

Abstract
We performed untargeted metabolomics in plasma of B6 mice with experimental autoimmune encephalitis (EAE) at the chronic phase of the disease in search of an altered metabolic pathway(s). Of 324 metabolites measured, 100 metabolites that mapped to various pathways (mainly lipids) linked to mitochondrial function, inflammation, and membrane stability were observed to be significantly altered between EAE and control (p < 0.05, false discovery rate <0.10). Bioinformatics analysis revealed six metabolic pathways being impacted and altered in EAE, including α-linolenic acid and linoleic acid metabolism (PUFA). The metabolites of PUFAs, including ω-3 and ω-6 fatty acids, are commonly decreased in mouse models of multiple sclerosis (MS) and in patients with MS. Daily oral administration of resolvin D1, a downstream metabolite of ω-3, decreased disease progression by suppressing autoreactive T cells and inducing an M2 phenotype of monocytes/macrophages and resident brain microglial cells. This study provides a proof of principle for the application of metabolomics to identify an endogenous metabolite(s) possessing drug-like properties, which is assessed for therapy in preclinical mouse models of MS.
Keywords: autoimmune disease, inflammation, metabolism, metabolomics, multiple sclerosis, neuroinflammation
Introduction
Multiple sclerosis (MS)4 is a chronic inflammatory and demyelinating disease of the CNS affecting more than 2.3 million people worldwide. It is a classic example of chronic disease leading to physical, psychological, emotional, and financial burden for patients and their loved ones. Based on the disease course, MS can be classified into four categories, including relapsing-remitting MS, secondary-progressive MS, primary-progressive MS, and progressive-relapsing. Despite various therapeutic options for treatment, there is no cure for MS. Studies suggest that the most destructive changes from MS in the brain occur very early in the disease process and may cause considerable damage even before symptoms appear. MS treatments are more effective during the early course of the disease when symptoms are mild. Therefore, early diagnosis of MS is important to initiate treatments. However, no treatments have been able to delay progression of the disease, even though they improve the quality of a patient's life (1) as a result of fewer exacerbations. Significant effort has been made to identify biomarkers from cerebrospinal fluid to diagnose MS, and this endeavor has proven to be challenging without success (2). The analysis of easily drawn bio-fluids, including blood for MS biomarkers, has only been minimally investigated but holds significant promise. The pathology of MS is associated with inflammatory response, glial activation, and scarring and demyelination that likely result in a systematic change in the circulatory metabolome. Thus, altered plasma metabolites may be a valuable approach as a surrogate for the disease.
Recently, metabolic aberrations have been defined in various disease processes, as either contributing to the disease as potential biomarkers or therapeutic targets (3). Metabolomics is the global exploration of endogenous small molecule metabolites of cellular processes in the biological system, including cell, tissue, organ, or organism (4, 5). Therefore, metabolic profiling can provide a window to the instantaneous physiological or pathological changes as a complement to transcriptomic and proteomic profiling for the systematic and functional study of living organisms (6, 7). Recent reports are implicating the importance of metabolomics in the possible identification of biomarkers in neurological disorders, including Alzheimer disease (8, 9), Parkinson disease (10), and in animal models of MS (11–14). We have previously performed a comprehensive analysis of plasma metabolites in the RR preclinical mouse model of MS (SJL-experimental autoimmune encephalitis (EAE)) (14) and identified 44 metabolites that distinguished the EAE from control mice. These metabolites were associated with alterations in lipid, amino acid, nucleotide, and xenobiotic metabolism (14).
This study examined plasma from a chronic B6 mouse model of MS to determine the metabolic changes occurring in chronic EAE compared with the control B6 group. Bioinformatics analysis has revealed that metabolism of the PUFA pathway (ω-3 and ω-6 metabolism) is down-regulated in the plasma of EAE, which has also been reported in patients with MS (15–19). We therefore examined the therapeutic potential of resolvin D1 (RvD1; 7S,8R,17S-trihydroxy-docosa-4Z,9E,11E,13Z,15E,19Z-hexaenoic acid) (20), a downstream metabolite of ω-3 pathways in the EAE disease progression.
Experimental Procedures
Animals
Female B6 and SJL mice (10–12 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed in the pathogen-free animal facility of Henry Ford Hospital, Detroit, MI, according to the animal protocols approved by the Animal Care and Use Committee of Henry Ford Hospital.
Peptide and Reagents
Murine myelin-oligodendrocyte glycoprotein peptide (MOG(35–55)) and proteolipid protein peptide (PLP(139–151)) were synthesized at New England Peptide, Inc. (Gardner, MA). Complete Freund's adjuvant and mycobacterium tuberculosis lyophilized powder were purchased from Difco. Labeled antibodies to anti-mouse CD4 (FITC; RM4-5), CD25 (PE; PC61), CD80 (Percp/cy5.5; 16-10A1), CD206 (APC; C068C2), and class II (I-Ab; FITC; KH74) for FACS analysis were purchased from BioLegend (San Diego). Anti-CD11b (PE-Cy7; M1/70), CD45 (eFluor 450; 30-F11), CD25 (APC-Cy7; PC61.5), IL17a (PerCp-Cy5.5; eBio17B7), IL4 (PE; 11B11), CD4 (APC; RM4–5), and FoxP3 (PE-Cy5; FJK-16s) were purchased from eBioscience (San Diego). Anti-Gr1 (APC-Cy7; RB6–8C5), CD86 (AF700; GL1), IFNγ (PE-CF594; XMG1.2), and class II (I-A/I-E; M5/114.15.2) were purchased from BD Biosciences. Anti-mouse scavenger receptor (PE; 214012) was purchased from R&D Systems, Inc. (Minneapolis, MN). ELISA kits for IFNγ, IL17a, IL6, IL23p19, and IL4 were purchased from BioLegend. RvD1 and its ELISA kit were purchased from Cayman (Ann Arbor, MI). The iScript complementary DNA synthesis kit and SYBR Green PCR master mix were purchased from Bio-Rad.
EAE Induction, Treatment, and Recall Response
B6 and SJL mice (10–12 weeks old) were immunized on day 0 by subcutaneous injections in the flank region with a total 200 μl of emulsion containing MOG(35–55) peptide (300 μg/mouse) or PLP(139–151) (100 μg/mouse), along with killed Mycobacterium tuberculosis H37Ra (400 μg) as described before (21). B6 mice were given pertussis toxin (300 ng/mouse) in a volume of 200 μl in PBS intraperitoneally on days 0 and 2 post-immunization. Pertussis toxin was not injected in SJL mice. One set of mice was injected with complete Freund's adjuvant/pertussis toxin without peptide named as control. Clinical disease was monitored daily in a blinded fashion by measuring paralysis according to the following conventional grading system: 0, no disease; 1, complete loss of tail tonicity; 2, partial hind limb paralysis (uneven gate of hind limb); 3, complete hind limb paralysis; 4, complete hind and forelimb paralysis; and 5, moribund or dead. On day 45, blood was collected for plasma isolation for metabolomics. Cells isolated from lymph nodes (2 × 106/ml) of EAE and control mice were cultured in the presence or absence of peptide (20 μg/ml). Cell proliferation and the production of various cytokines (IFNγ and IL17a) were examined as described before (21). On the same day, blood was drawn from both groups to isolate plasma for metabolomics analysis (in case of B6-EAE). For RvD1 treatment, RvD1 was given orally using a 22-gauge needle with a 1.25-mm ball diameter or intraperitoneally at a dose of 100 ng/mouse in the volume of 100 μl in PBS. In the control EAE group, 100 μl of PBS was given as vehicle.
Histology and Pathology of the Spinal Cord
Following perfusion with Trump's fixative, spinal cords were processed for histology and pathology on day 45 post immunization as described before (14).
Metabolite Analysis
Metabolomic profiling analysis was performed by Metabolon Inc. (Durham, NC), as described previously (14, 22–25).
Sample Preparation for Global Metabolomics
Samples were stored at −80 °C until processed. Sample preparation was carried out as described previously (26) at Metabolon, Inc. Briefly, recovery standards were added prior to the first step in the extraction process for quality control purposes. To remove protein, dissociated small molecules were bound to protein or trapped in the precipitated protein matrix, and to recover chemically diverse metabolites, proteins were precipitated with methanol under vigorous shaking for 2 min (Glen Mills Genogrinder 2000) followed by centrifugation. The resulting extract was divided into five fractions as follows: one for analysis by ultra-high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS; positive ionization), one for analysis by UPLC-MS/MS (negative ionization), one for the UPLC-MS/MS polar platform (negative ionization), one for analysis by gas chromatography-mass spectrometry (GC-MS), and one sample was reserved for backup.
Three types of controls were analyzed in concert with the experimental samples as follows: samples generated from a small portion of each experimental sample of interest served as technical replicate throughout the data set; extracted water samples served as process blanks; and a mixture of standards added to every analyzed sample allowed instrument performance monitoring. Instrument variability was determined by calculating the median relative standard deviation (RSD) for the standards that were added to each sample prior to injection into the mass spectrometers (median RSDs were determined to be 4–5%, depending on the matrix tested; n ≥ 30 standards). Overall process variability was determined by calculating the median RSD for all endogenous metabolites (i.e. non-instrument standards) present in 100% of the pooled human plasma or client matrix samples (median RSD = 6–12%, depending on the matrix tested; n = several hundred metabolites). Experimental samples and controls were randomized across the platform run.
Mass Spectrometry Analysis
Non-targeted MS analysis was performed at Metabolon, Inc. Extracts were subjected to either GC-MS or UPLC-MS/MS (1). The chromatography was standardized, and once the method was validated, no further changes were made. As part of Metabolon's general practice, all columns were purchased from a single manufacturer's lot at the outset of experiments. All solvents were similarly purchased in bulk from a single manufacturer's lot in sufficient quantity to complete all related experiments. For each sample, vacuum-dried samples were dissolved in injection solvent containing eight or more injection standards at fixed concentrations, depending on the platform. The internal standards were used both to ensure injection and chromatographic consistency. Instruments were tuned and calibrated for mass resolution and mass accuracy daily.
The UPLC-MS/MS platform utilized a Waters Acquity UPLC with Waters UPLC BEH C18–2.1 × 100 mm, 1.7-μm columns and a Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer interfaced with a heated electrospray ionization (HESI-II) source and Orbitrap mass analyzer operated at 35,000 mass resolution. The sample extract was dried and then reconstituted in acidic or basic LC-compatible solvents, each of which contained eight or more injection standards at fixed concentrations to ensure injection and chromatographic consistency. The first aliquot was analyzed using acidic positive ion-optimized conditions, and the second aliquot was analyzed using basic negative ion-optimized conditions in two independent injections using separate dedicated columns (Waters UPLC BEH C18–2.1 × 100 mm, 1.7 μm). Extracts reconstituted in acidic conditions were gradient-eluted using water and methanol containing 0.1% formic acid, although the basic extracts, which also used water/methanol, contained 6.5 mm ammonium bicarbonate. The third aliquot was analyzed via negative ionization following elution from a HILIC column (Waters UPLC BEH amide 2.1 × 150 mm, 1.7 μm) using a gradient consisting of water and acetonitrile with 10 mm ammonium formate. The MS analysis alternated between MS and data-dependent MS2 scans using dynamic exclusion, and the scan range was from 80 to 1000 m/z.
The samples destined for analysis by GC-MS were dried under vacuum desiccation for a minimum of 18 h prior to being derivatized under dried nitrogen using bistrimethylsilyltrifluoroacetamide. Derivatized samples were separated on a 5% diphenyl, 95% dimethylpolysiloxane-fused silica column (20 m × 0.18 mm inner diameter; 0.18-μm film thickness) with helium as carrier gas and a temperature ramp from 60 to 340 °C in a 17.5-min period. All samples were analyzed on a Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole MS using electron impact ionization and operated at unit mass resolving power. The scan range was from 50 to 750 m/z.
Compound Identification, Quantification, and Data Curation
Metabolites were identified by automated comparison of the ion features in the experimental samples to a reference library of chemical standard entries that included retention time, molecular weight (m/z), preferred adducts, and in-source fragments as well as associated MS spectra and curated by visual inspection for quality control using software developed at Metabolon (23, 27). Identification of known chemical entities is based on comparison with metabolomic library entries of purified standards. Commercially available purified standard compounds have been acquired and registered into LIMS for distribution to the various UPLC-MS/MS platforms for determination of their detectable characteristics. Additional mass spectral entries have been created for biochemicals that have been identified by virtue of their recurrent nature (both chromatographic and mass spectral) and classical structural analysis but have not been confirmed with purified standards. Peaks were quantified using area-under-the-curve.
Determination of RvD1 Plasma Levels by ELISA
Mouse plasma (100 μl) from control and EAE (B6 or SJL) was mixed with 9 volumes of sodium acetate (0.1 m, pH 5.0) and passed through solid-phase extraction (SeP-Pak; C-18 columns, Water). Samples were eluted with methanol and were dried under nitrogen. Samples were suspended in 500 μl of buffer (1 m phosphate solution containing 1% BSA, 4 m NaCl, 10 mm EDTA, and 0.1% sodium azide), and 50 μl was used for ELISA as per the manufacturer's protocol (Cayman Chemicals).
Flow Cytometry
For staining surface markers, cells were incubated with fluorochrome-conjugated antibodies to CD4, CD11b, Gr1, CD206, scavenger receptor, CD45, CD25, FoxP3, CD80, CD86, and class II at the recommended dilution for antibodies for 30 min at 4 °C. To analyze antigen-specific T helper (Th)1 and Th17 cells, spleen cells or CNS-infiltrating mononuclear cells were stimulated with 20 μg/ml of PLP(139–151) peptide for 18 h, followed by treatment with GolgiPlug for 5 h. Cells were surface-stained with monoclonal antibodies against CD4, then cells were washed, fixed, and permeabilized with cytofix/cytoperm buffer, and intracellular cytokines were stained with antibodies against IFNγ and IL17A. Flow cytometric analysis was performed on FACSCalibur (BD Biosciences), and results were analyzed using FACSDiva software (BD Biosciences).
Adoptive Transfer of PLP-specific T Cells to SJL Mice
SJL mice (8–10 weeks old) were immunized subcutaneously with 100 μg of PLP(139–151) per mouse in complete Freund's adjuvant. Post-10 days, we pressed spleen and lymph node cells through a 100-μm mesh into cold PBS to isolate them, and then cells were pelleted at 450 × g for 10 min at 4 °C. Spleen/lymph node cells were resuspended in red blood cell lysis buffer. Post-5 min incubation, cells were washed three times with cold PBS and counted with the hemocytometer using 0.4% trypan blue (Sigma). Spleen/lymph node cells were cultured in a 24-well plate (Corning Inc.) at 5 million cells/ml in Roswell Park Memorial Institute 1640 medium supplemented with 10% FBS, 1.25% HEPES buffer, 1% sodium pyruvate, 1% penicillin/streptomycin, 1% glutamine, 0.01% 2 mercaptoethanol (Sigma) for 72 h with PLP (20 μg/ml). PLP(139–151)-primed cells were harvested, washed with cold PBS, and counted using trypan blue to check cell viability, and 10 million cells were injected in a volume of 200 μl i.p. in each recipient SJL mouse. Mice received 200 ng of pertussis toxin at days 0 and 2 and were observed daily for disease onset and severity.
Antigen-specific Response
CD4+ T cells were isolated from PLP(139–151)-immunized SJL mice using EasyStep mouse CD4+ve selection kit (92% pure) from Stemcell Technology (Vancouver, Canada). These cells (2 × 106/ml) were cocultured with adherent macrophages from RvD1-treated and untreated groups in a ratio of 1:5 and incubated for 72 h. Cell proliferation was examined using Wst1 reagent (Promega Corp., Madison, WI), and cell supernatant was used for IFNγ and IL-17 analysis using an ELISA kit (BioLegend).
Coculture Studies
Mouse brain microglial cells were cultured from 1- to 3-day-old pups, and microglia were isolated by shaking flasks of mixed glia at 100 rpm for 1 h at 37 °C. Suspended cells were 90% pure for macrophage/microglial cell-specific marker (Iba1). Primary microglial cells were treated with RvD1 (100 nm) or vehicle (0.1% ethanol) for 24 h, and cells were processed for the expression of arginase 1 and chitinase-3-like-3 (YM1)/2 by quantitative PCR, and data were normalized with ribosomal protein L27 (L27) housekeeping gene, whose expression did not change under any of the experimental conditions studied. For coculture study with oligodendrocyte progenitor cells, treated and untreated microglial cells with RvD1 for 24 h were washed and cocultured with rat oligodendrocyte progenitor cells. Post-24 h of coculture, cells were processed for expression of myelin genes, including myelin-associated glycoprotein, MOG, and myelin basic protein by quantitative PCR, and data were normalized to the control gene ribosomal L27 housekeeping gene.
Statistical Analysis of Metabolites
Metabolites with missing intensity scores, indicating low levels of the metabolite in the sample, were imputed with a small number (half of the minimum value for the study). Principal component analysis was used to detect outlying samples. Partial least squares discriminant analysis was used for assessment of separability of the samples. A z-score plot was drawn with the z-scores based on the mean and standard deviation of the control samples per metabolite. t tests, allowing unequal variance, were used to compare changes in mean expression per metabolite between the control and disease groups. An estimate of the false discovery rate (q-value) was calculated to take into account the multiple comparisons that occur in metabolomic studies. A metabolite was considered to be statistically different when p < 0.05 and q < 0.10. Those significant metabolites were included in a heat map using metabolite-level normalized data. Samples were clustered by complete linkage on Pearson's correlation, and rows are ordered by direction of change and then molecule type. Statistical analyses were conducted using log10-transformed data.
Pathway Analysis of Significantly Altered Metabolites Included the Following Approaches
Metaboanalyst was used to assess 82 murine-associated Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways against differential metabolites and also for metabolite enrichment. Both over-representation analysis using the hypergeometric test and the impact of metabolite changes based on the pathway topology using the relative betweenness centrality measure (28) were considered. Metabolites were mapped to the KEGG pathways using Human Metabolome Database numbers; n = 75 were retained, which showed significance compared with control.
Statistical Analysis for EAE Studies
We utilized GraphPad Prism software (GraphPad Software Inc.) for statistical analysis. Student's t tests were employed to analyze clinical disease score. Statistics for densitometric values comparison for proliferation and cytokine responses were analyzed with one-way multiple-range analysis of variance and Student's t test. A value of p < 0.05 was considered significant.
Results
Characterization of the Clinical Pathological State of Chronic-progressive EAE Model
To identify the global metabolic changes in the plasma of a chronic preclinical mouse model of MS, distinct from control mice, we employed the approach of untargeted global metabolomics. EAE was induced in B6 mice using MOG(35–55) peptide as described previously (21, 29). As shown in Fig. 1A, B6 mice displayed a chronic-progressive clinical course. At the effector/chronic phase of disease (day 45), blood for plasma was collected for metabolomic profiling. Spinal cords were also collected and processed for CNS pathology recording inflammation, demyelination, and axonal loss. The lymph nodes were processed for recall response to identify the relationship between altered metabolic changes, clinical pathology, and immune response in B6 mice with EAE. Histological analysis of spinal cords showed that the EAE group displayed extensive demyelination, whereas the control group looked completely normal (Fig. 1B). Upon recall response in lymph node cells, we observed the production of pro-inflammatory cytokines (IFNγ and IL17) (Fig. 1, C and D). Overall, this set of data characterizes the clinical and pathological state of EAE in B6 mice obtained at day 45 from disease induction.
FIGURE 1.
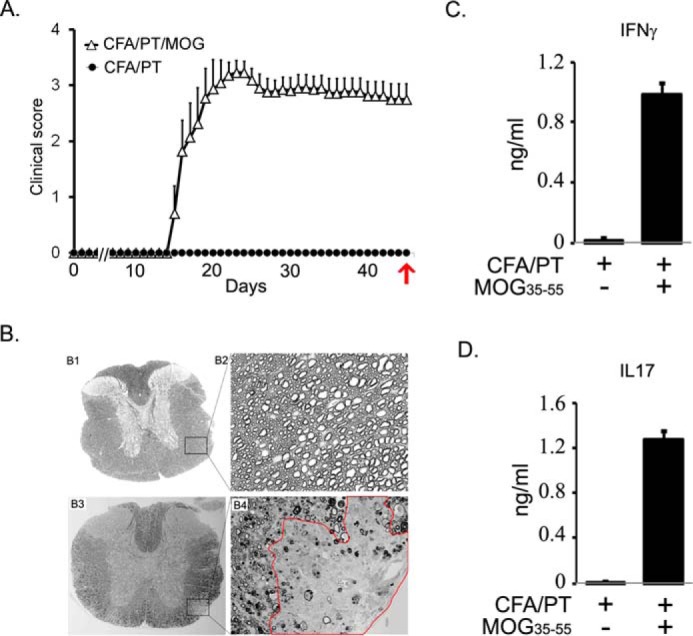
Characterization of clinical pathological state of EAE in chronic mouse model in B6. A, EAE was induced in C57B6 mice using MOG(35–55) peptide emulsified in CFA, and pertussis toxin was given on days 0 and 2 post-immunization. Clinical score was recorded daily (n = 10). The control group was given complete Freund's adjuvant/pertussis toxin without a peptide. On day 45, blood was drawn for analysis of isolated plasma. Red arrow indicates the time of sampling. B, at the end of the experiment, spinal cords were harvested from control (panels B1 and B2) and diseased mice (panels B3 and B4) to examine demyelination. Panels B1 and B3 show the whole mid-thoracic spinal cord section, and panels B2 and B4 show closer images from antero-lateral areas of the white matter. Completely normal and healthy axons can be appreciated on panel B2, and on panel B4 an area with clear demyelination is outlined in red. In addition, in areas that surround the demyelinating lesion, a patchy demyelination, dysmorphic and collapsed axons can be observed, too. C and D, recall response in cells isolated from lymph nodes, stimulated with 50 μg of MOG(35–55) for 72 h. Cell supernatant was used for measuring the levels of IFNγ and IL17 by ELISA (n = 4).
Cohort of Global Metabolomics
To characterize the differential plasma metabolite profiles of B6 EAE compared with controls, metabolomics profiling was performed on three independent instrument platforms, one GC-MS and two LC-tandem MS/MS platforms. A total of 324 structurally named biochemicals in plasma were identified and detected across both groups. Metabolite levels were calculated by automated comparison of the ion features in the experimental samples to a reference library of chemical standard entries. There was 1.5–4.3% missing data per sample, which were imputed with the minimum observed value for the experiment. Following log transformation (base 10), Welch's two-sample t test was used to identify biochemicals that differed significantly between experimental groups. In plasma, a total of 100 metabolites (30.9% of the 324 metabolites detected) was significantly altered; 13 metabolites were significantly increased, and 87 metabolites were significantly decreased in the EAE group compared with the controls (p < 0.05 with false discovery rate <0.10) (supplemental Table S1). Partial least squares discriminant analysis revealed a clear separation of EAE and control groups based on their metabolite profile (n = 5 per group; Fig. 2A). To discriminate these metabolite alterations, the intensities in the EAE samples (red dots) were plotted relative to the distribution of the intensities in the control groups (blue dots) (Fig. 2B). Each dot in the z-score plot represents one observation of one metabolite (rows) organized by major metabolic pathway. The magnitude is the number of standard deviations above (positive value) or below (negative value) the average value of the control group observation. To visualize the relationship between the 100 altered metabolites, a heat map was drawn with the metabolites arranged on the basis of relative change (up/down compared with control) and then by super pathway (Fig. 2C), and samples were ordered by hierarchical clustering. The chart in Fig. 2D enumerates these alterations indicating that the most altered metabolites belonged to the lipid (57%) and amino acid (19%) pathways, followed by the peptide (9%), xenobiotic (5%), carbohydrate (4%), nucleotide (4%), and energy (2%) pathways.
FIGURE 2.

Metabolomics profiling of plasma distinguishes EAE from the control group. A, metabolic profiling reveals clear separation between plasma from EAE (disease; D) and vehicle control mice (sham; S) by partial least squares discrimination analysis. B, z-score plot of EAE metabolite intensities (red, truncated at z-score −15 and +15) against control group metabolites (blue, taken as mean). Each dot represents one metabolite observation for one sample. C, heat map of significantly altered metabolites arranged according to direction of change and the super-metabolic pathway they appear. Yellow represents high, and blue represents the low intensity of the metabolite relative to its mean intensity (black). Each of the five replicates of plasma from vehicle control (S) and EAE (D) are arranged by hierarchical clustering. D, chart representing the number of altered metabolites within each superpathway of metabolism, specifically lipids, amino acids, peptides, xenobiotics, carbohydrates, nucleotides, and energy-related.
To understand the functional role of these alterations in plasma, the KEGG metabolic library was analyzed using Metaboanalyst (Fig. 3A) (30, 31). We assessed both a test for over-representation of altered metabolites within a pathway (hypergeometric tests) (32) and for the impact of the changed metabolites on the function of the pathway through alterations in critical junction points of the pathway (relative betweenness centrality) (28). The results of each of the 82 mouse pathways of KEGG were simultaneously plotted to show the most significant pathways in terms of hypergeometric test p values (vertical axis, shades of red) and impact (horizontal axis, circle diameter) (Fig. 3A). The top six pathways in plasma by p value (top four) or impact (top two) were indicated, including the following: 1) biosynthesis of unsaturated fatty acids (α-linolenic acid and linoleic acid metabolism); 2) glutamine and glutamate metabolism; 3) fatty acid biosynthesis; 4) pentose pathway; 5) tryptophan metabolism; and 6) arachidonic acid metabolism (Fig. 3B). Alteration in these pathways in plasma during chronic EAE disease suggests that perturbation of certain central metabolites could have an impact on multiple metabolic pathways that are interconnected.
FIGURE 3.

Metaboanalyst analysis of altered metabolites in plasma isolated from EAE compared with vehicle controls. A, metaboanalyst analysis of the KEGG metabolic library. Both the over-representation of altered metabolites within the pathway (hypergeometric test) and the impact of the changed metabolites on the function of the pathway through alterations in critical junction points of the pathway (relative betweenness centrality) were assessed. Results of each of the 82 mouse pathways of KEGG are simultaneously plotted to show the most significant pathways in terms of hypergeometric test p value (vertical axis as −log(p), shades of red) and impact (horizontal axis, circle diameter). B, top six pathways that arise with low p values (−log(p) >15) or with “high” impact (impact >0.3).
Most altered metabolites were categorized as lipids, suggesting lipid metabolism was heavily perturbed in the chronic model of EAE (Figs. 2D and 4A). A pathway enrichment overview (Metaboanalyst 3.0) of altered metabolites highlights α-linolenic acid and linoleic acid metabolism (PUFA) as being significantly enriched in quantitative metabolomics of plasma in EAE versus control mice (Fig. 4B). This pathway was also heavily impacted in the RR-EAE model (14). Moreover, metabolites of PUFAs were found to be significantly lower in the mouse chronic model of EAE (Fig. 4C and supplemental Table S1) and in patients with MS (15–19), suggesting the importance of PUFA in MS. α-Linolenic acid is the true essential ω-3 fatty acid found in plants. It is similar to the ω-3 fatty acids in fish oil, called eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). In the human body, α-linolenic acid is metabolized into EPA and DHA, which are enzymatically metabolized into eicosanoids, and ultimately to anti-inflammatory mediators, such as resolvins and protectins. Resolvins and protectins are known to have potent anti-inflammatory and pro-resolving actions in inflammatory models (33). We decided to use RvD1, a downstream metabolite of EPA-DHA, to examine its therapeutic potential in the EAE model. RvD1 has been proven to be very potent in treating a number of inflammation-associated human diseases, including peritonitis (34), dextran sulfate sodium-induced colitis (35), sepsis (36), adjuvant-induced arthritis (37), asthma (38), and immune complex-induced inflammation (39). There are no reports examining the effect of RvD1 in EAE disease progression.
FIGURE 4.

Visualization of significantly altered lipid pathway changes in EAE. A, lipids found significantly down-regulated in EAE (p < 0.05) are shown as blue nodes, and up-regulated lipids are shown in yellow. Unchanged and undetected lipids are shown as white and gray nodes, respectively. B, metabolite enrichment pathway (Metaboanalyst 3.0) overview highlights PUFAs being significantly enriched in the metabolomic profile of plasma of EAE versus control. C, ω-3 and ω-6 PUFAs are highlighted, and key metabolites in the plasma of vehicle control and EAE groups are presented as a bar graph (n = 5). p < 0.05 is considered significant.
RvD1 Treatment Attenuates the Disease Progression in EAE Model
We examined the therapeutic potential of RvD1 in EAE. The level of RvD1 in plasma of both EAE mouse models, including RR-EAE and chronic EAE, was found to be significantly reduced during disease compared with the control group in both models (Fig. 5A). Next, we examined whether supplementation of RvD1 in an RR-EAE mouse model could affect the progression of the disease. For this, we induced EAE in SJL mice and started oral administration of RvD1 at a dose of 100 ng/mouse on day 0 of immunization. Dose and route of administration of RvD1 was chosen based on a recent study where after oral administration, RvD1 (100 ng/mouse) rapidly accumulated in plasma and significantly reduced leukocyte infiltration in zymosan A-induced acute peritonitis (40). In EAE, RvD1 treatment did not show any effect on disease onset; however, there was a significant reduction in disease severity at the peak of the disease course compared with the vehicle-treated group (Fig. 5B and Table 1). Vehicle-treated groups exhibited two courses of relapses, whereas the RvD1 treatment group showed only one relapse with a lesser degree of severity (Fig. 5B). We also examined its therapeutic potential in a chronic model of EAE, induced in B6 mice by immunization with MOG(35–55) (21, 29). Oral administration of RvD1 treatment (100 ng/mouse) provided significant protection in the clinical symptoms in the chronic EAE model when given on day 7 post-immunization (Fig. 5C and Table 1). It reduced severity at the onset (0.4 ± 0.27) and the peak (1.1 ± 0.63) of the disease compared with the vehicle-treated group (1.4 ± 0.44 and 3.1 ± 0.12, respectively) (Fig. 5C). We also examined the effect of RvD1 given by a different route, i.e. intraperitoneally, and we found it equally effective as oral in terms of reducing clinical symptoms in the chronic EAE model (Fig. 5C and Table 1). Overall, RvD1 treatment significantly suppressed the severity of the disease in both models of EAE. Histological examination at day 22 post-immunization demonstrated that the RvD1-treated group had reduced CNS infiltration and demyelination as compared with RR-EAE mice treated with vehicle (Fig. 5D).
FIGURE 5.

Oral administration of RvD1 attenuates EAE disease progression. A, levels of RvD1 in plasma of RR- and chronic EAE by enzyme immunoassay (n = 9–10). B, SJL mice were immunized with PLP(139–151) on day 0 in complete Freund's adjuvant. One set of the group was given daily RvD1 from day 0 by gavage and another set was given vehicle in the same manner. Red line indicates the duration of treatment. Clinical score was taken until the end of the study (n = 10). C, B6 mice were immunized with MOG(35–55) on day 0 in complete Freund's adjuvant. One set of the group were given daily RvD1 from day 7 by gavage or intraperitoneally, and another set was given vehicle in the same manner. Clinical score was measured daily until end of the study (n = 8). Red line indicates the duration of treatment. D, photographs of spinal cord sections show inflammation (H&E; ×4 magnification) and demyelination (Luxol fast blue-periodic acid-Schiff; LFB/PAS, ×10 magnification) in both groups. Cells were stimulated with phorbol 12-myristate 13-acetate/ionomycin for 4 h in the presence of GolgiPlug. E, spleen cells were harvested and stimulated with PLP(139–151). Post-96 h, cell supernatants were used for ELISA (n = 4). F and G, for quantitative PCR, cells were harvested at 24 h post-stimulation with peptide, and expression of target genes was examined using quantitative PCR after normalizing with ribosomal protein L27 (n = 4). H, at 96 h of peptide stimulation, cells were stimulated with phorbol 12-myristate 13-acetate/ionomycin for 4 h in the presence of GolgiPlug. IL17a-, IFNγ-, and regulatory T cells (CD4+CD25+FoxP3)-expressing cells were measured by intracellular staining on a CD4+ gate. ***, p < 0.001; **, p < 0.01, and *, p < 0.05 compared with EAE.
TABLE 1.
Efficacy of RvD1 in EAE
** indicates p < 0.01 compared with vehicle-treated EAE (unpaired t test using GraphPad Prism software).
| Groups | Treatment | No. | Incidence | EAE day onset | EAE score at peak | EAE maximum score | EAE cumulative score |
|---|---|---|---|---|---|---|---|
| RR-EAE (SJL) | EAE- vehicle | 10 | 10 | 15 | 3.1 ± 0.12 | 3.1 ± 0.12 | 49 ± 11.2 |
| RvD1-treated; oral | 10 | 8 | 16 | 1.1 ± 0.63** | 1.5 ± 0.67** | 13.35 ± 14.6 | |
| Chronic-EAE (B6) | EAE- vehicle | 6 | 6 | 13 | 2.9 ± 0.4 | 3.1 ± 0.325 | 30.6 ± 7.2 |
| RvD1-treated; oral | 6 | 6 | 13 | 1.2 ± 0.41** | 1.3 ± 0.41** | 11.8 ± 5.9 | |
| Chronic-EAE (B6) | EAE-vehicle | 8 | 6 | 14 | 3.1 ± 0.12 | 3.2 ± 0.3 | 33 ± 6.4 |
| RvD1-treated (i.p.) | 8 | 6 | 14 | 1.2 ± 0.41** | 1.4 ± 0.27** | 13.3 ± 6.7 | |
| Adoptive transfer | |||||||
| Chronic-EAE (B6) | Vehicle | 4 | 4 | 22 | 1.67 ± 0.5 | 1.67 ± 0.5 | 70 ± 13.1 |
| RvD1-treated | 4 | 2 | 25 | 0.25 ± 0.14** | 0.5 ± 0.28** | 18 ± 5.2 | |
To evaluate whether RvD1 treatment altered EAE by modulating the autoreactive T cell immune response required for development of the disease, we isolated spleen cells from PLP(139–151) immunized groups either treated with or without RvD1 on day 22 post-immunization and stimulated them with PLP(139–151) (20 μg/ml). Post-72 h of stimulation, the status of pro- and anti-inflammatory cytokines was profiled in the supernatant by ELISA. RvD1-treated groups displayed significantly reduced levels of IFNγ, IL17a, IL23p19, and IL6 with significantly higher levels of IL5 (Fig. 5E). However, no change in the levels of IL4 was detected (data not shown). Consistent with protein level, the mRNA expression levels of IFNγ, IL17, IL23p19, and IL6 were down-regulated in the RvD1-treated group compared with EAE (Fig. 5F). We also examined the expression of signature-transcription factors, including T-box transcription factor (T-bet) and retinoic acid receptor-related orphan receptor γ (RORγ) for Th1 and Th17, respectively. We observed that spleen cells isolated from RvD1-treated groups exhibited significantly lower expression of T-bet and RORγ without affecting GATA3 expression, which explained the decreased levels of IFNγ and IL17, with no effect on IL4, when compared with the vehicle-treated EAE group (Fig. 5G). PLP(139–151)-primed CD4 T cells from RvD1-treated mice showed a 70- and 3.4-fold reduction in IFNγ and IL17 intracellular staining, respectively, compared with the EAE group; however, there was ∼1.9-fold induction in regulatory T cells (CD4+CD25+FoxP3+) in the treated group (Fig. 5H).
To examine whether RvD1 treatment could affect the encephalitogenic property of T cells, we immunized SJL mice with PLP(139–151), and one set of mice was given RvD1 (100 ng/mouse in 200 μl of PBS, daily by gavage), and another set received vehicle (1 μl of ethanol in 200 μl of PBS). Post-10 days, we isolated spleen and lymph node cells from both sets of mice and restimulated with PLP(139–151) (20 μg/ml). After 96 h of incubation, PLP(139–151)-primed T cells (107 cells per mouse intraperitoneally) from vehicle and RvD1-treated EAE groups were adoptively transferred into healthy SJL mice. Mice receiving PLP(139–151)-primed T cells from the RvD1-treated group exhibited a significantly less severe degree of clinical symptoms compared with the adoptive transfer of PLP(139–151)-primed T cells from the vehicle-treated group (Fig. 6A and Table 1). These results suggest that RvD1 treatment altered the generation of encephalitogenic T cells.
FIGURE 6.

Oral administration of RvD1 attenuates CNS inflammation. A, spleen cells were isolated from RvD1-treated and -untreated EAE groups and cultured with PLP(139–151). Post-72 h, PLP(139–151) primed T cells were harvested and washed, and 10 million cells were adoptively transferred in a volume of 200 μl via an intraperitoneal route in recipient mice (SJL). Mice were given 300 ng of pertussis toxin at days 0 and 2 and were observed every day for disease onset and disease (n = 4). *, p < 0.05 compared with adoptive transfer of vehicle-treated PLP(139–151)-primed T cells to SJL mice. B, BILs (brain and spinal cord) were isolated using a Percoll gradient as described under “Experimental Procedures.” The percentage of Th1/Th17/Th2 cells in the CD4 subset was analyzed by FACS through intracellular staining of IFNγ and IL17a in BILs isolated from RvD1-treated and -untreated EAE groups on day 22 post-immunization following restimulation with PLP(139–151) peptide for 18 h. C, on day 22 post-immunization, spinal cords were isolated from both groups, and the expression of IFNγ and IL17α was examined using quantitative PCR after normalizing with ribosomal protein L27 (n = 4).
We further examined the Th1, Th17, and Th2 subpopulations through intracellular staining for specific cytokines, including IFNγ, IL17, and IL4 in infiltrated mononuclear cells. As shown in Fig. 6B, infiltrated mononuclear cells from RvD1-treated EAE showed 2-fold reduction of IFNγ- and IL17-producing CD4+ve cells compared with the vehicle-treated EAE group, with no change in IL4-producing CD4 cells. These observations were further supported by quantitative PCR of IL17a and IFNγ in spinal cords isolated from RvD1-treated and vehicle EAE groups (Fig. 6C), further suggesting that RvD1 treatment reduced the pro-inflammatory environment in the CNS of treated EAE.
RvD1 Induced M2 Type Phenotype and Inhibited Immune Response
RvD1 has been reported to induce macrophage polarization toward an M2-like phenotype (41–45) known to be involved in tissue repair by producing extracellular matrix molecules and anti-inflammatory cytokines (46, 47). The importance of the M2 macrophage in MS has been highlighted by recent studies demonstrating that approved drugs for MS, including glatiramer acetate and laquinimod, reversed EAE by promoting development of M2-activated monocytes (48, 49). To examine whether RvD1-mediated protection in EAE could be due to polarization of monocytes/macrophages into the M2 type, we assessed the monocyte/macrophage polarization status during EAE disease. We isolated adherent monocytes/macrophages after panning on plastic plates (<85% CD11b+ F4/80+) and were treated with lipopolysaccharide and IFNγ for 6 h to induce an M1 response and with IL4 for 24 h to induce an M2 response. We observed that adherent monocytes/macrophages from the RvD1-treated EAE group displayed significantly lower levels of M1 markers, including inducible NOS and IL6 expression; however, RvD1 treatment significantly induced M2 markers (IL10, arginase 1, resistin-like-a (FIZZ1) and YM1) suggesting that RvD1 treatment altered the monocytes/macrophages phenotype (Fig. 7A). RvD1 treatment also attenuated the expression of costimulatory molecules (CD80 and CD86) and major histocompatibility class II molecules on adherent monocytes/macrophages isolated from the treated EAE group compared with the vehicle-treated EAE group (Fig. 7B).
FIGURE 7.

RvD1 induces M2-type phenotype and inhibits class II and co-stimulatory molecular expression in macrophages. A and B, expression of M1- and M2-specific genes, class II and co-stimulatory molecules (CD86 and CD60) were examined in adherent macrophages isolated from spleen cells of RvD1-treated and -untreated EAE groups after panning on a plastic dish and treated with lipopolysaccharide/IFNγ for 6 h (n = 4). C, SJL mice were immunized with PLP(139–151) in complete Freund's adjuvant as described before. Post-10 days, CD4 cells were isolated from spleens/lymph nodes and mixed with adherent monocytes/macrophages isolated from RvD1-treated and -untreated EAE groups at a ratio of 1:5. After 72 h of incubation, cell proliferation was examined using WST-1 reagent (n = 4). Cell supernatant was processed for IFNγ and IL17 analysis by ELISA (BioLegend) (n = 4). NS, not significant; ***, p < 0.001. D, cells were stimulated with phorbol 12-myristate 13-acetate/ionomycin for 4 h in the presence of GolgiPlug. IL17A- and IFNγ-expressing cells were measured by intracellular staining on a CD4+ gate (n = 2).
We next examined whether monocytes/macrophages isolated from the RvD1-treated mice have the ability to alter the antigen-specific T cell response. For this, PLP(139–151)-primed CD4 T cells were cocultured with monocytes/macrophages isolated from RvD1-treated and untreated EAE groups in the presence of PLP(139–151) (20 μg/ml) at a ratio of 1:5. Coculture of PLP(139–151)-primed CD4 T cells with monocytes/macrophages isolated from the RvD1-treated EAE group did not show any effect on CD4 T cell proliferation compared with CD4 T cells cocultured with monocytes/macrophages isolated from the vehicle-treated EAE group (Fig. 7C). However, production of IL17A and IFNγ was significantly attenuated in the coculture condition when PLP(139–151)-primed CD4 cells were cultured with monocytes/macrophages isolated from the RvD1-treated EAE group compared with the vehicle-treated EAE group (Fig. 6C). These observations were further supported by intracellular staining of CD4+ IL17A+ and -IFNγ+ cells (Fig. 7D), strongly suggesting that RvD1 treatment regulates the immunomodulatory properties of monocytes/macrophages and alters the antigen-specific immune response of CD4 T cells.
RvD1 Promoted M2-type Phenotype in CNS
Our finding that oral administration of RvD1 protected EAE disease progression by modulating T cell response, monocytes/macrophage polarization, and restricted IL17+ve and IFNγ+ve CD4 cells in the CNS compared with the vehicle-treated group suggests a less pro-inflammatory environment in the CNS. To capture the overall picture of inflammation, RNA was isolated from spinal cords of control, RvD1-, and vehicle-treated RR-EAE groups on day 22 post-immunization to detect the levels of inducible NOS, TNFα, IL1β, IL6 (M1 markers), and FIZZ1 (M2 marker). The vehicle-treated EAE group showed significantly (p < 0.001) higher expression of inducible NOS, TNFα, IL1β, and IL6, which were attenuated by the RvD1 treatment (Fig. 8A). Moreover, expression of FIZZ1 was significantly down-regulated in EAE groups (Fig. 8A), which was restored by RvD1 treatment, suggesting that the RvD1 treatment attenuated the pro-inflammatory environment and induced an M2 repair-mechanism response associated with tissue repair (46, 47). Demyelination in EAE is associated with reduced expression of myelin genes due to mature oligodendrocyte dysfunction and inability of oligodendrocyte progenitor cells to develop into mature oligodendrocytes. The expression of myelin genes, including MOG, PLP, and myelin basic protein, were significantly reduced in the spinal cords of the EAE group, which was restored/protected by the RvD1 treatment (Fig. 8A). These data were well supported by the expression of neurotrophic factors like leukemia inhibitory factor and glial cell line-derived neurotrophic factor, which are known to promote myelination (Fig. 8A). RvD1 treatment did not affect the expression of ciliary neurotrophic factor and PDGFα (data not shown).
FIGURE 8.

RvD1 potently attenuates M1 inflammatory markers while increasing the functional M2 marker. A, on day 22 post- immunization, spinal cords were isolated from both groups, and the expression of pro-inflammatory M1 cytokines, M2 gene, myelin genes, and neurotrophic factors was analyzed and normalized against the L27 housekeeping gene using quantitative PCR (n = 4). B, infiltrating mononuclear cells were isolated from RvD1-treated and -untreated groups, and the cellular M2 phenotype (CD206+ and scavenger receptor-positive) was profiled using FACS analysis for activated myeloid cells, including monocytes/macrophages (CD11b+CD45hi); C, resident microglia (CD11b+CD45low) (n = 4). D, infiltrating mononuclear cells were profiled for myeloid-derived suppressor cells (CD45hi CD11b+ Gr1+) and their M2 phenotype (CD206+ and scavenger receptor-positive) using FACS analysis (n = 4). E, rat mixed glial culture was treated with RvD1 (100 nm) for 2 h followed by treatment with pro-inflammatory cytokine combination (TNFα and IFNγ; 20 ng/ml). Post-24 h of incubation, RNA was isolated, and expression of inducible NOS (iNOS) and myelin genes (MOG and PLP) was normalized with L27 as a housekeeping gene (n = 4). F, panel i, primary mouse brain microglial cells were treated with vehicle (0.001% ethanol) or RvD1 (100 nm). Post-24 h of incubation, RNA was isolated, and expression of arginase 1 and chitinase-3-like-3 (YM1/2) was normalized with L27 as a housekeeping gene. F, panel ii, for the coculture study, treated and untreated microglial cells with RvD1 were washed three times with complete culture media, and primary rat oligodendrocyte progenitor cells (OPCs) were cocultured. After 24 h of incubation, cells were processed for expression of myelin genes, including myelin-associated glycoprotein (MAG), myelin oligodendrocyte glycoprotein (MOG), and myelin basic protein (MBP) by quantitative PCR, and data were normalized to the control gene ribosomal L27 housekeeping gene. ***, p < 0.001, and **, p < 0.01.
To address whether RvD1 treatment altered the phenotype of brain resident microglia and infiltrated monocytes/macrophages during disease, we isolated infiltrated mononuclear cells from RvD1-treated and vehicle-treated EAE groups on day 22 post-immunization. Flow cytometry analysis revealed a 5.8- and 3.1-fold increase in the number of CD206- and scavenger receptor-positive myeloid cells (CD45high CD11b+), respectively, in the CNS of the RvD1-treated EAE group (Fig. 8B). The RvD1-treated group also showed a 3.3-fold increase in CD204-positive resident microglia (CD45low CD11b+) (Fig. 8C), suggesting an induction of the M2 phenotype in resident microglia and infiltrated monocytes/macrophages during EAE disease under RvD1 treatment. We also observed an 8.5-fold increase in the number of myeloid-derived suppressor cells (MDSCs; CD45high CD11b+ Gr1+), which are known to alter immune response in autoimmune diseases, like EAE (50, 51), and in cancer (52). Interestingly, MDSCs in RvD1-treated groups displayed the M2 phenotype as they expressed higher levels of CD206 and scavenger receptor compared with the vehicle-treated EAE group (Fig. 8D).
Using an in vitro mixed glial cell model, we examined whether RvD1 treatment could affect the expression of inflammatory mediator and myelin genes under inflammation. Mixed glial cell cultures were treated with RvD1 (100 nm) in the presence or absence of a combination of pro-inflammatory cytokines (TNFα and IFNγ) (20 ng/ml) to create an inflammatory environment. As shown in Fig. 8E, cytokines significantly induced inducible NOS expression compared with the untreated cultures. Pretreatment of cells with RvD1 attenuated cytokine-induced inducible NOS gene expression (Fig. 8E). Under a similar experimental condition, we found that cytokine treatment significantly reduced expression of myelin genes (MOG and PLP), and RvD1 treatment restored or protected their expression. These findings further support the observation that RvD1 mediates attenuation of inflammation and induction of myelin genes in spinal cords of treated EAE groups. We next examined whether RvD1-treated polarized microglia could promote oligodendrocyte differentiation in the absence of an inflammatory condition. We treated brain microglial cells with or without RvD1 (100 nm) for 24 h and observed that RvD1 treatment induced the expression of arginase 1 and YM1/2, suggesting M2 polarization of the microglia (Fig. 8F, panel i). Treated and untreated microglial cells were washed and cocultured with oligodendrocyte precursor cells, and after 24 h of coculture, cells were processed for expression of myelin genes. As shown in Fig. 8F, panel ii, oligodendrocyte precursor cells cocultured with RvD1-polarized microglia significantly induced the expression of myelin genes, including myelin-associated glycoprotein, MOG, and myelin basic protein, suggesting that RvD1 promotes myelin expression by modulating M2-type microglia under inflammatory and non-inflammatory conditions.
Discussion
Thirteen disease-modifying drugs have been approved by the United States Food and Drug Administration for MS. All of these agents reduce relapse rate, have a beneficial effect on a variety of magnetic resonance imaging measures, and have been reported to delay disability in short term clinical trials. Most of them are relatively new, and their safety and efficacy in patients has yet to be determined for a longer therapeutic regimen. It is well accepted that treatment of MS is more effective during the early course of the disease with milder symptoms. Recent efforts in search of biomarkers in the cerebrospinal fluid have had limited success. Blood, a more accessible bio-fluid, could be easily drawn for biomarker analysis if such were available for diagnosis of MS at an early stage of the disease. The main goal of this study was to identify potential metabolite(s) and altered pathway(s) in plasma of the chronic mouse model of MS using a global metabolomics approach. We identified 100 metabolites that were significantly altered between control and EAE animals, and these molecules were classified into lipid, amino acid, peptide, xenobiotic, carbohydrate, nucleotide, and energy pathways. The most intriguing part of this study was the observation that most of the changes were found in the lipids. In humans having MS, being a presumed autoimmune disease, the role of lipids metabolism is understudied. A few key studies have clearly demonstrated roles for specific lipids, including CDP-choline (53), lactosylceramide (54), sulfatide (55), and fatty acids (56, 57), in altering disease outcome in animal models of MS, suggesting the importance of lipids in disease pathology. It has yet to be established whether the changes in lipids are the cause or the consequences of the disease pathology.
Recently, we reported a comprehensive analysis of plasma metabolites in the RR preclinical mouse model of MS (SJL-EAE) (14), and we identified 44 metabolites that distinguished RR-EAE from control mice. These metabolites were drawn from various metabolic pathways that correlated well with the severity of the RR form of EAE disease. The important findings of our plasma metabolomics data in RR-EAE and progressive chronic EAE models are the distinct signature and the affected pathways. In the RR model, we identified 44 metabolites of which 72% were increased and 28% were decreased. However, in the case of the chronic model, 100 metabolites were significantly altered of which 13% were increased and 87% were reduced. Using KEGG, only one pathway was overlapping (i.e. PUFA metabolism) suggesting that other pathways identified either in B6 or SJL animals might be specific for chronic or RR models, respectively. We carefully looked into metabolite signatures of both models, and the number of metabolites was similarly altered in both. These are citrulline, α-ketoglutarate, trigonelline (N-methynicotinate), and hippurate. More strikingly, only kynurenate was differentially altered in both models indicating the possible biomarker for distinguishing chronic versus RR. We have to keep in mind that altered levels of kynurenate in both models may be due to genetic variation in SJL and B6 mice; therefore, this observation needs validation. Moreover, this pathway is altered in neurodegenerative diseases (58, 59). Although some of the observed metabolite changes in RR and chronic models could easily be developed as biomarkers of the disease, the key to translating metabolomics into therapeutics requires figuring out the central altered metabolic pathway(s).
Although the main emphasis of our metabolomics study was to identify new diagnostic biomarkers of disease, we found that information related to the perturbed pathways enhances the understanding of disease mechanisms, thereby highlighting plausible endogenous metabolite(s) that have drug-like properties. Using KEGG and enrichment pathway analyses, we observed that α-linolenic acid and linoleic acid PUFA metabolism (ω-3 and ω-6) were altered in both murine EAE models (RR (14) and chronic (supplemental Table S1 and Fig. 5B)), and the levels of its metabolites were found to be low in patients with MS (15–19), suggesting that this pathway could potentially be a therapeutic target for MS. There are two major sources of ω-3 fatty acids: α-linolenic acid, which is found in plants, and EPA and DHA, which are found in fish. In mammals, α-linolenic acid is converted via elongation and desaturation to EPA, subsequently to DHA, and ultimately to anti-inflammatory mediators, such as resolvins and protectins that are local-acting mediators with both anti-inflammatory and proresolving actions (60–63). An intermediate in the conversion of EPA to DHA is ω-3 docosapentaenoic acid (64). Both ω-3 and ω-6 docosapentaenoic acid and n3 EPA were found to be significantly lower in the plasma of EAE compared with control. A recent study clearly demonstrated that docosapentaenoic acid is converted during resolution of inflammation in mice and by human leukocytes to n-3 products congenerous to D-series resolvins, protectins, and masresins, called specialized pro-resolving mediators (65).
Several epidemiological studies suggest that n-3 PUFA supplementation is linked with improved clinical outcomes in patients with MS (66–68). Beneficial effects of n-3 PUFA have been shown in EAE by altering the immunomodulatory effect on dendritic cells (69) and by inducing remyelination and inducing the M2 microglial phenotype in vivo (70). A number of clinical trials have been conducted by supplementing higher doses of EPA and DHA in patients with MS (68, 71). Although the level of ω-3 (EPA and DHA) could be restored by supplementation in patients with MS, most likely there is a failure of production of adequate amounts of resolution-inducing molecules, like resolvins and protectins. This would result in inappropriate inflammation and a delay in the healing/repair process, so neuronal damage continues. This resolution pathway is also found to be dysfunctional in Alzheimer disease (72, 73). Moreover, ω-3 supplementation or RvD1 prevents cognitive decline in animal models (74), improves amyloid-β phagocytosis, and regulates inflammation in Alzheimer patients (75–77).
We decided to use RvD1, a downstream metabolite of EPA-DHA, to examine its therapeutic potential in the EAE model. RvD1 has been reported to have a number of favorable properties including the following: 1) limiting excessive leukocyte infiltration and attenuating the production of pro-inflammatory cytokines (38); 2) inducing macrophage polarization toward an M2-like phenotype (41–45); 3) promoting IL-4-induced alternative activation via STAT6 and peroxisome proliferator-activated receptor γ signaling pathways in microglia (78); and 4) being effective at low doses, including 100 ng/mouse (5 μg/kg body weight) (40). Oral administration of RvD1 at a dose of 100 ng/mouse was very effective in attenuating EAE disease progression in our model compared with the vehicle group without affecting disease onset. These data support the hypothesis that proresolution and anti-inflammation are distinct processes. Resolution required blocking infiltration of mononuclear cells and clearance of lymphocytes at the site by macrophage-mediated phagocytosis.
Pro-inflammatory microglia and CNS-infiltrating monocytes/macrophages, recruited and activated in response to factors present in the inflamed CNS environment, are thought to contribute to the pathogenesis of MS and other neurodegenerative diseases (79, 80). RvD1 has been reported to enhance the macrophage phagocytosis (38, 81) and polarized macrophage toward an M2-like phenotype (41–45). These reports are in accordance with our finding of the M2 phenotype in treated mice, both at the periphery as well as in the CNS. RvD1 treatment did not affect the number of infiltrating monocytes/macrophages in the CNS, but it did induce the M2 phenotype. Moreover, resident microglia exhibited the M2 phenotype upon RvD1 treatment compared with the vehicle-treated EAE group. Although we have not examined the possibility of clearance of lymphocytes in the CNS, either by induction of apoptosis or inhibition of clonal expansion, we did observe a 33% reduction in CD4-positive cells in the RvD1-treated group in the CNS. Moreover, inhibition of IL17+ve and IFNγ+ve CD4 cells in the treated group suggests an inhibition of antigen-specific proliferation of CD4 cells. RvD1 also polarized macrophage into M2-like phenotype in a number of pathological conditions, including adipose tissue inflammation (45), smoke-induced lung inflammation (42), smoke-exposed human macrophage (41), obesity-induced steatohepatitis (43), and endotoxic uveitis (44), and it provided protection.
Most of the Food and Drug Administration-approved MS therapies including, interferon β, glatiramer acetate, dimethyl fumarate, laquinimod, and fingolimod, are anti-inflammatory in action (82, 83). Glatiramer acetate and laquinimod have been shown to induce type II monocytes and reverse the EAE disease by modulating T cell response (49, 84). Similar action of RvD1 was observed in EAE; however, it has yet to be investigated whether these drugs modulate the ω-3 metabolism, thereby altering the levels of pro-resolving mediators and downstream resolving actions. The dose used for RvD1, 100 ng/mouse, was effective and potent (5 μg/kg body weight) compared with other MS-approved therapies used in EAE models (48, 49, 85, 86).
RvD1-mediated protection in EAE involves multiple mechanisms, including induction of regulatory T cells, polarization of monocytes/macrophages and microglia into the M2 phenotype, and an increase in MDSCs. The role of MDSCs as regulators or inducers of inflammation in EAE is quite controversial. In general, these cells represent an important class of immunoregulatory cells that can be activated to suppress T cell function (52). We found increased levels of MDSCs (CD11b+ Gr1+) cells in the CNS of the treated group, which correlates well with the decreased CD4+ve immune response. We did not examine the nature of these MDSCs to determine whether they are granulocytic (CD11+Ly6ClowLy6G+) or monocytic (CD11+Ly6C+Ly6G−). Recently, it was reported that monocytic and granulocytic MDSCs are potent suppressors of T cells through nitric oxide (87) and by expressing high levels of programmed cell death ligand 1 (51), respectively. However, one study reported that CD11b+Gr1+ cells isolated from mice with EAE inhibited T cell response but induced Th17 cell differentiation (50). We made an interesting observation that MDSCs in the CNS of the treated mice displayed the M2 phenotype (CD206+ and scavenger receptor positive). MDSCs with M2-like phenotype have been identified in cancer with pro-tumoral and immunosuppressive activities (88, 89). It remains to be investigated whether RvD1 treatment could enhance the M2 phenotype in MDSCs in general.
This study demonstrates that a metabolomic approach can suggest new therapeutic targets or potential endogenous metabolites that may have drug-like properties that could easily be tested in MS (Fig. 9). RvD1 is one of the candidates that has been reported previously for treating a number of inflammatory diseases (34, 35, 37–39, 70). Here, we found it to be significantly effective in attenuating clinical symptoms in the preclinical mouse model of MS. This study provided a proof of principle for how a metabolomic approach could lead to the identification of small molecules that could be used for therapy.
FIGURE 9.

Schematic flow of implication of untargeted metabolomic approach in EAE.
Author Contributions
S. G. conceived, designed, performed, analyzed the data, coordinated, and wrote the paper. L. M. P. and I. D. analyzed the metabolomics data, wrote, and finalized the paper. H. S., J. S., A. S., and R. R. performed experiments, contributed to figure preparation, and finalized the paper. A. D. and M. R. performed the pathology and finalized the paper. K. L., M. N. H., A. K., M. C., S. E., and A. K. M. provided intellectual input, contributed to figure preparation, and finalized the paper. R. P. M. analyzed the metabolomics data, provided intellectual input, edited, and finalized the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Louisa Papke, Laurie Zoecklein, Mabel Pierce, and LaToya Jackson for brain and spinal cord staining and Stephanie Stebens for assistance with preparing and editing the manuscript.
This work was supported by Henry Ford Hospital Internal Funding Grant A20020, in part by National Multiple Sclerosis Society Research Grants RG 4311A4/4 and RG 5112A5/2 (to S. G.), and by National Institutes of Health Grant R01 GM092993 (to M. R.). R. M. is an employee of Metabolon, Inc., and as such has affiliations with or financial involvement with Metabolon, Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table S1.
- MS
- multiple sclerosis
- EAE
- experimental autoimmune encephalomyelitis
- RvD1
- resolvin D1
- RR
- relapsing-remitting
- UPLC-MS/MS
- ultrahigh performance liquid chromatography-tandem mass spectrometry
- KEGG
- Kyoto Encyclopedia of Genes and Genomics
- EPA
- eicosapentaenoic acid
- DHA
- docosahexaenoic acid
- MDSC
- myeloid-derived suppressor cells
- MOG
- myelin-oligodendrocyte glycoprotein
- PLP
- proteolipid protein
- Th
- T helper
- RSD
- relative standard deviation.
References
- 1. Miller J. R. (2004) The importance of early diagnosis of multiple sclerosis. J. Manag. Care Pharm. 10, S4–S11 [PubMed] [Google Scholar]
- 2. Lourenço A. S., Baldeiras I., Grãos M., and Duarte C. B. (2011) Proteomics-based technologies in the discovery of biomarkers for multiple sclerosis in the cerebrospinal fluid. Curr. Mol. Med. 11, 326–349 [DOI] [PubMed] [Google Scholar]
- 3. Zhang A., Sun H., Yan G., Wang P., and Wang X. (2015) Metabolomics for biomarker discovery: moving to the clinic. BioMed Res. Int. 2015, 354671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nicholson J. K., and Lindon J. C. (2008) Systems biology: metabonomics. Nature 455, 1054–1056 [DOI] [PubMed] [Google Scholar]
- 5. Zhang A. H., Sun H., and Wang X. J. (2013) Recent advances in metabolomics in neurological disease, and future perspectives. Anal. Bioanal. Chem. 405, 8143–8150 [DOI] [PubMed] [Google Scholar]
- 6. Johnson C. H., and Gonzalez F. J. (2012) Challenges and opportunities of metabolomics. J. Cell Physiol. 227, 2975–2981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mamas M., Dunn W. B., Neyses L., and Goodacre R. (2011) The role of metabolites and metabolomics in clinically applicable biomarkers of disease. Arch. Toxicol. 85, 5–17 [DOI] [PubMed] [Google Scholar]
- 8. Sato Y., Suzuki I., Nakamura T., Bernier F., Aoshima K., and Oda Y. (2012) Identification of a new plasma biomarker of Alzheimer's disease using metabolomics technology. J. Lipid Res. 53, 567–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trushina E., Nemutlu E., Zhang S., Christensen T., Camp J., Mesa J., Siddiqui A., Tamura Y., Sesaki H., Wengenack T. M., Dzeja P. P., and Poduslo J. F. (2012) Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer's disease. PLoS One 7, e32737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johansen K. K., Wang L., Aasly J. O., White L. R., Matson W. R., Henchcliffe C., Beal M. F., and Bogdanov M. (2009) Metabolomic profiling in LRRK2-related Parkinson's disease. PLoS One 4, e7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lutz N. W., Fernandez C., Pellissier J. F., Cozzone P. J., and Béraud E. (2013) Cerebral biochemical pathways in experimental autoimmune encephalomyelitis and adjuvant arthritis: a comparative metabolomic study. PLoS One 8, e56101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gebregiworgis T., Massilamany C., Gangaplara A., Thulasingam S., Kolli V., Werth M. T., Dodds E. D., Steffen D., Reddy J., and Powers R. (2013) Potential of urinary metabolites for diagnosing multiple sclerosis. ACS Chem. Biol. 8, 684–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noga M. J., Dane A., Shi S., Attali A., van Aken H., Suidgeest E., Tuinstra T., Muilwijk B., Coulier L., Luider T., Reijmers T. H., Vreeken R. J., and Hankemeier T. (2012) Metabolomics of cerebrospinal fluid reveals changes in the central nervous system metabolism in a rat model of multiple sclerosis. Metabolomics 8, 253–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mangalam A., Poisson L., Nemutlu E., Datta I., Denic A., Dzeja P., Rodriguez M., Rattan R., and Giri S. (2013) Profile of circulatory metabolites in a relapsing-remitting animal model of multiple sclerosis using global metabolomics. J. Clin. Cell Immunol. 4, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holman R. T., Johnson S. B., and Kokmen E. (1989) Deficiencies of polyunsaturated fatty acids and replacement by nonessential fatty acids in plasma lipids in multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 86, 4720–4724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gul S., Smith A. D., Thompson R. H., Wright H. P., and Zilkha K. J. (1970) Fatty acid composition of phospholipids from platelets and erythrocytes in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 33, 506–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aupperle R. L., Denney D. R., Lynch S. G., Carlson S. E., and Sullivan D. K. (2008) ω-3 fatty acids and multiple sclerosis: relationship to depression. J. Behav. Med. 31, 127–135 [DOI] [PubMed] [Google Scholar]
- 18. Nightingale S., Woo E., Smith A. D., French J. M., Gale M. M., Sinclair H. M., Bates D., and Shaw D. A. (1990) Red blood cell and adipose tissue fatty acids in mild inactive multiple sclerosis. Acta Neurol. Scand. 82, 43–50 [DOI] [PubMed] [Google Scholar]
- 19. Cunnane S. C., Ho S. Y., Dore-Duffy P., Ells K. R., and Horrobin D. F. (1989) Essential fatty acid and lipid profiles in plasma and erythrocytes in patients with multiple sclerosis. Am. J. Clin. Nutr. 50, 801–806 [DOI] [PubMed] [Google Scholar]
- 20. Serhan C. N., Hong S., Gronert K., Colgan S. P., Devchand P. R., Mirick G., and Moussignac R. L. (2002) Resolvins: a family of bioactive products of ω-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 196, 1025–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nath N., Khan M., Paintlia M. K., Singh I., Hoda M. N., and Giri S. (2009) Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J. Immunol. 182, 8005–8014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boudonck K. J., Mitchell M. W., Német L., Keresztes L., Nyska A., Shinar D., and Rosenstock M. (2009) Discovery of metabolomics biomarkers for early detection of nephrotoxicity. Toxicol. Pathol. 37, 280–292 [DOI] [PubMed] [Google Scholar]
- 23. Evans A. M., DeHaven C. D., Barrett T., Mitchell M., and Milgram E. (2009) Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 81, 6656–6667 [DOI] [PubMed] [Google Scholar]
- 24. Lawton K. A., Berger A., Mitchell M., Milgram K. E., Evans A. M., Guo L., Hanson R. W., Kalhan S. C., Ryals J. A., and Milburn M. V. (2008) Analysis of the adult human plasma metabolome. Pharmacogenomics 9, 383–397 [DOI] [PubMed] [Google Scholar]
- 25. Sreekumar A., Poisson L. M., Rajendiran T. M., Khan A. P., Cao Q., Yu J., Laxman B., Mehra R., Lonigro R. J., Li Y., Nyati M. K., Ahsan A., Kalyana-Sundaram S., Han B., Cao X., et al. (2009) Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457, 910–914 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 26. Evans A. M., Bridgewater B. R., Liu Q., Mitchell M. W., Robinson R. J., Dai H., Stewart S. J., DeHaven C. D., and Miller L. A. (2014) High resolution mass spectrometry improves data quantity and quality as compared to unit mass resolution mass spectrometry in high-throughput profiling metabolomics. Metabolomics 4, 132 [Google Scholar]
- 27. Dehaven C. D., Evans A. M., Dai H., and Lawton K. A. (2010) Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J. Cheminform. 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aittokallio T., and Schwikowski B. (2006) Graph-based methods for analysing networks in cell biology. Brief. Bioinform. 7, 243–255 [DOI] [PubMed] [Google Scholar]
- 29. Nath N., Khan M., Rattan R., Mangalam A., Makkar R. S., de Meester C., Bertrand L., Singh I., Chen Y., Viollet B., and Giri S. (2009) Loss of AMPK exacerbates experimental autoimmune encephalomyelitis disease severity. Biochem. Biophys. Res. Commun. 386, 16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xia J., Mandal R., Sinelnikov I. V., Broadhurst D., and Wishart D. S. (2012) Metaboanalyst 2.0–a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 40, W127–W133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xia J., Psychogios N., Young N., and Wishart D. S. (2009) Metaboanalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 37, W652–W660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goeman J. J., and Bühlmann P. (2007) Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics 23, 980–987 [DOI] [PubMed] [Google Scholar]
- 33. Seki H., Sasaki T., Ueda T., and Arita M. (2010) Resolvins as regulators of the immune system. ScientificWorldJournal 10, 818–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spite M., Summers L., Porter T. F., Srivastava S., Bhatnagar A., and Serhan C. N. (2009) Resolvin D1 controls inflammation initiated by glutathione-lipid conjugates formed during oxidative stress. Br. J. Pharmacol. 158, 1062–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bento A. F., Claudino R. F., Dutra R. C., Marcon R., and Calixto J. B. (2011) ω-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J. Immunol. 187, 1957–1969 [DOI] [PubMed] [Google Scholar]
- 36. Chen F., Fan X. H., Wu Y. P., Zhu J. L., Wang F., Bo L. L., Li J. B., Bao R., and Deng X. M. (2014) Resolvin D1 improves survival in experimental sepsis through reducing bacterial load and preventing excessive activation of inflammatory response. Eur. J. Clin. Microbiol. Infect. Dis. 33, 457–464 [DOI] [PubMed] [Google Scholar]
- 37. Lima-Garcia J. F., Dutra R. C., da Silva K., Motta E. M., Campos M. M., and Calixto J. B. (2011) The precursor of resolvin D series and aspirin-triggered resolvin D1 display anti-hyperalgesic properties in adjuvant-induced arthritis in rats. Br. J. Pharmacol. 164, 278–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rogerio A. P., Haworth O., Croze R., Oh S. F., Uddin M., Carlo T., Pfeffer M. A., Priluck R., Serhan C. N., and Levy B. D. (2012) Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J. Immunol. 189, 1983–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tang H., Liu Y., Yan C., Petasis N. A., Serhan C. N., and Gao H. (2014) Protective actions of aspirin-triggered (17R) resolvin D1 and its analogue, 17R-hydroxy-19-para-fluorophenoxy-resolvin D1 methyl ester, in C5a-dependent IgG immune complex-induced inflammation and lung injury. J. Immunol. 193, 3769–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Recchiuti A., Codagnone M., Pierdomenico A. M., Rossi C., Mari V. C., Cianci E., Simiele F., Gatta V., and Romano M. (2014) Immunoresolving actions of oral resolvin D1 include selective regulation of the transcription machinery in resolution-phase mouse macrophages. FASEB J. 28, 3090–3102 [DOI] [PubMed] [Google Scholar]
- 41. Croasdell A., Thatcher T. H., Kottmann R. M., Colas R. A., Dalli J., Serhan C. N., Sime P. J., and Phipps R. P. (2015) Resolvins attenuate inflammation and promote resolution in cigarette smoke-exposed human macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 10.1152/ajplung.00125.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hsiao H. M., Sapinoro R. E., Thatcher T. H., Croasdell A., Levy E. P., Fulton R. A., Olsen K. C., Pollock S. J., Serhan C. N., Phipps R. P., and Sime P. J. (2013) A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS One 8, e58258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rius B., Titos E., Morán-Salvador E., López-Vicario C., García-Alonso V., González-Périz A., Arroyo V., and Clària J. (2014) Resolvin D1 primes the resolution process initiated by calorie restriction in obesity-induced steatohepatitis. FASEB J. 28, 836–848 [DOI] [PubMed] [Google Scholar]
- 44. Rossi S., Di Filippo C., Gesualdo C., Potenza N., Russo A., Trotta M. C., Zippo M. V., Maisto R., Ferraraccio F., Simonelli F., and D'Amico M. (2015) Protection from endotoxic uveitis by intravitreal resolvin D1: involvement of lymphocytes, miRNAs, ubiquitin-proteasome, and M1/M2 macrophages. Mediators Inflamm. 2015, 149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Titos E., Rius B., González-Périz A., López-Vicario C., Morán-Salvador E., Martínez-Clemente M., Arroyo V., and Clària J. (2011) Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J. Immunol. 187, 5408–5418 [DOI] [PubMed] [Google Scholar]
- 46. Parsa R., Andresen P., Gillett A., Mia S., Zhang X. M., Mayans S., Holmberg D., and Harris R. A. (2012) Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes 61, 2881–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Verreck F. A., de Boer T., Langenberg D. M., van der Zanden L., and Ottenhoff T. H. (2006) Phenotypic and functional profiling of human proinflammatory type-1 and anti-inflammatory type-2 macrophages in response to microbial antigens and IFN-γ- and CD40L-mediated costimulation. J. Leukocyte Biol. 79, 285–293 [DOI] [PubMed] [Google Scholar]
- 48. Schulze-Topphoff U., Shetty A., Varrin-Doyer M., Molnarfi N., Sagan S. A., Sobel R. A., Nelson P. A., and Zamvil S. S. (2012) Laquinimod, a quinoline-3-carboxamide, induces type II myeloid cells that modulate central nervous system autoimmunity. PLoS One 7, e33797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weber M. S., Prod'homme T., Youssef S., Dunn S. E., Rundle C. D., Lee L., Patarroyo J. C., Stüve O., Sobel R. A., Steinman L., and Zamvil S. S. (2007) Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat. Med. 13, 935–943 [DOI] [PubMed] [Google Scholar]
- 50. Lin L., Huang Z., Gao Y., Yan X., Xing J., and Hang W. (2011) LC-MS based serum metabonomic analysis for renal cell carcinoma diagnosis, staging, and biomarker discovery. J. Proteome Res. 10, 1396–1405 [DOI] [PubMed] [Google Scholar]
- 51. Okajima K., Inoue M., and Morino Y. (1985) Studies on the mechanism for renal elimination of N-acetylphenylalanine: its pathophysiologic significance in phenylketonuria. J. Lab. Clin. Med. 105, 132–138 [PubMed] [Google Scholar]
- 52. Gabrilovich D. I., and Nagaraj S. (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Skripuletz T., Manzel A., Gropengiesser K., Schäfer N., Gudi V., Singh V., Salinas Tejedor L., Jörg S., Hammer A., Voss E., Vulinovic F., Degen D., Wolf R., Lee D. H., et al. (2015) Pivotal role of choline metabolites in remyelination. Brain 138, 398–413 [DOI] [PubMed] [Google Scholar]
- 54. Mayo L., Trauger S. A., Blain M., Nadeau M., Patel B., Alvarez J. I., Mascanfroni I. D., Yeste A., Kivisäkk P., Kallas K., Ellezam B., Bakshi R., Prat A., Antel J. P., Weiner H. L., and Quintana F. J. (2014) Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 20, 1147–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jahng A., Maricic I., Aguilera C., Cardell S., Halder R. C., and Kumar V. (2004) Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J. Exp. Med. 199, 947–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ho P. P., Kanter J. L., Johnson A. M., Srinagesh H. K., Chang E. J., Purdy T. M., van Haren K., Wikoff W. R., Kind T., Khademi M., Matloff L. Y., Narayana S., Hur E. M., Lindstrom T. M., He Z., et al. (2012) Identification of naturally occurring fatty acids of the myelin sheath that resolve neuroinflammation. Sci. Transl. Med. 4, 137ra173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Haghikia A., Jörg S., Duscha A., Berg J., Manzel A., Waschbisch A., Hammer A., Lee D. H., May C., Wilck N., Balogh A., Ostermann A. I., Schebb N. H., Akkad D. A., Grohme D. A., et al. (2015) Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 43, 817–829 [DOI] [PubMed] [Google Scholar]
- 58. Campbell B. M., Charych E., Lee A. W., and Möller T. (2014) Kynurenines in CNS disease: regulation by inflammatory cytokines. Front. Neurosci. 8, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stone T. W., and Darlington L. G. (2013) The kynurenine pathway as a therapeutic target in cognitive and neurodegenerative disorders. Br. J. Pharmacol. 169, 1211–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Serhan C. N., Chiang N., and Dalli J. (2015) The resolution code of acute inflammation: novel pro-resolving lipid mediators in resolution. Semin. Immunol. 27, 200–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Serhan C. N. (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Buckley C. D., Gilroy D. W., and Serhan C. N. (2014) Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 40, 315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Spite M., Clària J., and Serhan C. N. (2014) Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 19, 21–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. De Caterina R. (2011) n-3 fatty acids in cardiovascular disease. N. Engl. J. Med. 364, 2439–2450 [DOI] [PubMed] [Google Scholar]
- 65. Dalli J., Colas R. A., and Serhan C. N. (2013) Novel n-3 immunoresolvents: structures and actions. Sci. Rep. 3, 1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jelinek G. A., Hadgkiss E. J., Weiland T. J., Pereira N. G., Marck C. H., and van der Meer D. M. (2013) Association of fish consumption and ω3 supplementation with quality of life, disability and disease activity in an international cohort of people with multiple sclerosis. Int. J. Neurosci. 123, 792–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Weinstock-Guttman B., Baier M., Park Y., Feichter J., Lee-Kwen P., Gallagher E., Venkatraman J., Meksawan K., Deinehert S., Pendergast D., Awad A. B., Ramanathan M., Munschauer F., and Rudick R. (2005) Low fat dietary intervention with ω-3 fatty acid supplementation in multiple sclerosis patients. Prostaglandins Leukot. Essent. Fatty Acids 73, 397–404 [DOI] [PubMed] [Google Scholar]
- 68. Nordvik I., Myhr K. M., Nyland H., and Bjerve K. S. (2000) Effect of dietary advice and n-3 supplementation in newly diagnosed patients with MS. Acta Neurol. Scand. 102, 143–149 [DOI] [PubMed] [Google Scholar]
- 69. Kong W., Yen J. H., and Ganea D. (2011) Docosahexaenoic acid prevents dendritic cell maturation, inhibits antigen-specific Th1/Th17 differentiation and suppresses experimental autoimmune encephalomyelitis. Brain. Behav. Immun. 25, 872–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen S., Zhang H., Pu H., Wang G., Li W., Leak R. K., Chen J., Liou A. K., and Hu X. (2014) n-3 PUFA supplementation benefits microglial responses to myelin pathology. Sci. Rep. 4, 7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Torkildsen O., Wergeland S., Bakke S., Beiske A. G., Bjerve K. S., Hovdal H., Midgard R., Lilleås F., Pedersen T., Bjørnarå B., Dalene F., Kleveland G., Schepel J., Olsen I. C., and Myhr K. M. (2012) ω-3 fatty acid treatment in multiple sclerosis (OFAMS Study): a randomized, double-blind, placebo-controlled trial. Arch. Neurol. 69, 1044–1051 [DOI] [PubMed] [Google Scholar]
- 72. Wang X., Zhu M., Hjorth E., Cortés-Toro V., Eyjolfsdottir H., Graff C., Nennesmo I., Palmblad J., Eriksdotter M., Sambamurti K., Fitzgerald J. M., Serhan C. N., Granholm A. C., and Schultzberg M. (2015) Resolution of inflammation is altered in Alzheimer's disease. Alzheimer's Dementia 11, 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lukiw W. J., Cui J. G., Marcheselli V. L., Bodker M., Botkjaer A., Gotlinger K., Serhan C. N., and Bazan N. G. (2005) A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 115, 2774–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Koivisto H., Grimm M. O., Rothhaar T. L., Berkecz R., Lütjohann D. D., Giniatullina R., Takalo M., Miettinen P. O., Lahtinen H. M., Giniatullin R., Penke B., Janáky T., Broersen L. M., Hartmann T., and Tanila H. (2014) Special lipid-based diets alleviate cognitive deficits in the APPswe/PS1dE9 transgenic mouse model of Alzheimer's disease independent of brain amyloid deposition. J. Nutr. Biochem. 25, 157–169 [DOI] [PubMed] [Google Scholar]
- 75. Fiala M., Halder R. C., Sagong B., Ross O., Sayre J., Porter V., and Bredesen D. E. (2015) ω-3 supplementation increases amyloid-β phagocytosis and resolvin D1 in patients with minor cognitive impairment. FASEB J. 29, 2681–2689 [DOI] [PubMed] [Google Scholar]
- 76. Mizwicki M. T., Liu G., Fiala M., Magpantay L., Sayre J., Siani A., Mahanian M., Weitzman R., Hayden E. Y., Rosenthal M. J., Nemere I., Ringman J., and Teplow D. B. (2013) 1α,25-dihydroxyvitamin D3 and resolvin D1 retune the balance between amyloid-β phagocytosis and inflammation in Alzheimer's disease patients. J. Alzheimers Dis. 34, 155–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang X., Hjorth E., Vedin I., Eriksdotter M., Freund-Levi Y., Wahlund L. O., Cederholm T., Palmblad J., and Schultzberg M. (2015) Effects of n-3 FA supplementation on the release of proresolving lipid mediators by blood mononuclear cells: the OmegAD study. J. Lipid Res. 56, 674–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li L., Wu Y., Wang Y., Wu J., Song L., Xian W., Yuan S., Pei L., and Shang Y. (2014) Resolvin D1 promotes the interleukin-4-induced alternative activation in BV-2 microglial cells. J. Neuroinflammation 11, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. David S., and Kroner A. (2011) Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 12, 388–399 [DOI] [PubMed] [Google Scholar]
- 80. Murray P. J., and Wynn T. A. (2011) Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chiang N., Fredman G., Bäckhed F., Oh S. F., Vickery T., Schmidt B. A., and Serhan C. N. (2012) Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484, 524–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Johnston J., and So T. Y. (2012) First-line disease-modifying therapies in paediatric multiple sclerosis: a comprehensive overview. Drugs 72, 1195–1211 [DOI] [PubMed] [Google Scholar]
- 83. Melzer N., and Meuth S. G. (2014) Disease-modifying therapy in multiple sclerosis and chronic inflammatory demyelinating polyradiculoneuropathy: common and divergent current and future strategies. Clin. Exp. Immunol. 175, 359–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schulze-Topphoff U., Shetty A., Varrin-Doyer M., Molnarfi N., Sagan S. A., Sobel R. A., Nelson P. A., and Zamvil S. S. (2012) Laquinimod, a quinoline-3-carboxamide, induces type II myeloid cells that modulate central nervous system autoimmunity. PLoS ONE 7, e33797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Choi J. W., Gardell S. E., Herr D. R., Rivera R., Lee C. W., Noguchi K., Teo S. T., Yung Y. C., Lu M., Kennedy G., and Chun J. (2011) FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. U.S.A. 108, 751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schilling S., Goelz S., Linker R., Luehder F., and Gold R. (2006) Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin. Exp. Immunol. 145, 101–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhu B., Bando Y., Xiao S., Yang K., Anderson A. C., Kuchroo V. K., and Khoury S. J. (2007) CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J. Immunol. 179, 5228–5237 [DOI] [PubMed] [Google Scholar]
- 88. Ochando J. C., and Chen S. H. (2012) Myeloid-derived suppressor cells in transplantation and cancer. Immunol. Res. 54, 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Umemura N., Saio M., Suwa T., Kitoh Y., Bai J., Nonaka K., Ouyang G. F., Okada M., Balazs M., Adany R., Shibata T., and Takami T. (2008) Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J. Leukocyte Biol. 83, 1136–1144 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.