

Abstract
WW domain-containing oxidoreductase (WWOX), originally marked as a likely tumor suppressor gene, has over the years become recognized for its role in a much wider range of cellular activities. Phenotypic effects displayed in animal studies, along with resolution of WWOX's architecture, fold, and binding partners, point to the protein's multifaceted biological functions. Results from a series of complementary experiments seem to indicate WWOX's involvement in metabolic regulation. More recently, clinical studies involving cases of severe encephalopathy suggest that WWOX also plays a part in controlling CNS development, further expanding our understanding of the breadth and complexity of WWOX behavior. Here we present a short overview of the various approaches taken to study this dynamic gene, emphasizing the most recent findings regarding WWOX's metabolic- and CNS-associated functions and their underlying molecular basis.
Keywords: animal model, cell metabolism, central nervous system (CNS), protein-protein interaction, tumor suppressor gene, SDR domain, WW domain, WWOX, common fragile site, epilepsy, tumor suppressor, animal models
Introduction
The WWOX gene initially became a research target due to its localization in a region of considerable chromosomal instability (16q23.2), at common fragile site (CFS)3 FRA16D, which immediately marked it as a possible tumor suppressor gene (TSG) (1, 2). Inactivation of TSGs at CFSs has long been known as a hallmark of cancer (3). CFSs are evolutionarily conserved genomic regions that are sensitive to replicative stress (4). In 2010, Beroukhim et al. (5) demonstrated that deletion of TSGs spanning CFSs is among the most significant driver events in cancer. Although highly recombinogenic, CFSs are rarely involved in oncogenic activation, suggesting that CFSs are mainly hot spots for inactivation of tumor suppressors (3). A number of animal studies were conducted on WWOX, and in keeping with the above paradigm, many insights were gained into WWOX's association with cancer development (reviewed elsewhere (6–10)). However, a few surprising results led the research in other unanticipated directions, linking WWOX to regulation of metabolic function, as well as neuronal development (Fig. 1). These animal model studies were complemented by studies on the cellular and molecular levels, modeling WWOX structure and identifying its potential binding partners, thus generating a comprehensive functional model of WWOX activity.
FIGURE 1.
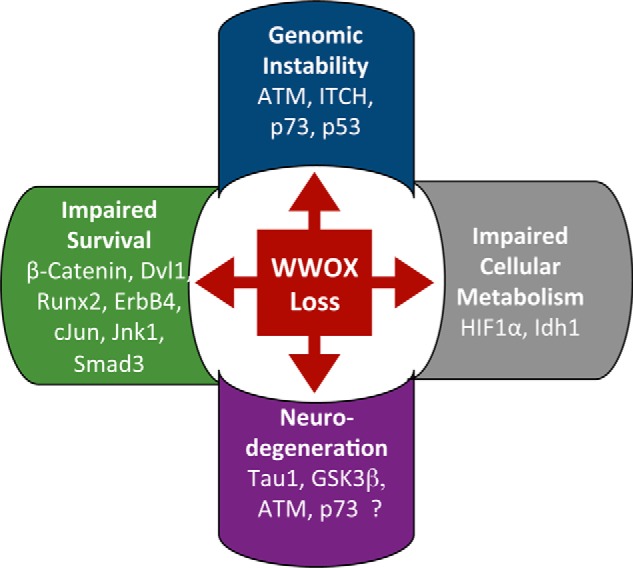
Overview of the biological effects of WWOX loss on signaling pathways. WWOX is involved in several biological functions through its ability to interact physically and functionally with many proteins. For example, upon DNA damage, WWOX interacts with ITCH and ATM, leading to ATM activation, and consequent activation of γH2AX, MDC1, and CHK2. Moreover, WWOX enhances apoptosis through interaction with p53 and p73. At the same time, WWOX inhibits cell growth by inhibiting the Wnt/β-catenin complex through Dvl1 inhibition. Besides, WWOX modulates cellular metabolism by interacting physically with HIF1α and functionally with Idh1. Finally, WWOX inhibits GSK3β activity to phosphorylate Tau that is crucial for proper neurodevelopment. Whether DDR proteins such as ATM and p73 are important for the neurodegeneration phenotype upon WWOX loss is unknown.
This review sketches out how our view of WWOX has evolved over the years, with emphasis on the recent findings that link WWOX to metabolic regulation and CNS homeostasis. We begin with an overview of the original WWOX animal model studies, followed by an outline of subsequent WWOX research performed at the cellular and molecular levels. We review potential WWOX binding partners and their associated cellular pathways. We further define WWOX structure and architecture, expounding its behavior in the context of its two tandem WW domains and expansive short-chain dehydrogenase/reductase (SDR) region. Taken together, we generate a functional model of WWOX that highlights the versatility of this protein and suggests possible new directions for further study of WWOX's complex nature.
Animal Models
Phylogenetic analysis of WWOX protein sequence revealed evolutionary conservation from insects to humans (11), including WWOX's functionally important tandem domains WW1 and WW2, which are followed by an SDR domain (Fig. 2). As such, various animal models have been engineered, not only to determine WWOX association with cancer, but also to provide an optimal framework for researching WWOX function in humans in general.
FIGURE 2.

WWOX protein structure and WWOX mutations in CNS disorders. A, the architecture of the WWOX protein. WWOX contains two N-terminal WW domains (green and magenta, respectively) that are separated by a linker with a nuclear localization sequence (NLS, yellow), and a C-terminal SDR domain composed of an N-terminal (blue), central (orange), and C-terminal (cyan) parts. WW2 stands out by its inability to bind to peptide motifs (e.g. PPXY) when compared with classical WW domains. Features that are usually not observed in classical SDR domains include a long inserted loop adjacent to the catalytic site (gray), as well as a C-terminal extension (cyan). MTS, mitochondrial transmission signal; P, proline. B, mutations in WWOX genes of patients with CNS disorders. Changes include mutations in both WW and SDR domains, ranging from point mutations that lead to non-synonymous amino acid changes in the protein, stop codons (indicated by an asterisk), to skipped exons. The two alleles of each of the reported patients (59, 63) are presented below the architecture scheme, together with their phenotype; S stands for SCAR12, and W stands for WOREE. Patient i, Refs. 20, 63, and 75; patient ii, Refs. 20 and 63); patient iii, Ref. 65; patient iv, Refs. 61 and 63; patient v, Ref. 62; and patients vi–ix, Ref. 63. Phenotypes for all patients except patient v were defined and reviewed in Refs. 63 and 65, whereas patient v has been identified by the authors according to the phenotype descriptions given by Refs. 63 and 65. The exact genotype for patent iii (V202*) is still unknown (65). Its unresolved portion is represented by a dotted line.
Rodent Models: Initial Studies on WWOX and Cancer, and Beyond
Because murine WWOX shares 94% sequence identity with its human homologue, and, notably, a CFS (FRA8E1) (12), Wwox-null mice can be assumed to display phenotypic effects similar to those seen in connection with human WWOX loss. In 2007, we generated the first of these models on congenic B6-129 mixed genetic background, leading to observations of focal lesions resembling osteosarcomas in offspring homozygous for Wwox deletion, as well as enhanced spontaneous and induced tumor incidence and multiplicity in heterozygous offspring (13–15). The Aldaz group (16) also reported that Wwox hypomorphic mice display increased incidence of lymphomas. This nicely fits the theory that WWOX is indeed anti-oncogenic.
However, we also found signs of growth retardation and premature death due to severe metabolic defect exhibiting signs of hypoglycemia, hypolipidemia, and hypocalcemia (15). WWOX deficiency was also associated with osteopenia and bone metabolic defect and steroidogenesis impairment (17, 18). These unexpected results suggested a causative effect not only of WWOX deletion in cancer onset, but also in metabolic malfunction (19). Furthermore, these same Wwox-null mice exhibited spontaneous and audiogenic seizures (20),4 which, in retrospect, is a finding remarkably consistent with recent studies linking WWOX and neurodegenerative diseases described later in this review. These phenotypes were recapitulated in other mouse models carrying a conditional loxp site when bred with a general delete EllA-Cre (21, 22), confirming WWOX in vivo requirement for these phenotypes.
Of important note, in 2009, Suzuki et al. (23) reported a 13-bps deletion in exon 9 of the Wwox gene in Lde/Lde rats (lethal dwarfism with epilepsy), a frameshift that changes its C-terminal protein sequence starting from position 371. They demonstrated that it is associated with seizures, lethal dwarfism, and epilepsy (23). Nevertheless, no tumor phenotype was observed in the Lde/Lde rat model, perhaps suggesting the significance of genetic background or differences of tumor dynamics. Another valid possibility is that WWOX could be more important for tumor progression, requiring another hit for tumor initiation. Alternatively, distinct regions in WWOX could be responsible for distinct functions. Altogether, these findings suggest that the different rodent models are useful for understanding multiple WWOX functions in vivo.
Drosophila melanogaster: Connecting WWOX to Metabolism
Sharing only ∼50% sequence identity with human WWOX (11), fruit fly models containing the dWwox orthologue have their limitations. Even if the fundamental processes between human and fly are somewhat similar, their implementation is often very different (24). Analysis of complex processes and modeling multifactorial human diseases is a challenge in D. melanogaster, although it could serve as a good initial screen that requires further validation in human settings. Drosophila models have in particular contributed to the further appreciation of WWOX's role in regulating mitochondrial metabolism (discussed in detail later in this review). By demonstrating the association of WWOX loss or its reduced expression (using siRNA) with enhanced aerobic glycolysis and reduced mitochondrial function (11), the D. melanogaster model supports the association between WWOX and metabolic function that was noted earlier on in the rodent models described above (15, 17, 22). Furthermore, altered levels of WWOX in D. melanogaster were recently shown to eliminate tumorigenic cells in the fly through TNFα/Egr modulation leading to caspase-3 activity alteration and cell death promotion (25). In contrast, little is known about neurological related phenotypes in dWWOX loss in D. melanogaster. Because the fruit fly genetic model is easy to manipulate genetically, further work should be pursued with these models to study context- and tissue-specific WWOX deletion, e.g. in neurons.
Zebrafish: Another Potential Model Organism for the Study of WWOX
Most recently, Tsuruwaka et al. (26) have demonstrated that depletion of WWOX in zebrafish (70% sequence identity with the human WWOX protein), using siRNA or antisense morpholino, is associated with reduced calcium levels, similar to the hypocalcemia displayed in Wwox-null mice described above. These knockdown fish displayed additional changes associated with growth retardation, postnatal lethality, altered calcium dynamics, and bone defects: phenotypes that very much resemble the Wwox-null mice (15, 17, 22). Thus, the zebrafish could serve as an additional vertebrate model for understanding molecular mechanisms of WWOX loss in human diseases (27). Altogether, it is very obvious that modeling WWOX loss using animal models significantly contributed to our understanding of WWOX functions, not only in cancer but also in homeostasis.
Expanded Search: The Many Faces of WWOX Unraveled by Its Partners
Early animal models made clear the eclectic nature of WWOX. Phenotypic effects of altered WWOX expression marked the protein not only in association with cancer onset, but metabolic regulation and CNS development as well. In an effort to explain WWOX involvement in these three divergent systems, our group sought to identify potential partner proteins of WWOX belonging to cancer-, metabolic-, or CNS-related pathways. Identification of a WWOX partner protein in one of these pathways would implicate WWOX as an acting member of the respective system. By defining the WWOX interactome, we assumed that we would uncover WWOX's putative roles and explain many of the phenotypes observed in animal models and human patients.
WWOX contains two WW domains and an expansive SDR region. WW domains mediate many regulatory interactions by recognizing PPXY and other proline-rich motifs on their partners (28, 29). We therefore conducted pulldown experiments to identify and categorize all WWOX WW domain binding partners. In 2004, we reported the first interacting partner of WWOX, p73 (30). Our strategy was based on “fishing” PPXY-containing proteins known to interact with WW domains of WWOX expressed as GST fusion proteins in an ELISA-like assay (30, 31). A list of putative interacting proteins was identified with variable affinities. In vitro GST pulldown and immunoprecipitation assays confirmed these interactions and validated WWOX interaction with p73 (30), ErbB4 (32), AP2 (33), and others (reviewed in Ref. 34). Some of these interactions were clinically relevant (35, 36). In parallel, the Aldaz group also reported selective interaction of WWOX with PPXY-containing proteins (37) such as SMAD3 (38).
Considering the number and variety of WWOX's PPXY-containing partners, our group took a closer look at the molecular mechanism employed by WWOX's tandem WW domains. Studies have shown that tandem WW domains utilize a variety of strategies to integrate information from binding events (39, 40). We wanted to determine the strategy specific to WWOX's tandem WW domains. Our group applied a combination of proteomic analysis, GST pulldown, and immunoprecipitation experiments to demonstrate that the main interacting module of WWOX is its WW1 domain (41). These observations were supported by the work of the Farooq group (42, 43), demonstrating that the WW2 domain of WWOX shows very low affinity to PPXY partners. Sequence analysis of WW1 and WW2 revealed that the change of a single, highly conserved residue in WW2, Tyr-85, is responsible for WW2's low affinity to the ligand. Indeed, a Y85W mutant is enough to re-establish binding of WW2 to partner PPXY motifs (44). Thus, it has been suggested that the specific mechanism used by WWOX's WW domains is that of a chaperone effect, with WW2 regulating binding of WW1 to its partner, either by stabilizing WW1 and thereby increasing binding affinity (44) or by binding to WW1 in an orientation that occludes the PPXY binding site, generating an internal interaction that can compete with interactions with partners (42, 43).
Whatever its strategy, it is clear that WWOX's tandem WW domains provide the protein with the ability to bind a wide range of different partners, as is evidenced by our pulldown experiments, along with the divergent systems affected by its altered expression. In keeping with findings from earlier animal studies, several of WWOX's putative interacting proteins have been identified as members of CNS-, metabolic-, or cancer related pathways, confirming the link between WWOX expression alteration and the phenotypic effects noted above. This will be elucidated in the following sections.
Complementary Findings: Toward a Comprehensive Picture
WWOX and Metabolism
Reports of functional interactions of dWWOX in the animal models described earlier in this review have shown that it functionally interacts with mitochondrial proteins including isocitrate dehydrogenase (IDH) and superoxide dismutase (SOD1) (45). These results led to the proposal that WWOX plays a role in regulating reactive oxygen species (ROS) and mitochondrial metabolism. Using microarray and proteomic analyses on dWwox-manipulated flies, the Richards lab (45) demonstrated that WWOX functionally interacts with mitochondrial proteins and modulates ROS levels. Importantly, WWOX levels change dramatically in different metabolic settings, with high levels during oxidative phosphorylation, and inversely, very low levels in conditions in which cells rely on glycolysis, such as exposure to hypoxic conditions (46). Indeed, we recently reported that WWOX, via its WW1 domain, physically and functionally interacts with HIF1α and regulates its transactivation function not only in vitro but also in vivo (47). These observations suggest that WWOX not only contributes to intracellular metabolic changes, but that in turn its expression is responsive to the metabolic status of the cell, highlighting its critical role in these settings (48). The fact that changes in WWOX levels might affect glycolysis and ROS levels could be very relevant to its role as a tumor suppressor. Importantly, genetic or pharmacologic depletion of HIF1α reverses WWOX-mediated phenotypes associated with its loss, including tumorigenesis (47). Screening for expression of WWOX and GLUT1, a direct target of HIF1α, in a breast cancer tissue microarray revealed an inverse relationship between these two proteins, confirming that WWOX loss is associated with increased glucose uptake and aerobic glycolysis, likely contributing to metabolism rewiring in cancer cells (47, 48). Moreover, WWOX's SDR domain, which contains a catalytic site and an NADP binding site (Fig. 2), may contribute to WWOX's metabolic role, especially through its enzymatic activity in hormone-secreting organs that show high expression of WWOX. Nonetheless, so far no biochemical studies have investigated the enzymatic activity of human WWOX or its SDR domain function. A recent clue came from studying dWWOX, showing that the SDR enzymatic activity of dWWOX is required for its cellular response to metabolic and mitochondrial defects (49).
WWOX and DNA Damage Response
The association with ROS links WWOX to regulation of the integrity and stability of genetic material. In addition to ROS, integrity is challenged by internal factors such as replication errors, as well as environmental factors, including ionizing irradiation, UV radiation, and chemicals (50). Our lab uncovered a central role of WWOX in maintaining genomic stability (51). The first clue came from increased expression of WWOX immediately after DNA damage including DNA double strand breaks (51) and single strand breaks.5 Proteomic analysis of WW1 domain partners revealed that the ubiquitin E3 ligase ITCH physically associates and functionally mediates Lys-63-linked ubiquitination of WWOX, leading to its stabilization and nuclear localization (41). In the nucleus, WWOX could interact with a number of proteins, including ATM and p73, to mediate efficient DNA damage response such as apoptosis and DNA repair. Our studies clearly demonstrate that WWOX interacts with and activates ATM, leading to increased phosphorylation of ATM substrates including H2AX, MDC1, CHK2, and KAP1 (51). Upon loss of WWOX, ATM function is hampered, leading to accumulation of DNA double strand breaks and consequently rendering the genome less stable (50). These observations and others led us to propose that WWOX, itself a product of a fragile region, FRA16D, functions to protect the genome from damage, and only upon its loss is an increase in DNA damage is observed (52). Because WWOX loss could be associated with unstable genomes, it would be of considerable interest to examine the consequences of treating WWOX-negative tumors with poly(ADP-ribose) polymerase (PARP) inhibitor and/or platinum-based drugs.
It is still a mystery why a gene responsible for maintenance of genome stability should itself be located in a fragile region. Could it be that DNA damage alerts WWOX, and likely other genes spanning CFSs, to activate DDR, but if the damage is too severe, these genes are lost, facilitating elimination of these “bad” cells? In this scenario, WWOX and other products of CFSs may function as electric fuses that “blow up” upon overcurrent to protect from further damage. Although this is a possibility, the picture seems to be more complicated as loss of WWOX is also associated with aggressive traits of cancer cells. This could be dependent on tissue context, as expression of CFS has been shown recently to be cell type-specific (53, 54). Alternatively, other hits inactivating effectors of DDR, such as p53, could also be involved. Moreover, WWOX up-regulation upon DNA damage could facilitate elimination of damaged cells by activating the apoptotic cascade, or decreasing growth rate. For example, WWOX physically interacts with p73 and functionally promotes apoptosis (30). The Chang group (55, 56) has also shown that WWOX could interact with p53 and Jnk1 in some cells, mediating cellular apoptosis. In addition, WWOX was reported to inhibit Wnt/β-catenin complex, inhibiting cell proliferation (57). Altogether, these observations suggest WWOX as a central player in DNA damage repair and apoptosis.
WWOX in the CNS: Patients and Structures
Early clues that linked WWOX with neurological disorders came from animal models demonstrating that WWOX loss is associated with epilepsy and ataxia (20, 23). Supporting evidence came from high expression levels of the WWOX protein in the CNS (15, 59, 60), further indicating a critical role of WWOX in CNS homeostasis. A number of recent studies have demonstrated several WWOX mutations in human patients characterized by early onset of epilepsy, developmental retardation, intellectual deficiency, and neurological manifestations of ataxia (20, 61–65) (Fig. 2). The patients described span a wide range of neurological severity, ranging from seizure disorder associated with global developmental delay, progressive microcephaly, bilateral optic atrophy, and spastic quadriplegia in very young age children (as early as 1.5 months) (recently termed WOREE phenotype, for WWOX-related epileptic encephalopathy), to non-progressive microcephaly and less severe phenotype in adolescence-adult with later onset at 9–12 months (associated with spinocerebellar ataxia type 12 (SCAR12)) (63, 66). This wide range of phenotypic abnormalities could be due to the nature of these mutations. Relatively milder phenotypes seem to originate from missense point mutations (P47T and P47R, G372R), whereas severe manifestations were observed in nonsense mutations (R54*, K297*, and W335*) or partial/complete deletions (Fig. 2). Therefore, WWOX genotypes might correlate with the reported phenotypes, although analysis of more patients would be required to further support this relationship.
The mechanism underlying these neurological abnormalities is poorly understood. The Chang group was a pioneer in demonstrating a functional link between WWOX and effectors of the CNS related to Alzheimer disease such as GSK3β and the cytoskeletal protein Tau-1 (reviewed in Ref. 67). Tau is a microtubule-associated protein that has an important role in microtubule assembly and stability. GSK3β has a vital role in Tau hyperphosphorylation at its microtubule binding domains. Hyperphosphorylated Tau has a low affinity for microtubules, thus disrupting microtubule stability. It has been shown that WWOX can inhibit GSK3β and thus in its absence result in Tau hyperphosphorylation (68, 69). Whether these effectors are important for the observed SCAR12 and WOREE phenotypes is still unknown. The spectrum of mutations in these diseases suggests that the WW1 domain, the nuclear localization sequence between the two WW domains, as well as the SDR domain are involved. The fact that patients harboring WWOX deletions or truncations exhibit a more severe phenotype suggests that the SDR domain indeed plays a critical role. We propose that the WW1 domain might facilitate interaction with partner protein(s) that in turn allow and modulate SDR function(s). It is also possible that WWOX loss of function associated with unstable genome or impaired ROS levels contributes to the observed epileptic encephalopathies.
Similar to Wwox-null mutant animals that died by 3–4 weeks (17), most severe WOREE patients died prematurely (63). However, no bone phenotypes or steroidogenesis impairment were reported or noted. Whether these WWOX mutations would also predispose patients to tumor is also unknown, because no tumor phenotype has yet been reported or noticed for them (20, 61–64). This is likely due to the short life span of these patients or because other mutations would be required for the appearance of the multistep process of tumor formation. Nevertheless, a genetic variant of the WWOX locus (CNV-67048), which is associated with reduced WWOX expression, has been recently correlated with predisposition to lung cancer and gliomas (70–72). Larger studies would be required, and careful analysis of genome integrity needs to be done to further analyze loss of WWOX in cancer predisposition in WOREE and SCAR12 patients.
Further Structural Indications
To the C-terminal side of WW1 and WW2, WWOX contains a third domain, the SDR domain, spanning the majority of the protein chain (Fig. 2). Included in the SDR are a catalytic site, a proton acceptor site, an NADP nucleotide binding site, a region that mediates targeting to the mitochondria, and a C-terminal extension specific to WWOX and a few other SDR-containing proteins only. Interactions mediated by the SDR domain of WWOX have also been reported (reviewed in Ref. 8). Of particular interest is the interaction of WWOX with Tau and GSK3β and its functional outcome in Alzheimer disease (68, 69). Collectively, the picture emerging from these interactions reveals WWOX as an adapter protein regulating transcription and stability as well as localization of its partners (34).
Structural Hints through the Lens of Mutations
The structural analyses described throughout this review demonstrate how identification of sequence-based structural elements, particularly domains, within a protein can indicate the protein's cellular activity and molecular mechanism. Along the same lines, recognition of sequence irregularities within these domains can further signify critical aspects of protein behavior. Recently, several CNS-related WWOX point mutations have been identified. Mapping of these neurological mutations together with known cancer-related point mutations onto the domain architecture of our structural model provides even greater insight into WWOX's molecular mechanism.
Although very few WWOX mutations in cancer patients have been located in the WW domain region, recent studies have linked mutations of Pro-47 of WWOX WW1 to the development of severe, early-onset encephalopathy (20, 63) (Figs. 2 and 3A). Cases of mutation at position 47 were found in two (unrelated) consanguineous families: a homozygous mutation P47T (20), and a heterozygous mutation P47R with a WWOX exon 1 frameshift allele (63). Pro-47 is one of the well conserved residues within WW domains. Its role as a molecular “buckle” that maintains the stability of the WW domain's three β-strand fold was revealed by the first structure of the domain. Interestingly, this proline interacts with the first conserved W to form the stabilizing “buckle” (73). Thus, although Pro-47 has not been determined as directly important to facilitating WWOX interaction with its partner ligands, substitution of this highly conserved residue is expected to cause marked destabilization of the protein core (Fig. 3A), which could in turn lead to changes in the positioning of those residues that are critical for WWOX interaction (20). Overall, point mutations are associated with less severe phenotypes when compared with the stop codon introduced after the first WW domain (R54*) (61), and mutations that lead to exon skipping or even to the overall loss of WWOX protein expression (see above and Fig. 2) (63, 64).
FIGURE 3.

Structural models of the different domains of WWOX. A, the first WW domain WW1 (generated based on the structure of WW3 domain of ITCH, Protein Data Bank (PDB) ID 4n7f (76), using Rosetta with the following constraints: fixed backbone design, constrained relaxation, and minimization (77)). Pro-47 is mutated in several patients with reported CNS-related diseases, reducing the stability of WW and consequently its binding to peptide targets (white). The central position in the core of Pro-47 (in magenta) and its interacting residues (in brown) are highlighted as sticks. B, a structural model of the SDR region (generated by the RosettaCM protocol as implemented by the Robetta server (78), starting from PDB ID 3rd5 (58) as a template structure) shows an extended central β sheet formed by Rossmann folds. A sizeable loop insertion located in the central domain and common to both WWOX and the hypothetical protein of structure 3rd5 is colored gray. Catalytic residues identified by alanine scanning are shown as sticks. Left panel: view from the top of the β sheet onto the catalytic site, with cancer mutation positions (according to Ref. 6) highlighted as spheres. Right panel: G372R, the only point mutation associated with CNS-related diseases (highlighted as spheres) is located at the beginning of the C-terminal extension, which is suspected to play a central role in proper neurodevelopment, as suggested by epilepsy and seizures characterizing lde/lde rats lacking that extension due to a frameshift mutation (23). Basic coloring is kept as in Fig. 2.
Regarding the WWOX SDR domain, those mutations linked to cancer onset (TCGA cBioPortal database) appear to be distributed throughout the whole SDR, both in the conserved Rossmann fold as well as in the α-helix extension that is characteristic of a smaller subgroup of SDR homologues (Fig. 3B). Within the Rossmann fold are highly conserved elements including the protein NADP binding site and the protein catalytic site, whose identifying motif YXXXK was confirmed by alanine substitution of residues Tyr-293 and Lys-297. Recently, the Richards group (49) provided evidence that Tyr-288 in dWWOX (equivalent of Tyr-293 in human WWOX sequence) is important for WWOX's cellular response to mitochondrial defects, highlighting the significance of the SDR enzymatic activity in metabolic reactions. The WWOX SDR domain also features a non-conserved insert of a long loop proximal to its catalytic site (residues 265–289). Whether this loop stabilizes the interaction with the C-terminal extension and/or regulates access to the NADP binding site, it is clear that it is critical to WWOX function; the detrimental mutation at Ser-281 is located in this loop, as is Tyr-287, which upon phosphorylation by Ack1 leads to rapid degradation of WWOX (74). Functional disruption leading to cancer development can easily be surmised from the close proximity of these mutations to any of these functional regions (Fig. 3B).
In contrast, the sole point mutation in the SDR region linked to CNS disease, G372R (20), seems to fall on a less constrained part of the protein. Its location on a seemingly non-functional surface region in the divide between the Rossmann fold and the C-terminal extension leaves many questions regarding the effect of the mutation in causing encephalopathy. However, together with the previously reported mutation in the WWOX gene leading to a frameshift starting from position 371 in Lde/Lde rats that is associated with epilepsy and seizures (23), this emphasizes the importance of this C-terminal tail for proper neural development.
Conclusion
WWOX, an emerging tumor suppressor, is commonly deleted in cancer, marking an advantage of neoplastic cell growth. Recent evidence, however, has linked WWOX function with metabolism and normal CNS development and disease. Over the years, the advantage of using complementary approaches to research a protein as complex as WWOX has repeatedly been made clear. Studies of WWOX at every level, from sequence to structure to clinical implication, provide a bigger picture of this versatile protein's biological function. However, many questions have gone unanswered, and still more have been raised. The binding partner motifs identified by pulldown experiments demand explanation for WWOX WW1's particular binding affinity. Lab experiments and homology modeling still have a way to go in exposing the exact mechanism behind WWOX activity. Also, studies of animal models challenge the association of WWOX with cancer development altogether, bringing our research full circle by re-examining the most basic, underlying assumptions with which we began. However, despite these loose ends, there is considerable overall complementation and confirmation between studies. Several animal models for WWOX have been generated and are being intensively studied in different settings. WWOX loss is a hallmark of aggressive tumors and specific forms of CNS pathologies. We will continue following WWOX along its many cellular pathways.
The authors declare that they have no conflicts of interest with the contents of this article.
R. I. Aqeilan et al., unpublished data.
M. Abu-Odeh, N. Heerema, and R. I. Aqeilan, unpublished data.
- CFS
- common fragile site
- WWOX
- WW domain-containing oxidoreductase
- TSG
- tumor suppressor gene
- SDR
- short-chain dehydrogenase/reductase
- ROS
- reactive oxygen species
- HIF1α
- hypoxia-inducible factor α
- DDR
- DNA damage response
- SCAR12
- spinocerebellar ataxia type 12
- WOREE
- WWOX-related epileptic encephalopathy
- ATM
- ataxia telangiectasia-mutated
- GSK3β
- glycogen synthase kinase 3β
- dWWOX
- Drosophila WWOX.
References
- 1. Bednarek A. K., Laflin K. J., Daniel R. L., Liao Q., Hawkins K. A., and Aldaz C. M. (2000) WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Res. 60, 2140–2145 [PubMed] [Google Scholar]
- 2. Ried K., Finnis M., Hobson L., Mangelsdorf M., Dayan S., Nancarrow J. K., Woollatt E., Kremmidiotis G., Gardner A., Venter D., Baker E., and Richards R. I. (2000) Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum. Mol. Genet. 9, 1651–1663 [DOI] [PubMed] [Google Scholar]
- 3. Gao G., and Smith D. I. (2014) Very large common fragile site genes and their potential role in cancer development. Cell. Mol. Life Sci. 71, 4601–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Durkin S. G., and Glover T. W. (2007) Chromosome fragile sites. Annu. Rev. Genet. 41, 169–192 [DOI] [PubMed] [Google Scholar]
- 5. Beroukhim R., Mermel C. H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J. S., Dobson J., Urashima M., Mc Henry K. T., Pinchback R. M., Ligon A. H., Cho Y. J., Haery L., Greulich H., Reich M., Winckler W., Lawrence M. S., Weir B. A., Tanaka K. E., Chiang D. Y., Bass A. J., Loo A., Hoffman C., Prensner J., Liefeld T., Gao Q., Yecies D., Signoretti S., Maher E., Kaye F. J., Sasaki H., Tepper J. E., Fletcher J. A., Tabernero J., Baselga J., Tsao M. S., Demichelis F., Rubin M. A., Janne P. A., Daly M. J., Nucera C., Levine R. L., Ebert B. L., Gabriel S., Rustgi A. K., Antonescu C. R., Ladanyi M., Letai A., Garraway L. A., Loda M., Beer D. G., True L. D., Okamoto A., Pomeroy S. L., Singer S., Golub T. R., Lander E. S., Getz G., Sellers W. R., and Meyerson M. (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aldaz C. M., Ferguson B. W., and Abba M. C. (2014) WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim. Biophys. Acta 1846, 188–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aqeilan R. I., and Croce C. M. (2007) WWOX in biological control and tumorigenesis. J. Cell. Physiol. 212, 307–310 [DOI] [PubMed] [Google Scholar]
- 8. Chang N. S., Hsu L. J., Lin Y. S., Lai F. J., and Sheu H. M. (2007) WW domain-containing oxidoreductase: a candidate tumor suppressor. Trends Mol. Med. 13, 12–22 [DOI] [PubMed] [Google Scholar]
- 9. Del Mare S., Salah Z., and Aqeilan R. I. (2009) WWOX: its genomics, partners, and functions. J. Cell. Biochem. 108, 737–745 [DOI] [PubMed] [Google Scholar]
- 10. Gardenswartz A., and Aqeilan R. I. (2014) WW domain-containing oxidoreductase's role in myriad cancers: Clinical significance and future implications. Exp. Biol. Med. (Maywood) 239, 253–263 [DOI] [PubMed] [Google Scholar]
- 11. Richards R. I., Choo A., Lee C. S., Dayan S., and O'Keefe L. (2015) WWOX, the chromosomal fragile site FRA16D spanning gene: Its role in metabolism and contribution to cancer. Exp. Biol. Med. (Maywood) 240, 338–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krummel K. A., Denison S. R., Calhoun E., Phillips L. A., and Smith D. I. (2002) The common fragile site FRA16D and its associated gene WWOX are highly conserved in the mouse at Fra8E1. Genes Chromosomes Cancer 34, 154–167 [DOI] [PubMed] [Google Scholar]
- 13. Abdeen S. K., Salah Z., Maly B., Smith Y., Tufail R., Abu-Odeh M., Zanesi N., Croce C. M., Nawaz Z., and Aqeilan R. I. (2011) Wwox inactivation enhances mammary tumorigenesis. Oncogene 30, 3900–3906 [DOI] [PubMed] [Google Scholar]
- 14. Aqeilan R. I., Hagan J. P., Aqeilan H. A., Pichiorri F., Fong L. Y., and Croce C. M. (2007) Inactivation of the Wwox gene accelerates forestomach tumor progression in vivo. Cancer Res. 67, 5606–5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aqeilan R. I., Trapasso F., Hussain S., Costinean S., Marshall D., Pekarsky Y., Hagan J. P., Zanesi N., Kaou M., Stein G. S., Lian J. B., and Croce C. M. (2007) Targeted deletion of Wwox reveals a tumor suppressor function. Proc. Natl. Acad. Sci. U.S.A. 104, 3949–3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ludes-Meyers J. H., Kil H., Nuñez M. I., Conti C. J., Parker-Thornburg J., Bedford M. T., and Aldaz C. M. (2007) Wwox hypomorphic mice display a higher incidence of B-cell lymphomas and develop testicular atrophy. Genes Chromosomes Cancer 46, 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aqeilan R. I., Hassan M. Q., de Bruin A., Hagan J. P., Volinia S., Palumbo T., Hussain S., Lee S. H., Gaur T., Stein G. S., Lian J. B., and Croce C. M. (2008) The WWOX tumor suppressor is essential for post-natal survival and normal bone metabolism. J. Biol. Chem. 283, 21629–21639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aqeilan R. I., Hagan J. P., de Bruin A., Rawahneh M., Salah Z., Gaudio E., Siddiqui H., Volinia S., Alder H., Lian J. B., Stein G. S., and Croce C. M. (2009) Targeted ablation of the WW domain-containing oxidoreductase tumor suppressor leads to impaired steroidogenesis. Endocrinology 150, 1530–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abu-Remaileh M., and Aqeilan R. I. (2015) The tumor suppressor WW domain-containing oxidoreductase modulates cell metabolism. Exp. Biol. Med. (Maywood) 240, 345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mallaret M., Synofzik M., Lee J., Sagum C. A., Mahajnah M., Sharkia R., Drouot N., Renaud M., Klein F. A., Anheim M., Tranchant C., Mignot C., Mandel J. L., Bedford M., Bauer P., Salih M. A., Schüle R., Schöls L., Aldaz C. M., and Koenig M. (2014) The tumour suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Brain 137, 411–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ludes-Meyers J. H., Kil H., Parker-Thornburg J., Kusewitt D. F., Bedford M. T., and Aldaz C. M. (2009) Generation and characterization of mice carrying a conditional allele of the Wwox tumor suppressor gene. PLoS ONE 4, e7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abdeen S. K., Del Mare S., Hussain S., Abu-Remaileh M., Salah Z., Hagan J., Rawahneh M., Pu X. A., Russell S., Stein J. L., Stein G. S., Lian J. B., and Aqeilan R. I. (2013) Conditional inactivation of the mouse Wwox tumor suppressor gene recapitulates the null phenotype. J. Cell. Physiol. 228, 1377–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suzuki H., Katayama K., Takenaka M., Amakasu K., Saito K., and Suzuki K. (2009) A spontaneous mutation of the Wwox gene and audiogenic seizures in rats with lethal dwarfism and epilepsy. Genes Brain Behav. 8, 650–660 [DOI] [PubMed] [Google Scholar]
- 24. Pandey U. B., and Nichols C. D. (2011) Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 63, 411–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Keefe L. V., Lee C. S., Choo A., and Richards R. I. (2015) Tumor suppressor WWOX contributes to the elimination of tumorigenic cells in Drosophila melanogaster. PLoS ONE 10, e0136356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsuruwaka Y., Konishi M., and Shimada E. (2015) Loss of wwox expression in zebrafish embryos causes edema and alters Ca2+ dynamics. PeerJ. 3, e727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bandmann O., and Burton E. A. (2010) Genetic zebrafish models of neurodegenerative diseases. Neurobiol Dis 40, 58–65 [DOI] [PubMed] [Google Scholar]
- 28. Chen H. I., and Sudol M. (1995) The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc. Natl. Acad. Sci. U.S.A. 92, 7819–7823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sudol M., Bork P., Einbond A., Kastury K., Druck T., Negrini M., Huebner K., and Lehman D. (1995) Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J. Biol. Chem. 270, 14733–14741 [DOI] [PubMed] [Google Scholar]
- 30. Aqeilan R. I., Pekarsky Y., Herrero J. J., Palamarchuk A., Letofsky J., Druck T., Trapasso F., Han S. Y., Melino G., Huebner K., and Croce C. M. (2004) Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc. Natl. Acad. Sci. U.S.A. 101, 4401–4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hu H., Columbus J., Zhang Y., Wu D., Lian L., Yang S., Goodwin J., Luczak C., Carter M., Chen L., James M., Davis R., Sudol M., Rodwell J., and Herrero J. J. (2004) A map of WW domain family interactions. Proteomics 4, 643–655 [DOI] [PubMed] [Google Scholar]
- 32. Aqeilan R. I., Donati V., Palamarchuk A., Trapasso F., Kaou M., Pekarsky Y., Sudol M., and Croce C. M. (2005) WW domain-containing proteins, WWOX and YAP, compete for interaction with ErbB-4 and modulate its transcriptional function. Cancer Res. 65, 6764–6772 [DOI] [PubMed] [Google Scholar]
- 33. Aqeilan R. I., Palamarchuk A., Weigel R. J., Herrero J. J., Pekarsky Y., and Croce C. M. (2004) Physical and functional interactions between the Wwox tumor suppressor protein and the AP-2γ transcription factor. Cancer Res. 64, 8256–8261 [DOI] [PubMed] [Google Scholar]
- 34. Salah Z., Aqeilan R., and Huebner K. (2010) WWOX gene and gene product: tumor suppression through specific protein interactions. Future Oncol 6, 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aqeilan R. I., Donati V., Gaudio E., Nicoloso M. S., Sundvall M., Korhonen A., Lundin J., Isola J., Sudol M., Joensuu H., Croce C. M., and Elenius K. (2007) Association of Wwox with ErbB4 in breast cancer. Cancer Res. 67, 9330–9336 [DOI] [PubMed] [Google Scholar]
- 36. Guler G., Iliopoulos D., Guler N., Himmetoglu C., Hayran M., and Huebner K. (2007) Wwox and Ap2γ expression levels predict tamoxifen response. Clin. Cancer Res. 13, 6115–6121 [DOI] [PubMed] [Google Scholar]
- 37. Ludes-Meyers J. H., Kil H., Bednarek A. K., Drake J., Bedford M. T., and Aldaz C. M. (2004) WWOX binds the specific proline-rich ligand PPXY: identification of candidate interacting proteins. Oncogene 23, 5049–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ferguson B. W., Gao X., Zelazowski M. J., Lee J., Jeter C. R., Abba M. C., and Aldaz C. M. (2013) The cancer gene WWOX behaves as an inhibitor of SMAD3 transcriptional activity via direct binding. BMC Cancer 13, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Salah Z., Alian A., and Aqeilan R. I. (2012) WW domain-containing proteins: retrospectives and the future. Front. Biosci. 17, 331–348 [DOI] [PubMed] [Google Scholar]
- 40. Dodson E. J., Fishbain-Yoskovitz V., Rotem-Bamberger S., and Schueler-Furman O. (2015) Versatile communication strategies among tandem WW domain repeats. Exp. Biol. Med. (Maywood) 240, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abu-Odeh M., Bar-Mag T., Huang H., Kim T., Salah Z., Abdeen S. K., Sudol M., Reichmann D., Sidhu S., Kim P. M., and Aqeilan R. I. (2014) Characterizing WW domain interactions of tumor suppressor WWOX reveals its association with multiprotein networks. J. Biol. Chem. 289, 8865–8880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McDonald C. B., Buffa L., Bar-Mag T., Salah Z., Bhat V., Mikles D. C., Deegan B. J., Seldeen K. L., Malhotra A., Sudol M., Aqeilan R. I., Nawaz Z., and Farooq A. (2012) Biophysical basis of the binding of WWOX tumor suppressor to WBP1 and WBP2 adaptors. J. Mol. Biol. 422, 58–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Farooq A. (2015) Structural insights into the functional versatility of WW domain-containing oxidoreductase tumor suppressor. Exp. Biol. Med. (Maywood) 240, 361–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schuchardt B. J., Bhat V., Mikles D. C., McDonald C. B., Sudol M., and Farooq A. (2013) Molecular origin of the binding of WWOX tumor suppressor to ErbB4 receptor tyrosine kinase. Biochemistry 52, 9223–9236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Keefe L. V., Colella A., Dayan S., Chen Q., Choo A., Jacob R., Price G., Venter D., and Richards R. I. (2011) Drosophila orthologue of WWOX, the chromosomal fragile site FRA16D tumour suppressor gene, functions in aerobic metabolism and regulates reactive oxygen species. Hum. Mol. Genet. 20, 497–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dayan S., O'Keefe L. V., Choo A., and Richards R. I. (2013) Common chromosomal fragile site FRA16D tumor suppressor WWOX gene expression and metabolic reprograming in cells. Genes Chromosomes Cancer 52, 823–831 [DOI] [PubMed] [Google Scholar]
- 47. Abu-Remaileh M., and Aqeilan R. I. (2014) Tumor suppressor WWOX regulates glucose metabolism via HIF1α modulation. Cell Death Differ. 21, 1805–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abu-Remaileh M., Seewaldt V. L., and Aqeilan R. I. (2014) WWOX loss activates aerobic glycolysis. Mol. Cell. Oncol. 2, e965640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Choo A., O'Keefe L. V., Lee C. S., Gregory S. L., Shaukat Z., Colella A., Lee K., Denton D., and Richards R. I. (2015) Tumor suppressor WWOX moderates the mitochondrial respiratory complex. Genes Chromosomes Cancer 54, 745–761 [DOI] [PubMed] [Google Scholar]
- 50. Aqeilan R. I., Abu-Remaileh M., and Abu-Odeh M. (2014) The common fragile site FRA16D gene product WWOX: roles in tumor suppression and genomic stability. Cell. Mol. Life Sci. 71, 4589–4599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Abu-Odeh M., Salah Z., Herbel C., Hofmann T. G., and Aqeilan R. I. (2014) WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc. Natl. Acad. Sci. U.S.A. 111, E4716–E4725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hazan I., Abu Odeh M., Hofmann T. G., and Aqeilan R. I. (2015) WWOX guards genome stability by activating ATM. Mol. Cell. Oncol. 2, e1008288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Letessier A., Millot G. A., Koundrioukoff S., Lachagès A. M., Vogt N., Hansen R. S., Malfoy B., Brison O., and Debatisse M. (2011) Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470, 120–123 [DOI] [PubMed] [Google Scholar]
- 54. Le Tallec B., Millot G. A., Blin M. E., Brison O., Dutrillaux B., and Debatisse M. (2013) Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 4, 420–428 [DOI] [PubMed] [Google Scholar]
- 55. Chang N. S., Doherty J., and Ensign A. (2003) JNK1 physically interacts with WW domain-containing oxidoreductase (WOX1) and inhibits WOX1-mediated apoptosis. J. Biol. Chem. 278, 9195–9202 [DOI] [PubMed] [Google Scholar]
- 56. Chang N. S., Doherty J., Ensign A., Schultz L., Hsu L. J., and Hong Q. (2005) WOX1 is essential for tumor necrosis factor-, UV light-, staurosporine-, and p53-mediated cell death, and its tyrosine 33-phosphorylated form binds and stabilizes serine 46-phosphorylated p53. J. Biol. Chem. 280, 43100–43108 [DOI] [PubMed] [Google Scholar]
- 57. Bouteille N., Driouch K., Hage P. E., Sin S., Formstecher E., Camonis J., Lidereau R., and Lallemand F. (2009) Inhibition of the Wnt/β-catenin pathway by the WWOX tumor suppressor protein. Oncogene 28, 2569–2580 [DOI] [PubMed] [Google Scholar]
- 58. Baugh L., Phan I., Begley D. W., Clifton M. C., Armour B., Dranow D. M., Taylor B. M., Muruthi M. M., Abendroth J., Fairman J. W., Fox D. 3rd, Dieterich S. H., Staker B. L., Gardberg A. S., Choi R., Hewitt S. N., Napuli A. J., Myers J., Barrett L. K., Zhang Y., Ferrell M., Mundt E., Thompkins K., Tran N., Lyons-Abbott S., Abramov A., Sekar A., Serbzhinskiy D., Lorimer D., Buchko G. W., Stacy R., Stewart L. J., Edwards T. E., Van Voorhis W. C., and Myler P. J. (2015) Increasing the structural coverage of tuberculosis drug targets. Tuberculosis (Edinb.) 95, 142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nunez M. I., Ludes-Meyers J., and Aldaz C. M. (2006) WWOX protein expression in normal human tissues. J. Mol. Histol 37, 115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen S. T., Chuang J. I., Wang J. P., Tsai M. S., Li H., and Chang N. S. (2004) Expression of WW domain-containing oxidoreductase WOX1 in the developing murine nervous system. Neuroscience 124, 831–839 [DOI] [PubMed] [Google Scholar]
- 61. Abdel-Salam G., Thoenes M., Afifi H. H., Körber F., Swan D., and Bolz H. J. (2014) The supposed tumor suppressor gene WWOX is mutated in an early lethal microcephaly syndrome with epilepsy, growth retardation and retinal degeneration. Orphanet J. Rare Dis. 9, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ben-Salem S., Al-Shamsi A. M., John A., Ali B. R., and Al-Gazali L. (2015) A novel whole exon deletion in WWOX gene causes early epilepsy, intellectual disability and optic atrophy. J. Mol. Neurosci. 56, 17–23 [DOI] [PubMed] [Google Scholar]
- 63. Mignot C., Lambert L., Pasquier L., Bienvenu T., Delahaye-Duriez A., Keren B., Lefranc J., Saunier A., Allou L., Roth V., Valduga M., Moustaïne A., Auvin S., Barrey C., Chantot-Bastaraud S., Lebrun N., Moutard M. L., Nougues M. C., Vermersch A. I., Héron B., Pipiras E., Héron D., Olivier-Faivre L., Guéant J. L., Jonveaux P., and Philippe C. (2015) WWOX-related encephalopathies: delineation of the phenotypical spectrum and emerging genotype-phenotype correlation. J. Med. Genet. 52, 61–70 [DOI] [PubMed] [Google Scholar]
- 64. Valduga M., Philippe C., Lambert L., Bach-Segura P., Schmitt E., Masutti J. P., François B., Pinaud P., Vibert M., and Jonveaux P. (2015) WWOX and severe autosomal recessive epileptic encephalopathy: first case in the prenatal period. J. Hum. Genet. 60, 267–271 [DOI] [PubMed] [Google Scholar]
- 65. Tabarki B., AlHashem A., AlShahwan S., Alkuraya F. S., Gedela S., and Zuccoli G. (2015) Severe CNS involvement in WWOX mutations: description of five new cases. Am. J. Med. Genet A, 10.1002/ajmg.a.37363 [DOI] [PubMed] [Google Scholar]
- 66. Tabarki B., Al Mutairi F., and Al Hashem A. (2015) The fragile site WWOX gene and the developing brain. Exp. Biol. Med. (Maywood) 240, 400–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chang H. T., Liu C. C., Chen S. T., Yap Y. V., Chang N. S., and Sze C. I. (2014) WW domain-containing oxidoreductase in neuronal injury and neurological diseases. Oncotarget 5, 11792–11799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sze C. I., Su M., Pugazhenthi S., Jambal P., Hsu L. J., Heath J., Schultz L., and Chang N. S. (2004) Down-regulation of WW domain-containing oxidoreductase induces Tau phosphorylation in vitro: a potential role in Alzheimer's disease. J. Biol. Chem. 279, 30498–30506 [DOI] [PubMed] [Google Scholar]
- 69. Wang H. Y., Juo L. I., Lin Y. T., Hsiao M., Lin J. T., Tsai C. H., Tzeng Y. H., Chuang Y. C., Chang N. S., Yang C. N., and Lu P. J. (2012) WW domain-containing oxidoreductase promotes neuronal differentiation via negative regulation of glycogen synthase kinase 3β. Cell Death Differ. 19, 1049–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yang L., Liu B., Huang B., Deng J., Li H., Yu B., Qiu F., Cheng M., Wang H., Yang R., Yang X., Zhou Y., and Lu J. (2013) A functional copy number variation in the WWOX gene is associated with lung cancer risk in Chinese. Hum. Mol. Genet. 22, 1886–1894 [DOI] [PubMed] [Google Scholar]
- 71. Yu K., Fan J., Ding X., Li C., Wang J., Xiang Y., and Wang Q. S. (2014) Association study of a functional copy number variation in the WWOX gene with risk of gliomas among Chinese people. Int. J. Cancer 135, 1687–1691 [DOI] [PubMed] [Google Scholar]
- 72. Yang L., Qiu F., Fang W., Zhang L., Xie C., Lu X., Huang D., Guo Y., Pan M., Zhang H., Zhou Y., and Lu J. (2015) The Functional Copy Number Variation-67048 in WWOX Contributes to Increased Risk of COPD in Southern and Eastern Chinese. COPD 12, 494–501 [DOI] [PubMed] [Google Scholar]
- 73. Macias M. J., Hyvönen M., Baraldi E., Schultz J., Sudol M., Saraste M., and Oschkinat H. (1996) Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature 382, 646–649 [DOI] [PubMed] [Google Scholar]
- 74. Mahajan N. P., Whang Y. E., Mohler J. L., and Earp H. S. (2005) Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 65, 10514–10523 [DOI] [PubMed] [Google Scholar]
- 75. Gribaa M., Salih M., Anheim M., Lagier-Tourenne C., H'mida D., Drouot N., Mohamed A., Elmalik S., Kabiraj M., Al-Rayess M., Almubarak M., Bétard C., Goebel H., and Koenig M. (2007) A new form of childhood onset, autosomal recessive spinocerebellar ataxia and epilepsy is localized at 16q21-q23. Brain 130, 1921–1928 [DOI] [PubMed] [Google Scholar]
- 76. Qi S., O'Hayre M., Gutkind J. S., and Hurley J. H. (2014) Structural and biochemical basis for ubiquitin ligase recruitment by arrestin-related domain-containing protein-3 (ARRDC3). J. Biol. Chem. 289, 4743–4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Das R., and Baker D. (2008) Macromolecular modeling with rosetta. Annu. Rev. Biochem. 77, 363–382 [DOI] [PubMed] [Google Scholar]
- 78. Song Y., DiMaio F., Wang R. Y., Kim D., Miles C., Brunette T., Thompson J., and Baker D. (2013) High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]