

Background: Cyclooxygenase-2 (COX-2) and sphingosine 1-phosphate receptor 2 (S1PR2) are highly expressed in human cholangiocarcinoma (CCA) and taurocholate (TCA) promotes CCA cell growth via S1PR2.
Results: TCA-mediated activation of S1PR2 contributed to COX-2 expression and CCA cell growth.
Conclusion: S1PR2 plays a critical role in TCA-induced COX-2 expression and CCA growth.
Significance: S1PR2 represents a novel therapeutic target for CCA.
Keywords: bile acid, cell biology, cyclooxygenase (COX), epidermal growth factor receptor (EGFR), gene regulation, metabolism, signal transduction, sphingosine-1-phosphate (S1P)
Abstract
Cholangiocarcinoma (CCA) is a rare, but highly malignant primary hepatobiliary cancer with a very poor prognosis and limited treatment options. Our recent studies reported that conjugated bile acids (CBAs) promote the invasive growth of CCA via activation of sphingosine 1-phosphate receptor 2 (S1PR2). Cyclooxygenase-2 (COX-2)-derived prostaglandin E2 (PGE2) is the most abundant prostaglandin in various human malignancies including CCA. Previous studies have indicated that COX-2 was highly expressed in CCA tissues, and the survival rate of CCA patients was negatively associated with high COX-2 expression levels. It has also been reported that CBAs induce COX-2 expression, whereas free bile acids inhibit COX-2 expression in CCA mouse models. However, the underlying cellular mechanisms and connection between S1PR2 and COX-2 expression in CCA cells have still not been fully elucidated. In the current study, we examined the role of S1PR2 in conjugated bile acid (taurocholate, (TCA))-induced COX-2 expression in a human HuCCT1 CCA cell line and further identified the potential underlying cellular mechanisms. The results indicated that TCA-induced invasive growth of human CCA cells was correlated with S1PR2-medated up-regulation of COX-2 expression and PGE2 production. Inhibition of S1PR2 activation with chemical antagonist (JTE-013) or down-regulation of S1PR2 expression with gene-specific shRNA not only reduced COX-2 expression, but also inhibited TCA-induced activation of EGFR and the ERK1/2/Akt-NF-κB signaling cascade. In conclusion, S1PR2 plays a critical role in TCA-induced COX-2 expression and CCA growth and may represent a novel therapeutic target for CCA.
Introduction
Cholangiocarcinoma (CCA)5 is the most common fatal biliary malignancy and accounts to 10 to 15% of primary hepatic cancer originating from the epithelial lining of bile ducts (1–4). Recent epidemiological studies indicate that the incidence of CCA is increasing worldwide, although the risk factors differ geographically (3). Unlike other gastrointestinal carcinomas, CCA is relatively rare and has not been extensively studied. Certain environment-linked risk factors, such as liver fluke infection and toxic chemicals, may play a critical role in the disease development and progression of CCA in some patients. Recent studies have identified several interacting molecular pathways as vital promoting factors in the development of CCA, such as cholestasis, chronic inflammation, oxidative stress, and dysregulation of the cell injury repair processes (1–3, 5, 6). However, the role of conjugated bile acids (CBAs) in CCA pathogenesis remain largely unknown. The current available therapeutic options are mainly limited to surgical resection or liver transplantation (7).
Cyclooxygenase-2 (COX-2)-derived prostaglandins (PG) have been implicated in various carcinogenesis including cholangiocarcinogenesis (9). Overexpression of COX-2 promoted CCA cell growth and survival, whereas depletion or pharmacological inhibition of COX-2 reduced CCA cell proliferation (9–11). COX-2-derived PGE2 is the most abundant prostanoid and mediates many physiological and pathological effects via activating different G protein-coupled receptors (GPCRs) EP1–4. Both enhanced expression of COX-2 and increased production of PGE2 have been associated with various cancers including CCA (12–14). Occurrence of CCA is often associated with chronic cholestasis, and the accumulation of bile acids, especially CBAs, in the liver and bile duct is linked to the disease progression of CCA. It also has been reported that CBAs induce COX-2 expression via the epidermal growth factor receptor (EGFR) and NF-κB pathways in CCA cells (15, 16). In CCA patients, the composition of taurine- and glycine-conjugated cholic acids is significantly increased when compared with patients with benign biliary diseases (17). Our recent studies showed that CBAs promoted the proliferation and invasion of both rodent and human CCA cells through the activation of sphingosine 1-phosphate receptor 2 (S1PR2) (18). S1PR2 was the predominant S1PRs expressed in CCA cell and tissues. Although several recent studies reported that S1PR2-mediated signaling pathways were involved in promoting tumor growth and progression, the underlying cellular/molecular mechanisms remain to be elucidated (19). Whether CBA-mediated activation of S1PR2 contributes to the up-regulation of COX-2 expression, PGE2 production in CCA cells has not been determined and is the major focus of this study.
In the current study, we examined the effect of TCA on COX-2 expression and elucidated the role of S1PR2 in TCA-induced COX-2 expression and further identified potential signaling pathways in human CCA cells.
Experimental Procedures
Materials
S1P and JTE-013 were purchased from Cayman Chemical (Boston, MA). pcDNA3.1–3xHA-tagged human S1PR2 (N terminus) was obtained from UMR cDNA Resource Center (Rolla, MO); Celecoxib, U0126, and MK-2206 were purchased from Selleck Chemicals (Boston, MA). Taurocholate (TCA), dimethyl sulfoxide (DMSO), and monoclonal anti-HA antibody were obtained from Sigma. BD Biocoat Matrigel Invasion Chamber was purchased from BD Biosciences. A QIAzol lysis reagent was from Qiagen Sciences (Valencia, CA). Thermo Hygreen supermix reagent was purchased from Thermo (Waltham, MA). RIPA lysis buffer and nuclear protein isolation kit were obtained from Beyotime (Nanjing, Jiangsu, China). Bio-Rad protein assay reagent and 4× sample buffer were from Bio-Rad. Primary antibodies against COX-2, p-ERK1/2, ERK1, ERK2, p-Akt, Akt, p-IKKα/β, IKKα/β, p-NF-κB p65, NF-κB p65, p-EGFR, EGFR, β-actin, and lamin B, as well as HRP-conjugated secondary antibodies for Western blot were from Santa Cruz Biotechnology (Santa Cruz, CA). Alexa Fluor® 488-conjugated secondary antibodies for immunofluorescence, High-capacity cDNA Reverse Transcription Kit, Lipofectamine 2000, cell cultures medium, and supplement components were all from Life Technologies (Grand Island, NY).
Cell Culture and Treatment
The human HuCCT1 CCA cell line was obtained from Guangzhou Jenniobio Biotechnology Co., Ltd. (Guangzhou, China) and cultured in RPMI 1640 medium, supplemented with 10% FBS, 2 mm of l-glutamine, and 50 μg/ml of gentamicin. TCA was dissolved in serum-free culture medium. S1P, JTE-013, Celecoxib, U0126, and MK-2206 were prepared in DMSO to the desired concentrations. Drugs were delivered by directly adding into cell culture medium. For the cell invasion assay, drugs were added into medium in the upper chamber of the Matrigel pre-coated transwell.
Transfection of shRNA for Down-regulating S1PR2
The stem loop sequences of short hairpin RNA (shRNA) specifically targeting human S1PR2 and the scrambled control shRNA plasmid were gifts from Dr. Murthy Karnam (Department of Physiology and Biophysics, Virginia Commonwealth University, Richmond, VA). HuCCT1 cells were transiently transfected with the control shRNA or S1PR2 shRNA plasmids using Lipofectamine 2000, as described previously (20).
Western Blot Analysis
Total cellular proteins were lysed with RIPA lysis buffer (Beyotime, China). Nuclear and cytosol proteins were isolated using a nuclear protein isolation kit (Beyotime, China). 50 μg of the proteins were resolved on a 10% SDS-PAGE gel and transferred to nitrocellulose membranes. Membranes were blocked for 1 h at room temperature with 5% BSA in TBS buffer and then incubated with specific primary antibodies for overnight at 4 °C. Immunoreactive bands were detected using horseradish peroxidase-conjugated secondary antibodies and ECL reagents (Thermo Scientific). Images were recorded using Bio-Rad ChemiDoc XRS imaging system (Bio-Rad) and further analyzed using Quantity One (Bio-Rad).
RNA Isolation and Real-time RT-PCR
Total cellular RNA was isolated using TRIzol reagent (Qiagen, Valencia, CA) and reverse transcribed into first-strand cDNA using the High-capacity cDNA Reverse Transcription Kit (Life Technologies) as described previously (18). IL-6, TNF-α, matrix metalloproteinase-2 (MMP2), and MMP9 mRNA levels were determined by real-time PCR using Thermo Hygreen supermix reagents and normalized using GAPDH as an internal control as described previously (18, 20). Primer sequences will be provided upon request.
Enzyme-linked Immunosorbent Assays (ELISA) of PGE2
Following a different treatment, cell culture medium was collected and centrifuged to remove cell debris. PGE2 levels in the supernatant were determined using a high sensitivity ELISA kit (Enzo Life Sciences) according to the manufacturer's instruction. Safire2 microplate reader (Tecan, Switzerland) was used to detect the absorbance at 405 nm.
Cell Invasion Assay
HuCCT1 cells and HuCCT1 cells transfected with control shRNA or S1PR2 shRNA were seeded in the upper chamber of the BD Biocoat Matrigel Invasion Chamber (BD Bioscience) and cultured in complete medium with 1% FBS. Cells were pretreated with JTE-013 (10 μm), Celecoxib (40 μm), or DMSO (vehicle control) for 1 h, then treated with TCA (100 μm), S1P (100 nm), or DMSO and incubated at 37 °C for 24 h. At the end of treatment, noninvasive cells were removed from the top surface of the upper chamber membrane. Invasive cells that migrated to lower surface were fixed with 3.7% paraformaldehyde and stained with 0.1% crystal violet solution. Invasive cells were counted and the invasion index was analyzed as previously described (18).
Immunofluorescence Staining of NF-κB p65
HuCCT1 cells were seeded into 96-well plates with glass bottoms (Matrical Bioscience, USA) and cultured overnight in serum-free medium. After treatment with TCA or S1P with or without JTE-013, cells were fixed with 3.7% formaldehyde in PBS for 45 min, permeabilized with 0.1% Triton X-100 for 3 min, and blocked with 3% BSA for 45 min. Cells were then incubated with rabbit anti-NF-κB p65 antibody (1:100 dilution) overnight at 4 °C, followed by incubation with Alexa Fluor 488-labeled goat anti-rabbit secondary antibody for 30 min at room temperature. After washing with PBS, staining of the intracellular and nuclear NF-κB p65 was visualized under an Olympus IX81 motorized inverted fluorescence microscope with a ×60 oil objective (Olympus, Japan).
Detection of S1PR2 Internalization
HEK293 cells were plated into 6-well plates with coverslips. Cells were cultured overnight to 60% confluence and transfected with 1 μg of plasmid DNA (pcDNA3.1–3xHA-tagged S1PR2) using PolyJetTM DNA In Vitro Transfection Reagent according to the manufacturer's instruction. After 24 h of transfection, cells were cultured in serum-free medium overnight, then stimulated with TCA (100 μm), S1P (100 nm), or vehicle control for 10 min. Cells were fixed with 3.7% formaldehyde in PBS for 15 min. The expression and localization of S1PR2 were monitored using immunofluorescence staining with anti-HA antibody and Alexa Fluor 488-labeled goat anti-mouse secondary antibody. The images were recorded using Zeiss Axio Imager A1 fluorescence microscope with a ×100 oil objective (Jenamed, Carl Zeiss, Germany).
Statistical Analysis
All data were expressed as the mean ± S.E. One-way ANOVA analysis of variance and Student's t test were employed to analyze the differences between sets of data. Statistical analysis was performed using Prism 5.0 (GraphPad, San Diego, CA) as described previously (18, 20). A value of p < 0.05 was considered statistically significant.
Results
TCA Induces COX-2 Expression and Chronic Inflammation via Activation of S1PR2
COX-2 is a key enzyme involved in production of prostaglandins and has been implicated in various cell transformations including cholangiocytes (21–25). Previous studies reported that CBAs induced COX-2 expression and promoted growth in human CCA cells in culture (15, 16). Our recent studies showed that CBA (TCA) promoted invasive cell growth via activation of S1PR2 in both rat and human CCA cell lines (18). However, whether activation of S1PR2 also contributes to CBA-mediated expression of COX-2 and PG synthesis remained unknown. Therefore, we first examined the effect of TCA on COX-2 expression in human HuCCT1 cells. As shown in Fig. 1, A and B, TCA significantly increased COX-2 protein levels in a time-dependent manner, peaking at 8 h. TCA-induced COX-2 expression was also dose-dependent (Fig. 1, C and D). To define the role of S1PR2 in TCA-induced COX-2 expression, a chemical antagonist of S1PR2, JTE-013, was used. As shown in Fig. 2, A and B, both S1P- and TCA-induced COX-2 expression was markedly inhibited by JTE-013. Similarly, as shown in Fig. 2, C–E, down-regulation of S1PR2 using a S1PR2 shRNA significantly blocked TCA-induced COX-2 expression.
FIGURE 1.

The effect of TCA on COX-2 expression in HuCCT1 cells. Cells were cultured in serum-free medium overnight and then treated with either 100 μm TCA for different time periods (0–24 h) or different concentrations of TCA (0, 25, 50, 100, or 200 μm) for 8 h. At the end of each treatment, cells were harvested and protein levels of COX-2 were detected by Western blot analysis. A and C, representative images of the immunoblots for COX-2 and actin are shown. B and D, relative densities of COX-2 were analyzed using Quantity One software using actin as a loading control. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: **, p < 0.01; ***, p < 0.001.
FIGURE 2.

The role of S1PR2 in TCA-induced COX-2 expression in HuCCT1 cells. Cells were cultured in serum-free medium overnight and then treated with S1P (100 nm) or TCA (100 μm) with or without JTE-013 (10 μm) for 8 h. At the end of treatment, total protein was isolated to determine COX-2 protein levels using Western blot analysis. A, representative images of the immunoblots for COX-2 and actin are shown. B, relative densities of COX-2 were determined using actin as a loading control. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: *, p < 0.05; ***, p < 0.001; statistical significance relative to the corresponding treatment group without JTE-013: #, p < 0.05; ###, p < 0.001. C, cells were transfected with either a control scrambled shRNA or S1PR2-specifc shRNA expression vector for 48 h. Relative mRNA levels of S1PR2 were detected by real-time RT-PCR and normalized using GAPDH as an internal control as described under “Experimental Procedures.” D, cells were treated with vehicle control or TCA (100 μm) for 8 h after transfection with control or S1PR2 shRNA for 48 h. Protein levels of COX-2 were determined by Western blot analysis. Representative images of the immunoblots for COX-2 and actin are shown. E, relative densities of COX-2 were determined using actin as a loading control. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: *, p < 0.05; statistical significance relative to the TCA-treated group with control shRNA: #, p < 0.05. F, cells were pretreated with JTE-013 (10 μm) for 1 h before being treated with TCA (100 μm) for 8 h. At the end of the treatment, cell culture medium was collected for detection of PGE2 secretion as described under “Experimental Procedures.” Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: **, p < 0.01; relative to the TCA treatment group; #, p < 0.05.
PGE2, the final enzymatic product of COX-2, has been implicated in various chronic inflammation-related cell transformation including CCA (14). Previous study from the laboratory of Dr. Wu reported that COX-2-derived PGE2 regulates human CCA cell growth (9). Our study also showed that TCA significantly increased PGE2 production in HuCCT1 cells (Fig. 2F). Furthermore, TCA-induced PGE2 production was significantly inhibited by JTE-013. However, both S1P and TCA had no effect on COX-1 protein expression in HuCCT1 cells at the protein level (Fig. 3, A and B). In addition, we examined the effect of S1P and TCA on the mRNA expression of two inflammatory cytokines, IL-6 and TNF-α. As shown in Fig. 3, C and D, both S1P and TCA significantly induced mRNA expression of IL-6 and TNF-α, which was attenuated by JTE-013. These results suggest that S1PR2 may be a key regulator in TCA-mediated COX-2 expression, PGE2 production, and chronic inflammation.
FIGURE 3.

A and B, effect of TCA on COX-1 expression. HuCCT1 cells were cultured in serum-free medium overnight and then treated with S1P (100 nm) or TCA (100 μm) with or without JTE-013 (10 μm) for 8 h. At the end of treatment, total proteins were isolated. The protein level of COX-1 was detected by Western blot analysis. A, representative images of immunoblots for COX-1 and Actin are shown. B, relative protein levels of COX-1 were analyzed using Actin as a loading control. Values represent the mean ± S.E. of three independent experiments. C and D, effect of TCA on inflammatory cytokines expression. HuCCT1 cells were cultured in serum-free medium overnight and then treated with S1P (100 nm) or TCA (100 μm) with or without JTE-013 (10 μm) for 8 h. At the end of treatment, total cellular RNA was isolated. Relative mRNA levels of IL-6 (A) and TNF-α (B) were determined by real-time RT-PCR and normalized using GAPDH as an internal control. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control; *, p < 0.05; **, p < 0.01; ***, p < 0.001; relative to S1P or TCA treatment group: #, p < 0.05; ##, p < 0.01; ###, p < 0.001.
TCA Enhances Invasion of HuCCT1 Cells through S1PR2-dependent COX-2 Activation
Although several studies have reported that CBAs promote CCA cell growth and invasion, the contribution of TCA-induced S1PR2 activation and COX-2 expression on CCA cell invasion has not been examined (15, 26, 27). Our recent studies showed that S1P- and TCA-induced invasive growth was blocked by inhibition of S1PR2 activation in rat CCA cells (18). To further verify the effect of TCA on the invasion of human CCA cells, HuCCT1 cells were plated in the upper chamber of Matrigel pre-coated transwell inserts and treated with TCA (100 μm), S1P (100 nm, as a positive control), or vehicle control, in the presence or absence of JTE-013 (10 μm). Similar to our findings in rat CCA cells, the invasion indexes of TCA and S1P groups were significantly increased in human HuCCT1 cells, which were significantly inhibited by JTE-013 (Fig. 4, A and B). Down-regulation of S1PR2 expression using a shRNA also blocked the TCA-induced increase of the invasion index (Fig. 4, C and D). To further define the role of TCA-induced S1PR2 activation and COX-2 expression in CCA cell invasion, a specific chemical inhibitor of COX-2, Celecoxib, was used. As shown in Fig. 4, E and F, in the presence of Celecoxib (40 μm), a TCA-induced increase of the invasion index was also markedly inhibited in HuCCT1 cells. These results suggest that TCA promotes CCA cell invasion via activation of S1PR2, increased COX-2 expression, and synthesis of PGE2.
FIGURE 4.

The effect of S1PR2 activation and COX-2 expression on TCA-induced invasiveness of HuCCT1 cells. A and B, cells were cultured inside of the Matrigel pre-coated transwell inserts and pretreated with JTE-013 (10 μm) for 1 h and then treated with either S1P (100 nm) or TCA (100 μm) for 24 h. C and D, cells were transfected with either control shRNA or S1PR2 shRNA for 48 h. Transfected cells were cultured inside of the Matrigel pre-coated transwell inserts and then treated with TCA (100 μm) for 24 h. E and F, cells were cultured inside of the Matrigel pre-coated transwell inserts and treated with TCA (100 μm) for 24 h with or without Celecoxib (40 μm). At the end of treatment, the number of invasive cells and invasive index were analyzed as described under “Experimental Procedures.” Representative images for each treatment from three independent experiments are shown. Statistical significance relative to the vehicle control: *, p < 0.05; **, p < 0.01; ***, p < 0.001; relative to the corresponding S1P or TCA treatment group: #, p < 0.05; ##, p < 0.01; ###, p < 0.001.
TCA Activates NF-κB via Activation of S1PR2
The transcription factor NF-κB is a well known evolutionarily conserved signaling molecule with many biological activities. NF-κB can be activated by cell signaling pathways that activate IκB kinase (IKKα/β). Activated IKKα/β further phosphorylates IκB and leads to degradation of IκB and nuclear translocation of NF-κB p65 (28). In addition, IKKα/β are also involved in the direct phosphorylation of NF-κB p65. Phosphorylation of NF-κB p65 not only enhances the efficiency of DNA binding, but also provides an additional interaction site for transcriptional co-activator CBP/p300 (29). Previous studies have shown that bile acids activate NF-κB signaling pathways in cancer cells (15, 30). To determine whether TCA also can activate the NF-κB signaling pathways, we examined the protein levels of phosphorylated IKKα/β (p-IKKα/β) and phosphorylated NF-κB p65 (p-NF-κB p65). As shown in Fig. 5, TCA significantly increased protein levels of p-IKKα/β and p-NF-κB p65 in both a time-dependent and dose-dependent manner. In addition, TCA significantly increased nuclear translocation of NF-κB p65 (Fig. 6). To further determine whether the NF-κB activation depends on TCA-mediated activation of S1PR2, we examined the effect of JTE-013 on TCA-induced activation of NF-κB in HuCCT1 cells. As shown in Figs. 7 and 8, JTE-013 not only inhibited the TCA-induced increase of p-IKKα/β and p-NF-κB p65, but also blocked TCA-induced nuclear translocation of NF-κB p65.
FIGURE 5.

The effect of TCA on activation of IKKα/β-NF-κB pathways in HuCCT-1 cells. A–C, time course of TCA-induced activation of IKKα/β-NF-κB pathways. Cells were cultured in serum-free medium overnight and then treated with TCA (100 μm) for different treatment periods (0–24 h). The protein levels of phosphorylated IKKα/β (p-IKKα/β), total IKKα/β, phosphorylated NF-κB p65 (p-NF-κB p65), and total NF-κB p65 were determined by Western blot analysis. D–F, TCA-induced dose-dependent activation of IKKα/β-NF-κB pathways. Cells were cultured in serum-free medium overnight and then treated with different concentrations of TCA (0, 25, 50, 100, or 200 μm) for 4 h. At the end of each treatment, cells were harvested, and total protein was isolated. The protein levels of p-IKKα/β, total IKKα/β, p-NF-κB p65, and total NF-κB p65 were determined by Western blot analysis. A and D, representative images of p-IKKα/β, total IKKα/β, p-NF-κB p65, and total NF-κB p65 are shown. B, C, E, and F, relative protein levels of p-IKKα/β/IKKα/β and p-NF-κB p65/NF-κB p65 were determined. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to vehicle control: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 6.

The effect of TCA on nuclear translocation of NF-κB. HuCCT1 cells were cultured in serum-free medium overnight and then treated with TCA (100 μm) for different treatment periods (0, 2, 4, 8, or 24 h). At the end of each treatment, cytosol and nuclear proteins were isolated as described under “Experimental Procedures.” Protein levels of NF-κB p65 were determined by Western blot analysis. Lamin B and actin were used as loading controls for nuclear and cytosol protein, respectively. A, representative images of the immunoblots for NF-κB p65, lamin B, and actin are shown. B, relative protein levels of NF-κB p65 in the nucleus and cytosol. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: ***, p < 0.001. C and D, immunofluorescence staining of NF-κB p65. HuCCT1 cells were cultured in serum-free medium overnight and treated with either 100 μm TCA for different treatment periods (0, 2, 4, or 8 h) or different concentrations of TCA (0, 25, 50, or 100 μm) for 4 h. At the end of treatment, immunofluorescence staining was performed to detect subcellular location of NF-κB p65. Representative images for each group are shown.
FIGURE 7.

The effect of chemical antagonist of S1PR2 on S1P- and TCA-induced activation of IKKα/β-NF-κB pathway. HuCCT1 cells were cultured in serum-free medium overnight and pre-treated with JTE-013 (10 μm) for 1 h, and then treated with S1P (100 nm) or TCA (100 μm) for 4 h. A and B, total protein was isolated, and protein levels of p-IKKα/β, total IKKα/β, p-NF-κB p65, and total NF-κB p65 were determined by Western blot analysis. The relative densities of p-IKKα/β/total IKKα/β and p-NF-κB p65/total NF-κBp65 were determined. C and D, cytosol and nuclear proteins were isolated as described under “Experimental Procedures.” Protein levels of NF-κB p65, lamin B, and actin were determined by Western blot analysis. Lamin B and actin were used as a loading control for nuclear and cytosol protein, respectively. A and C, representative images of the immunoblots for p-IKKα/β, total IKKα/β, p-NF-κB p65, and total NF-κB p65 are shown. B, relative protein levels of p-IKKα/β/total IKKα/β and p-NF-κB p65/total NF-κB p65. D, relative protein levels of nuclear NF-κB p65/lamin B and cytosol NF-κB p65/actin. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: *, p < 0.05; **, p < 0.01; ***, p < 0.001; relative to S1P or TCA treatment group: #, p < 0.05; ##, p < 0.01; ###, p < 0.001.
FIGURE 8.

The effect of JTE-013 on S1P- and TCA-induced nuclear translocation of NF-κB. HuCCT1 cells were cultured in serum-free medium overnight and then treated with either 100 μm TCA for different treatment periods (0, 2, 4, 8, or 24 h) or different concentrations of TCA (0, 25, 50, or 100 μm) for 4 h. At the end of treatment, immunofluorescence staining was performed to detect subcellular location of NF-κB p65. Representative images for each group are shown.
TCA Activates EGF Receptor via S1PR2
Previous studies reported that bile acids transactivate the EGF receptor in cholangiocytes via a TGF-α-dependent mechanism (31). Bile acid-induced activation of EGFR/ERK1/2 also induced COX-2 expression in CCA cells (16). In human CCA, EGFR expression level is significantly associated with tumor progression and poor prognosis (32). To further understand the relationship between S1PR2 and EGFR/ERK1/2 activation, we first examined the effect of TCA on the level of EGFR phosphorylation in HuCCT1 cells. As shown in Fig. 9, TCA rapidly and dose-dependently induced phosphorylation of EGFR. The presence of JTE-013 or down-regulation of S1PR2 using gene-specific shRNA completely blocked TCA-induced activation of EGFR, but had no effect on total protein expression (Fig. 10). One of the most common mechanisms of GPCR-mediated activation of EGFR is the release of the mature EGFR ligands from transmembrane precursors by MMPs. MMP2 and MMP9 are members of the MMP family reported to mediate EGFR activation through GPCRs (33). We further examined whether activation of S1PR2 had any effect on the expression of MMP2 and MMP9 in HuCCT1 cells. As shown in Fig. 11, both TCA and S1P significantly increased the mRNA levels as well as the protein levels of MMP2 and MMP9, but these increases were blocked by JTE-013.
FIGURE 9.

The effect of TCA on EGFR transactivation in HuCCT1 cells. HuCCT1 cells were cultured in serum-free medium overnight and then treated with either 100 μm TCA for different time periods (0, 0.17, 0.5, 1, 2, 4, or 8 h), or with different concentrations of TCA (0, 25, 50, 100, or 200 μm) for 15 min. At the end of treatment, total protein was isolated. Protein levels of phosphorylated EGFR (p-EGFR) and total EGFR (t-EGFR) were detected by Western blot analysis. A and C, representative images of the immunoblots for p-EGFR and t-EGFR are shown. B and D, relative protein levels of p-EGFR/t-EGFR were calculated from three independent experiments. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: *, p < 0.05; **, p < 0.01; ***, p < 0.001; relative to S1P or TCA treatment group: ##, p < 0.01; ###, p < 0.001.
FIGURE 10.

The role of S1PR2 in TCA-induced EGFR transactivation in HuCCT1 cells. A and B, cells were cultured in serum-free medium and pretreated with JTE-013 (10 μm) for 1 h, and then treated with S1P (100 nm) or TCA (100 μm) for 15 min. C and D, HuCCT1 cells were transfected with control shRNA or S1PR2 shRNA for 48 h and then treated with TCA (100 μm) for 15 min. At the end of treatment, total protein was isolated. Protein levels of p-EGFR and t-EGFR were detected by Western blot analysis. A and C, representative images of the immunoblots for p-EGFR and t-EGFR are shown. B and D, relative protein levels of p-EGFR/t-EGFR were calculated from three independent experiments. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: **, p < 0.01; ***, p < 0.001; relative to the S1P or TCA treatment group: ##, p < 0.01; ###, p < 0.001.
FIGURE 11.

The effect of TCA on MMP-2 and MMP9 expression. HuCCT1 cells were cultured in serum-free medium overnight and then treated with S1P (100 nm) or TCA (100 μm) with or without JTE-013 (10 μm) for 8 h. At the end of treatment, total cellular RNA and protein lysates were isolated. Relative mRNA levels of (A) MMP-2 and (B) MMP-9 were determined by real-time RT-PCR and normalized using GAPDH as an internal control. Protein levels of MMP2 and MMP9 were detected by Western blot analysis. Representative images of the immunoblots for MMP2 and MMP9 are shown (C and E). Relative protein levels of MMP2/Actin and MMP9/Actin were calculated from three independent experiments. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: **, p < 0.01; ***, p < 0.001; relative to S1P or TCA treatment group: #, p < 0.05; ##, p < 0.01.
Role of S1PR2 in TCA-mediated ERK1/2 and Akt Activation in HuCCT1 Cells
Bile acid-induced EGFR activation is linked to the activation of ERK1/2 and Akt (34). Our previous studies also showed that TCA-induced ERK1/2 and Akt activation via S1PR2 (18, 20). To confirm the role of S1PR2 activation in TCA-induced ERK1/2 and Akt activation in HuCCT1 cells, we examined the protein levels of p-ERK1/2 and p-Akt in TCA-treated HuCCT1 cells. As shown in Fig. 12, A and B, TCA rapidly induced ERK1/2 and Akt activation in a time-dependent manner, peaking at 4 h. Similar to TCA-induced EGFR activation, TCA also dose-dependently induced ERK1/2 and Akt activation (Fig. 12, C and D). Furthermore, both TCA- and S1P-induced ERK1/2 and Akt activation was markedly inhibited by JTE-013 (Fig. 12, E and F).
FIGURE 12.

The effect of TCA on ERK and Akt activation in HuCCT1 cells. A and B, time course of TCA-induced ERK and Akt activation. HuCCT1 cells were cultured in serum-free medium overnight and then treated with TCA (100 μm) for different treatment periods (0, 0.5, 1, 2, 4, 8, or 24 h). C and D, TCA-induced dose-dependent activation of ERK and Akt. HuCCT1 cells were cultured in serum-free medium overnight and then treated with different concentrations of TCA (0, 25, 50, or 100 μm) for 1 h. E and F, the effect of JTE-013 on TCA-/S1P-induced ERK and Akt activation. HuCCT1 cells were cultured in serum-free medium overnight and pre-treated with JTE-013 (10 μm) for 1 h and then treated with S1P (100 nm) or TCA (100 μm) for 1 h. At the end of each treatment, cells were harvested and total protein was isolated. The protein levels of phosphorylated ERK1/2 (p-ERK), total ERK1/2 (t-ERK), phosphorylated Akt1/2/3 (p-Akt), and total Akt1/2/3 (t-Akt) were detected by Western blot analysis. A, C, and E, representative images of the immunoblots for p-ERK, t-ERK, p-Akt, and t-Akt are shown. B, D, and F, relative levels of p-ERK/t-ERK and p-Akt/t-Akt. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: *, p < 0.05; **, p < 0.01; ***, p < 0.001; relative to S1P or TCA treatment groups: ##, p < 0.01; ###, p < 0.001.
Effect of TCA-induced ERK1/2 and Akt Activation on COX-2 Expression
It has been well documented that COX-2 expression is regulated by ERK1/2 and Akt in various cell types (8, 35–38). To further delineate the role of S1PR2-mediated ERK1/2 and Akt activation in TCA-induced COX-2 expression in HuCCT1 cells, selective chemical inhibitors of MEK1/2 and Akt were used. As shown in Fig. 13, A and B, in the presence of U0126 (10 μm), both TCA- and S1P-induced ERK1/2 activation was completely blocked. TCA- and S1P-induced COX-2 expression was also significantly inhibited by U0126 (Fig. 14. A and B). Similarly, both TCA- and S1P-induced Akt activation and COX-2 expression were markedly inhibited by MK2206, a highly selective inhibitor of Akt1/2/3 (Figs. 13, C and D, and 14, C and D).
FIGURE 13.

Inhibition of TCA-induced ERK1/2 and Akt activation by chemical inhibitors in HuCCT1 cells. HuCCT1 cells were cultured in serum-free medium overnight and pretreated with a selective inhibitor of MEK1/2, U0126 (10 μm), or a selective inhibitor of Akt, MK2206 (3 μm) for 1 h and then treated with S1P (100 nm) or TCA (100 μm) for 1 h. At the end of treatment, total protein was isolated. Protein levels of p-ERK1/2, total ERK1/2, p-Akt, and t-Akt were detected by Western blot analysis. A and C, representative images of the immunoblots for p-ERK1/2, t-ERK1/2, p-Akt, and t-Akt are shown. B and D, relative protein levels of p-ERK1/2/t-ERK1/2 and p-Akt/t-Akt were calculated from three independent experiments. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to vehicle control: **, p < 0.01, ***, p < 0.001; relative to S1P or TCA treatment group: ##, p < 0.05; ###, p < 0.001.
FIGURE 14.

Effect of TCA-induced ERK1/2 and Akt activation on COX-2 expression in HuCCT1 cells. Cells were cultured in serum-free medium overnight and pretreated with a selective inhibitor of MEK1/2, U0126 (10 μm) for 1 h, and then treated with S1P (100 nm) or TCA (100 μm) for 1 h. At the end of treatment, total protein was isolated and protein levels of COX-2 and actin were detected by Western blot analysis. A and C, representative images of the immunoblots for COX-2 and actin are shown. B and D, relative protein levels of COX-2/Actin were calculated from three independent experiments. Values represent the mean ± S.E. of three independent experiments. Statistical significance relative to the vehicle control: *, p < 0.05; **, p < 0.01; relative to the S1P or TCA treatment group: #, p < 0.05; ###, p < 0.01.
S1P and TCA Induce S1PR2 Internalization
Our previous studies and current study strongly indicated that TCA activates S1PR2. To delineate whether TCA is able to directly interact with S1PR2, we examined the effect of TCA on S1PR2 internalization in HEK293 cells. Cells were transfected with HA-tagged human S1PR2 and then treated with TCA (100 μm) or S1P (100 nm) or vehicle control for 10 min at 37 °C. To stop the ligand-mediated internalization process, cells were fixed with 3.7% formaldehyde at 4 °C. The expression and localization of HA-S1PR2 were monitored by immunofluorescence staining using a specific anti-HA antibody. As shown in Fig. 15, both TCA and S1P rapidly induced S1PR2 internalization. These results suggest that TCA directly interact with S1PR2 as previously suggested from molecular modeling experiments (20).
FIGURE 15.
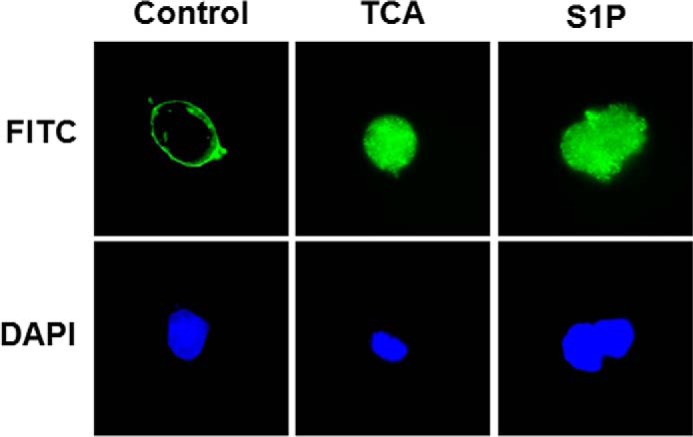
Effect of TCA and S1P on S1PR2 internalization. HEK293 cells were transfected with 3×HA-tagged human S1PR2 as described under “Experimental Procedures.” Cells were stimulated with vehicle control or TCA (100 μm) or S1P (100 nm) for 10 min, the internalization process was stopped by fixing with 3.7% formaldehyde at 4 °C. The localization of S1PR2 was monitored by immunofluorescence staining. Representative images of each treatment group are shown.
Discussion
CCA is very difficult to diagnose at early stages due to its anatomic location. Delayed diagnosis precludes many CCA patients from surgical intervention, the treatment option with the best prognosis (1). Concurrently, the outcomes of current conventional chemotherapies and radiation are also very poor (3). Therefore, identification of novel therapeutic targets for CCA is extremely urgent. Although a significant amount of effort has been put in understanding CCA development and progression during the last two decades, the etiopathogenesis of this cancer are still largely unknown.
Accumulation of bile acids in bile ducts and liver is associated with an increased incidence of CCA (39). However, the exact role of bile acids in cholangiocarcinogenesis is still unclear. Several studies suggested that bile acid-mediated activation of the nuclear receptor FXR (farnesoid X receptor) and the GPCR, TGR5, might be involved in regulating the proliferation and survival of cholangiocytes (40). However, our recent studies also identified that CBA-mediated activation of S1PR2 plays a critical role in invasive growth of CCA cells in culture (18). Impaired bile flow is often accompanied with chronic inflammation and a high basal level of COX-2 expression in the biliary tracts (1), whereas the underlying cellular mechanisms are still not fully identified. Inflammation is a key stimulator of carcinogenesis, promoting tumor cell invasion and migration in various cancers including CCA (41–43). Recently, there has been significant progress in identifying the critical role of COX-2 in tumorgenesis of various cancers including CCA (4, 9, 44), and PGE2, the enzymatic product of COX-2, has been identified as a strong promoter for mitogenesis, cell proliferation, and invasion of CCA (9, 45, 46).
The principal findings of the current study directly link CBA-mediated activation of S1PR2 to both COX-2 expression and PGE2 production in human CCA cells. The results indicated that TCA-induced activation of S1PR2 is responsible for activation of EGFR/ERK1/2/Akt signaling pathways, which further activates NF-κB and increases COX-2 expression in CCA cells. A schematic diagram of potential pathways involved in TCA-induced COX-2 expression in CCA cells is illustrated in Fig. 16. The current results provide novel mechanisms underlying CBA-mediated chronic inflammation in CCA.
FIGURE 16.
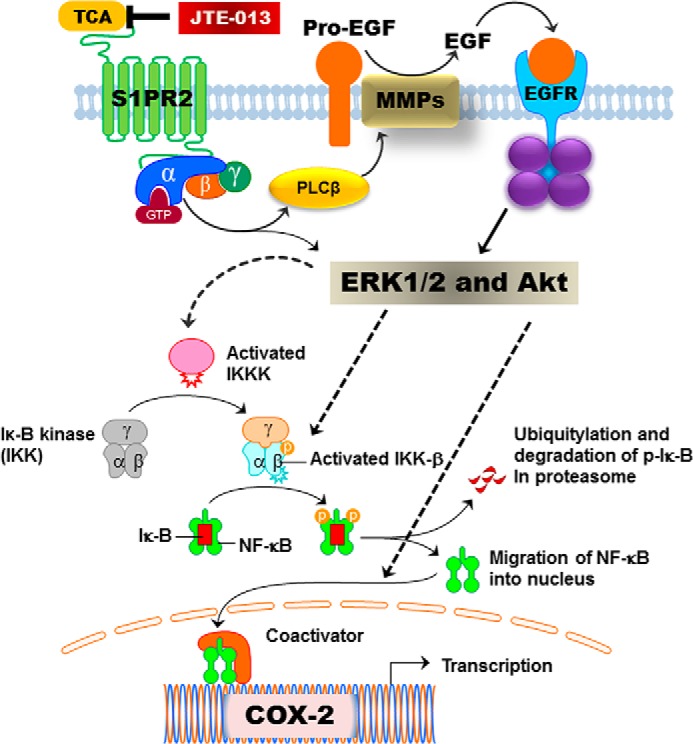
Proposed signaling pathways involved in TCA-induced COX-2 expression in human CCA cells. Activation of S1PR2 by TCA directly activates ERK1/2/AKt via activated G proteins or indirectly activates ERK1/2/Akt via EGFR. Activated ERK1/2/Akt further activate NF-κB and COX-2 expression by inducing phosphorylation of IKKα/β and IκB, ubiquitination and degradation of p-IκB, and nuclear translocation of NF-κB.
S1PR2 is one of the five S1PRs identified so far and has been implicated in various physiological and pathological conditions (19). A recent study reported that extracellular S1P induced COX-2 expression and PGE2 production via activation of S1PR2, as well as subsequent activation of ERK1/2 in rat renal mesangial cells (47). It also has been reported that S1PR2 signaling is involved in macrophage retention and inflammatory secretions in the atherosclerotic lesions (48). However, the role of S1PR2 in tumor growth and progression is controversial in different types of cancers, probably due to its coupling to different G proteins in different types of cells (19). In this study, we demonstrated that TCA dose-dependently induced expression of COX-2 via activation of S1PR2 in human HuCCT1 cells. The protein level of COX-2 was significantly increased by TCA even at a concentration of 25 μm (Fig. 1). Inhibition of S1PR2 activation by a chemical antagonist or down-regulation of S1PR2 with gene-specific shRNA completely blocked TCA-induced COX-2 expression and CCA cell invasion (Figs. 2 and 4). Previous studies reported that bile acids induced COX-2 expression via activation of EGFR/MAPK signaling pathways in CCA cells (16). We have also reported that bile acids activated MAPK via EGFR in hepatocytes (34). In this study, we found that TCA also significantly activated the EGFR/MAPK signaling pathways in HuCCT1 cells (Fig. 10). A recent study reported that taurolithocholic acid promotes CCA cell growth through the activation of the muscarinic acetylcholine receptor and the EGFR/ERK1/2 signaling pathway (8). However, the concentration of taurolithocholic acid is relatively low compared with conjugated primary bile acids in CCA as cholestasis decreases the formation of secondary bile acids. Clinical studies have shown that the concentrations of conjugated primary bile acids (cholic and chenodeoxycholic acid) are significantly higher in the bile of CCA patients than in those with the benign disease (17). In addition, we have reported that S1PR2 can be activated by TCA, and S1PR2 is the predominant S1PR expressed in CCA cells and human tumor tissues (18, 20). Our current study further confirmed that TCA rapidly induced S1PR2 internalization, indicating that TCA may directly activate S1PR2 (Fig. 15). Therefore, S1PR2 appears to play a key role in CBA-mediated COX-2 expression and PGE2 synthesis in CCA cells.
It has been shown that activation of Src kinase or GPCR-mediated activation of Gi protein can increase the expression of MMPs, which play an important role in forming active EGFR ligands (16, 31, 33, 46). Similarly, in this study, we also found that both S1P and TCA significantly increased the expression of MMP2 and MMP9 in HuCCT1 cells, which was markedly inhibited by JTE-013 (Fig. 11). These results suggest that TCA-induced activation of S1PR2 contributes to the increase of MMPs and subsequent activation of EGFR/ERK/Akt signaling pathways. In addition, S1PR2 can also directly activate ERK and Akt directly through Gαi (18). It has been well established that activation of ERK/Akt signaling pathways is essential for activation of NF-κB. NF-κB is a key transcription factor involved in the regulation of various inflammatory factors including COX-2. Consistent with previous reports, we also found that TCA-induced S1PR2/EGFR/ERK/Akt activation was linked to NF-κB activation in CCA cells (Figs. 5–10), and JTE-013 inhibited not only TCA-/S1P-induced NF-κB activation, but also the nuclear translocation of NF-κB. By using specific chemical inhibitors of MEK1 and Akt, we further demonstrated that TCA-induced activation of S1PR2/ERK1/2/Akt is responsible for COX-2 expression in CCA cells (Fig. 14). Inhibition of ERK1/2/Akt also blocked TCA-induced nuclear translocation of NF-κB (data not shown). Furthermore, S1P- and TCA-induced expression of inflammatory cytokines (IL-6 and TNF-α) was also blocked by JTE-013 (Fig. 3).
In summary, as illustrated in Fig. 16, we determined that S1PR2 is a key player in TCA-induced COX-2 expression in HuCCT1 cells. Activation of S1PR2 by TCA activates ERK1/2 and Akt signaling pathways via G proteins and EGFR. Activated ERK1/2 and Akt further leads to the activation of NF-κB and subsequent COX-2 expression and PGE2 synthesis. Our study provides further evidence indicating that S1PR2 represents a novel therapeutic target for CCA.
Author Contributions
R. L., P. B. H., Z. J., L. Z., and H. Z. conceived the original ideas, designed the study, analyzed the data, and wrote the manuscript; R. L., X. L., L. L., and X. Q. carried out the experiments and data analysis.
This work was supported by National Natural Science Foundation of China Grants 81320108029 (to L. Z.), 81070245 and 81270489 (to H. Z.), China National 12th Five-year Plan “Major Project of Science and Technologies for Significant Drug Discovery” Grant 2012ZX09504001-001 (to L. Z.), National Institutes of Health Grant R01 DK-057543 (to P. B. H. and H. Z.), Veterans Affairs Merit Awards (to H. Z.), and Virginia Commonwealth University Massey Cancer Pilot Grant A35362 (to H. Z.). The authors declare no competing financial interest with the contents of this article.
- CCA
- cholangiocarcinoma
- Akt
- protein kinase B
- CBA
- conjugated bile acid
- EGFR
- epidermal growth factor receptor
- EP
- prostaglandin E receptor
- IκB
- inhibitor of κB
- IKK α/β
- IκB kinase α/β
- MMP-2/9
- matrix metalloproteinases 2/9
- NF-κB
- nuclear factor κ light chain enhancer of activated B cells
- PGE2
- prostaglandin E2
- S1P
- sphingosine 1-phosphate
- S1PR2
- sphingosine 1-phosphate receptor 2
- TCA
- taurocholate
- GPCR
- G protein-coupled receptor
- DMSO
- dimethyl sulfoxide.
References
- 1. Gores G. J. (2003) Cholangiocarcinoma: current concepts and insights. Hepatology 37, 961–969 [DOI] [PubMed] [Google Scholar]
- 2. Sirica A. E. (2005) Cholangiocarcinoma: molecular targeting strategies for chemoprevention and therapy. Hepatology 41, 5–15 [DOI] [PubMed] [Google Scholar]
- 3. Al-Bahrani R., Abuetabh Y., Zeitouni N., and Sergi C. (2013) Cholangiocarcinoma: risk factors, environmental influences and oncogenesis. Ann. Clin. Lab. Sci. 43, 195–210 [PubMed] [Google Scholar]
- 4. Yeh C. N., Chiang K. C., Juang H. H., S Pang J. H., Yu C. S., Lin K. J., Yeh T. S., and Jan Y. Y. (2013) Reappraisal of the therapeutic role of celecoxib in cholangiocarcinoma. PloS One 8, e69928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kiguchi K. (2014) Molecular aspects of cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 21, 371–379 [DOI] [PubMed] [Google Scholar]
- 6. Aleksandrova K., Boeing H., Nöthlings U., Jenab M., Fedirko V., Kaaks R., Lukanova A., Trichopoulou A., Trichopoulos D., Boffetta P., Trepo E., Westhpal S., Duarte-Salles T., Stepien M., Overvad K., Tjønneland A., Halkjaer J., Boutron-Ruault M. C., Dossus L., Racine A., Lagiou P., Bamia C., Benetou V., Agnoli C., Palli D., Panico S., Tumino R., Vineis P., Bueno-de-Mesquita B., Peeters P. H., Gram I. T., Lund E., Weiderpass E., Quirós J. R., Agudo A., Sánchez M. J., Gavrila D., Barricarte A., Dorronsoro M., Ohlsson B., Lindkvist B., Johansson A., Sund M., Khaw K. T., Wareham N., Travis R. C., Riboli E., and Pischon T. (2014) Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer. Hepatology 60, 858–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blechacz B., Komuta M., Roskams T., and Gores G. J. (2011) Clinical diagnosis and staging of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 8, 512–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amonyingcharoen S., Suriyo T., Thiantanawat A., Watcharasit P., and Satayavivad J. (2015) Taurolithocholic acid promotes intrahepatic cholangiocarcinoma cell growth via muscarinic acetylcholine receptor and EGFR/ERK1/2 signaling pathway. Int. J. Oncol. 46, 2317–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu T. (2005) Cyclooxygenase-2 and prostaglandin signaling in cholangiocarcinoma. Biochim. Biophys. Acta 1755, 135–150 [DOI] [PubMed] [Google Scholar]
- 10. Nzeako U. C., Guicciardi M. E., Yoon J. H., Bronk S. F., and Gores G. J. (2002) COX-2 inhibits Fas-mediated apoptosis in cholangiocarcinoma cells. Hepatology 35, 552–559 [DOI] [PubMed] [Google Scholar]
- 11. Sirica A. E., Lai G. H., Endo K., Zhang Z., and Yoon B. I. (2002) Cyclooxygenase-2 and ERBB-2 in cholangiocarcinoma: potential therapeutic targets. Semin. Liver Dis. 22, 303–313 [DOI] [PubMed] [Google Scholar]
- 12. Reader J., Holt D., and Fulton A. (2011) Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 30, 449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Surh I., Rundhaug J., Pavone A., Mikulec C., Abel E., and Fischer S. M. (2011) Upregulation of the EP1 receptor for prostaglandin E2 promotes skin tumor progression. Mol. Carcinog. 50, 458–468 [DOI] [PubMed] [Google Scholar]
- 14. Jongthawin J., Chusorn P., Techasen A., Loilome W., Boonmars T., Thanan R., Puapairoj A., Khuntikeo N., Tassaneeyakul W., Yongvanit P., and Namwat N. (2014) PGE2 signaling and its biosynthesis-related enzymes in cholangiocarcinoma progression. Tumour Biol. 35, 8051–8064 [DOI] [PubMed] [Google Scholar]
- 15. Dai J., Wang H., Dong Y., Zhang Y., and Wang J. (2013) Bile acids affect the growth of human cholangiocarcinoma via NF-κB pathway. Cancer Invest. 31, 111–120 [DOI] [PubMed] [Google Scholar]
- 16. Yoon J. H., Higuchi H., Werneburg N. W., Kaufmann S. H., and Gores G. J. (2002) Bile acids induce cyclooxygenase-2 expression via the epidermal growth factor receptor in a human cholangiocarcinoma cell line. Gastroenterology 122, 985–993 [DOI] [PubMed] [Google Scholar]
- 17. Jusakul A., Khuntikeo N., Haigh W. G., Kuver R., Ioannou G. N., Loilome W., Namwat N., Bhudhisawasdi V., Pugkhem A., Pairojkul C., and Yongvanit P. (2012) Identification of biliary bile acids in patients with benign biliary diseases, hepatocellular carcinoma and cholangiocarcinoma. Asian Pac. J. Cancer Prev. 13, 77–82 [PubMed] [Google Scholar]
- 18. Liu R., Zhao R., Zhou X., Liang X., Campbell D. J., Zhang X., Zhang L., Shi R., Wang G., Pandak W. M., Sirica A. E., Hylemon P. B., and Zhou H. (2014) Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology 60, 908–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adada M., Canals D., Hannun Y. A., and Obeid L. M. (2013) Sphingosine-1-phosphate receptor 2. FEBS J. 280, 6354–6366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Studer E., Zhou X., Zhao R., Wang Y., Takabe K., Nagahashi M., Pandak W. M., Dent P., Spiegel S., Shi R., Xu W., Liu X., Bohdan P., Zhang L., Zhou H., and Hylemon P. B. (2012) Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 55, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bocca C., Ievolella M., Autelli R., Motta M., Mosso L., Torchio B., Bozzo F., Cannito S., Paternostro C., Colombatto S., Parola M., and Miglietta A. (2014) Expression of Cox-2 in human breast cancer cells as a critical determinant of epithelial-to-mesenchymal transition and invasiveness. Expert Opin. Ther. Targets 18, 121–135 [DOI] [PubMed] [Google Scholar]
- 22. Fukazawa E. M., Baiocchi G., Soares F. A., Kumagai L. Y., Faloppa C. C., Badiglian-Filho L., Coelho F. R., Gonçalves W. J., Costa R. L., and Góes J. C. (2014) Cox-2, EGFR, and ERBB-2 expression in cervical intraepithelial neoplasia and cervical cancer using an automated imaging system. Int. J. Gynecol. Pathol. 33, 225–234 [DOI] [PubMed] [Google Scholar]
- 23. Generali D., Buffa F. M., Deb S., Cummings M., Reid L. E., Taylor M., Andreis D., Allevi G., Ferrero G., Byrne D., Martinotti M., Bottini A., Harris A. L., Lakhani S. R., and Fox S. B. (2014) COX-2 expression is predictive for early relapse and aromatase inhibitor resistance in patients with ductal carcinoma in situ of the breast, and is a target for treatment. Br. J. Cancer 111, 46–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ishii Y., Sasaki T., Serikawa M., Minami T., Okazaki A., Yukutake M., Ishigaki T., Kosaka K., Mouri T., Yoshimi S., Shimizu A., Tsuboi T., and Chayama K. (2013) Elevated expression of cyclooxygenase-2 and microsomal prostaglandin E synthase-1 in primary sclerosing cholangitis: iotamplications for cholangiocarcinogenesis. Int. J. Oncol. 43, 1073–1079 [DOI] [PubMed] [Google Scholar]
- 25. Aishima S., Mano Y., Tanaka Y., Kubo Y., Shirabe K., Maehara Y., and Oda Y. (2013) Different roles of inducible nitric oxide synthase and cyclooxygenase-2 in carcinogenesis and metastasis of intrahepatic cholangiocarcinoma. Hum. Pathol. 44, 1031–1037 [DOI] [PubMed] [Google Scholar]
- 26. Dai J., Wang H., Shi Y., Dong Y., Zhang Y., and Wang J. (2011) Impact of bile acids on the growth of human cholangiocarcinoma via FXR. J. Hematol. Oncol. 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han C., Leng J., Demetris A. J., and Wu T. (2004) Cyclooxygenase-2 promotes human cholangiocarcinoma growth: evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 64, 1369–1376 [DOI] [PubMed] [Google Scholar]
- 28. Ghosh S., and Hayden M. S. (2012) Celebrating 25 years of NF-κB research. Immunol. Rev. 246, 5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayden M. S., and Ghosh S. (2004) Signaling to NF-κB. Genes Dev. 18, 2195–2224 [DOI] [PubMed] [Google Scholar]
- 30. Glinghammar B., Inoue H., and Rafter J. J. (2002) Deoxycholic acid causes DNA damage in colonic cells with subsequent induction of caspases, COX-2 promoter activity and the transcription factors NF-κB and AP-1. Carcinogenesis 23, 839–845 [DOI] [PubMed] [Google Scholar]
- 31. Werneburg N. W., Yoon J. H., Higuchi H., and Gores G. J. (2003) Bile acids activate EGF receptor via a TGFα-dependent mechanism in human cholangiocyte cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G31–36 [DOI] [PubMed] [Google Scholar]
- 32. Yoshikawa D., Ojima H., Iwasaki M., Hiraoka N., Kosuge T., Kasai S., Hirohashi S., and Shibata T. (2008) Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br. J. Cancer 98, 418–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roelle S., Grosse R., Aigner A., Krell H. W., Czubayko F., and Gudermann T. (2003) Matrix metalloproteinases 2 and 9 mediate epidermal growth factor receptor transactivation by gonadotropin-releasing hormone. J. Biol. Chem. 278, 47307–47318 [DOI] [PubMed] [Google Scholar]
- 34. Rao Y. P., Studer E. J., Stravitz R. T., Gupta S., Qiao L., Dent P., and Hylemon P. B. (2002) Activation of the Raf-1/MEK/ERK cascade by bile acids occurs via the epidermal growth factor receptor in primary rat hepatocytes. Hepatology 35, 307–314 [DOI] [PubMed] [Google Scholar]
- 35. Eo S. H., Cho H. S., and Kim S. J. (2014) Resveratrol regulates type II collagen and COX-2 expression via the ERK, p38 and Akt signaling pathways in rabbit articular chondrocytes. Exp. Ther. Med. 7, 640–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ma Y., Li H., Yue Z., Guo J., Xu S., Xu J., Jia Y., Yu N., Zhang B., Liu S., Liu M., Shao W., Chen S., and Liu P. (2014) Cryptotanshinone attenuates cardiac fibrosis via downregulation of COX-2, NOX-2, and NOX-4. J. Cardiovasc. Pharmacol. 64, 28–37 [DOI] [PubMed] [Google Scholar]
- 37. Kim H. G., Kim Y. R., Park J. H., Khanal T., Choi J. H., Do M. T., Jin S. W., Han E. H., Chung Y. H., and Jeong H. G. (2015) Endosulfan induces COX-2 expression via NADPH oxidase and the ROS, MAPK, and Akt pathways. Arch. Toxicol. 89, 2039–2050 [DOI] [PubMed] [Google Scholar]
- 38. Mercau M. E., Astort F., Giordanino E. F., Martinez Calejman C., Sanchez R., Caldareri L., Repetto E. M., Coso O. A., and Cymeryng C. B. (2014) Involvement of PI3K/Akt and p38 MAPK in the induction of COX-2 expression by bacterial lipopolysaccharide in murine adrenocortical cells. Mol. Cell. Endocrinol. 384, 43–51 [DOI] [PubMed] [Google Scholar]
- 39. Lozano E., Sanchez-Vicente L., Monte M. J., Herraez E., Briz O., Banales J. M., Marin J. J., and Macias R. I. (2014) Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol. Cancer Res. 12, 91–100 [DOI] [PubMed] [Google Scholar]
- 40. Xia X., Francis H., Glaser S., Alpini G., and LeSage G. (2006) Bile acid interactions with cholangiocytes. World J. Gastroenterol. 12, 3553–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mathema V. B., and Na-Bangchang K. (2015) Current insights on cholangiocarcinoma research: a brief review. Asian Pac. J. Cancer Prev. 16, 1307–1313 [DOI] [PubMed] [Google Scholar]
- 42. Ghouri Y. A., Mian I., and Blechacz B. (2015) Cancer review: cholangiocarcinoma. J. Carcinogen. 14, 1, 10.4103/1477-3163.151940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rizvi S., and Gores G. J. (2014) Molecular pathogenesis of cholangiocarcinoma. Dig. Dis. 32, 564–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nuñez Martinez O., Clemente Ricote G., and Garcia Monzón C. (2003) Role of cyclooxygenase-2 in the pathogenesis of chronic liver diseases. Med. Clin. (Barc.) 121, 743–748 [DOI] [PubMed] [Google Scholar]
- 45. Zhang L., Jiang L., Sun Q., Peng T., Lou K., Liu N., and Leng J. (2007) Prostaglandin E2 enhances mitogen-activated protein kinase/Erk pathway in human cholangiocarcinoma cells: involvement of EP1 receptor, calcium and EGF receptors signaling. Mol. Cell. Biochem. 305, 19–26 [DOI] [PubMed] [Google Scholar]
- 46. Sun B., Rong R., Jiang H., Zhang H., Wang Y., Bai X., Zhang M., Ma J., Xia S., Shu W., Zhang L., and Leng J. (2013) Prostaglandin E2 receptor EP1 phosphorylate CREB and mediates MMP2 expression in human cholangiocarcinoma cells. Mol. Cell. Biochem. 378, 195–203 [DOI] [PubMed] [Google Scholar]
- 47. Völzke A., Koch A., Meyer Zu Heringdorf D., Huwiler A., and Pfeilschifter J. (2014) Sphingosine 1-phosphate (S1P) induces COX-2 expression and PGE2 formation via S1P receptor 2 in renal mesangial cells. Biochim. Biophys. Acta 1841, 11–21 [DOI] [PubMed] [Google Scholar]
- 48. Skoura A., Michaud J., Im D. S., Thangada S., Xiong Y., Smith J. D., and Hla T. (2011) Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 31, 81–85 [DOI] [PMC free article] [PubMed] [Google Scholar]