2A protein (Weissman et al., 2011), which could increase the risk of antigen exposure and potentially detrimental NR2A-specific autoreactivity. As cognitive decline is common to stroke survivors and mediated by B cells in animal models of stroke (Doyle et al., 2015), future studies should determine whether an NMDA-specific autoimmune response limits functional recovery following stroke.
What is more in line with the correlation of neuronal autoreactivity to better stroke recovery in the clinical data (Planas et al., 2012a) is the high autoreactive response to MAP2 for both T and B cells from the spleens of mice with low to moderate CNS injury. MAP2 is a cytoskeletal protein found in dendrites that is crucial to both dendritic arborization and pruning during post-stroke neuronal plasticity that occurs in both the ischemic and the uninjured hemispheres during behavioral recovery (Hsu and Jones, 2006). Specifically, the upregulation of MAP2 associates with reduced infarct volumes and better functional recovery (Zhang et al., 2012). In stroke patients, also with better functional recovery, CD68+ macrophages isolated from palatine tonsils and cervical lymph nodes colocalized with MAP2 (Planas et al., 2012a), which indicates an innate immune response to MAP2 that may precede a MAP2-directed autoimmune CD4 T cell response. While the time frame between our data (4 days) and the clinical data (90 days) is greatly different, the correlation between high autoreactivity to MAP2 and less severe CNS injury suggests that autoimmune activation of MAP2 may either preserve neurons at-risk of injury in the ischemic brain, or promote recovery mechanisms within the injured cortex.
Unfortunately, the data regarding myelin autoreactivity on stroke outcome are ambiguous. Both MBP antigen presentation prior to stroke (Becker et al., 1997) and a sustained regulatoryTcell response to MBP (Kala et al., 2011) were associated with improved long-term recovery in mice. But the determination of whether myelin autoimmune responses are detrimental or beneficial after stroke in patients is confounded by post-stroke co-morbidities with infections. A clinical study of 114 patients showed that at 90 days after stroke onset, the severity of functional recovery correlated with enhanced CD4 T cell responses to MBP, with autoreactivity exacerbated by infection and fever within the first 15 days after stroke (Becker et al., 2011). The authors thus concluded that autoimmune responses to myelin were associated with worse outcome. A Letter to the Editor in response to this assumption pointed out that the autoimmune responses to MBP, PLP, and even GFAP, at either 3 or 7 days after stroke onset had no correlation with long-term functional outcome at 90 days (Urra et al., 2012). Only the 90-day immune response correlated with the 90-day severity of stroke. These data highlight the complex interplay between autoantigen presentation, injury, and recovery in the ischemic cortex and begs the question: is the higher autoimmune response to myelin contributing to the loss of functional recovery, or is the inability to recover lost function driving a myelin-derived — and potentially neuronal-derived — autoimmune response in an effort to promote plasticity during continued recovery?
Several future studies should address limitations in our data. First, we observed a profound reduction in autoreactivity at day 8 in the cervical lymph nodes, which at least visually rebounded by day 10. The day 8 group had an n=5 for tMCAo, though this experiment was duplicated on two separate days. Also, global immunoreactivity from T cell splenocytes did not reach significance compared to shams at days 8 and 10 post stroke. Therefore, it will be necessary to add replicates in order to confirm the day 8 loss of autoreactivity, though this did not simultaneously occur in the spleen. If a loss of autoreactivity at day 8 is confirmed, these data may be explained by either a loss of active brain injury, a mobilization of autoreactive immune cells to other tissues, or post-stroke immunosuppression, possibilities which will be investigated in future studies. As also mentioned above, long-term autoreactive CD4 T cell responses were associated with poor outcome in stroke patients (Becker et al., 2011). CNS-directed post-stroke autoreactivity was investigated in a 2010 study with several genetic lines of mice previously used in mouse models of multiple sclerosis (Kleinschnitz et al., 2010). Restriction of autoreactivity to a non-CNS peptide in both CD8 and CD4 T cells, and restriction of autoreactivity to only MOG35-55, did not contribute to T cell-mediated growth of infarct volumes. However, injury was evaluated only at 24 hours after stroke onset, within a timeframe that precedes activation of the adaptive immune system in the injured CNS (Kamel and Iadecola, 2012).
Future studies should first adoptively transfer myelin, or more importantly NR2A or MAP2-stimulated T and B cells, at least 1 day after stroke to determine the long-term contribution of neuronal autoreactivity to post-stroke neuronal plasticity and functional recovery. Therapeutic strategies could eventually be aimed at suppressing unfavorable immune activation through immune modulating drugs, or augmenting favorable immune responses through cell-based immunotherapy or vaccinations, to accelerate neurological recovery in stroke patients. Secondly, future studies should also address the possibility that the memory aspect of an autoimmune response may play a significant role in secondary stroke severity or complications, particularly with regard to a long-term circulation of CNS-directed lymphocytes. Finally, NR2F is an orphan nuclear receptor that mediates brain development (Lin et al., 2011) and contributes to post-stroke angiogenesis (Li et al., 2013). Originally intended to be a neuronal protein control for NR2A and MAP2 identified in the clinical study (Planas et al., 2012b), future studies should investigate the specific ramifications of an autoimmune response to a neuronal nuclear protein, especially as autoreactivity is thus far confined to mice with larger infarct volumes.
Conclusions
Although the immune system does not play a role in the initiation of stroke, there is a growing perception that stroke-induced immune activation may potentiate either neuropathology or neurorepair. Elucidating novel neuronal surface antigens that are targets for immune cells offers the first unique insights into potential cellular and systemic consequences of autoimmunity during post-stroke neuronal plasticity. There is a dire need to further investigate if the autoreactive immune responses contribute to secondary brain injury, if they predispose an individual to other neurological diseases, or if there is a role for autoreactivity in successful neurorepair. By observing similar types of adaptive immune responses in mice that are mirrored in patient studies, we now have a specific and sensitive immune-based assay that can be used to elucidate the mechanisms of neuronal autoreactivity following ischemic brain injury.
Acknowledgments
The authors would like to acknowledge grant funding from NIH/NINDS (NS088555), the American Heart Association (14SDG1841002, 14POST20480373), and the Beatrice Menne Haggerty Center for Brain Injury and Repair (UTSW). The authors would like to thank Neha Methani, Uma Maheswari Selvaraj, and Dr. Erik Plautz from the Neuromodels Facility, UT Southwestern. We also wish to thank Drs. Ding Chen, Mark Goldberg, and Jerry Niederkorn for helpful discussions.
Footnotes
Disclosure
The authors have no conflicts of interest to disclose.
References
- Becker KJ. Sensitization and tolerization to brain antigens in stroke. Neuroscience. 2009;158(3):1090–1097. doi: 10.1016/j.neuroscience.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KJ, Kalil AJ, Tanzi P, Zierath DK, Savos AV, Gee JM, Hadwin J, Carter KT, Shibata D, Cain KC. Autoimmune responses to the brain after stroke are associated with worse outcome. Stroke. 2011;42(10):2763–2769. doi: 10.1161/STROKEAHA.111.619593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KJ, Mccarron RM, Ruetzler C, Laban O, Sternberg E, Flanders KC, Hallenbeck JM. Immunologic tolerance to myelin basic protein decreases stroke size after transient focal cerebralischemia. Proc Natl Acad Sci U S A. 1997;94(20):10873–10878. doi: 10.1073/pnas.94.20.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien CG, Bauer J, Deckwerth TL, Wiendl H, Deckert M, Wiestler OD, Schramm J, Elger CE, Lassmann H. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen's encephalitis. Ann Neurol. 2002;51(3):311–318. doi: 10.1002/ana.10100. [DOI] [PubMed] [Google Scholar]
- Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. IL-10-producing B-cells limit CNS inflammation and infarct volume in experimental stroke. Metab Brain Dis. 2013;28(3):375–386. doi: 10.1007/s11011-013-9413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brea D, Agulla J, Rodriguez-Yanez M, Barral D, Ramos-Cabrer P, Campos F, Almeida A, Davalos A, Castillo J. Regulatory T cells modulate inflammation and reduce infarct volume in experimental brain ischaemia. J Cell Mol Med. 2014;18(8):1571–1579. doi: 10.1111/jcmm.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro Á , Meisel A, Planas AM, Urra X, Van De Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8(7):401–410. doi: 10.1038/nrneurol.2012.98. [DOI] [PubMed] [Google Scholar]
- Chu HX, Kim HA, Lee S, Moore JP, Chan CT, Vinh A, Gelderblom M, Arumugam TV, Broughton BRS, Drummond GR, Sobey CG. Immune cell infiltration in malignant middle cerebral artery infarction: comparison with transient cerebral ischemia. J Cereb Blood Flow Metab. 2014;34(3):450–459. doi: 10.1038/jcbfm.2013.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt RE, Price DA, Stastny P, Douek DC, Koup RA, Racke MK, Karandikar NJ. High prevalence of autoreactive, neuroantigen-specific CD8+ T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood. 2004;103(11):4222–4231. doi: 10.1182/blood-2003-11-4025. [DOI] [PubMed] [Google Scholar]
- Doyle KP, Quach LN, Sole M, Axtell RC, Nguyen TV, Soler-Llavina GJ, Jurado S, Han J, Steinman L, Longo FM, Schneider JA, Malenka RC, Buckwalter MS. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci. 2015;35(5):2133–2145. doi: 10.1523/JNEUROSCI.4098-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba P, Klausberger T, Karayannis T, Suen WY, Huck J, Tomioka R, Rockland K, Capogna M, Studer M, Morales M, Somogyi P. Expression of COUP-TFII nuclear receptor in restricted GABAergic neuronal populations in the adult rat hippocampus. J Neurosci. 2010;30(5):1595–1609. doi: 10.1523/JNEUROSCI.4199-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40(5):1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127(1):e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotta JC, Jacobs TP, Koroshetz WJ, Moskowitz MA. Stroke program review group: an interim report. Stroke. 2008;39(4):1364–1370. doi: 10.1161/STROKEAHA.107.510776. [DOI] [PubMed] [Google Scholar]
- Hong J, Li H, Chen M, Zang YCQ, Skinner SM, Killian JM, Zhang JZ. Regulatory and pro-inflammatory phenotypes of myelin basic protein-autoreactive T cells in multiple sclerosis. Int Immunol. 2009;21(12):1329–1340. doi: 10.1093/intimm/dxp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JE, Jones TA. Contralesional neural plasticity and functional changes in the less-affected forelimb after large and small cortical infarcts in rats. Exp Neurol. 2006;201(2):479–494. doi: 10.1016/j.expneurol.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Hussain RZ, Hayardeny L, Cravens PC, Yarovinsky F, Eagar TN, Arellano B, Deason K, Castro-Rojas C, Stüve O. Immune surveillance of the central nervous system in multiple sclerosis - Relevance for therapy and experimental models. J Neuroimmunol. 2014;276(1-2):9–17. doi: 10.1016/j.jneuroim.2014.08.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Jin X, Ueno M, Tanabe S, Kubo T, Serada S, Naka T, Yamashita T. Adoptive transfer of Th1-conditioned lymphocytes promotes axonalremodeling and functional recovery after spinalcordinjury. Cell Death Dis. 2012;3:e363. doi: 10.1038/cddis.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87(5):779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kala M, Miravalle A, Vollmer T. Recent insights into the mechanism of action of glatiramer acetate. J Neuroimmunol. 2011;235(1-2):9–17. doi: 10.1016/j.jneuroim.2011.01.009. [DOI] [PubMed] [Google Scholar]
- Kamel H, Iadecola C. Brain-immune interactions and ischemic stroke: clinical implications. Arch Neurol. 2012;69(5):576–581. doi: 10.1001/archneurol.2011.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami N, Odoardi F, Ziemssen T, Bradl M, Ritter T, Neuhaus O, Lassmann H, Wekerle H, Flügel A. Autoimmune CD4+ T cell memory: lifelong persistence of encephalitogenic T cell clones in healthy immune repertoires. J Immunol. 2005;175(1):69–81. doi: 10.4049/jimmunol.175.1.69. [DOI] [PubMed] [Google Scholar]
- Kidd PM. Integrated brain restoration after ischemic stroke–medical management, risk factors, nutrients, and other interventions for managing inflammation and enhancing brain plasticity. Altern Med Rev. 2009;14(1):14–35. [PubMed] [Google Scholar]
- Kim BJ, Takamoto N, Yan J, Tsai SY, Tsai MJ. Chicken Ovalbumin Upstream Promoter-Transcription Factor II (COUP-TFII) regulates growth and patterning of the postnatal mouse cerebellum. Dev Biol. 2009;326(2):378–391. doi: 10.1016/j.ydbio.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipnis J, Mizrahi T, Yoles E, Ben-Nun A, Schwartz M. Myelin specific Th1 cells are necessary for post-traumatic protective autoimmunity. J Neuroimmunol. 2002;130(1-2):78–85. doi: 10.1016/s0165-5728(02)00219-9. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, Austinat M, Nieswandt B, Wiendl H, Stoll G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115(18):3835–3842. doi: 10.1182/blood-2009-10-249078. [DOI] [PubMed] [Google Scholar]
- Laguna Goya R, Busch R, Mathur R, Coles AJ, Barker RA. Human fetal neural precursor cells can up-regulate MHC class I and class II expression and elicit CD4 and CD8 T cell proliferation. Neurobiol Dis. 2011;41(2):407–414. doi: 10.1016/j.nbd.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Lazar-Molnar E, Tebo AE. Autoimmune NMDA receptor encephalitis. Clin Chim Acta. 2015;438:90–97. doi: 10.1016/j.cca.2014.08.010. [DOI] [PubMed] [Google Scholar]
- Li Y, Xia Y, Wang Y, Mao L, Gao Y, He Q, Huang M, Chen S, Hu B. Sonichedgehog (Shh) regulates the expression of angiogenic growth factors in oxygen-glucose-deprived astrocytes by mediating the nuclear receptor NR2F2. Mol Neurobiol. 2013;47(3):967–975. doi: 10.1007/s12035-013-8395-9. [DOI] [PubMed] [Google Scholar]
- Liesz A, Karcher S, Veltkamp R. Spectratype analysis of clonal T cell expansion in murine experimental stroke. J Neuroimmunol. 2013;257(1-2):46–52. doi: 10.1016/j.jneuroim.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15(2):192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Lin FJ, Qin J, Tang K, Tsai SY, Tsai MJ. Coup d’Etat: an orphan takes control. Endocr Rev. 2011;32(3):404–421. doi: 10.1210/er.2010-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehlen J, Schröder HD, Klareskog L, Olsson T, Kristensson K. Axotomy induces MHC class I antigen expression on rat nerve cells. Neuroscience Letters. 1988;92(1):8–13. doi: 10.1016/0304-3940(88)90733-1. [DOI] [PubMed] [Google Scholar]
- Miller SD, Karpus WJ, Davidson TS. Experimental Autoimmune Encephalomyelitis in the Mouse. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2001. [Google Scholar]
- Monson NL, Ireland SJ, Ligocki AJ, Chen D, Rounds WH, Li M, Huebinger RM, Munro Cullum C, Greenberg BM, Stowe AM, Zhang R. Elevated CNS inflammation in patients with preclinical Alzheimer’s disease. J Cereb Blood Flow Metab. 2014a;34(1):30–33. doi: 10.1038/jcbfm.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monson NL, Ortega SB, Ireland SJ, Meeuwissen AJ, Chen D, Plautz EJ, Shubel E, Kong X, Li MK, Freriks LH, Stowe AM. Repetitive hypoxic preconditioning induces an immunosuppressed B cell phenotype during endogenous protection from stroke. J Neuroinflam. 2014b;11:22. doi: 10.1186/1742-2094-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega SB, Kashi VP, Tyler AF, Cunnusamy K, Mendoza JP, Karandikar NJ. The disease-ameliorating function of autoregulatory CD8 T cells is mediated by targeting of encephalitogenic CD4 T cells in experimental autoimmune encephalomyelitis. J Immunol. 2013;191(1):117–126. doi: 10.4049/jimmunol.1300452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas AM, Gomez-Choco M, Urra X, Gorina R, Caballero M, Chamorro A. Brain-derived antigens in lymphoid tissue of patients with acute stroke. J Immunol. 2012a;188(5):2156–2163. doi: 10.4049/jimmunol.1102289. [DOI] [PubMed] [Google Scholar]
- Planas AM, Gomez-Choco M, Urra X, Gorina R, Caballero M, Chamorro A. Brain-Derived Antigens in Lymphoid Tissue of Patients with Acute Stroke. J Immunol. 2012b;188(5):2156–2163. doi: 10.4049/jimmunol.1102289. [DOI] [PubMed] [Google Scholar]
- Radjavi A, Smirnov I, Kipnis J. Brain antigen-reactive CD4+ T cells are sufficient to support learning behavior in mice with limited T cell repertoire. Brain Behav Immun. 2014;35:58–63. doi: 10.1016/j.bbi.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi F, Fassina L, Venturini L, Inguscio M, Guerriero F, Rolfo E, Ricevuti G. Alzheimer’s disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmun Rev. 2011;11(2):149–153. doi: 10.1016/j.autrev.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Moalem G. Beneficial immune activity after CNS injury: prospects for vaccination. J Neuroimmunol. 2001;113(2):185–192. doi: 10.1016/s0165-5728(00)00447-1. [DOI] [PubMed] [Google Scholar]
- Stowe AM, Altay T, Freie AB, Gidday JM. Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann Neurol. 2011;69(6):975–985. doi: 10.1002/ana.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Goverman JM. Passive induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1(4):1952–1960. doi: 10.1038/nprot.2006.284. [DOI] [PubMed] [Google Scholar]
- Urra X, Planas AM, Chamorro A. Letter by Urra et al regarding article, “Autoimmune responses to the brain after stroke are associated with worse outcome”. Stroke. 43(2):e26. doi: 10.1161/STROKEAHA.111.643460. author reply e27-28, 2012. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Murray V, Berge E, Del Zoppo G, Sandercock P, Lindley RL, Cohen G. Recombinant tissueplasminogenactivator for acute ischaemic stroke: an updated systematic review and meta-analysis. Lancet. 2012;379(9834):2364–2372. doi: 10.1016/S0140-6736(12)60738-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman JD, Khunteev GA, Heath R, Dambinova SA. NR2 antibodies: risk assessment of transient ischemic attack (TIA)/stroke in patients with history of isolated and multiple cerebrovascular events. J Neurol Sci. 2011;300(1-2):97–102. doi: 10.1016/j.jns.2010.09.023. [DOI] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113(17):2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- York NR, Mendoza JP, Ortega SB, Benagh A, Tyler AF, Firan M, Karandikar NJ. Immune regulatory CNS-reactive CD8+T cells in experimental autoimmune encephalomyelitis. J Autoimmun. 2010;35(1):33–44. doi: 10.1016/j.jaut.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang Y, Zhu P, Wang X, Lv M, Feng H. siRNA-mediated silence of protease-activated receptor-1 minimizes ischemic injury of cerebral cortex through HSP70 and MAP2. J Neurol Sci. 2012;320(1-2):6–11. doi: 10.1016/j.jns.2012.05.040. [DOI] [PubMed] [Google Scholar]