

Abstract
LepB is a key membrane component of the cellular secretion machinery, which releases secreted proteins into the periplasm by cleaving the inner membrane-bound leader. We showed that LepB is also an essential component of the machinery hijacked by the tRNase colicin D for its import. Here we demonstrate that this non-catalytic activity of LepB is to promote the association of the central domain of colicin D with the inner membrane before the FtsH-dependent proteolytic processing and translocation of the toxic tRNase domain into the cytoplasm. The novel structural role of LepB results in a stable interaction with colicin D, with a stoichiometry of 1:1 and a nanomolar Kd determined in vitro. LepB provides a chaperone-like function for the penetration of several nuclease-type bacteriocins into target cells. The colicin-LepB interaction is shown to require only a short peptide sequence within the central domain of these bacteriocins and to involve residues present in the short C-terminal Box E of LepB. Genomic screening identified the conserved LepB binding motif in colicin-like ORFs from 13 additional bacterial species. These findings establish a new paradigm for the functional adaptability of an essential inner-membrane enzyme.
Keywords: bacteriocin, endonuclease, membrane, microbiology, protease, protein import, toxicity, LepB, colicin
Introduction
The type I signal peptidase LepB (36 kDa) is an essential inner membrane enzyme of Escherichia coli and an integral part of the protein export machinery. LepB is a specialized serine protease that uses a Ser-90/Lys-145 catalytic dyad to perform the highly specialized task of removing the short N-terminal leader signal peptide of 15–30 amino acids from the N terminus of many secreted and membrane proteins (1, 2). LepB spans the inner membrane twice with a large C-terminal domain protruding into the periplasmic space (3).
Colicins are antibacterial toxins produced and released under stress conditions that selectively kill non-colicin producing strains. Colicin D is a toxic tRNase known to kill target cells by cleaving the anticodon loop of all four isoaccepting tRNAArg (4). A specific spontaneous mutation N274K of LepB was shown to result in the loss of sensitivity of E. coli target cells to colicin D (5). Nuclease colicins (DNase or RNase) must be translocated across both the outer and inner membranes to reach their targets in the cytoplasm (6, 7). In this scenario LepB is specifically required for a late import step of colicin D (i.e. after the translocation step across the outer membrane). Moreover, the catalytic activity of LepB is not required for colicin D import and toxicity (8, 9).
Colicin producers are protected against both endogenous and exogenous toxin molecules by the constitutive expression of an immunity (Imm) protein, which forms a neutral complex with nuclease colicins (6, 10, 11). Most colicins (e.g. colicins E) have structurally identifiable N-terminal, central, and C-terminal domains. During early steps of import, the first two domains are required for receptor binding and translocation across the outer membrane, respectively, whereas the C-terminal domain is needed for killing activity (12). The colicin D protein has an unusual tripartite organization. The N-terminal domain is required for both the binding of the colicin to the high affinity, iron-siderophore receptor, FepA, and for its subsequent translocation across the outer membrane (13, 14). The passage of colicin D through the outer membrane is only partially understood, but it is dependent on the proton motive force of the cytoplasmic membrane, transduced by the TonB/ExbB-D import pathway (7, 15). The 277-residue central domain (CD)3 of colicin D is essential for cell killing. It plays dual roles in TonB-mediated import and formation of the colicin D immunity complex (16).
We demonstrated that, in the case of nuclease-type colicins, an endoproteolytic processing step is essential to transfer the cytotoxic C-terminal domain across the inner membrane into the cytoplasm (9, 17). The inner-membrane protease FtsH (18) had previously been shown to be necessary for the toxicity of nuclease colicins (19), and we showed this requirement was at the level of colicin translocation across the inner membrane and that it is presumably responsible for processing of RNase or DNase colicins (9, 20). This hijacked function of FtsH during colicin import is coherent with its usual biological role of degrading misassembled or damaged membrane proteins (21, 22). In the case of colicin D, the productive FtsH-dependent processing may be mediated by LepB. This unexpected function of LepB was suggested to involve in vitro an interaction with the central domain of colicin D (9).
In the present work we demonstrate in vitro that LepB forms a specific and highly stable complex with colicin D, and this interaction is essential for its import. We show that processing and translocation steps are dependent upon the colicin interaction with LepB. We report also that other RNase cytotoxins from two different species of Klebsiella need the novel non-catalytic activity of LepB to kill target cells. Genomic comparisons suggest that this LepB role is presumably required in the case of 10 other bacteriocins.
Experimental Procedures
Bacterial Strains and Plasmids
E. coli K12 DH5α and XL1-blue were used as wild-type host strains for cloning and mutagenesis, respectively. All other strains and plasmids used or constructed are listed in Table 1.
TABLE 1.
Strains and plasmid construction
| Strain or plasmid | Genetic description | Source |
|---|---|---|
| E. coli strains | ||
| DH5α | F- Φ80lacZΔM15Δ (lacZYA-argF) U169 recA1 | Invitrogen |
| BL21(DE3) | ΔompT gal dcm lon hsdSB (rB− mB−)λ (DE3) | (50) |
| A38a | C600 lepB (N274K) | (5) |
| AD202 | ompT1000::kan, KmR | (51) |
| Plasmids | ||
| E. coli | ||
| pLEPB1-wt | pET23 (tag-His6)lepB, AmpR | (32) |
| pLEPB(N274K) | pET23 (tag-His6)lepB (N274K), AmpR | (9) |
| pLEPB1-mutb | pET23 (tag-His6)lepB(mutb), AmpR | This work |
| pColD1c | pUC18 cda-cdi-cdl, (EcoRI and BamHI), AmpR | (52) |
| pColDIc | pET11a cda-cdi(tag-His6), (NdeI/BamHI), AmpR | (9) |
| pColDI-mutc,d | pET11a cda(mutd)-cdi(tag-His6), (NdeI/BamHI), AmpR | This work |
| pColDI(H611A)c | pET11a cda-cdi(H611A; tag-His6), (NdeI/BamHI), AmpR | This work |
| pColD-CD(Glu-314--Met-590) | pET11a cda (tag-H6)Glu-314--Met-590, (NdeI/BamHI), AmpR | (9) |
| pColD-A(Glu-314--Leu-697) | pET11a cda(Glu-314--Leu-697)-cdi(tag-His6), (NdeI/BamHI), AmpR | This work |
| pColD-B(Met-1--Met-590) | pET11a cda (tag-His6)Met-1--Met-590, (NdeI/BamHI), AmpR | This work |
| pColD-E(Met-1--Met-590) | pACYC-Duet1 cda Met-1--Met-590, (NdeI/BglII), CmR | This work |
| pColD-Del(1–14) | pET11c truncated cda(tag-His6), (NdeI/BamHI), AmpR | This work |
| K. pneumoniae | ||
| pMC04-KlebC2e | pBSK+ kcp-kca-kci (BamHI), AmpR | (31) |
| pKlebCI | pACYC-Duet1 kca-kci(tag-His6), (NdeI/BglII), CmR | This work |
| K. oxytoca | ||
| pKlebDf | pBSK+ kdp-kda-kdi, (EcoRI), AmpR | (31) |
a Strain sensitive to pore-forming colicin B but resistant to tRNase colicin D.
b Mutations in LepB: N274A, N274D, N277A, N277K, G272A, or A279G.
c cda and cdi are genes encoding colicin D and its immunity protein; cdl encodes the bacteriocin-release protein.
d Mutations in colicin D: G413A, A422D, A422V, G423D, S426A, P430A, G431D, G431A, R432A.
e kca and kci are genes encoding klebicin C and its immunity protein; kcp and kdp are genes for phage tail fiber-like proteins in Klebsiella strains.
f kda and kdi are genes encoding klebicin D and its immunity protein.
Genetic Constructs and Mutagenesis
Construction of N- or C-terminally truncated colicin D derivatives
The 17 truncated colicin D derivatives (pColD -CD, -A, -B, -Del; Table 1) were created by PCR amplifications employing plasmid pColDI or pColDI(H611A) (carrying an inactivated catalytic domain) as template. The upstream oligonucleotide contained an NdeI restriction site including the ATG start codon in-frame with nucleotides of the cda (colicin D) coding sequence. The downstream oligonucleotide is complementary to cda and ended by an in-frame stop codon followed by a BamHI restriction site. A His6 tag was added to the 5′- or 3′-primer used in each amplification. PCR products after digestion were cloned into plasmid pET11c (Table 1) and transformed into DH5α strain.
Colicins D Constructs Mutated in the Central Domain, LepB Constructs Mutated in the Conserved Box E
Using as template the plasmids pColDI, point mutations within the N-terminal part of the colicin D central domain (between positions 413–454) were generated by QuikChange site-directed mutagenesis method (Stratagene) to give the series of pColDI-mut (Table 1). Similarly, LepB mutants within the C-terminal conserved Box E (Gly-272–Arg-282) were generated by using as template the plasmid pLEPB1-wt (pLEPB1-mut; Table 1). Two oligonucleotide primers containing the desired mutation were used, each complementary to opposite strands of the plasmid, to generate the mutant derivatives. The mutated DNAs were subsequently transformed into XL1-Blue super-competent cells, prepared in the presence of 10 mm RbCl.
Construction of Chimeric Colicin/Klebicin Variants
Chimeric colicin D(A317)/klebicin D(E321) variant was created by the three-step PCR amplifications method, modified from Mora et al. (16). Employing plasmid pColD1 (Table 1) as template, the first PCR used an upstream 113-mer oligonucleotide (containing a modified tac promoter and the cda ribosomal binding site, gctgaaatgagctgttgacaattaatcatcggctcgtataatgtgtggaattgtgagcggataacaatttcacacaaagaggtgttttt, followed by 30 nucleotides of the cda (colicin D) coding sequence starting from the fMet codon and a downstream oligonucleotide, complementary to cda and starting at A317, located upstream of the central domain. The second PCR amplification used an upstream oligonucleotide that is in part complementary to the downstream oligonucleotide used in the first PCR and that is in-frame with the codon E321 of kda (located at the start of the central domain of klebicin D) together with a downstream oligonucleotide, which is complementary to the stop codon of kdi (the immunity protein of klebicin D), using the plasmid pKlebD as template (Table 1), carrying the klebicin D operon. A denaturation-renaturation step of the mixed first two PCR products led to an annealing between complementary strands of the common amplified central part. This annealed DNA mix was employed as template for the third PCR amplification using the upstream oligo (first PCR) and the downstream oligo (second PCR), and this led to the amplification of a linear double-stranded chimeric colicin D/klebicin D DNA. The third PCR product was used directly as a template for in vitro protein synthesis. The same three-step PCR amplification technique was used to create hybrid colicin D(A317)/klebicin C(E223). Here the second PCR amplification used an upstream oligonucleotide that is in part complementary to the downstream oligonucleotide used in the first PCR (as described above), and it is in-frame with the codon E223 of kca (located upstream of the central domain of klebicin C) together with a downstream oligonucleotide complementary to the stop codon of kci (the immunity protein of klebicin C) by employing the plasmid pMC04-KlebC2 (Table 1) as template. Wild-type and hybrid constructs were expressed in vitro in a coupled transcription-translation, Zubay-S30 system from E. coli, optimized for linear DNA templates (Promega) at 37 °C for 60 min.
Wild-type and Recombinant Protein Purification
Full-size wild-type colicin D in complex with the immunity protein carrying a C-terminal His6 tag was produced as previously described (9) from BL21(DE3) cells carrying the plasmid pColDI. The complex was denaturated in 9 m urea, and then the His-tagged ImmD was retained by two passages over a Ni-NTA-agarose column (Protino, Macherey-Nagel), whereas the wild-type colicin D freed of ImmD present in the flow-through was concentrated after dialysis by ultrafiltration (Amicon Ultra-4 10K NMWL; Millipore).
The 17 truncated, His-tagged colicin D derivatives (Table 1), including the whole or part of the central domain, and mutated colicin D-Imm(His6) (pColDI-mut; Table 1) complexes were expressed and eluted by Ni-NTA affinity chromatography at 200 mm imidazole. Truncated colicin Derivatives that were insoluble (for instance the CD) were purified from inclusion bodies. Crude cell extracts were centrifuged, and the pellet was resuspended in 20 mm Tris, pH 7.5, 0.2 m NaCl containing 200 μg of DNase, antiprotease mixture tablet, and lysozyme 1 mg/ml, incubated 30 min at 4 °C, and then sonicated (4 × 1 min). After centrifugation, the pellet was resuspended for 30 min in 50 mm Tris, pH 7.5, 0.2 m NaCl, and 9 m urea at 20 °C. After centrifugation, the supernatant was loaded onto Ni-NTA column, eluted as described above, dialyzed against 20 mm Tris, pH 7.5 (renaturation), and concentrated by ultrafiltration.
His-tagged LepB, wild type or mutated, was produced from BL21(DE3) cells containing plasmid pLEPB1-wt, pLEPB(N274K), or pLEPB1-mut, respectively (Table 1). At A600 = 0.5 the expression of the LepB was induced by isopropyl 1-thio-β-d-galactopyranoside (1 mm), and incubation was continued for 3 h at 37 °C. LepB proteins were purified by Ni-NTA affinity chromatography (adapted from Paetzel et al. (23)). After centrifugation the cells were resuspended, incubated for 30 min at 4 °C in loading buffer (25 mm Tris, pH 7.6, 0.1 m NaCl, 1% Triton X-100, 10 mm imidazole containing 200 μg of DNase and antiprotease mixture tablet), and then broken by sonication. The supernatant was loaded onto a 1-ml Ni-NTA-agarose column and washed with 25 mm Tris, pH 7.6, 0.1 m NaCl, and 0.25% reduced Triton X-100 (a non-ionic and non-UV absorbing surfactant). The His-tagged solubilized LepB was eluted with 200 mm imidazole and then dialyzed against the same buffer. The D273A, R275A, and S281A mutant LepB could not be analyzed due to either their poor expression or the difficulty in extracting them from the detergent-solubilized membrane fraction.
Size Exclusion Chromatography (SEC) of in Vitro Mixed LepB and Colicin D
Purified, full-size (FS), wild-type colicin D, and LepB(His6) alone were analyzed individually by SEC. In addition, purified FS colicin D and a slight excess of His-tagged LepB were mixed in 25 mm Tris, pH 7.5, 0.1 m NaCl, and 0.25% reduced Triton X-100, incubated for 1 h at 25 or 37 °C, and then analyzed by SEC. Purified proteins or protein mixes were run on a Superdex 200GL 10/300 column (GE Healthcare) monitored by an AKTA purifier FPLC system (GE Healthcare). 500-μl fractions were collected for detection of the LepB-colicin D complex and comparison of the SEC profile of the mixed proteins with that of colicin D or LepB analyzed alone. Proteins in the elution fractions were separated on 15% SDS-PAGE.
Real-time Surface Plasmon Resonance Assays
The SPR biosensor real-time experiments were performed on a Biacore 2000 instrument (GE Healthcare) equilibrated at 25 or 37 °C with running buffer (25 mm Tris, pH 7.5, 0.1 m NaCl, 0.3 mm EDTA, 0.05% reduced Triton X-100). The SEC purified, His-tagged LepB protein (encoded by plasmid pLEPB1-wt or pLEPB(N274K)) was captured on Ni-NTA sensor chip (GE Healthcare) surface at an immobilization level of 150 resonance units (pg.mm−2). Colicin D was then injected at different concentrations with a flow rate of 20 μl min−1 for 750 s. The dissociation of the colicin D-LepB complexes was monitored for a 10-min flow of the running buffer without colicin D. The association and dissociation profiles were double-referenced, i.e. both the nonspecific background signals from a streptavidine-His6 reference surface and those from blank experiments (using running buffer without protein) were subtracted. The experimental data of six independent injections per concentration were overlaid and analyzed using the BIAevaluation 4.1 software (GE Healthcare) considering a 1:1 Langmuir binding model. The experimental SPR signals were normalized against the fitted maximum binding signal (Rmax), and the figures thus show the proportion of LepB molecules that are complexed on the y axis (“LepB site occupancy %”). Between binding assays, the surface was regenerated by successive injections of EDTA 0.35 m (2 min), NaOH 50 mm (1 min) and SDS 0.1% (1 min) followed by washing with the running buffer.
To test the interaction of the N-terminally His-tagged CD of colicin D with the His-tagged LepB, the latter was covalently immobilized in an oriented fashion onto a nitrilotriacetic acid-derivatized sensorchip according to Jomain et al. (24) with different surface densities. Immobilized recombinant VEGF (at resonance units = 3000) receptor was used as a non-relevant control protein for the interaction. Colicin D CD was injected with a flow of 20 μl min−1 for 240 s and followed then by two regeneration injections of Gly-HCl 10 mm, pH 2.5.
Detection of LepB Interaction with Colicin D Derivatives by Far Western Blotting
Purified truncated colicin D derivatives (2–5 μg) were separated on a 15% SDS-PAGE, transferred to nitrocellulose membrane, and then stained by Amido Black. A denaturation-renaturation step according to Wu et al. (25) was performed to refold the proteins. After incubation for 1 h at 37 °C with purified wild-type LepB (1 μg/ml) and successive washing steps, the binding of LepB to colicins was analyzed by an anti-LepB antiserum and ECL. Data were collected in triplicate, and the LepB-binding rates averaged.
Killing Activity Test of Bacteriocins
The cytotoxicity of wild-type purified colicin D or colicin D molecules carrying point mutations in the central domain as well as hybrid colicin-klebicins synthesized in vitro (Zubay-S30) was analyzed in vivo by growth inhibition (halo) assay. Undiluted colicins (and 10-fold serial dilutions (5 μl) in the case of purified colicins) were spotted onto a lawn of sensitive AD202 or mutant A38 (carrying the LepB(N274K) mutation) cells on LB plates and then incubated overnight. To increase the binding of in vitro synthesized hybrid colicin Derivatives to the outer-membrane receptor FepA, the killing activity test was performed in the presence of the iron chelator 2,2′-dipyridyl (200 μm). Wild-type klebicin D was found to be non-toxic onto E. coli strains.
In Vivo Detection of Colicin D Processing
50-ml cultures of wild-type or mutant E. coli strains were grown in LB to A600 = 0.4 at 37 °C in the presence of 0.2 mm 2,2′-dipyridyl, which increases the expression of the FepA receptor (26), then 1 mg of wild-type or mutant colicin D-ImmD complex was added to the cultures for 45 min. The cells were harvested, washed three times, resuspended in phosphate buffer, pH 7.4 (1× PBS, Euromedex), and treated with proteinase K at 100 μg/ml for 1 h at 37 °C to hydrolyze residual colicin molecules bound to the cell surface. After four additional washings the colicin- and proteinase K-treated cells were resuspended in 2 ml of PBS buffer with a protease inhibitor complex (Complete EDTA-free; Roche Applied Science) and then disrupted by sonication. Proteins of the S100 cytoplasmic fraction were separated on 15% SDS-PAGE. The processed colicin D form (PF) was detected by Western blot analysis using an antiserum to the colicin D (9).
In Silico Identification of Conserved Amino Acid Motifs at and Near to the LepB Binding Sites (LBS) Located in the Central Domain of E. coli Colicin D
The plasmid-encoded colicin D (pColD CA23) protein originating from E. coli, the colicin D central domain (Glu-314–Met-590), and a shorter peptide segment centered on the LBS were used independently as bait to detect homologous ORFs. The 3241 completely sequenced bacterial chromosomes provided by the NCBI repository were analyzed locally with a C# script using TBLASTN on the SYNTTAX genomic database (27). Colicin D-like ORFs were identified in Gram- Klebsiella pneumoniae (AY578793), Klebsiella oxytoca (AY578792), K. pneumoniae (AAD39262), Erwinia tasmaniensis Et1 99 (bp 59,029), Serratia sp. FS14 (bp 198,771) (the ORF carried by Serratia sp. FS14 contains an internal frameshift mutation suggesting a presumable genetic drift), Serratia marcescens WW4 (bp 188,478), Serratia sp. SCBI (bp91109), Serratia proteamaculans 568 (bp 58,725), Serratia plymuthica S13 (bp 210,642), Serratia liquefaciens ATCC 27592 (bp 212,306), Enterobacter aerogenes KCTC 2190 (bp 68,103), Rahnella aquatilis CIP 78 65 ATCC 33071 (bp 86,855), Pseudomonas aeruginosa PA7 (bp 58,627), and Gram+ Paenibacillus mucilaginosus (WP_041618719.1). Consensus peptide motifs were deduced from chromosomally encoded or plasmid-borne colicin D-like ORFs aligned with Clustal Omega Program.
Results
LepB and Colicin D Form a Stable Complex in Vitro
We became interested in the hijacked function of LepB in colicin D import because of one intriguing possibility that the colicin-LepB interaction replaces or completes the electrostatic associations previously reported between several E-type, nuclease colicins, and the inner membrane (19). Although such a charge-dependent interaction has been proposed as essential to initiate the late import steps, this was not found to be relevant for colicin D (9, 28).
As a first step in this study, we sought to ascertain the existence of a complex formed by LepB and colicin D. We attempted to reconstitute in vitro the complex from separately purified wild-type LepB(His6) and FS colicin D. Both partners were purified, mixed together, and then incubated at 25 or 37 °C for 1 h. The presence of an in vitro assembled complex was assayed by SEC analysis (Fig. 1A). Free FS colicin D ran as a single peak, with an elution volume of 14.2 ml, close to that for free LepB (14 ml). The apparent molecular mass of ∼145 kDa indicated that in solution colicin D (75 kDa) has a dimeric structure, like RNase colicin E3 (29). The membrane protein LepB (36 kDa) in solution is bound to the micelle formed by the detergent Triton X-100 (∼90 kDa; Ref. 30). The apparent molecular weight of LepB (∼145 kDa) presumably indicates a monomeric structure. At both temperatures we obtained the same dichotomic elution profiles (Fig. 1A) with the mixed proteins (i.e. peaks 1 and 2, with elution volumes of 11.9 ml and 14.2 ml, respectively). The molecular weight of the LepB-ColD heterodimer, including Triton binding in solution, could be expected to be ∼220 kDa (145 + 75 kDa). However, our SEC elution data were consistent with the presence of a LepB-ColD complex in peak 1, corresponding to an apparent molecular mass of ∼430 kDa, implying that in vitro the complex may behave as a dimer of heterodimers. More complex was formed at 37 °C, the area under the peak 1 was ∼1.5–2-fold larger at 37 °C than at 25 °C, whereas peak 2, corresponding to isolated LepB and FS colicin D, was diminished by about a factor of 2. The analysis of SEC fractions of peak 1 by SDS-PAGE indicated that both partners assembled into a stable complex and supported the presence of LepB and colicin D in a 1:1 stoichiometry both at 25 °C and at 37 °C (Fig. 1B), as suggested by the SEC analysis.
FIGURE 1.

Complex formation between LepB and colicin D in vitro and formation of the stable LepB-ColD complex after the co-expression of partners, characterized by SEC. A, SEC profiles of LepB(His6) (blue curve), FS colicin D (red curve), and an equimolar ratio of the two proteins mixed in vitro and incubated for 1 h at 25 °C (upper panel) or at 37 °C (bottom panel) are shown. The co-eluted LepB-FS ColD complex separated from both the free LepB and colicin D were solved as peaks 1 and 2 of the chromatogram (black curve), respectively. The Mr calibration curve was obtained with a series of commercial, soluble, standard proteins (ferritin 440 kDa, Stokes radius 61 Å; aldolase 158 kDa, Stokes radius 48.1 Å; conalbumin 75 kDa, Stokes radius 36.4 Å; carbonic anhydrase 29 kDa, Stokes radius 23 Å, and ribonuclease 13.7 kDa, Stokes radius 16.4 Å); for each the constant Kav is plotted against the log Mr (inset). Vertical arrows indicate the apparent Mr for the two peaks. mAU, milliabsorbance units. B, proteins of fractions collected during the SEC elution of the mixed complex, preincubated at 25 or 37 °C were analyzed by 15% SDS-PAGE. SEC fractions corresponding to the LepB-ColD complex (Peak 1) are boxed ahead of those of free partners (Peak 2). C, pLEPB1-wt expressing His-tagged LepB, carrying its intact transmembrane domains and pColD-E expressing C-terminally truncated colicin D (Met-1–Met-590) were co-expressed in BL21(DE3) cells (Table 1). The sonication of cells was followed by 1 h of incubation at 25 °C and then solubilized LepB was co-purified by Ni-NTA affinity chromatography with colicin D (Met-1–Met-590) as shown by SDS-PAGE. Two elution fractions together were subsequently analyzed by SEC and gave two peaks (black curve). The LepB-ColD (Met-1–Met-590) complex, eluted as peak A, had an apparent molecular mass of ∼430 kDa. Peak B was close to the single elution peak of the free monomeric LepB chromatogram (blue curve). The monomeric LepB with the micelle formed by the detergent Triton X-100 (90 kDa) had an apparent molecular mass of ∼145 kDa, as also shown in A. D, the SEC elution fractions were analyzed by SDS-PAGE. The stoichiometry of the LepB-ColD complex appeared to support a 1:1 molar ratio of the two partners (boxed fractions; see also B). The free LepB was enriched in peak B (indicated by a horizontal arrow).
We confirmed the existence of the LepB-ColD complex after the co-expression of His-tagged LepB and C-terminally truncated, non-toxic colicin D (Met-1–Met-590) that included both the N-terminal- and central domains but not the toxic tRNase domain. Colicin D (Met-1–Met-590) was co-purified with solubilized LepB(His6) by Ni-NTA affinity chromatography, and the complex was found to be stable by SEC (Fig. 1, C and D).
In Vitro Binding Affinity of Colicin D to LepB Peptidase
The formation of a stable complex between the wild-type partners shown above by SEC (Fig. 1) prompted us to determine by surface plasmon resonance (SPR) the kinetic constants characterizing the in vitro binding of FS colicin D to LepB. Representative real-time association-dissociation profiles corresponding to a large range of colicin D concentrations injected over wild-type LepB(His6), immobilized by nickel chelation on the sensor chip surface, showed a stable and specific interaction between the partners at both 25 and 37 °C (Fig. 2, A and B). The SPR response corresponding to the LepB site occupancy by colicin D is proportionally increased, with higher colicin D concentrations. Remarkably, the koff (2.6 ± 0.4 × 10−4 s−1) was found to be identical at both temperatures, which indicated that the complex once formed is stable (t½ = 45 min) and that its dissociation was not dependent on the temperature. In contrast, the kon (2.2 ± 0.4 × 104 m−1 s−1 at 25 °C and 15.1 ± 3.0 × 104 m−1 s−1 at 37 °C) increased with temperature. This may reflect variations in the detergent-mediated micelle form of free LepB in vitro, which in turn delays LepB recognition by colicin D at 25 °C. Colicin D binds the peptidase LepB with high affinity (Kd = 12.5 ± 4 × 10−9 m or 2.0 ± 0.4 × 10−9 m at 25 and 37 °C, respectively); thus this interaction should presumably be sufficient in vivo to localize colicin D close to the inner-membrane surface once it is present in the periplasmic space. A higher binding affinity at 37 °C than 25 °C is consistent with the more extensive complex formation detected by SEC at 37 °C (Fig. 1A).
FIGURE 2.

Kinetic measurement of the interaction of wild-type colicin D or the central domain of colicin D with LepB by real-time SPR. A, wild-type LepB(His6) was immobilized by chelation on the surface of a Ni-NTA sensorchip. Different concentrations of wild-type colicin D were injected (t = 0) on the chip surface for 750 s at 25 °C. Color-coded experimental profiles were deduced from the LepB-site occupancy by colicin molecules in real time, whereas fitted curves are shown in gray. Relevant kinetic and equilibrium parameters were determined from the association and dissociation profiles (fitted curves). B, the same experiment was performed at 37 °C. C, the association and dissociation SPR profiles with mutant LepB(N274K) protein, captured on the sensor chip, were obtained with wild-type colicin D injected at 25 °C on the ship surface. D, SPR profiles of the purified colicin D central domain (encoded by plasmid pColD-CD(Glu-314–Met-590); Table 1) injected at 25 °C (380 μg/ml) over different densities (resonance units (RU)= 750–6100) of covalently immobilized wild-type LepB(His6) or VEGF receptor (resonance units = 3000; used as a non-relevant protein) are shown. E, wild-type LepB(His6) was covalently immobilized on the surface of sensor chip. Serial dilutions of purified colicin D CD were injected for 450 s at 25 °C. The corresponding color-coded association profiles, deduced from the LepB-site occupancy by colicin molecules in real time, are shown.
Under the same conditions, the injection of colicin D at different concentrations on immobilized, His-tagged LepB(N274K) protein (from the A38 mutant (Table 1), which is resistant to colicin D killing), showed no detectable interaction at either temperatures (Fig. 2C). It is striking that the absence of an interaction is not the consequence of a global structural unfolding in LepB due to the N274K mutation, as the catalytic function was shown to be unaffected in A38 strain (5). Thus, the absence of interaction may be attributable either directly to the replacement of the Asn-274 residue by a Lys or indirectly to local conformational changes induced by the point mutation. Additional SPR investigations confirmed previous Far Western blot analyses showing that the N-terminally His-tagged central domain of colicin D (CD, purified from pColD-CD; Table 1) specifically recognized the immobilized wild-type LepB, covalently coupled to the sensor chip. No interaction was detected with a non-relevant VEGF-receptor protein (Fig. 2D). Experiments conducted in parallel showed that the rate of interaction increased proportionally with the concentration of the CD of colicin D injected on the covalently immobilized LepB (Fig. 2E), as also observed with the full-size colicin D (Fig. 2A). Further analysis of the binding affinity of CD to LepB could not be investigated by SPR, as the precise oligomerization state of the purified, isolated CD could not be determined unambiguously. However, the global structural integrity of the purified CD could be confirmed by circular dichroism spectroscopy (Fig. 3A).
FIGURE 3.
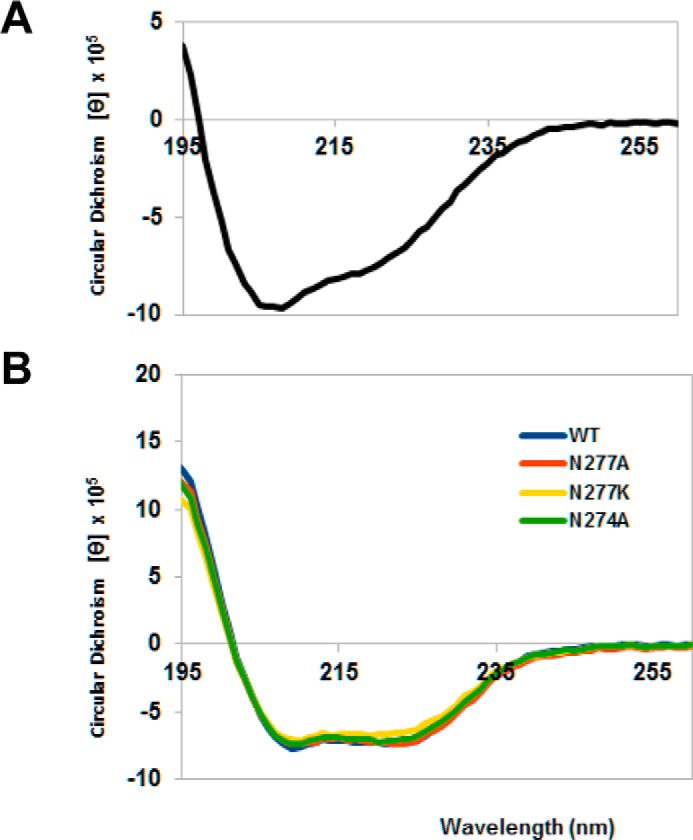
Circular dichroism spectra of the central domain of colicin D and LepB (wild-type and mutant proteins). Far ultraviolet spectra were measured in 20 mm phosphate buffer, pH 7.5, using a Chirascan Spectrometer (Applied Photophysics Ltd., Leatherhead, UK) and recorded at 20 °C from 260 to 195 nm in a 1.0-mm optical path length quartz cell. Spectra were an average of three scans and were corrected for the buffer baseline. Concentrations of each proteins were determined using molar extinction coefficients at 280 nm (49,515 cm−1 m−1 and 20,970 cm−1 m−1 for LepB proteins and the central domain of colicin D, respectively), and the circular dichroism spectra obtained in millidegrees were converted to mean residue molar ellipticity ([θ], MRV are deg cm2 dmol−1). A, the spectrum of colicin D central domain showed characteristic double minima at 208 and 222 nm, indicating the presence of α-helices. B, the spectra of mutated LepB, measured in the presence of 0.25% reduced (non-UV absorbing) Triton X-100, were found to be comparable in pattern and intensity to wild-type (WT) LepB, indicating that the point mutations did not alter the secondary structure of the proteins.
LepB Recognizes Specifically a Small Peptide Motif Inside of the Colicin D CD
17 truncated, colicin D-derived constructs were obtained by PCR amplification and subsequent cloning into a pET11 vector, yielding pColD-A, -B, -CD, and pColD-Del(1–14) (Table 1). The purified colicin derivatives, shown in Fig. 4A, were analyzed by Far Western blotting for their ability to bind to LepB. As expected, only colicin D derivatives carrying the CD were found to be the specific target of LepB binding (Fig. 4C). Neither the presence of the N- nor C-terminal domains affected the binding (Fig. 4, A and C; ColD compared with the derivatives A or B or CD). No interaction was detected with derivatives either starting at or downstream of position 444 (derivatives 2, 3, and 5) or those located before position 410 (derivatives 9 and 13) (Fig. 4C). In contrast, colicin D derivatives 12 or 6, starting at position 410 and ending at position 444, respectively (Fig. 4, A and C), exhibited an interaction with LepB. Accordingly, with all other truncated colicin derivatives straddling the region 410–444 (constructs 1, 4, 7, and 8), binding with LepB was observed. The importance of the CD peptide motif 410–444 in LepB recognition is highlighted by the observation that truncated derivatives starting (derivative 10) or ending (derivative 11) at position 422 showed no interaction with LepB. However, the derivative 14 containing residues 410–437 exhibited a full interaction signal with LepB. This 28 amino acid-long peptide motif is hereafter referred to as the LepB binding site, LBS (Fig. 4A).
FIGURE 4.

Physical maps of truncated/deleted derivatives of colicins D and hybrid colicin/klebicin derivatives. In vitro interaction of the LepB signal peptidase with truncated colicins D or partially deleted central-domain derivatives. A, the domain structure of wild-type (FS) colicin D (formed by the import domain (I), CD, the toxic tRNase domain) is shown in different colors. Regions present in the 17 colicin D derivatives (CD, A (CD+C), B (I+CD), and those numbered 1–14) are indicated. Amino acid positions at the start and stop of the truncated proteins are indicated on the map of colicin D. The small black rectangles at the N or C terminus indicate the location of the His6 tag in each colicin derivative used for protein purification. The location of the LBS is delineated, and the 28-amino acid-long sequence is highlighted in blue. Colicin D constructs carrying the LBS sequence are shown in red. The equivalent putative LBS sequences in tRNase klebicin D (KlebD) and RNase klebicin C (KlebC) are shown. Identical residues in the three LBS sequences are written in red letters. B, color-coded domain structures of wild-type colicin D, klebicins D and C, and the two hybrid colicin/klebicins constructed in this work are shown. In hybrid colicin/klebicin molecules, the intact N-terminal import domain of colicin D is followed by the central and catalytic domains of klebicin D (starting at positions 321) or those of klebicin C (starting at positions 225). The positions of residues delimiting each domain in wild-type molecules or the position of in-frame fusion points in the hybrids are given. The location of the LBS consensus motif, with the positions of the first amino acid, is given for each wild-type or hybrid colicin molecules (yellow boxes). C, polypeptides of full-size colicin D (ColD), colicin D derivatives, or klebicin D (KlebD) were separated on a 15% SDS-PAGE, transferred to a nitrocellulose membrane, and detected by Amido Black staining (AB). Those interacting with purified wild-type LepB were detected with anti-LepB antiserum (Far Western blotting (FW)). Molecular masses (MW) of reference proteins are given in kDa. The numbering of derivatives is the same as in A. D, analysis of the LepB binding to the central domain of colicin D in the presence of competing colicin D derivatives including the LBS (constructs A, 6, and 14) or not (construct 5). Proteins were separated on a 15% SDS-PAGE and treated as above except that LepB was preincubated with the competing colicin derivatives for 30 min before incubation with the membrane. The effect of the preincubation on the efficiency of LepB interaction as measured with anti-LepB antiserum was quantified and expressed as a percentage of the value measured with wild-type LepB without preincubation with any colicin derivative (100%).
To confirm that the recognition of LBS by LepB was responsible for its stable interaction with colicin D, we tested whether extra copies of LBS-containing colicin D sequences could compete for binding LepB with colicin D derivatives. Thus, wild-type purified LepB preincubated with the truncated colicin derivative 6 or 14 (both containing the LBS sequence; Fig. 4A) was shown by Far Western blotting to have a strongly reduced ability (12–20% of free LepB) to bind the same and other truncated colicin derivatives separated on SDS-PAGE (Fig. 4D). In contrast, the intensity of the wild-type interaction signal observed with LepB was unchanged when it was preincubated with the truncated colicin D derivative 5, which does not contain the LBS sequence. Thus, the non-catalytic role of LepB required for import (9) is presumably based upon the site-specific interaction with the incoming colicin D.
The Structural Function of LepB Is Also Required for the Toxicity of Other RNase-type Cytotoxins
Klebicins D and C produced by K. oxytoca and K. pneumoniae, respectively, have a ribonuclease activity and a central domain like colicin D (16, 31) (Fig. 4B). We identified a conserved peptide motif within the LBS sequence present in all three bacteriocins (AG-GSD-VPGR; named LBS consensus motif) (Fig. 4A). This observation suggested that colicin D may not constitute the only substrate that requires a stable structural interaction with LepB. The N-terminal import domains (I) of klebicins C and D are not homologous to colicin D (Fig. 4B). Klebicin D shows no toxicity against wild-type E. coli, suggesting that it uses a Klebsiella-specific import pathway. Klebicin C is toxic for E. coli, but its translocation across the outer membrane of E. coli does not require the FepA receptor, unlike colicin D, but remains dependent on the energy transducing TonB-system (Refs. 7 and 15 and data not shown). To investigate a possible LepB requirement in klebicin import into E. coli target cells, we sought to dissociate the klebicin-specific translocation step across the outer membrane from the putative LepB-dependent crossing of the cytoplasmic membrane. To this aim, we constructed hybrid colicin D-klebicin D (or -klebicin C) in which the intact N-terminal import domain of klebicin D or C was replaced with that of colicin D (Fig. 4B). Thus, translocation of hybrid derivatives into the periplasm became dependent on FepA and TonB, as for colicin D. As shown by a growth inhibition test (Fig. 5A), the in vitro expressed wild-type and hybrid bacteriocins exhibited a strong toxicity on wild-type E. coli. The same bacteriocins all completely lost their toxicity on the LepB(N274K) mutant strain (Fig. 5A). We also verified that an FtsH-deficient strain is fully insensitive to purified klebicin C and the hybrid colicins, like colicin D (data not shown). The in vivo toxicity test provided evidence that the LepB protein also contributes to the toxicity of these two other distinct nuclease cytotoxins, implying that LepB may have a wider, non-catalytic role in bacteriocin import. To investigate this hypothesis, we tried to identify by TBLASTN any ORFs carrying a central region similar to that of colicin D in entirely sequenced bacterial genomes and plasmids. Our inspection showed that, in addition to colicin D and klebicins C and D, the LBS consensus motif (G-GSD-VPGRD) was present in many ORFs encoded by chromosomal or plasmid-borne cda-like genes, present in 10 other species, all belonging to Gram- Gammaproteobacteria (Fig. 6). A less conserved LBS consensus was identified in the phylogenetically distant P. aeruginosa. The genomic analysis also identified in all colicin D-like ORFs a second highly conserved peptide motif (459V-PVRG465 in E. coli, 26 amino downstream of the LBS consensus) (Fig. 6). However, this conserved sequence was not required for the interaction of colicin D with LepB, as truncated peptides missing this sequence (i.e. constructs 6 and 14) still interacted with LepB (Fig. 4, A and C).
FIGURE 5.
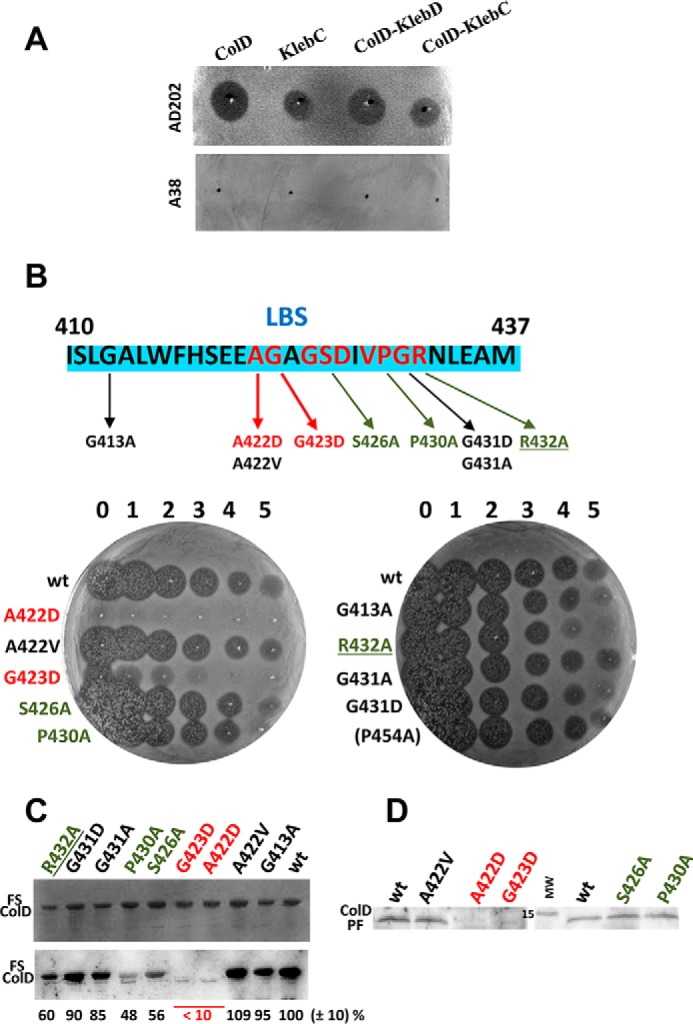
Cell killing by hybrid colicin/klebicin derivatives, effect of point mutations in the LBS on toxicity, interaction with LepB in vitro and processing. A, comparison of the cytotoxicity of in vitro synthesized (Zubay-S30 system), wild-type colicin D (ColD), klebicin C (KlebC), hybrid colicin D/klebicin D (ColD-KlebD), and colicin D/klebicin C (ColD-KlebC) by growth inhibition test is shown. Undiluted aliquots were spotted, as indicated by black points, onto a lawn of AD202 wild-type or A38 LepB(N274K) mutant E. coli strain. Dark halos indicate a clear zone of growth inhibition (toxicity), and no clearing reveals the resistance to wild-type or hybrid colicins. B, nine point mutations to alanine, aspartate, or valine introduced into or close to the LepB-binding site (blue-boxed peptide sequence located in the colicin D central domain) are indicated. The killing activity of these purified mutated colicins D was quantified by spotting aliquots of undiluted colicin (numbered 0) and serial 10-fold dilutions up to 10−5 (numbered 1–5) directly onto a lawn of AD202 strain and analyzed as in A. Turbid zones indicate marginal toxicity. C, wild-type or mutated FS colicins (∼50 ng) were separated by 15% SDS-PAGE and detected by Amido Black staining (upper panel). Their interaction with purified wild-type LepB was detected in vitro by Far Western blotting (lower panel) with anti-LepB antiserum. The efficiency of interaction was quantified and expressed as a percentage of value measured with wild-type colicin D (wt, 100%). D, in vivo detection of the colicin D processed form (ColD PF, 12.4 kDa) from the S100 cytoplasmic fraction of an OmpT-deficient colicin D-sensitive strain treated by purified wild-type or mutated colicins D. Total S100 extracts were separated by 15% SDS-PAGE, and the ColD PF was detected by Western blotting. Mutations provoking a weak or an almost full defect on toxicity on the interaction with LepB and on the processing are indicated in green and red, respectively.
FIGURE 6.

Alignment of conserved amino acid motifs of various bacteriocins homologous to the central domain of colicin D. Alignment of the retrieved protein sequences detected two conserved motifs, the LBS consensus sequence (column 2) and another uncharacterized consensus sequence (column 3), deduced from chromosomally encoded or plasmid-borne (p) colicin D-like ORFs aligned with Clustal-Omega Program. The consensus motifs are separated by 26 amino acids not shown, indicated by (//). Strictly or mostly conserved residues of the consensus sequences are indicated by red and black boldface type capital letters, respectively. Conserved residues in each species are highlighted for each consensus in green (LBS) and blue, respectively. The protein functions, deduced from the peptide sequence comparisons of the C-terminal domains, are indicated (column 4). Sequence references (column 1) are given under “Experimental Procedures.” ORF retrieved from P. mucilaginosus is not homologous to colicin D central domain, although it contains a well conserved LBS-like consensus motif.
Function of LepB in Colicin D Import and Cell Killing
The loss of the interaction of mutated LepB(N274K) with colicin D showed above in vitro and the fact that this mutation specifically prevented both the colicin processing and the sensitivity of target cells to colicin D supported the structural role of LepB in colicin D cytotoxicity (9, 20). We attempted to analyze whether (i) the recognition of LepB may be specifically dependent on conserved amino acids belonging to the LBS of colicin D and (ii) the direct interaction between colicin D LBS and LepB is an indispensable prerequisite for the FtsH-dependent colicin processing and subsequent cell killing. Point mutations (mainly to alanine) were introduced to replace amino acids in six positions of the colicin D LBS consensus (Fig. 5B). The Ala replacements had no or only a slight effect on colicin D toxicity, as shown by a growth inhibition test (Fig. 5B). For instance, colicin D mutated at positions S426A, P430A, or R432A exhibited a weak ∼10-fold reduced toxicity. However, a charged aspartate introduced to replace Ala-422 or Gly-423 almost completely abolished the toxicity. As A422V showed no loss in toxicity, the extra negative charge is probably responsible for the loss in toxicity, at least in the case of A422D. A more graduated response was found by analyzing the interaction between the mutated colicin D proteins and wild-type LepB by Far Western blotting (Fig. 5C). Only with A422D or G423D was the interaction with LepB almost completely abolished (<10%), whereas S426A, P430A, and R432A exhibited an ∼50% reduction in interaction. Cell killing by nuclease colicins is known to require only a few copies of the nuclease domains to reach the cytoplasm (7). Thus, loss of 50% of the binding observed between these three mutated colicin D and LepB remains compatible with an efficient, only 10-fold decreased cell killing (Fig. 5, B and C).
Finally, we tested the level of processing in vivo of several mutated colicin D and observed a wild-type level of the processed colicin D tRNase form (PF; Ref. 9) in the cytoplasm of target cells (AD202) treated by all of them, except for A422D and G423D (Fig. 5D). No PF of these two mutated colicins was detected in the cytoplasm. The processing defect thus appeared to be the direct consequence of the absence of their productive interaction with LepB at the inner membrane. All these results indicated that the structural role of LepB is a key player in the process of colicin D uptake.
Involvement of the Box E Residues of LepB in the Interaction with Colicin D
Four strictly conserved residues of the LepB Box E (positions 272–282) are critical for maintaining a functional enzyme (32) (Fig. 7A). The analysis of the solvent accessibility using the ESPript program (33) showed that most Box E residues are buried near the catalytic center, whereas only Asp-276, Asn-277, and Ala-279 have intermediate or full accessibility to water molecules. In addition to the N274K mutation of strain A38, we have constructed several mutants at (and in the vicinity of) Asn-274. The interactions of these mutated LepB proteins with colicin D constructs (derivatives A and 6; Fig. 4A) were detected by Far Western blotting. The level of interaction was diminished by a factor of ∼2 in the case of N274A, whereas the N274K (and N274D, data not shown) mutants showed only a marginal interaction (<5%) with the central domain (derivative A) or the truncated colicin D derivative 6 (Fig. 7A). The mutation of partially exposed and non-catalytic Asn-277 to an Ala had only a weakly reduced effect (10%) on colicin D binding, whereas N277K showed a strongly reduced binding to the same colicin D derivatives (Fig. 7A). Even though Asn-274 is a strictly conserved residue in prokaryotes, its mutation either to alanine or to Lys had no effect on the enzymatic activity of LepB (5, 32). The buried Gly-272 (an essential residue for signal peptidase activity), when mutated to Ala also abolished the interaction with colicin D constructs (Fig. 7A). In contrast, the water-accessible Ala-279 mutated to Gly exhibited only a moderately reduced level of interaction (85% that of wild type). The global conformational integrity of mutated LepB, either at Box E-residues essential for the catalytic activity (Gly-272; Ref. 32) or at residues found to be relevant only for the interaction of LepB with colicin D (Asn-274 and Asn-277; Fig. 3B) was checked by circular dichroism spectroscopy. The study of these mutations on the three-dimensional structure of LepB (34) showed that the replacement of the asparagine at 274 or 277 by the smaller residue alanine does not introduce any significant steric interference (Fig. 7B). The introduction of the bulkier charged lysine residue at either Asn-274 or Asn-277 as well as the N274D change in LepB potentially impairs the local conformation. Thus, the interaction with colicin D may be abolished, as possibly a consequence of electrostatic or steric hindrance affecting the surface-exposed antiparallel β sheet 8 (amino acids 188–196) or β sheet 9 (amino acids 204–209) (35) close to the colicin D LBS would be located (Fig. 7B).
FIGURE 7.
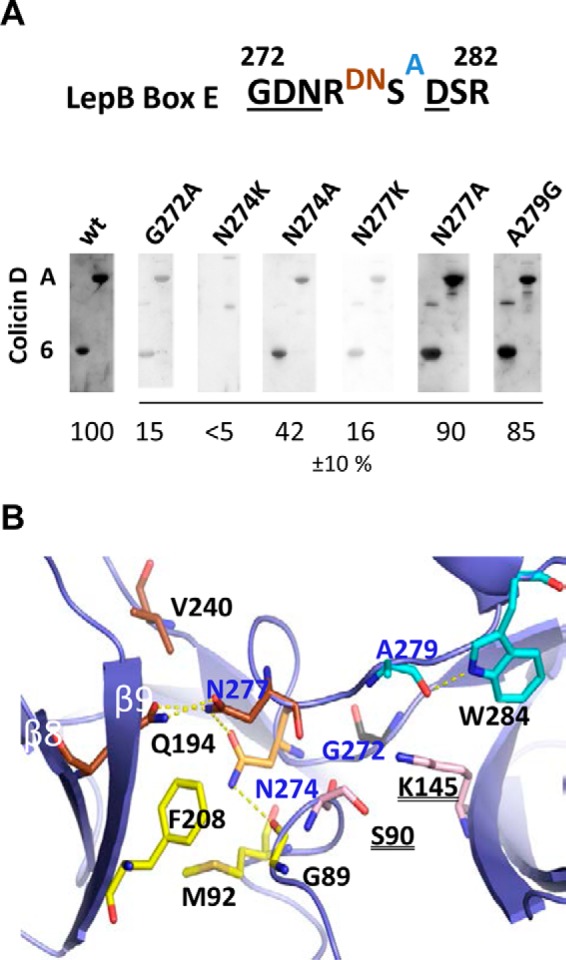
Interaction of the Box E residues of LepB with colicin D and their three-dimensional structure. A, conserved residues of the LepB Box E (positions 272–282; Ref. 32) are underlined. Residues buried near the catalytic center (black letters) in an intermediate position (brown letters) or with full accessibility to water molecules (blue letter) are indicated. The interactions of mutated LepB proteins with colicin D constructs (carrying the central domain (derivative A) or the LBS motif (derivative 6)) separated by 15% SDS-PAGE were detected by Far Western blotting with anti-LepB antiserum. The efficiency of interaction was quantified and expressed as a percentage of values measured with wild-type LepB (wt) and calculated from three sets of experiments. B, in the crystal structure representation of the region surrounding Box E of the E. coli LepB (PDB code 1KN9; Ref. 34) mutated residues Gly-272, Asn-274, Asn-277, and Ala-279 are shown in sticks and are indicated in blue (PyMOL Molecular Graphic System). LepB residues within interacting distance (<3.6 Å) of the mutated residues are indicated in black. Some relevant hydrogen bonds are shown as dotted yellow lines. Residues of the catalytic dyad (Ser-90, Lys-145) are represented in pink sticks.
Discussion
The E. coli LepB, a type I signal peptidase, is a key membrane-bound enzyme of the protein secretion process that has been identified in eubacteria, archaea, and eukaryotes (36). LepB cleaves the signal peptide and thereby releases the mature protein into the periplasm (35, 37). In addition to this canonical scenario, our current work has demonstrated a second function of LepB, which is of ensuring a stable association of the incoming colicin D with the inner membrane during its import. This step appeared to be necessary for the processing of colicin D by FtsH before the subsequent translocation of the toxic domain across the inner membrane into the cytoplasm (17). FtsH acts preferentially on unfolded proteins rather than unfolding the substrates themselves (21). Consequently, it is reasonable to assume that this novel LepB activity implies a structural destabilization of the colicin molecules so that the unstructured C-terminal tails can protrude into the catalytic center of FtsH.
Association of the positively charged and periplasmically exposed nuclease domains with the anionic phospholipids of the inner membrane has been demonstrated to be essential for the toxicity of several E-type nuclease colicins (19, 38, 39). Docking onto the inner membrane was proposed to drive the unfolding of the catalytic domain (28), thereby exposing it as a substrate for FtsH. The central domains of colicin D and klebicins C and D as well as the catalytic domains of colicin D and klebicin D do not exhibit an excess of positive charges, facilitating their electrostatically driven interaction with the inner membrane. Thus, in the case of these incoming bacteriocins, the interaction with LepB can be understood as a way of ensuring or at least improving their association with the cytoplasmic membrane.
In vitro experiments demonstrated the strong and site-specific interaction between LepB and the tRNase colicin D with a Kd in the nanomolar range. The high affinity binding of colicin D with LepB may subsequently make the catalytic domain of colicin D available to the FtsH protease for its subsequent processing. Attachment of the incoming colicin to membrane components appears to be essential throughout the colicin import process. Both the association with an outer membrane receptor (40) and the interaction of colicin D and other bacteriocins with the inner membrane LepB, as demonstrated here, seem to necessitate a high affinity interaction to initiate the translocation into the periplasm and cytoplasm, respectively.
Colicin D hydrolysis by the outer-membrane protease OmpT in vitro, reported as requiring the simultaneous attachment of the LepB protein to the substrate (9), supports the structural role of LepB. LepB binding, ∼200 residues upstream of the OmpT cleavage site, may introduce a local, structural unfolding into colicin D that thus exposes it to hydrolysis by OmpT. Thereby, LepB seems to function similarly to several chaperones or to other adaptor proteins that are reported as necessary for the degradation of various natural substrates by FtsH. For instance, such a chaperone scenario implying FtsH was reported in the case of DnaK/J-mediated degradation of σ32 factor at lower temperatures or for HflK/C-modulated protein quality control of the inner membrane (18, 41, 42).
In addition to Asn-274, two other nearby residues, Asn-277 and Gly-272, also located in the conserved C-terminal Box E of LepB (32), were shown in vitro to be essential for the interaction with colicin D. In the case of the first two residues a dramatic loss was only observed when the asparagine was changed to a bulkier positively charged lysine or by a negatively charged aspartate (at N274D). In contrast, the mutation of Gly-272 to alanine is sufficient to abolish the interaction. Although essential, the contribution of these partly “buried” residues for the interaction appeared to be possibly indirect with regard of the study of their mutations on the solved crystal structure of LepB. The interaction site on colicin D was shown to be located to a 28-amino acid-long LBS motif within the central domain. We also showed that the C-terminal half of the LBS was found to be conserved in the central domains of two other RNases bacteriocins encoded by Klebsiella species, whose toxicity is also strictly dependent on LepB and FtsH. The LBS consensus motif was identified by genomic screening in nuclease-type bacteriocins that carry various import and/or catalytic domains in 13 different bacterial species, suggesting that LepB is a common participant in their import. This observation is supported by the fact that the klebicin C operon was previously reported to have been inserted into a lambdoid prophage, which would explain klebicin diversification as a consequence of bacteriophage-mediated horizontal transfer (31). Thus, the requirement of LepB for colicin import seems to be linked to the spread of a colicin D-type central domain during diversification of bacteriocins by exchanges and mixing of functional domains either by recombination (31, 43, 44) or mediated by bacteriophages (31). It should be noted that when we used only the single LBS sequence (Fig. 4A) for the in silico analysis, the presence of an additional (and longer in comparison with E. coli) consensus sequence (G-GSD-VP-RN-EAM; Fig. 6) was identified near the adenylation domain of the bacitracin synthetase, a 9088-amino acid-long protein complex encoded by the Gram+ bacteria, P. mucilaginosus. The hypothetical role of LepB in the functioning of this key enzyme of the non-ribosomal synthesis of the antimicrobial polypeptide bacitracin (45) still remains elusive.
Although it is well established that colicin import involves the hijacking of various cellular functions, the present study shows that in the case of LepB the hijacking results in a stable interaction between various nuclease-type bacteriocins and the integral, innermembrane enzyme. Usually antibacterial proteins, and in particular colicins, “hijack” or more exactly they “exploit” or “borrow” the innate biological functions of membrane proteins (e.g. outer-membrane receptors-transporters) to permit their entry into target cells. In contrast, previous results showed that the catalytic activity of LepB was not required for colicin D sensitivity (9). Together with the results presented here suggesting that the LepB region required for the interaction with colicin D is located on surface-exposed β sheets rather than close to the catalytic site (Ser-90, Lys-145) as well as the absence of LBS consensus motif in endogenous substrates, this indicates that the leader-peptidase activity is independent of the colicin binding activity of LepB. Thus among many appropriated cellular processes required for colicin recognition, import and toxicity, the case of LepB appears to be unique because there is no functional relationship between its purely structural, presumably “chaperon-like” requirement in colicin import and its canonical signal peptidase activity in protein secretion.
This observation implies that LepB could be considered as a “multifunctional protein,” but in the absence of the identification of any other endogenous cell substrate, the chaperone-like activity of LepB is only a putative cellular function. However, it is interesting to wonder if LepB could be another example of a “moonlighting (multitasking) protein,” of which the chaperon-like function for colicin D is a manifestation (46, 47). Several examples of moonlighting proteins exist, and one example is particularly pertinent. The E. coli anti-oxidant protein thioredoxin (TrxA) is involved in the defense against oxidative damage. However, it was also shown that upon infection with T7 bacteriophage, this enzyme forms a complex with T7 DNA polymerase and thus enhances phage replication (48, 49). The anti-oxidant function of TrxA is fully independent of the T7 replication function, which leads to cell lysis. This is analogous to LepB's catalytic function, which is distinct from the colicin binding function and which also results in cell killing. In conclusion, LepB's exploitation as a structural requirement for bacteriocin import demonstrates an unexpected role of a canonical enzyme.
On the other hand, colicin released into the environment by colicin producers can re-invade producer cells as well as non-producers. In the case of producer cells, on reaching the cytoplasm the colicin is neutralized by the immunity protein. In this case the contact with LepB allows colicin entry into the cytoplasm and may constitute a positive evolutionary pressure for LepB for its colicin binding capacity. In this regard it is interesting to note that the Box E motif of LepB (32) is identical in all species carrying colicin D-like proteins (GDNRDNSADSR), as listed in Fig. 6, whereas there are several variations in the Box E residues in other bacterial species (32).4 Dissecting how this novel structural function of LepB in targeting FtsH processing of nuclease colicins is orchestrated during late import steps remains an exciting challenge for future.
Author Contributions
L. M. and M. de Z. conceived and designed the research. L. M., K. M., and P. E. performed the experiments. L. M., K. M., and M. de Z. analyzed the data. J. O. contributed to the in silico analysis. M. de Z. wrote the paper.
Acknowledgments
We are grateful to Jackie Plumbridge, Kyle Tanner, and Mathias Springer for helpful discussions and valuable comments on the manuscript.
This work was supported by the CNRS (France). The authors declare that they have no conflicts of interest with the contents of this article.
M. de Zamaroczy, unpublished data.
- CD
- central domain
- Ni-NTA
- nickel-nitrilotriacetic acid
- SEC
- size exclusion chromatography
- FS
- full-size
- PF
- processed form
- LBS
- LepB binding site
- SPR
- surface plasmon resonance
- ColD
- colicin D.
References
- 1. Dalbey R. E., Lively M. O., Bron S., and van Dijl J. M. (1997) The chemistry and enzymology of the type I signal peptidases. Protein Sci. 6, 1129–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paetzel M., and Dalbey R. E. (1997) Catalytic hydroxyl/amine dyads within serine proteases. Trends Biochem. Sci. 22, 28–31 [DOI] [PubMed] [Google Scholar]
- 3. Bilgin N., Lee J. I., Zhu H. Y., Dalbey R., and von Heijne G. (1990) Mapping of catalytically important domains in Escherichia coli leader peptidase. EMBO J. 9, 2717–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tomita K., Ogawa T., Uozumi T., Watanabe K., and Masaki H. (2000) A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc. Natl. Acad. Sci. U.S.A. 97, 8278–8283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Zamaroczy M., Mora L., Lecuyer A., Géli V., and Buckingham R. H. (2001) Cleavage of colicin D is necessary for cell killing and requires the inner membrane peptidase LepB. Mol. Cell 8, 159–168 [DOI] [PubMed] [Google Scholar]
- 6. Cascales E., Buchanan S. K., Duché D., Kleanthous C., Lloubès R., Postle K., Riley M., Slatin S., and Cavard D. (2007) Colicin biology. Microbiol. Mol. Biol. Rev. 71, 158–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Zamaroczy M., and Chauleau M. (2011) Colicin killing: foiled cell defense and hijacked cell functions. In Procaryotic Antimicrobial Peptides: from Genes to Applications (Drider D., and Rebuffat S., eds) pp. 255–288, Springer Science Media, Springer New York [Google Scholar]
- 8. de Zamaroczy M., and Buckingham R. H. (2002) Importation of nuclease colicins into E. coli cells: endoproteolytic cleavage and its prevention by the Immunity protein. Biochimie 84, 423–432 [DOI] [PubMed] [Google Scholar]
- 9. Chauleau M., Mora L., Serba J., and de Zamaroczy M. (2011) FtsH-dependent processing of RNase colicins D and E3 means that only the cytotoxic domains are imported into the cytoplasm. J. Biol. Chem. 286, 29397–29407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. James R., Kleanthous C., and Moore G. R. (1996) The biology of E colicins: paradigms and paradoxes. Microbiology 142, 1569–1580 [DOI] [PubMed] [Google Scholar]
- 11. Graille M., Mora L., Buckingham R. H., van Tilbeurgh H., and de Zamaroczy M. (2004) Structural inhibition of the colicin D tRNase by the tRNA-mimicking immunity protein. EMBO. J. 23, 1474–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kleanthous C. (2010) Swimming against the tide: progress and challenges in our understanding of colicin translocation. Nat. Rev. Microbiol. 8, 843–848 [DOI] [PubMed] [Google Scholar]
- 13. Braun V., Patzer S. I., and Hantke K. (2002) Ton-dependent colicins and microcins: modular design and evolution. Biochimie 84, 365–380 [DOI] [PubMed] [Google Scholar]
- 14. Hilsenbeck J. L., Park H., Chen G., Youn B., Postle K., and Kang C. (2004) Crystal structure of the cytotoxic bacterial protein colicin B at 2.5 Å resolution. Mol. Microbiol. 51, 711–720 [DOI] [PubMed] [Google Scholar]
- 15. Roos U., Harkness R. E., and Braun V. (1989) Assembly of colicin genes from a few DNA fragments. Nucleotide sequence of colicin D. Mol. Microbiol. 3, 891–902 [DOI] [PubMed] [Google Scholar]
- 16. Mora L., Klepsch M., Buckingham R. H., Heurgué-Hamard V., Kervestin S., and de Zamaroczy M. (2008) Dual roles of the central domain of colicin D tRNase in TonB-mediated import and in immunity. J. Biol. Chem. 283, 4993–5003 [DOI] [PubMed] [Google Scholar]
- 17. de Zamaroczy M., and Mora L. (2012) Hijacking cellular functions for processing and delivery of colicins E3 and D into the cytoplasm. Biochem. Soc. Trans. 40, 1486–1491 [DOI] [PubMed] [Google Scholar]
- 18. Ito K., and Akiyama Y. (2005) Cellular functions, mechanism of action, and regulation of FtsH protease. Annu. Rev. Microbiol. 59, 211–231 [DOI] [PubMed] [Google Scholar]
- 19. Walker D., Mosbahi K., Vankemmelbeke M., James R., and Kleanthous C. (2007) The role of electrostatics in colicin nuclease domain translocation into bacterial cells. J. Biol. Chem. 282, 31389–31397 [DOI] [PubMed] [Google Scholar]
- 20. Mora L., and de Zamaroczy M. (2014) In vivo processing of DNase colicins E2 and E7 is required for their import into the cytoplasm of target cells. PloS ONE 9, e96549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Herman C., Prakash S., Lu C. Z., Matouschek A., and Gross C. A. (2003) Lack of a robust unfoldase activity confers a unique level of substrate specificity to the universal AAA protease FtsH. Mol. Cell 11, 659–669 [DOI] [PubMed] [Google Scholar]
- 22. Ayuso-Tejedor S., Nishikori S., Okuno T., Ogura T., and Sancho J. (2010) FtsH cleavage of non-native conformations of proteins. J. Struct. Biol. 171, 117–124 [DOI] [PubMed] [Google Scholar]
- 23. Paetzel M., Strynadka N. C., Tschantz W. R., Casareno R., Bullinger P. R., and Dalbey R. E. (1997) Use of site-directed chemical modification to study an essential lysine in Escherichia coli leader peptidase. J. Biol. Chem. 272, 9994–10003 [DOI] [PubMed] [Google Scholar]
- 24. Jomain J. B., Tallet E., Broutin I., Hoos S., van Agthoven J., Ducruix A., Kelly P. A., Kragelund B. B., England P., and Goffin V. (2007) Structural and thermodynamic bases for the design of pure prolactin receptor antagonists: x-ray structure of Del1–9-G129R-hPRL. J. Biol. Chem. 282, 33118–33131 [DOI] [PubMed] [Google Scholar]
- 25. Wu Y., Li Q., and Chen X. Z. (2007) Detecting protein-protein interactions by Far Western blotting. Nat. Protoc. 2, 3278–3284 [DOI] [PubMed] [Google Scholar]
- 26. Higgs P. I., Larsen R. A., and Postle K. (2002) Quantification of known components of the Escherichia coli TonB energy transduction system: TonB, ExbB, ExbD, and FepA. Mol. Microbiol. 44, 271–281 [DOI] [PubMed] [Google Scholar]
- 27. Oberto J. (2013) SyntTax: a web server linking synteny to prokaryotic taxonomy. BMC Bioinformatics 14, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vankemmelbeke M., O Shea P., James R., and Penfold C. N. (2012) Interaction of nuclease colicins with membranes: insertion depth correlates with bilayer perturbation. PloS ONE 7, e46656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Soelaiman S., Jakes K., Wu N., Li C., and Shoham M. (2001) Crystal structure of colicin E3: implications for cell entry and ribosome inactivation. Mol. Cell 8, 1053–1062 [DOI] [PubMed] [Google Scholar]
- 30. Lichtenberg D., Robson R. J., and Dennis E. A. (1983) Solubilization of phospholipids by detergents: structural and kinetic aspects. Biochim. Biophys. Acta 737, 285–304 [DOI] [PubMed] [Google Scholar]
- 31. Chavan M., Rafi H., Wertz J., Goldstone C., and Riley M. A. (2005) Phage associated bacteriocins reveal a novel mechanism for bacteriocin diversification in Klebsiella. J. Mol. Evol. 60, 546–556 [DOI] [PubMed] [Google Scholar]
- 32. Klenotic P. A., Carlos J. L., Samuelson J. C., Schuenemann T. A., Tschantz W. R., Paetzel M., Strynadka N. C., and Dalbey R. E. (2000) The role of the conserved Box E residues in the active site of the Escherichia coli type I signal peptidase. J. Biol. Chem. 275, 6490–6498 [DOI] [PubMed] [Google Scholar]
- 33. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paetzel M., Dalbey R. E., and Strynadka N. C. (1998) Crystal structure of a bacterial signal peptidase in complex with a beta-lactam inhibitor. Nature 396, 186–190 [DOI] [PubMed] [Google Scholar]
- 35. Paetzel M. (2014) Structure and mechanism of Escherichia coli type I signal peptidase. Biochim. Biophys. Acta 1843, 1497–1508 [DOI] [PubMed] [Google Scholar]
- 36. Paetzel M., Karla A., Strynadka N. C., and Dalbey R. E. (2002) Signal peptidases. Chem. Rev. 102, 4549–4580 [DOI] [PubMed] [Google Scholar]
- 37. Economou A., and Wickner W. (1994) SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell 78, 835–843 [DOI] [PubMed] [Google Scholar]
- 38. Mosbahi K., Walker D., Lea E., Moore G. R., James R., and Kleanthous C. (2004) Destabilization of the colicin E9 Endonuclease domain by interaction with negatively charged phospholipids: implications for colicin translocation into bacteria. J. Biol. Chem. 279, 22145–22151 [DOI] [PubMed] [Google Scholar]
- 39. Mosbahi K., Walker D., James R., Moore G. R., and Kleanthous C. (2006) Global structural rearrangement of the cell penetrating ribonuclease colicin E3 on interaction with phospholipid membranes. Protein Sci. 15, 620–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kurisu G., Zakharov S. D., Zhalnina M. V., Bano S., Eroukova V. Y., Rokitskaya T. I., Antonenko Y. N., Wiener M. C., and Cramer W. A. (2003) The structure of BtuB with bound colicin E3 R-domain implies a translocon. Nat. Struct. Biol. 10, 948–954 [DOI] [PubMed] [Google Scholar]
- 41. Akiyama Y. (2009) Quality control of cytoplasmic membrane proteins in Escherichia coli. J. Biochem. 146, 449–454 [DOI] [PubMed] [Google Scholar]
- 42. Narberhaus F., Obrist M., Führer F., and Langklotz S. (2009) Degradation of cytoplasmic substrates by FtsH, a membrane-anchored protease with many talents. Res. Microbiol. 160, 652–659 [DOI] [PubMed] [Google Scholar]
- 43. Jakes K. S., Davis N. G., and Zinder N. D. (1988) A hybrid toxin from bacteriophage f1 attachment protein and colicin E3 has altered cell receptor specificity. J. Bacteriol. 170, 4231–4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kageyama M., Kobayashi M., Sano Y., and Masaki H. (1996) Construction and characterization of pyocin-colicin chimeric proteins. J. Bacteriol. 178, 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murphy T., Roy I., Harrop A., Dixon K., and Keshavarz T. (2007) Effect of oligosaccharide elicitors on bacitracin A production and evidence of transcriptional level control. J. Biotechnol. 131, 397–403 [DOI] [PubMed] [Google Scholar]
- 46. Piatigorsky J., and Wistow G. J. (1989) Enzyme/crystallins: gene sharing as an evolutionary strategy. Cell 57, 197–199 [DOI] [PubMed] [Google Scholar]
- 47. Jeffery C. J. (2003) Moonlighting proteins: old proteins learning new tricks. Trends Genet 19, 415–417 [DOI] [PubMed] [Google Scholar]
- 48. Bedford E., Tabor S., and Richardson C. C. (1997) The thioredoxin binding domain of bacteriophage T7 DNA polymerase confers processivity on Escherichia coli DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 94, 479–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huberts D. H., and van der Klei I. J. (2010) Moonlighting proteins: an intriguing mode of multitasking. Biochim. Biophys. Acta 1803, 520–525 [DOI] [PubMed] [Google Scholar]
- 50. Studier F. W., and Moffatt B. A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130 [DOI] [PubMed] [Google Scholar]
- 51. Akiyama Y., and Ito K. (1990) SecY protein, a membrane-embedded secretion factor of E. coli. is cleaved by the OmpT protease in vitro. Biochem. Biophys. Res. Commun. 167, 711–715 [DOI] [PubMed] [Google Scholar]
- 52. Mora L., Diaz N., Buckingham R. H., and de Zamaroczy M. (2005) Import of the transfer RNase colicin D requires site-specific interaction with the energy-transducing protein TonB. J. Bacteriol. 187, 2693–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]