

Abstract
Iron is an essential element in biological systems, but excess iron promotes the formation of reactive oxygen species, resulting in cellular toxicity. Several iron-related genes are highly expressed in the liver, a tissue in which hepatocyte nuclear factor 4α (HNF4α) plays a critical role in controlling gene expression. Therefore, the role of hepatic HNF4α in iron homeostasis was examined using liver-specific HNF4α-null mice (Hnf4aΔH mice). Hnf4aΔH mice exhibit hypoferremia and a significant change in hepatic gene expression. Notably, the expression of transferrin receptor 2 (Tfr2) mRNA was markedly decreased in Hnf4aΔH mice. Promoter analysis of the Tfr2 gene showed that the basal promoter was located at a GC-rich region upstream of the transcription start site, a region that can be transactivated in an HNF4α-independent manner. HNF4α-dependent expression of Tfr2 was mediated by a proximal promoter containing two HNF4α-binding sites located between the transcription start site and the translation start site. Both the GC-rich region of the basal promoter and the HNF4α-binding sites were required for maximal transactivation. Moreover, siRNA knockdown of HNF4α suppressed TFR2 expression in human HCC cells. These results suggest that Tfr2 is a novel target gene for HNF4α, and hepatic HNF4α plays a critical role in iron homeostasis.
Keywords: iron metabolism, liver, nuclear receptor, transcription regulation, transferrin, HNF4α, hepcidin, hypoferremia, transferrin receptor 2
Introduction
Iron is an essential metallic element in living organisms and is required for the transport of oxygen by hemoglobin in red blood cells and for redox reactions by catalase and various cytochrome P450s. Because most of the iron in the human body is contained in hemoglobin and is recycled from aged red blood cells in the reticuloendothelial system, only 1–2 mg of iron are absorbed at the duodenum, and the amount exactly matches that of iron loss (1, 2). Iron deficiency causes anemia, whereas iron overload has a cytotoxic effect due to the production of reactive oxygen species (ROS).2 Thus, the amount of iron in the organism must be tightly regulated by various processes such as absorption, transport, cellular uptake, and storage (1–3).
Liver is the most important organ for iron metabolism, and many iron-related genes encoding transferrin (TF), transferrin receptor 2 (TFR2), and hepcidin (HAMP), for example, are highly expressed in hepatocytes. TF is a plasma iron carrier, and iron-loaded TF is mainly endocytosed into most tissues through transferrin receptor 1 (TFR1) (2–4). Congenital apotransferrinemia, deficiency of serum TF, causes iron deficiency anemia (5). Unlike TFR2, TFR1 is ubiquitously expressed in many tissues and is required for iron delivery to cells (4, 6). Mutations of the TFR2 gene cause iron overload disease, hereditary hemochromatosis, and the affinity of TFR2 to holo-TF was 25-fold lower when compared with that of TFR1, indicating that TFR2 functions as an iron sensor unlike TFR1 (7–9). TFR2 is also an upstream regulator of HAMP, a peptide hormone secreted from liver (10). HAMP binds to iron exporter, ferroportin 1 (FPN1), and induces internalization and degradation of FPN1, resulting in decreased efflux of iron from the duodenum (11). In addition to the TFR2 gene, mutations of the HAMP and FPN1 genes also cause hereditary hemochromatosis (12). Thus, the HAMP-FPN1 axis is a central coordinator and an iron sensor in the body. Agonists and antagonists of these factors, including HAMP, are under development as therapeutic targets for iron overload disease and anemia of inflammation (1).
HNF4α is an orphan member of the nuclear receptor superfamily and positively regulates many genes involved in liver-specific functions (13, 14). HNF4α was shown to transactive at the TF gene through an HNF4α-binding site within the promoter region in the human hepatoma cell line (15). Moreover, an HNF4α-binding site in the Hamp promoter was essential for bone morphogenetic protein- and hemojuvelin-induced transactivation (16), and hepatic expression of Hamp mRNA was increased in liver-specific Hnf4a-null (Hnf4aΔH) mice (17). These results suggest that hepatic HNF4α could be involved in the control of iron metabolism through these target genes, but the in vivo relationship between HNF4α and iron metabolism in adult liver has not been explored. Because Hnf4aΔH mice exhibit many phenotypes that attenuate liver-specific functions such as lipid and ammonia metabolism and bile acid synthesis (18–20), dysregulation of iron metabolism was also suspected in Hnf4aΔH mice.
In this study, Hnf4aΔH mice were found to exhibit iron metabolism-related phenotypes, resulting in hypoferremia, but no iron deficiency anemia and hemochromatosis was observed. Gene expression profiling revealed that hepatic expression of Tfr2 mRNA was markedly decreased in Hnf4aΔH mice. Promoter analysis showed that the Tfr2 distal promoter region containing GC-rich sequences was required for basal transactivation of the Tfr2 gene. In addition, two HNF4α-binding sites in the proximal promoter region were essential for the maximal transactivation in combination with the proximal GC-rich sequences, revealing that Tfr2 is a novel HNF4α target gene in liver.
Experimental Procedures
Animal
Liver-specific Hnf4a-null (Hnf4aΔH) mice and liver-specific Cebpa-null (CebpaΔH) mice were described previously (18, 21). All experiments were performed with 45-day-old male Hnf4a-floxed (Hnf4af/f) and Hnf4aΔH mice and 2-month-old male Cebpa-floxed (Cebpaf/f) and CebpaΔH mice. Mice were housed in a pathogen-free animal facility under standard 12-h light/12-h dark cycle with water and chow ad libitum. All experiments with mice were carried out under Association for Assessment and Accreditation of Laboratory Animal Care guidelines with approval of the Animal Care and Use Committee of the NCI, National Institutes of Health, and Gunma University Animal Care and Experimentation Committee.
Serum and Liver Iron Measurements
Mice were anesthetized with 2.5% avertin and decapitated, and trunk blood was collected for the measurement of red blood cells (RBC), hemoglobin, hematocrit, and the mean cell volume, or collected in a serum separator tube (BD Biosciences). The serum was separated by centrifugation at 7,000 × g for 5 min and stored at −20 °C prior to analysis. The serum iron concentration and the unsaturated iron-binding capacity were determined colorimetrically using iron and the total iron-binding capacity kit (Sigma). The total iron-binding capacity was calculated by adding the serum iron concentration and the unsaturated iron-binding capacity. Transferrin saturation (%) was calculated as (serum iron concentration/total iron-binding capacity) × 100. Livers were perfused with phosphate-buffered saline (PBS) to remove serum iron, excised, and dried at 100 °C for 16 h. Dried liver samples were dissolved in acidic solution (3 n HCl, 0.6 m trichloroacetic acid) and incubated at 65 °C for 20 h. Extracted iron solution was reacted with chromogen solution (1.25 m acetic acid (pH 4.7), 0.008% bathophenanthroline, 0.08% glyceric acid) at room temperature for 30 min, and the liver iron was determined colorimetrically at 510 nm.
IronChip Analysis
For the cDNA microarray experiments, the “IronChip” (version 3.0) was used. Experimental details concerning the selection of the cDNA clones, the preparation of the microarray platform, the synthesis of fluorescent cDNA probes, prehybridization, and hybridization conditions of the microarrays as well as scanning and data analysis are described elsewhere (22–24). The symbols of the genes mentioned and RefSeq accession number in the text are summarized in Table 4.
TABLE 4.
Expression profiling of hepatic mRNA involved in iron metabolism in Hnf4aΔH mice
Microarray analysis was performed using pooled total RNA from liver tissues of Hnf4aΔH and Hnf4af/f mice. Ratio of expression levels between both of them was indicated as fold-change.
| Gene symbol | Ref. Seq. (NM_) | Fold-change (Hnf4aΔH/Hnf4af/f) | Gene symbol | Ref. Seq. (NM_) | Fold-change (Hnf4aΔH/Hnf4af/f) |
|---|---|---|---|---|---|
| Tf | 133977 | 0.17 | Irp1 | 007386 | 0.72 |
| Ltf | 008522 | 0.19 | Fth | 010239 | 0.73 |
| Mgst1 | 019946 | 0.29 | Zfp36 | 011756 | 0.82 |
| Tfr2 | 015799 | 0.30 | Fmo2 | 018881 | 1.31 |
| Atp7b | 007511 | 0.33 | Npm1 | 008722 | 1.38 |
| Selp | 011347 | 0.33 | Hif1a | 010431 | 1.39 |
| Cat | 009804 | 0.38 | Fmo1 | 010231 | 1.42 |
| Efna1 | 010107 | 0.44 | Mt3 | 013603 | 1.47 |
| Gpx1 | 008160 | 0.48 | Srsf3 | 013663 | 1.52 |
| Sod1 | 011434 | 0.48 | Por | 008898 | 1.53 |
| Ldh1 | 010699 | 0.50 | Cox17 | 001017429 | 1.55 |
| Rplp0 | 007475 | 0.51 | Arg1 | 007482 | 1.58 |
| Atp7a | 009726 | 0.52 | Maob | 172778 | 1.61 |
| Prodh | 011172 | 0.53 | Hamp | 032541 | 1.68 |
| Nr1i3 | 009803 | 0.56 | Hspa8 | 031165 | 1.71 |
| Ftl | 010240 | 0.58 | Cp | 007752 | 1.72 |
| Acss2 | 019811 | 0.58 | Hspa5 | 022310 | 1.72 |
| Slc31a1 | 175090 | 0.60 | Psap | 011179 | 1.72 |
| Hpx | 017371 | 0.63 | App | 007471 | 1.88 |
| Acox3 | 030721 | 0.63 | Gsn | 146120 | 1.89 |
| Atox1 | 009720 | 0.64 | Tfr1 | 011638 | 3.31 |
| Impa1 | 018864 | 0.64 | Mt1 | 013602 | 3.80 |
| Cyp3a13 | 007819 | 0.64 | Fmo3 | 008030 | 4.34 |
| Cyp1a1 | 009992 | 0.65 | Spp1 | 009263 | 5.05 |
| Txn1 | 011660 | 0.70 | Mt2 | 008630 | 6.19 |
| Nfs1 | 010911 | 0.70 | |||
RNA Extraction, Reverse Transcription, and Real Time PCR
Total RNA from the mouse livers and HepG2 cells was extracted using Tripure Isolation Reagent (Roche Applied Science, Tokyo, Japan). cDNA was transcribed using ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO, Osaka, Japan), and real time PCR was performed using FastStart SYBR Green Master Mix (Roche Applied Science) with the specific primers on LightCycler 480 System II (Roche Applied Science). Levels of mRNA expression were normalized relative to Gapdh mRNA as an internal control using the ΔΔCt method. The following primers were used for real time PCR: mouse Hnf4a (agaggttctgtcccagcagatc and cgtctgtgatgttggcaatc); human HNF4A (caggctcaagaaatgcttcc and ggctgctgtcctcatagctt); mouse Tfr2 (ctatctggtcctgatcaccct and tcagggttgacatcttcatcga); human TFR2 (gtggaccgacacgcactac and tgtaggggcagtagacgtcag); mouse Gapdh (gacttcaacagcaactcccac and tccaccaccctgttgctgta); human GAPDH (agccacatcgctcagacac and gcccaatacgaccaaatcc); mouse Tf (cgcagtcctcttgagaaagc and agcctgggcacagttgac); mouse Hjv (gccaacgctaccaccatc and tcaaaggctgcaggaagatt); mouse Ftl (ttttgatcgggatgacgtg and cgttctgaaactcgaggagac); mouse Fth (tggagttgtatgcctcctacg and tggagaaagtatttggcaaagtt); mouse Hpx (ggaagaatcccatcacctca and caggagggtacacccagact); mouse Cat (ccttcaagttggttaatgcaga and caagtttttgatgccctggt); mouse Sod1 (ccatcagtatggggacaataca and ggtctccaacatgcctctct); mouse Gpx1 (ggtttcccgtgcaatcagt and tcggacgtacttgagggaat); mouse Tfr1 (tggaatcccagcagtttctt and gctgctgtacgaaccatttg); mouse Hamp (gatggcactcagcactcg and ctgcagctctgtagtctgtctca); mouse Hfe (ggaaaaggaaggcttcagga and cctccaagtctttggctgag); mouse Fpn1 (tggccactctctctccactt and tgtcaagagaaggctgtttcc); mouse Heph (tctatacatgcccatggagttct and tgggatgttccactggtaagt); mouse Cp (gggccaatgaaaatatgcaa and tcaaacactgtgggaaacaagt); and mouse Cebpa (tggacaagaacagcaacgag and tcactggtcaactccagcac).
Cloning of Promoter Region of Mouse Tfr2 Gene
The −1972, −982, −396, −241, −116, −82, −66, −46/+67, −116, −46, +505/+1144, −46, −116/+504, and −116/+896 fragments from the transcription start site of the mouse Tfr2 promoter containing KpnI and XhoI sites were amplified with genomic DNA from mouse liver by PCR and cloned into the luciferase reporter vector, pGL4.11 (Promega, Madison, WI). HSV-TK mini and HSV-TK promoters were amplified with pGL4.74 (Promega) by PCR and cloned into EcoRV and BglII/HindIII sites of the pGL4.11 and named pGL4.11/TK and pGL4.11/TK mini, respectively. Mutations were introduced into GC-rich sequences and HNF4α-binding sites in the Tfr2 promoter by overlapped or inverted PCR-based site-directed mutagenesis. The following primers were used for amplification of HSV-TK mini promoter (ggcatagatctttcgcatattaaggtgacgcgtgtggcctc and ggcataagcttttaagcgggtcgctgcagggtcgctcggtg) and the TK promoter (gcataagcttaaatgagtcttcggacctcg and ggcataagcttttaagcgggtcgctgcagggtcgctcggtg). Similarly, the following primers were also used for PCR-based mutagenesis of GC-1 region (atttgttagtcctctggggcgg and aacaaatcgcacgtccttttct), GC-2 region (tttacatttgctgggggcgtgctct and caaatgtaaaagaggactccgcccc), GC-3 region (taaactaactctagtgggtgtgg and ttagtttacaggccccgccccag), the HNF4α-binding site at +182/+201 (tgacgctgtgatttcccattcacagctgc and acctcacccttacttctggtccagg), and the distal HNF4α-binding site at +830/+842 (cgcgtctgctctgaggtttaaaaaa and ggccaacttggttcccgtcacaggt). The induced mutations are indicated as bold and underlined.
Transient Transfection and Luciferase Assays
HepG2 and HEK293T cells were cultured at 37 °C in Dulbecco's modified Eagle's medium (WAKO, Osaka, Japan) containing 10% fetal bovine serum (HyClone, Logan, UT) and 100 units/ml penicillin/streptomycin (Invitrogen). For suspension transfection, wild type or mutated Tfr2 promoters cloned into pGL4.11 and pGL4.74 encoding Renilla luciferase regulated by the HSV-TK promoter as an internal control were transfected into HepG2 cells with polyethyleneimine max (Polyscience, Warrington, PA) as a transfection reagent. For co-transfection using HEK293T cells, HNF4α expression plasmid, pSG5/HNF4α, was used (19). After 48 h, the cells were washed with phosphate-buffered saline (PBS), and promoter activities were measured using Dual-Glo Luciferase Assay System (Promega).
Transfection of siRNA
Two kinds of siRNA for human HNF4α (10 nm, Sigma, Tokyo, Japan) were independently transfected into HepG2 cells with Lipofectamine RNAiMAX (Life Technologies, Inc.). After 48 h, cells were trypsinized and re-transfected with 10 nm of the same siRNA. After 48 h of re-transfection, cells were harvested, and total RNA was extracted using Tripure Isolation Reagent for RT quantitative PCR. Nucleotide sequences for the siRNA duplexes targeting human HNF4α are follows: GGCAGUGCGUGGUGGACAA for siHNF4α-1 and AGAGAUCCAUGGUGUUCAA for siHNF4α-2.
Western Blot
Liver samples from Hnf4af/f and Hnf4aΔH mice were homogenized in lysis buffer (7 m urea, 2 m thiourea, 1% Triton X-100) and allowed to sit on ice for 30 min. The homogenate was centrifuged at 12,000 × g for 30 min at 4 °C, and the supernatants were used as whole cell lysates. The whole cell lysates and serum protein (40 μg), determined by Quick StartTM Bradford Dye Reagent (Bio-Rad), were diluted with Laemmli sample buffer, incubated at 65 °C for 15 min, and fractionated by 10% SDS-PAGE. The gels were stained with Coomassie Brilliant Blue R-250 or transferred onto a PVDF membrane (GE Healthcare, Tokyo, Japan). The membrane was incubated for 1 h with PBS containing 0.1% Tween 20 and 5% skim milk and then incubated for 1 h with anti-transferrin (Santa Cruz Biotechnology, Dallas, TX), anti-transferrin receptor 2 (Abcam, Tokyo, Japan), and anti-γ-tubulin (TUBG) (Sigma) antibodies. After washing, the membrane was incubated for 1 h with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology), and the reaction product was visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce). Expression of TF and TFR2 proteins was quantified by densitometric analysis using ImageJ software and the expression in Hnf4aΔH was presented as expression differences relative to the Hnf4aff/f mice.
Gel Mobility Shift Analysis
Nuclear extracts from HepG2 cells were prepared using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific, Yokohama, Japan), and gel shift analysis was carried out using LightShift Chemiluminescent EMSA kit (Thermo Fisher Scientific). The following double-stranded probes were used (mutations are indicated as bold and underlined); the HNF4α-binding site at +182/+201 in the mouse Tfr2 promoter (wild type, gggtgaggtggaccctgaactttcccattcacagctgca and tgcagctgtgaatgggaaagttcagggtccacctcaccc; mutant, gggtgaggttgacgctgtgatttcccattcacagctgca and tgcagctgtgaatgggaaatcacagcgtcaacctcaccc); the HNF4α-binding site at +830/+842 in the mouse Tfr2 promoter (wild type, gaaccaagttggccaaagtctgctctgaggt and acctcagagcagactttggccaacttggttc; mutant, gaaccaagttggcccgcgtctgctctgaggt and acctcagagcagacgcgggccaacttggttc); and the HNF4α-binding site at −203/−192 in the mouse ornithine transcarbamylase (Otc) promoter as a positive control (gttaggcttaaagttcaagtg and cacttgaactttaagcctaac) (19). Nuclear extracts (3 μg) and the 5′-biotin-labeled probes of the HNF4α-binding sites for the Tfr2 promoter (wild type) were added, and the reaction mixture was incubated on ice for 10 min. For competition experiments, a 50-fold excess of unlabeled probe was added to the reaction mixture, and the mixture was incubated on ice for 10 min prior to the addition of the 5′-biotin-labeled probe. For supershift analysis, 1 μg of anti-HNF4α or anti-peroxisome proliferator-activated receptor α antibodies (Santa Cruz Biotechnology) was added to the reaction mixture, and the mixture was incubated on ice for 10 min after the addition of the 5′-biotin-labeled probe. DNA-protein complexes were fractionated by 7% PAGE containing 5% glycerol and blotted onto a Biodyne B nylon membrane (Pall, Tokyo, Japan). After washing, DNA-protein complexes were visualized using a detection module in the kit on an ImageQuant LAS4000.
Chromatin Immunoprecipitation
HepG2 cells cultured in a 10-cm dish were fixed in 0.5% formaldehyde for 10 min and then quenched with 125 mm glycine for 5 min at room temperature. After washing with ice-cold PBS, the cells were resuspended in 3 ml of Lysis buffer 1 (50 mm HEPES-KOH (pH 7.5), 140 mm NaCl, 1 mm EDTA, 10% glycerol, 0.5% Nonidet P-40, 0.25% Triton X-100, and protease inhibitor (Roche Applied Science)) on ice for 10 min and then centrifuged at 1,400 × g for 5 min. The cell pellet was resuspended in 3 ml of Lysis buffer 2 (10 mm Tris-HCl (pH 8.0), 200 mm NaCl, 1 mm EDTA, and 5 mm EGTA) for 10 min at room temperature and then centrifuged at 1,400 × g for 5 min. The cell pellet was resuspended in 1 ml of Lysis buffer 3 (10 mm Tris-HCl (pH 8.0), 300 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, and 0.1% sodium deoxycholate). Liver samples stored at −80 °C were ground to pieces with a pestle and mortar under liquid nitrogen and fixed in PBS containing 20 mm sodium butyrate, 1% formaldehyde, and protease inhibitor mixture for 10 min at room temperature. After centrifugation, the pellet was resuspended in Lysis buffer (50 mm Tris-HCl (pH 8.0), 10 mm EDTA, 1% SDS, and 20 mm sodium butyrate). The cell lysate from HepG2 cells and liver samples was disrupted by an ultrasonicator (UR-20P, TOMY, Tokyo, Japan) for 5 min on ice and then 1% Triton X-100 was added, followed by centrifuging at 20,000 × g for 10 min. A small volume of the supernatant was stored at 4 °C as the input samples. The remaining supernatant was pre-cleared by adding of 15 μl of a 50% slurry of protein G-Sepharose 4 Fast Flow (GE Healthcare) with sonicated salmon sperm DNA and rotated for 4 h at 4 °C, followed by centrifuging at 1,900 × g for 5 min. The supernatant was divided into two pieces and incubated with anti-HNF4α antibody (4 μg, Santa Cruz Biotechnology) or control normal goat IgG for 16 h at 4 °C. The reaction mixture was centrifuged at 1,900 × g for 5 min at 4 °C, and the pellet collected was washed for 5 min at 4 °C with 1 ml of RIPA-1 buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% SDS, and 0.1% sodium deoxycholate), RIPA-2 buffer (50 mm Tris-HCl (pH 8.0), 300 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% SDS, and 0.1% sodium deoxycholate), LiCl wash solution (10 mm Tris-HCl (pH 8.0), 0.25 m LiCl, 1 mm EDTA, 0.5% Nonidet P-40, and 0.5% sodium deoxycholate), and TE (10 mm Tris-HCl (pH 8.0) and 1 mm EDTA), respectively. Then the ChIP direct elution buffer (10 mm Tris-HCl (pH 8.0), 300 mm NaCl, 5 mm EDTA, and 0.5% SDS) was added to the pellet and incubated for 16 h at 65 °C for decross-linking. After treatment with RNase A for 30 min at 37 °C and proteinase K for 2 h at 55 °C, the DNA was purified and used for PCR and real time PCR. Enrichment of the HNF4α binding was normalized to the input samples using the ΔΔCt method and expressed as fold-enrichment as compared with the control normal IgG antibody. The following primers were used for amplification of the HNF4α-binding sites in the human TFR2 promoter (gggaactaggaggccaaagt and tctcccctgccaatctctc), the human MIR-194/192 gene without HNF4α-binding site as a negative control (ccttgtgagggcacaccttt and aaagccaggcagtcagtgct), the HNF4α-binding sites in the mouse Tfr2 promoter (caggaagaccggctaacg and tcttgggtctggttgctagg), and the mouse Hmgcs2 gene without HNF4α-binding site as a negative control (gatccctgggactcacaca and gaatgcacatttatggaggtca).
Statistical Analysis
All values are expressed as the mean ± S.D. All data were analyzed by the unpaired Student's t test for significant differences between the mean values of each group.
Results
Hnf4aΔH Mice Exhibit Hypoferremia
Hematological parameters are good indicators for alternation of iron homeostasis. Here, hepatic HNF4α was found to maintain serum iron levels (Table 1). Hnf4aΔH mice exhibited a 60% reduction in serum iron compared with control Hnf4af/f mice. Likewise, total iron binding capacity, an index of the total amount of serum iron that transferrin can bind, was also reduced in Hnf4aΔH mice, indicating a lower availability of transferrin protein in Hnf4aΔH mice. However, because no significant difference in transferrin saturation was observed in Hnf4aΔH mice, iron-binding transferrin is normal in Hnf4aΔH mice. Moreover, red blood cell counts (RBC), hemoglobin levels, the hematocrit, and the mean cell volume were unchanged in Hnf4aΔH mice compared with Hnf4aΔH mice (Table 2). Furthermore, no significant difference in liver iron content was detected in Hnf4aΔH mice (Table 3), suggesting that Hnf4aΔH mice do not develop liver iron deficiency disease nor hereditary hemochromatosis.
TABLE 1.
Serum iron content in Hnf4aΔH mice
Values are presented as mean ± S.D. (n = 8). TIBC is total iron-binding capacity. Significant differences are compared with Hnf4af/f mice.
| Mice | Iron | TIBC | Transferrin saturation |
|---|---|---|---|
| μg/dl | μg/dl | % | |
| Hnf4af/f | 235.7 ± 43.1 | 751.8 ± 223.1 | 34.6 ± 15.3 |
| Hnf4aΔH | 95.7 ± 22.8a | 336.9 ± 150.9a | 34.8 ± 19.8 |
a p < 0.005.
TABLE 2.
Hematological parameters in Hnf4aΔH mice
RBC is red blood cells; HGB is hemoglobin; MCV is mean corpuscular volume; HCT is hematocrit. All values are presented as means ± S.D. (n = 3–5).
| Mice | RBC | HGB | MCV | HCT |
|---|---|---|---|---|
| m/μl | g/dl | fl | % | |
| Hnf4af/f | 9.2 ± 0.1 | 14.3 ± 0.1 | 49.6 ± 2.0 | 45.7 ± 1.5 |
| Hnf4aΔH | 10.2 ± 0.2 | 14.8 ± 1.3 | 47.6 ± 2.8 | 49.1 ± 3.6 |
TABLE 3.
Liver iron content in Hnf4aΔH mice
Values are presented as mean ± S.D. (n = 5).
| Mice | Iron |
|---|---|
| μg/g liver | |
| Hnf4af/f | 235 ± 92 |
| Hnf4aΔH | 200 ± 108 |
Expression of Genes Involved in Iron Metabolism is Altered in Hnf4aΔH Mice
The expression profile of mRNAs encoded by iron metabolism-related genes was analyzed in Hnf4aΔH and Hnf4af/f mouse livers using IronChip microarray analysis (Table 4). Hepatic expression of many mRNAs significantly changed in Hnf4aΔH mice. Among the down-regulated mRNAs were transferrin (Tf), transferrin receptor 2 (Tfr2), hemojuvelin (Hjv), ferritin light chain (Ftl), ferritin heavy chain (Fth), hemopexin (Hpx), catalase (Cat), Cu-Zn superoxide dismutase (Sod1), and glutathione peroxidase 1 (Gpx1). Up-regulated mRNAs included transferrin receptor 1 (Tfr1), hepcidin (Hamp), and ceruloplasmin (Cp) mRNAs. Microarray data were validated by quantitative RT-PCR (Fig. 1A). Because atransferrinemia/hypotransferrinemia is caused by recessive mutations in the Tf gene resulting in iron-deficient hypochromic anemia in humans and mice (25, 26), reduced expression of TF may cause hypoferremia. Indeed, as expected from reduced Tf mRNA expression, serum TF protein in Hnf4aΔH mice was decreased by 66% compared with Hnf4af/f mice (Fig. 1B). These data show that misregulation of a previously validated HNF4α target gene is preserved in Hnf4aΔH mice (15). Thus, HNF4α-dependent regulation of Tf could partially contribute to the hypoferremia observed in this mouse model.
FIGURE 1.

Hepatic expression of iron metabolism-related genes in Hnf4aΔH mice. A, real time PCR for Hnf4a, Tf, Tfr2, Hjv, Ftl, Fth, Hpx, Cat, Sod1, and Gpx1 (upper panel), and Tfr1, Hamp, Hfe, Fpn1, Heph, and Cp (lower panel) mRNAs from total liver RNA of Hnf4aΔH and Hnf4af/f mice (n = 5 for each genotype). The normalized expression in Hnf4aΔH mice was presented relative to that in Hnf4af/f mice. B, Western blot of TF protein from serum of Hnf4aΔH and Hnf4af/f mice (n = 3 for each genotype) (upper panel). Coomassie Brilliant Blue stain of serum total protein of Hnf4aΔH and Hnf4af/f mice as loading controls (lower panel) is shown. The expression of TF protein was quantified by densitometric analysis using ImageJ software. The normalized TF expression in Hnf4aΔH was presented as relative to that in Hnf4af/f mice. C, Western blot of TFR2 (upper panel) and TUBG (lower panel) protein from livers of Hnf4aΔH and Hnf4af/f mice (n = 3 for each genotype). The TFR2 expression normalized by TUBG in Hnf4aΔH was presented as relative to Hnf4af/f mice. D, real time PCR for Cebpa and Tfr2 mRNAs from total liver RNA of CebpaΔH and Cebpaf/f mice (n = 5 for each genotype). The normalized expression in CebpaΔH mice was presented relative to that in Cebpaf/f mice. Data are mean ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
In addition, decreased expression of hepatic Tfr2 mRNA to 30% was observed in Hnf4aΔH mice, although expression of Tfr1 mRNA encoding a receptor with affinity to holo-TF 25-fold higher than TFR2 was increased to 4-fold (Fig. 1A) (8). TFR2 protein in Hnf4aΔH mice was also decreased by 19% compared with Hnf4af/f mice (Fig. 1C). Because TFR2 and HNF4α are highly expressed in the liver and mutations in the TFR2 gene cause type III hemochromatosis in humans (6, 7), further studies were focused on the Tfr2 gene. Previous reports suggest that the promoter activity of the Tfr2 gene was induced by C/EBPα expression (27). However, expression of hepatic Tfr2 was unchanged in liver-specific Cebpa-null (CebpaΔH) mice (Fig. 1D), indicating that C/EBPα is unlikely to be the main regulator of Tfr2 expression in the liver.
Promoter Analysis of the Distal Region of the Tfr2 Promoter
Promoter analysis of the mouse Tfr2 gene was performed to determine whether HNF4α directly regulates Tfr2 expression. Analysis of the distal promoter was carried out in HepG2 cells that highly express endogenous HNF4α and TFR2 (6) revealing significant promoter activities with the region between −116 and −46 from the transcription start site (Fig. 2B). Activity of the Otc promoter that has two HNF4α-binding sites was completely suppressed by HNF4α knockdown using siRNA; however, no repression of the promoter activities was detected in all Tfr2 promoters by siRNA against HNF4α (Fig. 2C), indicating that the distal promoter of the Tfr2 gene was activated in an HNF4α-independent manner. Searching the JASPER database for transcription factor-binding sites in the region between −116 and −46 revealed three GC boxes (GC-1, 2, and -3) that were predicted between −91 and −53 (Fig. 2D). Furthermore, the stepwise transactivation of the promoter activity was observed in the presence of these GC boxes (Fig. 2B). To determine which regions of these GC boxes are sufficient for transactivation, mutations were introduced into each GC box. Mutations of the GC-2 region significantly suppressed the promoter activity, and mutations of all GC regions further suppressed the activity (Fig. 2E), indicating that the GC-2 region is a central cis-element for transactivation, and the GC-1 and GC-3 regions are required for the maximal transactivation of the distal promoter.
FIGURE 2.

Promoter analysis of distal region of the mouse Tfr2 gene. A, schematic structure of the promoter region of the mouse Tfr2 gene. B, promoter activity of the distal region of the Tfr2 gene in HepG2 cells. C, promoter constructs were transfected into HepG2 cells with 20 nm siRNA for HNF4α (siHNF4α) or negative control (siControl). Otc promoter whose transactivation is dependent on HNF4α expression was used as a positive control. D, nucleotide sequences of the distal promoter of the Tfr2 gene. Three GC-rich sequences (GC-1, -2, and -3) are boxed. E, mutations were introduced into three GC-rich sequences (1–3) between −93 and −46 in the −241/+67 promoter (left panel). The promoter activities were measured in HepG2 cells (right panel). The normalized activity ± S.D. (n = 3) was presented as promoter activity based on pGL4.11.
Promoter Analysis of the Proximal Region of the Tfr2 Promoter
As described above, regions that respond to HNF4α were not identified in the distal promoter. No transactivation was detected in this promoter region (+505/+1144 and −46/+1144) as compared with the promoterless and the distal promoter (−116/+67, see Fig. 3A). Therefore, the proximal promoter was analyzed for the presence of an HNF4α-dependent enhancer. At first, the full-length proximal promoter (−46/+1144) had enhancer activity in the presence of the core Tk promoter (Fig. 3B). In addition, the shorter promoter regions (−46/+505 and −46/+1144) had weak enhancer activities, showing that these regions might cooperatively contribute to enhancer transactivation. Next, the −46/+504 and +505/+1144 proximal promoters were transactivated by HNF4α expression vector, and the full-length promoter was further transactivated by HNF4α in the presence of the Tk mini promoter (Fig. 3C). These results indicate that the proximal promoter contains at least two HNF4α-dependent elements located at −46/+504 and +505/+1144. In addition, the proximal promoters with the distal promoter (−116/+504, −116/+896, and −116/+1144) was also transactivated by HNF4α as compared with the distal promoter alone (−116/+67), with the result that HNF4α has the potential to transactivate the native Tfr2 promoter via two regions located at +67/+504 and +505/+896 (Fig. 3D). By searching with the JASPER database, three potential HNF4α-binding sites were found at +182/+194, +189/+201, and +830/+842, and the binding sites located at +182/+194 and +189/+201 overlapped (Fig. 3E). Of these, the binding site at +830/+842 was highly conserved among species (Fig. 4A). To determine whether these predicted HNF4α-binding sites could transactivate the Tfr2 gene in an HNF4α-dependent manner, mutations were introduced into the binding sites. Because the binding site at +182/+194 and +189/+201 overlapped, mutations were introduced into the middle region at +182/+201 so that HNF4α could not bind to both sites. Consequently, disruption of each binding site decreased HNF4α-dependent transactivation, and disruption of both binding sites further repressed the transactivation, indicating that these HNF4α-binding sites were transactivated in an HNF4α-dependent manner (Fig. 4B).
FIGURE 3.

Promoter and enhancer analysis of the proximal region of the mouse Tfr2 gene. A, promoter activity of the proximal region of the Tfr2 gene in HepG2 cells. B, enhancer activity of the proximal region of the Tfr2 gene with TK promoter. C, enhancer activity of the proximal region of the Tfr2 gene with TK mini promoter in HEK293T cells. Empty vector (None), or HNF4α expression vector (+HNF4α) was co-transfected. D, enhancer activity of the proximal region of the Tfr2 gene with the distal promoter in HEK293T cells. Empty vector or HNF4α expression vector was also co-transfected. E, nucleotide sequences of the proximal promoter of the Tfr2 gene. Predicted HNF4α-binding sites are boxed. The normalized activity ± S.D. (n = 3) is presented as promoter activity based on pGL4.11 (A and B) or empty vector-transfected control (C and D).
FIGURE 4.

Requirement of the minimal promoter of the Tfr2 gene for transactivation by HNF4α. A, sequence alignment of the proximal promoter of the Tfr2 gene in mouse, human, rat, rabbit, gorilla, chimpanzee, cow, dolphin, horse, panda, and elephant. Predicted HNF4α-binding site is boxed. Completely conserved nucleotides among the species are shown by asterisks. B, mutations were introduced into the HNF4α-binding sites in the proximal promoter of the Tfr2 gene. Empty vector or HNF4α expression vector was co-transfected with the mutated proximal promoter and the minimal distal promoter, and the enhancer activity was measured. C, mutations were introduced into the HNF4α-binding sites and GC-rich sequences in the Tfr2 promoter. Empty vector or HNF4α expression vector was co-transfected with the mutated promoter, and the enhancer activity was measured. The normalized activity ± S.D. (n = 3) is presented as promoter activity based on empty vector-transfected control.
To investigate whether the minimal promoter region in the distal promoter would be essential for transactivation by HNF4α, mutations were introduced into both the GC boxes and HNF4α-binding sites (Fig. 4C). In the presence of the HNF4α-binding sites in the proximal promoter, mutations of the GC-2 region (GC2/M) inhibited the transactivation by HNF4α by about 35% as compared with the wild-type promoter (−116/+1144WT). Mutations of each HNF4α-binding site with mutations of the GC-2 region (GC2/M-H4/M1 and GC2/M-H4/M2) markedly suppressed the transactivation by HNF4α, and further suppression was detected by mutations in both HNF4α-binding sites (GC2/M-H4/M1+M2). Additionally, mutations of all GC boxes (GC-all/M) further inhibited the transactivation by HNF4α when compared with that of the mutations of the GC-2 region alone (GC2/M). Transactivation by HNF4α was particularly inhibited by mutations in all GC regions and HNF4α-binding sites, indicating that the GC-2 region plays a critical role in transactivation of the Tfr2 gene by HNF4α.
Direct Binding of HNF4α to the Proximal Region of the Tfr2 Promoter
To determine whether HNF4α can directly bind to both HNF4α-binding sites in the Tfr2 promoter, gel mobility shift analysis was performed (Fig. 5A). Nuclear extracts from HepG2 cells bound to the distal (+182/+201) and proximal (+830/+842) HNF4α-binding sites (Fig. 5A, lane 2, lower arrows). This complex was diminished by the addition of excess amounts of unlabeled Tfr2 competitor and the Otc competitor that contains a bona fide HNF4α-binding site (Fig. 5A, lanes 3 and 4) (19) but not the competitor that has mutations in the HNF4α-binding site of the Tfr2 promoter (Fig. 5A, lane 5). Moreover, the complex was supershifted by anti-HNF4α antibody (Fig. 5A, lane 6, upper arrow) but not the unrelated anti-peroxisome proliferator-activated receptor α antibody (lane 7). These results indicate that HNF4α indeed binds to the distal and proximal regions of the Tfr2 promoter. Next, chromatin immunoprecipitation (ChIP) was used to determine whether HNF4α directly binds to the Tfr2 promoter in vivo. HNF4α bound to the predicted HNF4α-binding sites in HepG2 cells as compared with IgG control (Fig. 5B). In addition, ChIP analysis using the livers of Hnf4af/f and Hnf4aΔH mice indicated that hepatic HNF4α in Hnf4af/f mice significantly bound to the promoter region, and the binding was lower in Hnf4af/f mouse livers (Fig. 5C), suggesting that HNF4α directly and physiologically binds to the promoter region of the Tfr2 gene in human and mouse liver.
FIGURE 5.
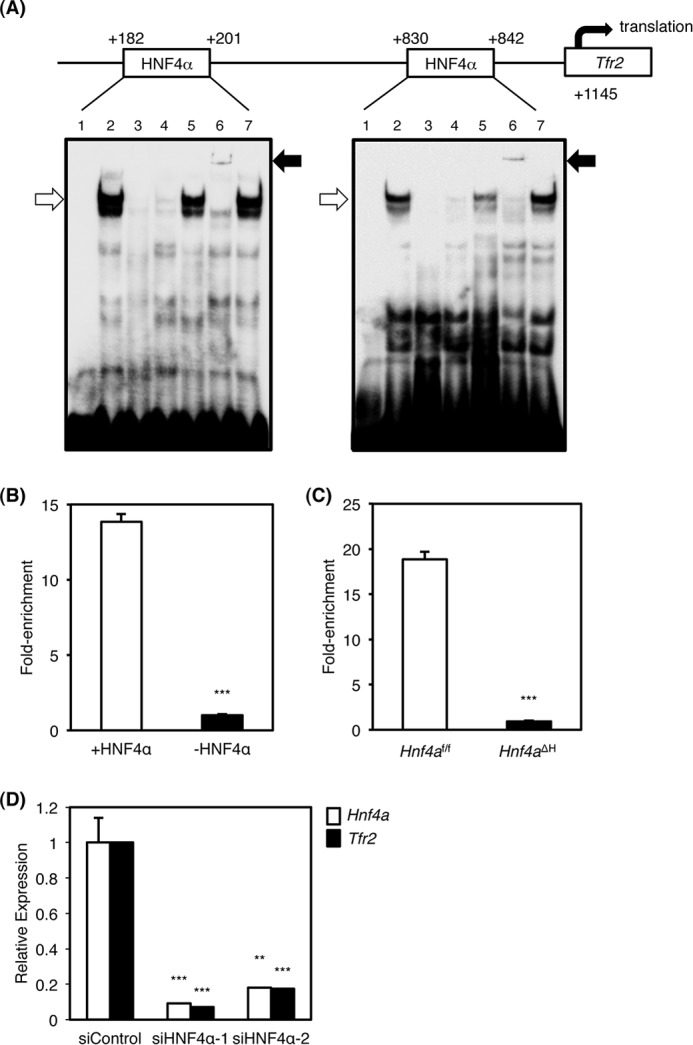
Identification of HNF4α-binding sites in the Tfr2 promoter and HNF4α-dependent transactivation of the Tfr2 gene. A, gel mobility shift analysis. Nuclear extracts from HepG2 cells were incubated with the probe carrying the distal (left panel) and proximal (right panel) HNF4α-binding sites in the Tfr2 promoter in the absence (lane 2) or presence of the unlabeled Tfr2, the Otc, and the mutated Tfr2 probes (lanes 3–5). For supershift analysis, anti-HNF4α and anti-PPARα antibodies were added (lanes 6 and 7). B, chromatin immunoprecipitation using HepG2 cells were performed with anti-HNF4α antibody and normal goat IgG. The regions with the proximal HNF4α-binding sites (+HNF4α) and the regions without HNF4α-binding site (−HNF4α) in the human MIR-194/192 gene were amplified. The data from real time PCR were normalized relative to the input and expressed as fold-enrichment. ***, p < 0.001. C, chromatin immunoprecipitation using the livers of Hnf4aΔH and Hnf4af/f mice with anti-HNF4α antibody and normal goat IgG. The regions between the proximal and the distal HNF4α-binding sites in the mouse Tfr2 promoter and the regions without HNF4α-binding site in the mouse Hmgcs2 gene were amplified by real time PCR. The data were normalized relative to the input and expressed as fold-enrichment over data from the IgG control. ***, p < 0.001. D, 10 nm siRNA for HNF4α (siHNF4α) or negative control (siControl) was transfected into HepG2 cells. Real time PCR for HNF4a (white bar) and TFR2 mRNA (black bar) from total RNA of siRNA-transfected cells (n = 3) is shown. Data are mean ± S.D. **, p < 0.01; ***, p < 0.001 compared with siControl-treated cells.
To determine whether regulation of hepatic Tfr2 expression by HNF4α observed in mice could be also maintained in a human hepatocyte-derived cell line, HNF4α knockdown by siRNAs was performed revealing that HNF4α was suppressed in HepG2 cells (Fig. 5D). Expression of TFR2 mRNA was decreased by HNF4α knockdown, indicating that expression of the TFR2 mRNA is positively correlated with HNF4α expression in HepG2 cells (Fig. 5D).
Discussion
The present data show that serum iron levels are decreased in Hnf4aΔH mice without significant changes in RBC, HBG, and hematocrit and no evidence for iron deficiency anemia. Changes in the expression of multiple genes in Hnf4aΔH mice make it difficult to ascribe a phenotype to loss of expression of a single gene. However, expression of hepatic Tf mRNA and serum TF protein was decreased in Hnf4aΔH mice, due to loss of direct transactivation of TF by HNF4α (15). Hypotransferrinemic mice carrying a mutation in the Tf gene die from iron deficiency anemia before weaning, but these mice can be rescued by injection of serum or purified TF (26). Thus, the reduced expression of TF would only partially cause hypoferremia in Hnf4aΔH mice. Serum TF protein still remains about 70% of normal as revealed by Western blot data, suggesting that the symptoms in Hnf4aΔH mice would not be severe enough to develop iron deficiency anemia. Because serum TF protein has a relatively long half-life (8–10 days), the majority of serum TF protein may remain in Hnf4aΔH mice despite that TF protein is mainly expressed in the liver, and hepatic expression of Tf mRNA in Hnf4aΔH mice was decreased to 20% compared with control mice. Furthermore, expression of Tfr1 mRNA was markedly increased in Hnf4aΔH mice. Tfr1 mRNA is post-transcriptionally regulated via iron-response elements in the 3′-untranslated regions by iron-regulatory proteins during low iron levels, resulting in Tfr1 mRNA stabilization and increased TFR1 protein (3, 28). However, both liver iron content and expression of IRP-1 and -2 remained unchanged in Hnf4aΔH mice, suggesting that TFR1 protein expression would also be increased in Hnf4aΔH mice as well as Tfr1 mRNA without post-transcriptional regulation. Thus, increased iron uptake into the liver by increased TFR1 may cause the hypoferremia found in Hnf4aΔH mice.
The other cause of hypoferremia in Hnf4aΔH mice might be due to up-regulation of Hamp expression. Excess HAMP degrades or internalizes duodenal FPN1 and inhibits iron export into the bloodstream, leading to induction of hypoferremia, and thus the HAMP-FPN1 axis is the most important regulator of iron homeostasis (1, 12). Expression of HAMP is positively regulated at the transcriptional level through hemojuvelin, TFR2, and the pro-inflammatory IL6/STAT3 pathway (1, 2, 10, 29). However, despite the decreased expression of Hjv and Tfr2 mRNA, Hamp mRNA is elevated 3-fold in Hnf4aΔH mice. In contrast, Hnf4aΔH mice showed hepatosteatosis and increased serum alanine transaminase, an indicator of liver dysfunction and inflammation (18). Moreover, ROS are known to induce inflammation through the production of proinflammatory cytokines (30), and hepatic expression of CAT, SOD1, and GPX1 that degrade ROS was decreased in Hnf4aΔH mice (31). These data indicate that the accumulation of ROS could lead to liver inflammation, and the resultant up-regulation of Hamp in Hnf4aΔH mice could be caused by enhanced inflammatory signaling.
Even though reduced expression of hepatic Tfr2 that causes hereditary hemochromatosis when mutated in humans (7), Hnf4aΔH mice did not show evidence of this disorder. Because hepatic Hamp expression in both the complete and liver-specific Tfr2-null mice was decreased at a similar age as the Hnf4aΔH mice used in this study (32, 33) and because HAMP is the downstream regulator of TFR2, up-regulation of Hamp might be the main reason why Hnf4aΔH mice do not show hemochromatosis despite the down-regulation of Tfr2 expression. It is noteworthy that liver-specific Tfr2-null mice also exhibited elevated transferrin saturation (33), suggesting that absorption of iron from the duodenal FPN1 would be increased by reduced expression of Hamp. In contrast, Hnf4aΔH mice exhibited normal transferrin saturation. Thus, the phenotype of Hnf4aΔH mice does not overlap with those of liver-specific Tfr2-null mice, despite the fact that HNF4α is an upstream regulator of TFR2. A potential reason for this discrepancy is that lack of HNF4α may have a profound effect on hepatic iron metabolism compared with that of TFR2.
This study further shows that HNF4α binds and activates transcription of Tfr2. Previous studies showed that the mouse Tfr2 proximal and distal promoter was transactivated in the presence of GATA-1 and C/EBPα (27). Of these factors, GATA-1 would not be an essential factor to transactivate Tfr2 in the liver because GATA-1 is a master regulator for erythroid differentiation and transactivates erythroid-specific genes (34, 35). Moreover, C/EBPα is enriched in the liver and positively regulates many liver-enriched genes (36). Although the expression of hepatic Cebpa was decreased by 45% in Hnf4aΔH mice (37), no significant change of hepatic Tfr2 mRNA expression was detected in CebpaΔH mice. Additionally, the transactivation potential of C/EBPα alone for the Tfr2 promoter without GATA-1 is much lower as compared with that with GATA-1 (27), and the contribution of C/EBPα to Tfr2 regulation would be minor in the liver. Although details on transcriptional regulation of the Tfr2 gene in the liver are not clear, HNF4α could directly regulate the Tfr2 expression through at least two HNF4α-binding sites. Unlike the highly conserved binding site at +830/+842, the binding site at +182/+201 is not conserved among the species, and disruption of both binding sites did not completely inhibit the HNF4α-dependent transactivation. However, HNF4α knockdown greatly suppressed TFR2 expression in human hepatoma cells, indicating that there might be additional HNF4α-binding sites within the proximal promoter. This study revealed that GC-rich regions in the distal promoter were also important for maximal transactivation of the Tfr2 gene promoter. It was reported that cooperation of the HNF4α and GC-rich regions is important for transactivation of the human APOC3 gene and mouse Slc27a5 gene (38, 39), indicating that the GC-rich regions and the HNF4α-binding sites in the Tfr2 promoter can be the basal cis-elements and the liver-specific enhancer elements, respectively.
Although HNF4α was found to directly regulate Tfr2 expression, the mechanism for repressed expression of hepatic Hamp expression in Hnf4aΔH mice could not be determined in this study. Because HNF4α is a master regulator for maintenance of liver-specific functions and severe liver dysfunction and inflammation were actually observed in Hnf4aΔH mice, the possibility cannot be excluded that complicated secondary effects would transactivate Hamp expression and lower the effect of reduced expression of Tfr2 in Hnf4aΔH mice. Despite the fact that Tfr2 is a direct target for HNF4α, remarkable changes of gene expression override the potential effects and phenotypes caused by decreased expression of Tfr2 in Hnf4aΔH mice. Consequently, HNF4α is expected to transactivate Hamp through modulation of Tfr2 expression in the normal liver.
In conclusion, hepatic HNF4α significantly contributes to iron homeostasis and directly regulates Tfr2 expression. Thus, augmentation of hepatic HNF4α signaling could be of value for the treatment of hypoferremia and anemia.
Author Contributions
S. M., M. O., Y. M., S. S., and T. F. conducted most of the experiments and analyzed the results. M. U. M. conducted experiments of IronChip analysis. M. T. and N. A. conducted experiments of real time PCR and ChIP analysis. H. O. and M. S. conducted experiments of siRNA. Y. I. conceived the idea for the project and wrote the paper with F. J. G.
Acknowledgments
We thank Prof. K. Wakamatsu for encouragement in the early phases of this work. We also acknowledge R. Yoshida for exploratory work on this project.
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan Grant-in-aid for Scientific Research 25460490 (to Y. I.) and Kato Memorial Bioscience Foundation (to Y. I.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ROS
- reactive oxygen species
- HNF4α
- hepatocyte nuclear factor 4α
- TUBG
- γ-tubulin
- TF
- transferrin.
References
- 1. Ganz T. (2013) Systemic iron homeostasis. Physiol. Rev. 93, 1721–1741 [DOI] [PubMed] [Google Scholar]
- 2. Muñoz M., García-Erce J. A., and Remacha A. F. (2011) Disorders of iron metabolism. Part 1: molecular basis of iron homoeostasis. J. Clin. Pathol. 64, 281–286 [DOI] [PubMed] [Google Scholar]
- 3. Wang J., and Pantopoulos K. (2011) Regulation of cellular iron metabolism. Biochem. J. 434, 365–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aisen P. (2004) Transferrin receptor 1. Int. J. Biochem. Cell Biol. 36, 2137–2143 [DOI] [PubMed] [Google Scholar]
- 5. Hayashi A., Wada Y., Suzuki T., and Shimizu A. (1993) Studies on familial hypotransferrinemia: unique clinical course and molecular pathology. Am. J. Hum. Genet. 53, 201–213 [PMC free article] [PubMed] [Google Scholar]
- 6. Kawabata H., Yang R., Hirama T., Vuong P. T., Kawano S., Gombart A. F., and Koeffler H. P. (1999) Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem. 274, 20826–20832 [DOI] [PubMed] [Google Scholar]
- 7. Camaschella C., Roetto A., Calì A., De Gobbi M., Garozzo G., Carella M., Majorano N., Totaro A., and Gasparini P. (2000) The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 25, 14–15 [DOI] [PubMed] [Google Scholar]
- 8. West A. P. Jr., Bennett M. J., Sellers V. M., Andrews N. C., Enns C. A., and Bjorkman P. J. (2000) Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J. Biol. Chem. 275, 38135–38138 [DOI] [PubMed] [Google Scholar]
- 9. Worthen C. A., and Enns C. A. (2014) The role of hepatic transferrin receptor 2 in the regulation of iron homeostasis in the body. Front. Pharmacol. 5, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao J., Chen J., Kramer M., Tsukamoto H., Zhang A. S., and Enns C. A. (2009) Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 9, 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nemeth E., Tuttle M. S., Powelson J., Vaughn M. B., Donovan A., Ward D. M., Ganz T., and Kaplan J. (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 [DOI] [PubMed] [Google Scholar]
- 12. Yun S., and Vincelette N. D. (2015) Update on iron metabolism and molecular perspective of common genetic and acquired disorder, hemochromatosis. Crit. Rev. Oncol. Hematol. 95, 12–25 [DOI] [PubMed] [Google Scholar]
- 13. Sladek F. M., and Seidel S. D. (2001) in Nuclear Receptor and Genetic Disease (Burris T. P., and McCabe E., eds) pp. 309–361, Academic Press, San Diego [Google Scholar]
- 14. Schrem H., Klempnauer J., and Borlak J. (2002) Liver-enriched transcription factors in liver function and development. Part I: the hepatocyte nuclear factor network and liver-specific gene expression. Pharmacol. Rev. 54, 129–158 [DOI] [PubMed] [Google Scholar]
- 15. Schaeffer E., Guillou F., Part D., and Zakin M. M. (1993) A different combination of transcription factors modulates the expression of the human transferrin promoter in liver and Sertoli cells. J. Biol. Chem. 268, 23399–23408 [PubMed] [Google Scholar]
- 16. Truksa J., Lee P., and Beutler E. (2009) Two BMP responsive elements, STAT, and bZIP/HNF4/COUP motifs of the hepcidin promoter are critical for BMP, SMAD1, and HJV responsiveness. Blood 113, 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Courselaud B., Pigeon C., Inoue Y., Inoue J., Gonzalez F. J., Leroyer P., Gilot D., Boudjema K., Guguen-Guillouzo C., Brissot P., Loréal O., and Ilyin G. (2002) C/EBPα regulates hepatic transcription of hepcidin, an antimicrobial peptide and regulator of iron metabolism. J. Biol. Chem. 277, 41163–41170 [DOI] [PubMed] [Google Scholar]
- 18. Hayhurst G. P., Lee Y. H., Lambert G., Ward J. M., and Gonzalez F. J. (2001) Hepatocyte nuclear factor 4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell Biol. 21, 1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Inoue Y., Hayhurst G. P., Inoue J., Mori M., and Gonzalez F. J. (2002) Defective ureagenesis in mice carrying a liver-specific disruption of hepatocyte nuclear factor 4α (HNF4α). HNF4α regulates ornithine transcarbamylase in vivo. J. Biol. Chem. 277, 25257–25265 [DOI] [PubMed] [Google Scholar]
- 20. Inoue Y., Yu A. M., Yim S. H., Ma X., Krausz K. W., Inoue J., Xiang C. C., Brownstein M. J., Eggertsen G., Björkhem I., and Gonzalez F. J. (2006) Regulation of bile acid biosynthesis by hepatocyte nuclear factor 4α. J. Lipid Res. 47, 215–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Inoue Y., Inoue J., Lambert G., Yim S. H., and Gonzalez F. J. (2004) Disruption of hepatic C/EBPα results in impaired glucose tolerance and age-dependent hepatosteatosis. J. Biol. Chem. 279, 44740–44748 [DOI] [PubMed] [Google Scholar]
- 22. Muckenthaler M., Richter A., Gunkel N., Riedel D., Polycarpou-Schwarz M., Hentze S., Falkenhahn M., Stremmel W., Ansorge W., and Hentze M. W. (2003) Relationships and distinctions in iron-regulatory networks responding to interrelated signals. Blood 101, 3690–3698 [DOI] [PubMed] [Google Scholar]
- 23. Muckenthaler M., Roy C. N., Custodio A. O., Miñana B., deGraaf J., Montross L. K., Andrews N. C., and Hentze M. W. (2003) Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat. Genet. 34, 102–107 [DOI] [PubMed] [Google Scholar]
- 24. Richter A., Schwager C., Hentze S., Ansorge W., Hentze M. W., and Muckenthaler M. (2002) Comparison of fluorescent tag DNA labeling methods used for expression analysis by DNA microarrays. BioTechniques 33, 620–628 [DOI] [PubMed] [Google Scholar]
- 25. Bartnikas T. B. (2012) Known and potential roles of transferrin in iron biology. Biometals 25, 677–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bernstein S. E. (1987) Hereditary hypotransferrinemia with hemosiderosis, a murine disorder resembling human atransferrinemia. J. Lab. Clin. Med. 110, 690–705 [PubMed] [Google Scholar]
- 27. Kawabata H., Germain R. S., Ikezoe T., Tong X., Green E. M., Gombart A. F., and Koeffler H. P. (2001) Regulation of expression of murine transferrin receptor 2. Blood 98, 1949–1954 [DOI] [PubMed] [Google Scholar]
- 28. Wallander M. L., Leibold E. A., and Eisenstein R. S. (2006) Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim. Biophys. Acta 1763, 668–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Core A. B., Canali S., and Babitt J. L. (2014) Hemojuvelin and bone morphogenetic protein (BMP) signaling in iron homeostasis. Front. Pharmacol. 5, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rolo A. P., Teodoro J. S., and Palmeira C. M. (2012) Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 52, 59–69 [DOI] [PubMed] [Google Scholar]
- 31. Handy D. E., and Loscalzo J. (2012) Redox regulation of mitochondrial function. Antioxid. Redox Signal. 16, 1323–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wallace D. F., Summerville L., Lusby P. E., and Subramaniam V. N. (2005) First phenotypic description of transferrin receptor 2 knockout mouse, and the role of hepcidin. Gut 54, 980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wallace D. F., Summerville L., and Subramaniam V. N. (2007) Targeted disruption of the hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology 132, 301–310 [DOI] [PubMed] [Google Scholar]
- 34. Pevny L., Simon M. C., Robertson E., Klein W. H., Tsai S. F., D'Agati V., Orkin S. H., and Costantini F. (1991) Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 349, 257–260 [DOI] [PubMed] [Google Scholar]
- 35. Welch J. J., Watts J. A., Vakoc C. R., Yao Y., Wang H., Hardison R. C., Blobel G. A., Chodosh L. A., and Weiss M. J. (2004) Global regulation of erythroid gene expression by transcription factor GATA-1. Blood 104, 3136–3147 [DOI] [PubMed] [Google Scholar]
- 36. Schrem H., Klempnauer J., and Borlak J. (2004) Liver-enriched transcription factors in liver function and development. Part II: the C/EBPs and D site-binding protein in cell cycle control, carcinogenesis, circadian gene regulation, liver regeneration, apoptosis, and liver-specific gene regulation. Pharmacol. Rev. 56, 291–330 [DOI] [PubMed] [Google Scholar]
- 37. Wiwi C. A., Gupte M., and Waxman D. J. (2004) Sexually dimorphic P450 gene expression in liver-specific hepatocyte nuclear factor 4α-deficient mice. Mol. Endocrinol. 18, 1975–1987 [DOI] [PubMed] [Google Scholar]
- 38. Kardassis D., Falvey E., Tsantili P., Hadzopoulou-Cladaras M., and Zannis V. (2002) Direct physical interactions between HNF-4 and Sp1 mediate synergistic transactivation of the apolipoprotein CIII promoter. Biochemistry 41, 1217–1228 [DOI] [PubMed] [Google Scholar]
- 39. Inoue Y., Yu A. M., Inoue J., and Gonzalez F. J. (2004) Hepatocyte nuclear factor 4α is a central regulator of bile acid conjugation. J. Biol. Chem. 279, 2480–2489 [DOI] [PubMed] [Google Scholar]