

Abstract
Objective
Alpha-1-antitrypsin deficiency (A1AT) is a common genetic disease with unpredictable and highly variable course. The Childhood Liver Disease Research and Education Network (ChiLDREN) is an NIH, multi-center, longitudinal consortium studying pediatric liver diseases, with the objective of prospectively defining natural history and identifying disease modifiers.
Methods
Longitudinal, cohort study of A1AT patients birth through 25 years diagnosed with liver disease, type PIZZ or PISZ. Medical history, physical exam, laboratory, imaging, and standardized survey tool data were collected during the provision of standard of care.
Results
In this report of the cohort at baseline, 269 subjects were enrolled between Nov. 2008 and Oct. 2012 (208 with their native livers and 61 post-liver transplant). Subjects with mild disease (native livers and no portal hypertension [PHT]) compared to severe disease (with PHT or post-liver transplant) were not different in age at presentation. 57% of subjects with mild disease and 76% with severe disease were jaundiced at presentation (p=0.0024). 29% of subjects with native livers had PHT, but age at diagnosis and growth were not different between the no PHT and PHT groups (p>0.05). Subjects with native livers and PHT were more likely to have elevated bilirubin, ALT, AST, INR, and GGTP than the no PHT group (p≪0.001), but overlap was large. Chemistries alone could not identify PHT.
Conclusion
Many A1AT subjects presenting with elevated liver tests and jaundice improve spontaneously. Subjects with PHT have few symptoms and normal growth. Longitudinal cohort follow up will identify genetic and environmental disease modifiers. NCT00571272.
Keywords: cirrhosis, liver transplant, liver enzymes, metabolic liver disease, jaundice
Introduction
Alpha-1-antitrypsin (A1AT) deficiency is a common genetic liver disease occurring in 1 in 2,000-3,500 births (1, 2). It is associated with chronic liver disease and cirrhosis in children and adults. The Z mutant of the A1AT gene is a point mutation that encodes a single amino acid substitution, and is associated with the vast majority of A1AT liver disease, either as the classical form of homozygous ZZ A1AT deficiency, or as the SZ compound heterozygous form (1). The mechanism of the liver injury involves the accumulation within hepatocytes of the A1AT mutant Z protein, which triggers hepatocellular death, compensatory regeneration and hepatic fibrosis (1, 3-5). The clinical course of A1AT deficiency liver disease is highly variable. Some patients present with neonatal cholestasis, which often resolves spontaneously, although some patients develop cirrhosis later in infancy or childhood. Other patients may come to medical attention because of chronic hepatomegaly, splenomegaly, or elevated liver chemistries discovered during childhood evaluations. Older children and adults may present with cirrhosis or hepatocellular carcinoma, although many patients remain healthy and free of severe liver injury until late adulthood. The genetic and environmental modifiers of this wide variability are largely unknown (1, 2, 6, 7). In the early 1970s, a cohort of 127 ZZ A1AT deficient children was identified in Sweden by newborn screening(8-10). This cohort had a very low, (<5%), rate of life threatening liver disease in childhood, and no liver disease in survivors from the second through the fourth decades of life. This is in contrast to various single center, retrospective reports from more genetically mixed groups of patients in North America and Britain showing a wider range of liver complications and the progression of disease in some patients throughout childhood(1, 2, 11-15). It may be that some complications of liver disease are too infrequent to be detected in the Swedish cohort of 127 individuals, there may be ascertainment bias, or there may be a different set of disease modifiers associated with more severe disease are found outside of Sweden.
The Childhood Liver Disease Research and Education Network (ChiLDREN) is an NIH funded consortium of 16 pediatric tertiary care centers in North America focused on the study of rare pediatric liver diseases (16-19). This network includes the Longitudinal Observational Study of Genetic Causes of Intrahepatic Cholestasis (LOGIC), the goal of which is to describe the natural history and disease modifiers of a group of metabolic-cholestatic liver diseases, including A1AT deficiency. This is a non-interventional, observational protocol with retrospective and prospective components, however it is biased to referral centers and not a birth cohort. We hypothesized that the study of a large, well characterized, and multi-center cohort of patients will provide a better understanding of the natural history and clinical variability, and that construction of a database of patient information linked to DNA and biospecimens will allow identification of the genetic and environmental modifiers. This is the baseline enrollment report of the LOGIC subjects with A1AT deficiency with the aim of characterizing baseline history, physical exam and laboratory findings in those enrolled with severe disease (pre-existing portal hypertension or liver transplant) compared to those with mild disease (native liver with little or no fibrosis). Our hypothesis is that history, physical exam and laboratory findings associated with severe disease can be identified and will aid in diagnosis, disease prognostication and may give clues to areas of future treatments and research. Future reports will be made as the cohort is followed prospectively and as outcomes are linked to ongoing biospecimen analysis.
Methods
A1AT enrollment into LOGIC at baseline is reported Nov. 2008-Oct. 2012. IRB approvals and consents were obtained at each site. Eligibility for enrollment included PIZZ or PISZ serum protein phenotype or genotype, low serum level of A1AT protein, age birth to 25years, and the presence of or a history of liver disease as defined by one of the following (#1-6): (#1) neonatal cholestasis (serum direct or conjugated bilirubin >2mg/dL and >20% of total bilirubin), (#2) >1.25 × normal ALT, AST, or GGT, (#3) chronic hepatomegaly (clinically measured liver span >95%ile for age for at least 3mo.), (#4) clinical findings or complications of portal hypertension or cirrhosis (as determined by the clinical judgment of the investigator, including for example, esophageal variceal hemorrhage or hard hepatosplenomegaly in the context of imaging consistent with cirrhosis), (#5) impaired liver synthetic function, or (#6) abnormal liver biopsy histology showing liver injury, other than globular inclusions of A1AT (inflammation, fibrosis, cholestasis, bile duct paucity, or necrosis). Subjects were also enrolled if they had undergone liver transplant in the past for A1AT deficiency and had previously fulfilled these criteria.
At enrollment, medical history was obtained, including review of available medical records, and a physical exam was performed. Samples of serum, plasma, urine, and DNA were obtained during clinical laboratory draws at the baseline visit and at subsequent yearly visits, and stored in a repository. Updates in medical history and physical exam were documented at annual follow up visits. The results of imaging studies, surgery, and endoscopy, performed for standard of care indications, were collected. Quality of life measures were obtained annually (Pediatric Quality of Life Inventory, Version 4.0), and standardized neurodevelopmental assessments (1-2 years old: Bayley Scales of Infant Development-III, 3-5 years old Wechsler Preschool and Primary Scale of Intelligence-III, 6-16 years old Wechsler Intelligence Scale for Children-IV, 17-25 years old Wechsler Adult Intelligence Scale-III).
In this analysis, portal hypertension (PHT) was defined by previously validated and published criteria for patients with their native liver as “definite” when there was either a history of a complication of PHT (variceal hemorrhage, ascites, or hepatopulmonary syndrome) or clinical findings consistent with PHT (both splenomegaly [spleen > 2 cm below the costal margin] and thrombocytopenia [platelet count < 150,000/mm3])(19). PHT was denoted as “possible” if either splenomegaly or thrombocytopenia were present in the absence of a complication. PHT was considered to be absent if none of these criteria were met. For the analysis as noted below the “definite” and “possible” PHT were grouped together as “PHT” unless otherwise noted. For comparisons noted below, patients with “severe” liver disease were defined as any subject status-post liver transplant or any subject with their native liver with PHT. Patients with “mild” liver disease were defined as subjects with their native liver without PHT. It is also important to note that this is an observational cohort enrolled during the provision of clinical care at multiple centers. There were not standardized, protocol driven imaging studies, endoscopies, or biopsies, although the results of studies like these done as a part of clinical care were captured. Therefore, our eligibility and case definitions, as defined in the Methods, are based on available history, physical exam, and minimally invasive (blood draw) laboratory studies. As part of the data analysis, studies such as ultrasounds, as described below, were then compared to the existing, enrollment clinical case definitions.
For continuous outcomes, the mean, standard deviation, median, first and third quartiles, and minimum and maximum are reported by group. For categorical outcomes, the number and percentage are reported by group. Comparisons of the following subgroups were performed using Wilcoxon rank-sum tests for continuous outcomes and Fisher's exact tests for discrete outcomes: 1) The native liver group versus the post-transplant group (Table 1); 2) Subjects with mild liver disease versus subjects with severe liver disease (Table 2); and 3) Native liver subjects with PHT, versus native liver subjects without PHT (Table 3). Splenomegaly as determined by ultrasound was also available for many subjects in the cohort, defined as published by spleen size greater than 90th percentile for age (20). The two definitions of PHT, one using physical exam-based splenomegaly, and the other using ultrasound-based splenomegaly, were compared and are reported.
Table 1. Demographic Characteristics of A1AT Liver Disease Cohort, with Native Liver and Post-Transplant.
| Characteristic | A1AT Liver Disease Cohort1(N=269) | Native Liver Cohort2(N=208) | Post-Transplant Cohort(N=61) | p-value3 |
|---|---|---|---|---|
| Age, years | <0.0001 | |||
| N | 269 | 208 | 61 | |
| Mean (SD) | 8.2 (6.38) | 6.9 (5.65) | 12.9 (6.51) | |
| Median (Q1, Q3) | 6.8 (2.8, 12.9) | 5.5 (2.0, 9.8) | 13.7 (7.0, 18.4) | |
| Min, Max | 0,0, 24.9 | 0.0, 24.9 | 0.4, 24.6 | |
| Age at symptoms first noticed, years | 0.38 | |||
| N | 245 | 190 | 55 | |
| Mean (SD) | 1.3 (3.14) | 1.4 (3.33) | 0.9 (2.34) | |
| Median (Q1, Q3) | 0.1 (0, 0.9) | 0.1 (0, 1.0) | 0.2 (0.1, 0.4) | |
| Min, Max | 0, 20.2 | 0, 20.2 | 0, 12.6 | |
| Age at liver disease diagnosed, years | 0.76 | |||
| N | 255 | 199 | 56 | |
| Mean (SD) | 1.7 (3.29) | 1.8 (3.43) | 1.3 (2.71) | |
| Median (Q1, Q3) | 0.3 (0.1, 1.6) | 0.3 (0.1, 1.6) | 0.2 (0.2, 0.9) | |
| Min, Max | 0, 20.2 | 0, 20.2 | 0, 12.9 | |
| Gender, N (%) | 0.46 | |||
| Male | 164 (61.0%) | 124 (59.6%) | 40 (65.6%) | |
| Female | 105 (39.0%) | 84 (40.4%) | 21 (34.4%) | |
| Ethnicity, N (%) | 0.77 | |||
| Hispanic | 18 (6.8%) | 15 (7.4%) | 3 (4.9%) | |
| Non-Hispanic | 247 (93.2%) | 189 (92.6%) | 58 (95.1%) | |
| Race, N (%) | 0.49 | |||
| Native American | 1 (0.4%) | 1 (0.5%) | 0 (0%) | |
| Black | 3 (1.1%) | 1 (0.5%) | 2 (3.3%) | |
| Caucasian | 250 (92.9%) | 194 (93.3%) | 56 (91.8%) | |
| Multiracial | 14 (5.2%) | 11 (5.3%) | 3 (4.9%) | |
| Not Reported | 1 (0.4%) | 1 (0.5%) | 0 (0%) | |
| Genotype/phenotype, N (%) | 0.57 | |||
| SZ | 21 (7.8%) | 17 (8.2%) | 4 (6.6%) | |
| ZZ | 246 (91.5%) | 190 (91.4%) | 56 (91.8%) | |
| Missing | 2 (0.7%) | 1 (0.5%) | 1 (1.6%) |
A1AT Liver Disease Cohort includes subjects with A1AT who enroll in the study either with their native liver or after a liver transplant; they are assessed for liver disease severity at the time of their enrollment.
The Native Liver A1AT Cohort includes subjects with A1AT who enroll in the study with their native liver (pre-transplant) and are assessed for PHT at the time of their enrollment.
p-value compares the Native Liver and Post-transplant cohorts using Fisher's exact test for discrete data and the Wilcoxon rank-sum test for continuous data.
Table 2. Comparison of Demographic and Disease Characteristics by Liver Disease Severity Status at Enrollment Including; Native Liver and Post-Transplant A1AT Cohorts.
| Characteristics | Mild liver disease1(N = 148) | Severe liver disease2(N = 121) | p-value3 |
|---|---|---|---|
| Age, years | <0.0001 | ||
| N | 148 | 121 | |
| Mean (SD) | 6.6 (5.36) | 10.3 (6.94) | |
| Median (Q1, Q3) | 5.4 (2.1, 9.2) | 9.1 (4.5, 16..8) | |
| Min, Max | 0.3, 24.9 | 0.0, 24.6 | |
| Age at symptoms first noticed, years | 0.12 | ||
| N | 134 | 111 | |
| Mean (SD) | 1.1 (2.40) | 1.6 (3.85) | |
| Median (Q1, Q3) | 0.1 (0, 1.0) | 0.2 (0.1, 0.6) | |
| Min, Max | 0, 13.3 | 0, 20.2 | |
| Age at liver disease diagnosed, years | 0.55 | ||
| N | 143 | 112 | |
| Mean (SD) | 1.5 (2.60) | 1.9 (4.00) | |
| Median (Q1, Q3) | 0.3 (0.1, 1.7) | 0.2 (0.2, 1.2) | |
| Min, Max | 0, 18.3 | 0, 20.2 | |
| Gender, N (%) | 0.08 | ||
| Male | 83 (56.1%) | 81 (66.9%) | |
| Female | 65 (43.9%) | 40 (33.1%) | |
| Ethnicity, N (%) | 0.34 | ||
| Hispanic | 12 (8.3%) | 6 (5.0%) | |
| Non-Hispanic | 133 (91.7%) | 114 (95.0%) | |
| Race, N (%) | 0.13 | ||
| Native American | 0 (0%) | 1 (0.8%) | |
| Black | 0 (0%) | 3 (2.5%) | |
| Caucasian | 140 (94.6%) | 110 (90.9%) | |
| Multiracial | 8 (5.4%) | 6 (5.0%) | |
| Not Reported | 0 (0%) | 1 (0.8%) | |
| Genotype/phenotype, N (%) | 1.00 | ||
| SZ | 12 (8.1%) | 9 (7.4%) | |
| ZZ | 135 (91.2%) | 111 (91.7%) | |
| Missing | 1 (0.7%) | 1 (0.8%) |
Mild liver disease is defined as subjects with native liver who have not experienced PHT.
Severe liver disease is defined as subjects after liver transplant or those with native liver who have possible/definite PHT.
Wilcoxon rank-sum tests are used to compare liver severity groups for continuous outcomes and Fisher's exact tests are used to compare the groups for discrete outcomes.
Table 3. Comparison of Demographic and Disease Characteristics by PHT Status at Enrollment. A1AT Patients with Native Liver Only.
| Characteristics | No PHT1(N = 148) | PHT2(N = 60) | p-value3 |
|---|---|---|---|
| Age, years | 0.60 | ||
| N | 148 | 60 | |
| Mean (SD) | 6.6 (5.36) | 7.5 (6.31) | |
| Median (Q1, Q3) | 5.4 (2.1, 9.2) | 5.9 (1.8, 12.7) | |
| Min, Max | 0.3, 24.9 | 0, 21.3 | |
| Gender, N (%) | 0.12 | ||
| Male | 83 (56.1%) | 41 (68.3%) | |
| Female | 65 (43.9%) | 19 (31.7%) | |
| Ethnicity, N (%) | 0.56 | ||
| Hispanic | 12 (8.3%) | 3 (5.1%) | |
| Non-Hispanic | 133 (91.7%) | 56 (94.9%) | |
| Race, N (%) | 0.06 | ||
| Native American | 0 | 1 (1.7%) | |
| Black | 0 | 1 (1.7%) | |
| Caucasian | 140 (94.6%) | 54 (90.0%) | |
| Multiracial | 8 (5.4%) | 3 (5.0%) | |
| Not Reported | 0 | 1 (1.7%) | |
| Genotype/phenotype, N (%) | 1.00 | ||
| SZ | 12 (8.1%) | 5 (8.3%) | |
| ZZ | 135 (91.2%) | 55 (91.7%) | |
| Missing | 1 (0.7%) | 0 | |
| AST (u/l) | <0.0001 | ||
| N | 143 | 59 | |
| Mean (SD) | 85. (69.28) | 139.9 (91.00) | |
| Median (Q1, Q3) | 66 (43, 101) | 128 (67, 200) | |
| Min, Max | 14, 432 | 23, 435 | |
| ALT (u/l) | 0.03 | ||
| N | 143 | 59 | |
| Mean (SD) | 87.3 (71.83) | 100.3 (62.99) | |
| Median (Q1, Q3) | 62 (40, 110) | 89 (53, 131) | |
| Min, Max | 15, 386 | 22, 309 | |
| INR | 0.0001 | ||
| N | 89 | 55 | |
| Mean (SD) | 1.1 (0.39) | 1.2 (0.34) | |
| Median (Q1, Q3) | 1.0 (1.0, 1.1) | 1.2 (1.0, 1.3) | |
| Min, Max | 0.8, 4.6 | 0.9, 3.0 | |
| GGTP (u/l) | <0.0001 | ||
| N | 123 | 49 | |
| Mean (SD) | 124.1 (290.93) | 195.3 (358.33) | |
| Median (Q1, Q3) | 27 (18, 71) | 198 (72, 364) | |
| Min, Max | 6, 2443 | 15, 1786 | |
| WBC (× 10(x002C6)3/mm(x002C6)3) | |||
| N | 129 | 57 | 0.0036 |
| Mean (SD) | 8.7 (3.61) | 8.7 (8.85) | |
| Median (Q1, Q3) | 7.7 (6.4, 10.1) | 5.8 (3.8, 9.7) | |
| Min, Max | 3.8, 25.8 | 1.5, 56.0 | |
| AST/ALT ratio | |||
| N | 143 | 59 | <0.0001 |
| Mean (SD) | 1.1 (0.59) | 1.5 (0.64) | |
| Median (Q1, Q3) | 1.0 (0.8, 1.2) | 1.4 (1.0, 1.8) | |
| Min, Max | 0.4, 6.1 | 0.6, 3.9 |
Mild liver disease is defined as subjects with native liver who have not experienced PHT.
Severe liver disease is defined as subjects after liver transplant or those with native liver who have possible/definite PHT.
Wilcoxon rank-sum tests are used to compare liver severity groups for continuous outcomes and Fisher's exact tests are used to compare the groups for discrete outcomes.
Results
Demographics of the A1AT liver disease cohort
As of October 2012, there were 269 A1AT liver disease subjects in the cohort (Table 1). Of these, 208 had their native livers and 61 were post-liver transplant. Of the 269 subjects, 61% are male, 93% are white race, 1% African American, and 5% report multiracial. Ethnicity was >90% non-Hispanic white and 7% Hispanic, as consistent with the known genetics of A1AT deficiency, which is overwhelmingly found in Caucasians (1, 3). 246 subjects are homozygous ZZ and 21 are SZ. The mean age at which symptoms of liver disease were first noted was 1.3 years and the mean age at diagnosis of A1AT deficiency (these were usually determined retrospectively, prior to enrollment), was 1.7 years, (range 0 to 20.2 years) (1, 2, 8). There was no difference between the native liver and post liver transplant group in age when symptoms of liver disease were first noted, age at A1AT deficiency diagnosis, gender, race, ethnicity, or A1AT genotype.
Comparison of mild liver disease to severe liver disease
The range of severity of A1AT deficiency liver disease is very broad, although by definition of eligibility for study enrollment, all of the subjects in this cohort have a history of some form of liver disease. Therefore, in order to better understand the natural history and to better associate genetic and environmental factors with disease severity we have identified sub groups of mild liver disease (55% of the subjects) and severe liver disease (45% of the subjects) (Table 2 and Supplemental Data). The mild liver disease group is comprised of patients with their native livers and no evidence of portal hypertension (PHT), while the severe liver disease group includes all post-liver transplant subjects, and all subjects with their native livers and PHT (see methods for validated and published criteria)(18). 56% of the mild liver disease group are male, compared to 67% of the severe liver disease group (p=0.08). There was also no significant difference in race, ethnicity, or in the proportion of ZZ to SZ subjects between the groups. Mean age at which liver disease was first noted was 1.1 years in the mild group and 1.6 years in the severe group, which was not significantly different (Figure 1). This was felt to be an important observation, as some clinicians use early life events to make disease prognosis projections for families. Jaundice at presentation was common in all subjects, but was significantly more likely in the severe (76%) disease than the mild disease (57%) group (p=0.0024). Many subjects who presented with jaundice resolved spontaneously.
Figure 1. Age at diagnosis of the mild versus severe liver disease patients.
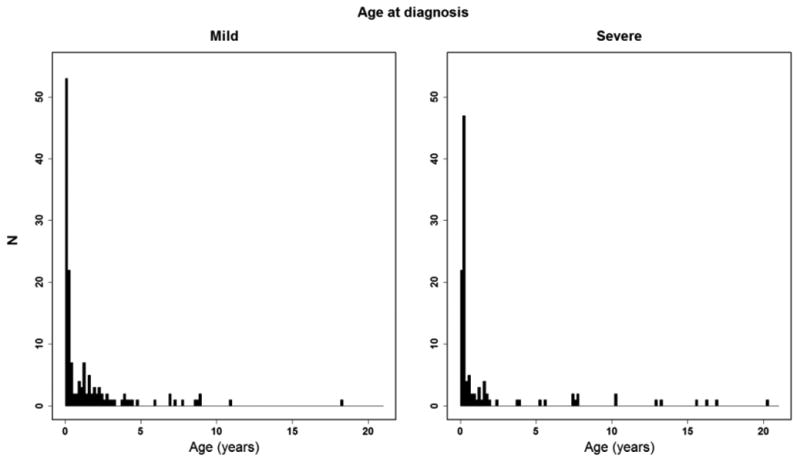
Relationship of age in years on the horizontal axis versus number of patients (N) at a given age at diagnosis on vertical axis for mild liver disease patients (left panel) and severe liver disease patients (right panel).
A history of ascites and gastrointestinal bleeding at disease presentation were significantly associated with the severe liver disease group, although they were uncommon in both groups. A history of pruritus, diarrhea, failure to thrive, bone fractures, rickets, vitamin E deficiency, and abnormal liver enzymes were not significantly different between these groups, but were also very uncommon. Jaundice, ascites, pruritus, failure to thrive and gastrointestinal bleeding when present at the time of definitive disease diagnosis, were significantly associated with the severe liver disease group (p<0.05). When signs, symptoms and complications over lifetime, including presentation and the time of definitive diagnosis, were examined, ascites, peritonitis, gastrointestinal bleeding, coagulopathy, and pruritus were all significantly associated with the severe liver disease group. It was also found that 4 subjects (3.4%) in the severe liver disease group had a history of pancreatitis, while none of the mild disease subjects were reported to have had pancreatitis (p=0.04). Pancreatitis has not been previously reported to be associated with A1AT deficiency in children.
Finally, we examined quality of life measures. Fewer parents rated their child's health as excellent in the severe liver disease group (32%) compared to the mild liver disease group (56%) (p<0.0001). There was also more likely to be “some” (49%) or “a great deal” (23%) of effect of liver disease on family life in the severe liver disease group (p<0.0001). The interesting aspect of these data is the fairly small impact on quality of life measures of this life-long and multi-organ genetic disease unless the liver disease became severe, and even then 29% of severe disease respondents report no effect on family life. When these data were examined in patients with their native livers, only, subjects without PHT had higher quality of life measures (p<0.0001) than those with PHT, but again a high quality was still present in most PHT subjects. 51.7% of those with PHT reported their health as either “very good” or “excellent” and only 10.3% answered “fair” or worse.
The role of portal hypertension in the A1AT subjects with native livers
In some parts of the analyses, for example, physical exam or liver chemistries at enrollment, it was not appropriate to compare the post-liver transplant subjects, that make up part of the severe liver disease group, to the mild liver disease with native liver subjects. It was also not possible to retrospectively determine when PHT developed in post-transplant patients. Therefore, to examine the role of portal hypertension in the cohort, we performed an analysis of the 208 subjects with native livers comparing the 148 (71%) with no PHT to the 60 (29%) with PHT (Table 3 and Supplemental Data). The data revealed no difference in the A1AT genotype, gender, race or ethnicity by PHT status. At the time of enrollment physical exam, vital signs of the two groups were not significantly different, except the mean diastolic blood pressure was mildly, but statistically significantly higher in the group with PHT. Z scores for weight, height, and head circumference, as well as BMI, were not significantly different from the normal population and not different based on PHT. There was no significant difference in the mean liver size, either span by percussion or liver edge distance from mid clavicular line, in the no PHT subjects compared to the subjects with PHT. However, a liver NOT palpable at the costal margin was found in 48% of the no PHT group and 68% of the PHT group (p<0.05). When the liver could be palpated it was statistically significantly more likely to have a hard or nodular texture in the group with PHT, but that was only found in 15% of subjects (p<0.0001). There was a small difference in the subjects with no PHT compared to those with PHT in functional heart murmur (2% versus 9%, respectively). Jaundice and icterus were rare in both groups, but icterus was associated with PHT. These data suggest that an examination focused on detecting splenomegaly, in conjunction with laboratory and imaging evaluation, is necessary to detect severe liver disease in A1AT deficiency.
Examination of laboratory parameters obtained as a part of routine care revealed frequent elevations of AST, ALT and GGT, as is common in A1AT deficiency (Table 3)(1, 3, 8-12). The mean levels were lower in the subjects with no PHT compared to those with PHT, although the overlap of the groups was large (Figure 2). The differences in magnitude of the GGT elevations associated with PHT were more clinically relevant. Examination of bilirubin revealed that the mean total and direct bilirubin levels were higher in subjects with PHT. However, most of the subjects had mild elevations that would not lead to clinical jaundice, and the overlap of the ranges of the groups was large. The median INR of the subjects with no PHT (1.1) was significantly different compared to subjects with PHT (1.2) (p=0.0001), but the group overlap was again large, and most subjects in both groups had normal INR. A1AT levels were not significantly different between the subjects with no PHT and those with PHT. The platelet count was strongly associated with the presence of PHT, as expected since it was part of the operational definition. We then reviewed past abdominal ultrasounds indicative of PHT and compared to our PHT formula. We found 79% agreement (kappa statistics) with our assignment by clinical formula (Table 4). No conclusion could be drawn from review of the large range of medications the subjects were taking, except to note that the use of ursodeoxycholic acid was found in 22% of subjects with no PHT and 50% of subjects with PHT (p<0.0001). Taken together, these data show that this cohort of A1AT subjects with liver disease has a burden of PHT, 29%, but that most of the subjects are normally grown and leading generally unrestricted lives regardless of their PHT status.
Figure 2. AST and GGTP Levels by PHT Status.
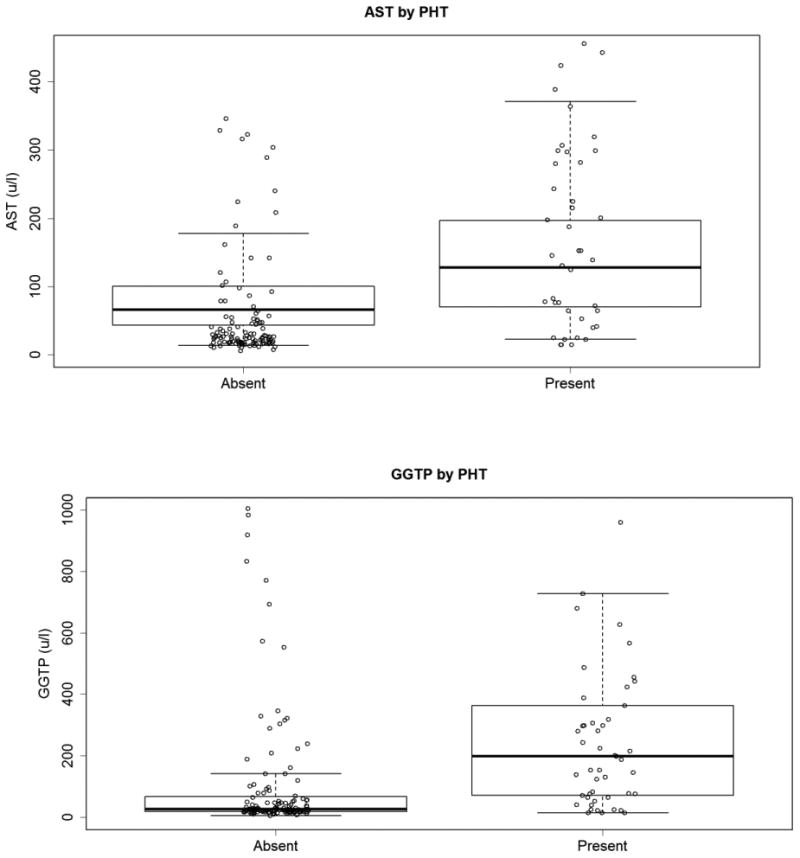
AST and GGTP levels in U/L on the vertical axis for groups with PHT absent or present. Mean values bold line, box 25th-75th quartiles (two points from the PHT Absent group [1005 and 2443 u/l] and two points from the PHT Present group [1566 and 1786 u/l] do not appear on this graph).
Table 4. Concordance of Ultrasound and Physical Examination Determination of PHT at Enrollment.
| Physical Examination Finding (Spleen > 2 cm below the costal margin) | |||
|---|---|---|---|
| Ultrasound finding of PHT based on splenomegaly | No PHT | PHT | Total |
| No PHT | 137 | 7 | 144 |
| PHT | 11 | 53 | 64 |
| Total | 148 | 60 | 208 |
| Kappa = 0.79 | |||
| 95% confidence interval = (0.70, 0.88) | |||
Discussion
The LOGIC study has been undertaken to provide previously unavailable natural history data for pediatric liver diseases in the North American population, and to identify previously unknown genetic and environmental disease modifiers. The LOGIC study includes the largest cohort of A1AT liver disease patients ever assembled. In this report of the baseline characteristics of the cohort, we show that a large proportion of the A1AT liver disease patients have fairly mild clinical involvement characterized by moderate enzyme elevations, normal liver synthetic function, normal somatic growth and development, and very little self-report of restricted activities. However, there is a significant burden of PHT which requires multifaceted examination to identify and to monitor, including careful physical examination focused on detecting splenomegaly, laboratory testing and the judicious use of imaging. Interestingly, while the mean levels of liver enzyme elevations are greater in patients with PHT, the overlap is so great that the clinical utility of simple AST, ALT, or even GGT measures to identify patients with severe liver disease is limited. These data suggest that successful management of A1AT patients, including avoidance of complications of PHT such as splenic trauma during sports, avoidance of NSAID use that would potentiate GI bleeding, identification and correction of vitamin deficiency, timing of endoscopic evaluation for esophageal varices, and possible planning for transplantation will require regular and careful follow up visits. Our data also show that a clinical identification of PHT, using the criteria described involving clinical history, splenomegaly, and platelet count, has good agreement with ultrasound determination of portal hypertension in these patients. As additional prospective data is gathered on the cohort, it may be possible to develop rational, data driven management guidelines for young patients with A1AT, which has never before been possible. Questions such as how often asymptomatic patients should be seen, both by specialists and by their primary care, and what schedule of lab and imaging should be used are often of concern to families and physicians, but have no clear answers at our present level of knowledge.
Another important observation from our analysis is that an early presentation of A1AT deficiency as neonatal cholestasis does not necessarily confer a bad prognosis in the first few years of life. This is similar to the reports from the Swedish birth cohort (8). While jaundice at disease presentation was statistically associated with severe disease, more than half of the mild liver disease subjects presenting with jaundice resolved spontaneously. Conversely, some of the severe liver disease subjects never had a history of cholestasis, and never had jaundice, but still progressed to PHT or liver transplant.
The major limitation of this study is that this is an observational, non-intervention cohort of A1AT subjects identified based on the presence of liver disease, and with a tertiary care center referral bias. Therefore, these data do not define the prevalence of liver disease, the prevalence of PHT, or the rate of liver transplant in all children with A1AT deficiency. It is likely, given the reports of the Swedish cohort and the limited reports from North American centers, that there are many asymptomatic A1AT deficient children with and without liver disease in the population that remain undiagnosed (1, 9, 10, 13). This is a challenge for both specialists and for primary care providers, as the relatively high frequency of this “rare” disease makes it certain that many young A1AT patients are being seen by physicians regularly but escaping detection. What changes in detection or management would benefit these patients remains undefined. We are also limited in that the standard of care for these children, which is the data available, does not usually include invasive assessments of PHT or cirrhosis in the absence of declining liver function or severe complications. Therefore, our assignment of PHT, while previously validated, does not include invasive investigations adults would be more commonly exposed to, such as standardized enrollment liver biopsy, endoscopy or hepatic wedge pressure.
In our continued evaluation of the ChiLDREN cohort we propose to document in the group of A1AT subjects any development of pulmonary symptoms, the rate at which patients with mild liver disease progress to PHT, the rate of progression to liver transplant, and the features of the history, physical exam and environment that are associated with progression of disease. Furthermore, we are establishing a rich repository of biospecimens and DNA, linked to the patient data, to fuel future biochemical and genetic studies into the molecular mechanisms of liver injury and the identification of genetic modifiers. A well-defined cohort such as this is also an attractive resource for future therapeutic trials, which in rare liver diseases are often limited by patient availability and the cost of data gathering. We plan continued enrollment, analysis, and data reporting from this patient cohort.
Supplementary Material
Summary Box.
A1AT deficiency is a relatively common genetic disease which often goes undiagnosed in childhood, but which can lead to significant liver morbidity and mortality in some patients.
Genetic and environmental modifiers are still being investigated.
We provide a baseline report of the ChiLDREN study, which is the largest collection of liver affected patients ever reported, and is being followed longitudinally.
Among other findings, we commonly see portal hypertension in the cohort, but a high quality of life.
Acknowledgments
Funding support: This work was supported by funding from the Alpha-1 Foundation (University of Colorado Denver and Saint Louis University School of Medicine) and by U01 grants from the National Institute of Diabetes, Digestive and Kidney Diseases and from the National Center for Advancing Translational Sciences (NCATS) UL1 grants: DK 62445 [Mt. Sinai School of Medicine], DK 62497 and UL1 TR000077 [Cincinnati Children's Hospital Medical Center, University of Cincinnati], DK 62470 [Baylor College of Medicine] DK 62470 [Children's Healthcare of Atlanta, Emory University], DK 62481 and UL1 TR000003[The Children's Hospital of Philadelphia, University of Pennsylvania], DK 62456 [University of Michigan], DK 84536 and UL1 TR000006 [Riley Hospital for Children, Indiana University], DK 84575 and UL1 TR000423[Seattle Children's Hospital, University of Washington], DK 62500 and UL1 TR000004 [UCSF Children's Hospital, University of California San Francisco], DK 62503 and UL1 TR000424 [Johns Hopkins School of Medicine], DK 62466 and UL1 UL1 TR000005 [Children's Hospital of Pittsburgh, University of Pittsburgh], DK 62453 and UL1 000154 [University of Colorado Denver, The Children's Hospital Denver], DK 62452 and UL1 TR000448[Washington University School of Medicine, St. Louis, St. Louis Children's Hospital], DK 84538 and UL1 TR000130 [Children's Hospital Los Angeles, University of Southern California], DK 62436 and UL1 TR000150 [Ann and Robert H. Lurie Children's Hospital of Chicago, Northwestern University].
Abbreviations
- A1AT
Alpha-1-antitrypsin
- PHT
Portal hypertension
- ChiLDREN
Childhood Liver Disease Research and Education Network
- LOGIC
Longitudinal Study of Genetic Causes of Intrahepatic Cholestasis
- NSAID
Non-steroidal anti-inflammatory drug
Footnotes
Clinical Trial Registry: ClinicalTrials.gov: Evaluating the Genetic Causes and Progression of Cholestatic Liver Diseases (LOGIC) NCT00571272.
The authors have no relevant conflicts of interest with the report
Contributor Information
Jeffrey H Teckman, Email: teckmanj@slu.edu.
Philip Rosenthal, Email: prosenth@peds.ucsf.edu.
Robert Abel, Email: abelr@umich.edu.
Lee M. Bass, Email: LBass@luriechildrens.org.
Sonia Michail, Email: smichail@chla.usc.edu.
Karen F. Murray, Email: karen.murray@seattlechildrens.org.
David A. Rudnick, Email: Rudnick_D@kids.wustl.edu.
Daniel W. Thomas, Email: dthomas@chla.usc.edu.
Cathie Spino, Email: spino@umich.edu.
Ronen Arnon, Email: ronen.arnon@mountsinai.org.
Paula M. Hertel, Email: phertel@bcm.edu.
James Heubi, Email: James.Heubi@cchmc.org.
Binita M. Kamath, Email: Binita.Kamath@sickkids.ca.
Wikrom Karnsakul, Email: wkarnsa1@jhmi.edu.
Kathleen M. Loomes, Email: LOOMES@email.chop.edu.
John C. Magee, Email: mageej@med.umich.edu.
Jean P. Molleston, Email: jpmolles@iupui.edu.
Rene Romero, Email: rromero@emory.edu.
Benjamin L Shneider, Email: Benjamin.Shneider@chp.edu.
Averell H Sherker, Email: averell.sherker@nih.gov.
Ronald J Sokol, Email: Ronald.Sokol@childrenscolorado.org.
References
- 1.American Thoracic Society/European Respiratory Society Statement: Standards for the Diagnosis and Management of Individuals with Alpha-1 Antitrypsin Deficiency. Am J Respir Crit Care Med. 2003;168:818–900. doi: 10.1164/rccm.168.7.818. [DOI] [PubMed] [Google Scholar]
- 2.Teckman JH. Alpha1-antitrypsin deficiency in childhood. Semin Liver Dis. 2007;27:274–281. doi: 10.1055/s-2007-985072. [DOI] [PubMed] [Google Scholar]
- 3.Lindblad D, Blomenkamp K, Teckman J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology. 2007;46:1228–1235. doi: 10.1002/hep.21822. [DOI] [PubMed] [Google Scholar]
- 4.Marcus NY, Blomenkamp K, Ahmad M, Teckman JH. Oxidative stress contributes to liver damage in a murine model of alpha-1-antitrypsin deficiency. Exp Biol Med (Maywood) 2012;237:1163–1172. doi: 10.1258/ebm.2012.012106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlmutter DH, Silverman GA. Hepatic fibrosis and carcinogenesis in alpha1-antitrypsin deficiency: a prototype for chronic tissue damage in gain-of-function disorders. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a005801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan S, Huang L, McPherson J, Muzny D, Rouhani F, Brantly M, Gibbs R, et al. Single nucleotide polymorphism-mediated translational suppression of endoplasmic reticulum mannosidase I modifies the onset of end-stage liver disease in alpha1-antitrypsin deficiency. Hepatology. 2009;50:275–281. doi: 10.1002/hep.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudnick DA, Shikapwashya O, Blomenkamp K, Teckman JH. Indomethacin increases liver damage in a murine model of liver injury from alpha-1-antitrypsin deficiency. Hepatology. 2006;44:976–982. doi: 10.1002/hep.21326. [DOI] [PubMed] [Google Scholar]
- 8.Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294:1316–1321. doi: 10.1056/NEJM197606102942404. [DOI] [PubMed] [Google Scholar]
- 9.Sveger T. The natural history of liver disease in alpha 1-antitrypsin deficient children. Acta Paediatr Scand. 1988;77:847–851. doi: 10.1111/j.1651-2227.1988.tb10767.x. [DOI] [PubMed] [Google Scholar]
- 10.Sveger T, Thelin T, McNeil TF. Young adults with alpha 1-antitrypsin deficiency identified neonatally: their health, knowledge about and adaptation to the high-risk condition. Acta Paediatr. 1997;86:37–40. doi: 10.1111/j.1651-2227.1997.tb08828.x. [DOI] [PubMed] [Google Scholar]
- 11.Mowat AP. Alpha 1-antitrypsin deficiency (PiZZ): features of liver involvement in childhood. Acta Paediatr Suppl. 1994;393:13–17. doi: 10.1111/j.1651-2227.1994.tb13200.x. [DOI] [PubMed] [Google Scholar]
- 12.Volpert D, Molleston JP, Perlmutter DH. Alpha1-antitrypsin deficiency-associated liver disease progresses slowly in some children. J Pediatr Gastroenterol Nutr. 2000;31:258–263. doi: 10.1097/00005176-200009000-00011. [DOI] [PubMed] [Google Scholar]
- 13.Wall M, Moe E, Eisenberg J, Powers M, Buist N, Buist AS. Long-term follow-up of a cohort of children with alpha-1-antitrypsin deficiency. J Pediatr. 1990;116:248–251. doi: 10.1016/s0022-3476(05)82882-3. [DOI] [PubMed] [Google Scholar]
- 14.Ibarguen E, Gross CR, Savik SK, Sharp HL. Liver disease in alpha-1-antitrypsin deficiency: prognostic indicators. J Pediatr. 1990;117:864–870. doi: 10.1016/s0022-3476(05)80123-4. [DOI] [PubMed] [Google Scholar]
- 15.Psacharopoulos HT, Mowat AP, Cook PJ, Carlile PA, Portmann B, Rodeck CH. Outcome of liver disease associated with alpha 1 antitrypsin deficiency (PiZ) Implications for genetic counselling and antenatal diagnosis Arch Dis Child. 1983;58:882–887. doi: 10.1136/adc.58.11.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pawlikowska L, Strautnieks S, Jankowska I, Czubkowski P, Emerick K, Antoniou A, Wanty C, et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol. 2010;53:170–178. doi: 10.1016/j.jhep.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sokol RJ. Reloading against rare liver diseases. J Pediatr Gastroenterol Nutr. 2010;50:9–10. doi: 10.1097/MPG.0b013e3181b47b49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamath BM, Piccoli DA, Magee JC, Sokol RJ. Pancreatic insufficiency is not a prevalent problem in alagille syndrome. J Pediatr Gastroenterol Nutr. 2012;55:612–614. doi: 10.1097/MPG.0b013e31825eff61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shneider BL, Abel B, Haber B, Karpen SJ, Magee JC, Romero R, Schwarz K, et al. Portal hypertension in children and young adults with biliary atresia. J Pediatr Gastroenterol Nutr. 2012;55:567–573. doi: 10.1097/MPG.0b013e31826eb0cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konus OL, Ozdemir A, Akkaya A, Erbas G, Celik H, Isik S. Normal liver, spleen, and kidney dimensions in neonates, infants, and children: evaluation with sonography. AJR Am J Roentgenol. 1998;171:1693–1698. doi: 10.2214/ajr.171.6.9843315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.