

Abstract
Porins, a major class of outer membrane proteins in Gram-negative bacteria, primarily act as transport channels. OmpU is one of the major porins of human pathogen, Vibrio cholerae. In the present study, we show that V. cholerae OmpU has the ability to induce target cell death. Although OmpU-mediated cell death shows some characteristics of apoptosis, such as flipping of phosphatidylserine in the membrane as well as cell size shrinkage and increased cell granularity, it does not show the caspase-3 activation and DNA laddering pattern typical of apoptotic cells. Increased release of lactate dehydrogenase in OmpU-treated cells indicates that the OmpU-mediated cell death also has characteristics of necrosis. Further, we show that the mechanism of OmpU-mediated cell death involves major mitochondrial changes in the target cells. We observe that OmpU treatment leads to the disruption of mitochondrial membrane potential, resulting in the release of cytochrome c and apoptosis-inducing factor (AIF). AIF translocates to the host cell nucleus, implying that it has a crucial role in OmpU-mediated cell death. Finally, we observe that OmpU translocates to the target cell mitochondria, where it directly initiates mitochondrial changes leading to mitochondrial membrane permeability transition and AIF release. Partial blocking of AIF release by cyclosporine A in OmpU-treated cells further suggests that OmpU may be inducing the opening of the mitochondrial permeability transition pore. All of these results lead us to the conclusion that OmpU induces cell death in target cells in a programmed manner in which mitochondria play a central role.
Keywords: bacterial pathogenesis, cell death, mitochondria, Vibrio cholerae, virulence factor, OmpU, porin, programmed cell death
Introduction
Classically, two major forms of cell death have been described: apoptosis and necrosis. Apoptosis is a form of programmed cell death that is characterized by sequential activation of events in a highly regulated manner, leading to the death of cells (1–3). Necrotic cell death is supposed to be sudden or abrupt and is mainly a passive event.
Apoptosis involves mainly two pathways, the extrinsic pathway and the intrinsic pathway (3, 4). Sometimes the extrinsic pathway initiates the intrinsic pathway, or the two pathways work in isolation. Both the extrinsic and the intrinsic pathway involve the activation of a specialized class of enzymes called cysteine-dependent aspartate-specific proteases or caspases, which are considered to be indispensable for apoptotic cell death. In the case of the extrinsic pathway, death receptors are involved. Upon death receptor activation, caspase-8 becomes activated initially, and sequential events ultimately lead to the activation of caspase-3 (5). Mitochondria play a central role in the intrinsic pathway of apoptosis.
For many years, the term apoptosis has been used synonymously with programmed cell death (PCD).2 However, recent years have witnessed an increase in the number reports describing different forms of PCD that occur independent of caspases (6, 7). Depending on the type of morphological and biochemical events that are initiated during programmed cell death, different terms have been proposed: apoptosis like PCD, necrosis like PCD, necroptosis, and many others (8, 9). Diverse sets of molecules and different organelles like lysosomes, endoplasmic reticulum, and mitochondria interact in a cell, leading to its demise (6).
Involvement of mitochondria in caspase-independent PCD has been established for some cell types in response to particular stimuli (10, 11). Any disruption in the normal bioenergetic state of mitochondria induced by stress generated in cells by various external or internal factors can lead to a loss of mitochondrial membrane potential. Generally, members of the Bcl-2 family, such as Bax, translocate to mitochondria and permeabilize the outer mitochondrial membrane (12–14). Certain stimuli can affect the integrity of the inner mitochondrial membrane, and this leads to the mitochondrial membrane permeability transition (MMPT), further resulting in the release of intermembrane space molecules like cytochrome c and apoptosis-inducing factor (AIF) into the cytosol (11). Cytochrome c may bind Apaf-1 in the presence of dATP and promote caspase-9 and caspase-3 activation (15–17). AIF plays a crucial role in caspase-independent pathways. It can directly translocate to the nucleus and cause DNA fragmentation independent of caspases (18). Therefore, mitochondria seem to play a key role in both caspase-dependent and -independent pathways of PCD.
Different types of PCD play a crucial role in host-pathogen interactions. Pathogens induce cell death in order to invade host tissues or to evade host immune responses (19). Gram-negative pathogenic bacteria use such processes to damage host tissues and cause sepsis by invading deeper into them (20). The host uses such mechanisms to prevent pathogenic infections by inducing the death of infected cells.
A number of bacterial molecules have been implicated in the induction of apoptosis or other forms of PCD in the host cells. During bacterial infections, some pathogens secrete toxins that may trigger cell death (21, 22). In addition, the mode of invasion of the bacteria, such as endocytosis, and the structural elements of the bacteria that help in invading the host cell can trigger the death of target cells. In Gram-negative bacteria like Neisseria gonorrhoeae and Pseudomonas aeruginosa, porins, a class of outer membrane integral proteins, induce apoptosis of the target cells (23, 24).
Porins are trans-membrane channels that allow selective uptake of nutrients, a requisite for bacterial cell survival (25). In addition, porins may act as pathogen-associated molecular patterns that are recognized by pattern recognition receptors on host cell surfaces and induce pro-inflammatory responses. Porins of Shigella, Salmonella, Vibrio cholerae, etc. are known to induce the production of pro-inflammatory cytokines (26–29). Some porins are also reported to be anti-apoptotic, such as PorB of Neisseria meningitidis (30).
OmpU, one of the major outer membrane proteins of V. cholerae, is porin in nature (31). In addition to its role as a porin, OmpU has been speculated to play a crucial role in the pathogenesis of V. cholerae. This speculation is mainly based on the fact that expression of OmpU is under the control of the ToxR regulon, which controls major virulence factors required for the pathogenesis of V. cholerae (32–34). Moreover, OmpU has been reported to facilitate intestinal colonization of the bacterium by conferring resistance against bile and anti-microbial peptides. It probably acts as an adhesin as well, although there are contrasting reports regarding its role in adhesion (35–37). Moreover, OmpU has been shown to possess the ability to down-regulate the LPS-mediated pro-inflammatory effect (28). Therefore, its regulation and reported functions imply that OmpU may have a major role in the bacterial pathogenesis process. However, the contribution of OmpU in the induction of cell death has not been evaluated.
To date, an effective vaccine against cholera is not available. OmpU is considered as a good candidate for vaccine generation mainly because of the fact that OmpU is present in most of the clinical isolates (38). Recently, a report suggested that OmpU can be used as a biomarker to distinguish between epidemic and non-epidemic strains (39). Therefore, it is very important to characterize OmpU for its role in the induction of multiple cellular processes in the host. Based on all of the above knowledge and the speculated role of OmpU in pathogenesis and weighing the importance of cell death responses in host-pathogen interactions, in the present paper, we have studied the role of OmpU in the induction of cell death in human cells and the possible mechanism involved in the process.
Experimental Procedures
Purification of Recombinant OmpU
Recombinant OmpU was purified as described previously by Khan et al. (40).
Detection of Endotoxin Contamination in Purified Protein Preparation
The presence of endotoxin in different batches of purified protein was measured by the limulus amebocyte lysate test using the E-TOXATETM kit (Sigma-Aldrich) as per the manufacturer's protocol.
Mammalian Cell Culture
The human monocytic cell line, THP-1 (National Centre for Cell Science, Pune, India), was maintained in RPMI 1640 (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Prior to each experiment, cells were conditioned in medium containing 5% FBS for 24 h, followed by 2% FBS for 12 h. The human embryonic kidney cell line, HEK 293 (ATCC), and human colon carcinoma cell line, Caco-2 (ATCC), were maintained in DMEM (Invitrogen) containing 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Similar to THP-1, prior to each experiment, HEK 293 cells and Caco-2 cells were conditioned in medium containing reducing concentrations of serum.
Experimental Design
For the majority of the experiments, THP-1 monocytes and Caco-2 cells were plated at a density of 1 × 106 cells/ml, and HEK 293 cells were plated at a density of 0.5 × 106 cells/ml, respectively, in media containing 2% serum and treated with either recombinant OmpU or buffer (protein buffer containing 10 mm Tris-HCl, 10 mm NaCl, and 0.5% lauryldimethylamine oxide, diluted in PBS + 0.5% lauryldimethylamine oxide). Lauryldimethylamine oxide was obtained from Sigma-Aldrich. Following incubations, cells were subjected to flow cytometry or Western blotting.
Most of the flow cytometry experiments were performed using a BD FACSCalibur cell analyzer (BD Biosciences) unless otherwise indicated. Acquisition for all of the experiments was done in CellQuest Pro, and analysis was done using FlowJo software (Tree Star). For Western blot analysis, blots were visualized in an ImageQuant LAS 4010 imager (GE Healthcare).
Detection of Early Morphological Changes
THP-1 monocytes were treated with OmpU (10 μg/ml) or buffer and incubated for 24 h. Cells were harvested and washed twice with phosphate-buffered saline (PBS). Finally, cells were resuspended in 500 μl of PBS and analyzed by flow cytometry under the parameters of forward scatter and side scatter.
Detection of Cell Viability Using MTT Assay
THP-1 monocytes, Caco-2 cells, and HEK 293 cells were plated in a 96-well plate format and were treated with different concentrations of OmpU (1.5, 3, 5, 7, and 10 μg/ml) or buffer and incubated for 24 h. Following incubations, cells were subjected to an MTT assay for cell viability. The MTT assay is based on the principle that it is a tetrazolium salt which is water-soluble and gives yellow color in solution. Active dehydrogenases of living cells can convert the tetrazolium ring into insoluble formazan crystals, which are purple in color. Dead cells are incapable of carrying out the conversion. Formazan crystals can be dissolved using acidified isopropyl alcohol. The color so obtained is measured spectrophotometrically. The concentration of the dye is proportional to the percentage of viable cells. The MTT assay was done using a kit (Sigma-Aldrich) as per the manufacturer's instructions. Briefly, solution provided in the kit was added to the cells at 10% (v/v) and incubated for 3 h. After incubation, formazan crystals were dissolved by adding an equal volume of acidified propanol (prepared by adding 0.1 n HCl in isopropyl alcohol). After the crystals were completely dissolved, absorbance was measured at 570 nm. Absorbance values obtained were employed to calculate the percentage of cell viability using the following formula: (treated cells − medium only) × 100/(untreated cells − medium only). Untreated cells in media alone were considered to be 100% viable.
Detection of Cell Cytotoxicity Using Lactate Dehydrogenase (LDH) Release Assay
THP-1 monocytes or Caco-2 cells were plated and treated with different concentrations of OmpU (5, 7, and 10 μg/ml) or buffer for 24 h. After 24 h, cells were subjected to an LDH release assay. It is based on the principle that upon permeabilization of plasma membrane, LDH, which is a cytosolic enzyme, is released in the culture supernatants. Upon the addition of substrates like tetrazolium salts, LDH present in the culture supernatant converts the salt into a colored product. Intensity of the color produced is proportional to the number of lysed cells and can be measured spectrophotometrically. The LDH release assay was performed using the CytoTox 96® non-radioactive cytotoxicity assay kit (Promega Corp., Madison, WI) as per the manufacturer's instructions. Briefly, cells were pelleted, and supernatant for each treated sample was collected separately. Substrate provided in the kit was added to the supernatant of different samples. Supernatant from cells treated with lysis solution provided in the kit served as a positive control. Supernatants were incubated with substrate for 10 min. Following incubation, stop solution was added, and absorbance was measured at 490 nm.
Detection of DNA Laddering Pattern by Agarose Gel Electrophoresis
THP-1 monocytes were plated and treated with 10 μg/ml OmpU for different time periods (24 and 36 h) or buffer for 36 h. Cells treated with staurosporine (1 μm) for 4 h were used as the positive control. Following respective incubations, cells were harvested and washed twice with 1× PBS. Cell pellets were resuspended in 50 μl of lysis buffer (1% Nonidet P-40 in 20 mm EDTA, 50 mm Tris-HCl, pH 7.5, 10 μl/106 cells, minimum) and centrifuged for 5 min at 1600 × g. Supernatant was collected, and extraction was repeated once. Then supernatants were combined, and SDS was added such that the final concentration of SDS was 1% (w/v). Then supernatant was treated with RNase A (final concentration 5 mg/ml) at 56 °C for 2 h, followed by treatment with proteinase K (final concentration 1 mg/ml) at 37 °C for 2 h. Following incubations, a half-volume of ammonium acetate was added, and DNA was precipitated with 2.5 volumes of absolute ethanol. After precipitation, pellets were dissolved in DNA loading buffer and separated on 1.5% agarose gel.
Detection of Phosphatidylserine Exposure by Annexin V-FITC Staining
THP-1 monocytes were treated with different doses of OmpU (1.5, 3, 5, 7, or 10 μg/ml) or buffer and incubated for 24 h. Following treatment, cells were harvested and stained using an annexin V-FITC and propidium iodide (PI) staining kit (BD Biosciences) according to the manufacturer's protocol. Following staining, cells were analyzed by flow cytometry. A quadrant gate was applied (according to unstained and single-stained controls) to all of the dot plots corresponding to the tests, and the percentages of cells in different quadrants were calculated using quadrant statistics in FlowJo.
For inhibitor study, cells were incubated with 20 μm total caspase inhibitor (Z-VAD-fmk; Sigma-Aldrich) for 1 h, followed by treatment with OmpU (10 μg/ml) or buffer for 24 h.
Analysis of DNA Fragmentation by TUNEL Assay
THP-1 cells were treated with OmpU (10 μg/ml) or buffer and incubated for different time periods (12, 24, or 36 h). HEK 293 cells were incubated with OmpU (10 μg/ml) or buffer for 24 h. In another set, cells were preincubated with Z-VAD-fmk (20 μm) for 1 h, followed by treatment with OmpU (10 μg/ml) or buffer for 24 h. Cells were harvested and fixed using 1% paraformaldehyde following the respective incubations. Fixed cells were washed with ice-cold PBS, resuspended in 70% ethanol, and incubated at −20 °C for 12–18 h. Finally, cells were stained using the APO-BRDU kit (Sigma-Aldrich) according to the manufacturer's protocol and subjected to flow cytometry.
Detection of Caspase-3 Activation
THP-1 monocytes were treated with OmpU (10 μg/ml) or buffer and incubated for 12, 24, and 36 h. As a positive control, cells were treated with 1 μm staurosporine (Sigma-Aldrich) and incubated for 4 h. Following respective incubations, cells were harvested and stained with FITC-conjugated rabbit anti-human active caspase-3 antibody using a staining kit (BD Biosciences) as per the manufacturer's protocol and analyzed by flow cytometry.
Analysis of Change in Mitochondrial Membrane Potential
THP-1 monocytes were treated with OmpU (10 μg/ml) or buffer and incubated for different time periods (4, 8, and 24 h). After the respective incubations, cells were harvested and stained with JC-1 dye (Sigma-Aldrich) according to the manufacturer's protocol. Changes in mitochondrial membrane potential were detected by flow cytometry.
Similarly, HEK 293 cells were incubated with OmpU (10 μg/ml) or buffer for 24 h. Following incubation, cells were stained with JC-1 dye and analyzed for changes in mitochondrial membrane potential.
For microscopic analysis, mitochondrial membrane potential-sensitive dye, MitoTracker Red CMXros (Invitrogen), was used. HEK 293 cells were seeded at a density of 0.1 × 106 cells/ml on coverslips and incubated overnight. Cells were treated with 10 μg/ml OmpU or buffer and incubated for 24 h. Cells treated with staurosporine (1 μm) for 4 h were taken as positive control. Following the respective incubations, cells were stained with a 30 nm solution of MitoTracker Red CMXros and incubated for 30 min at 37 °C. After incubation, cells were washed with warm medium (37 °C) four times. Following washing, cells were fixed with 4% paraformaldehyde and washed 3–4 times with PBS. Stained coverslips were mounted and observed under a confocal microscope. Imaging was done using a Zeiss LSM 780 (Carl Zeiss) confocal laser scanning microscope with a ×63 oil immersion objective having a 1.4 numerical aperture. Identical parameters were used for all of the samples. ImageJ (National Institutes of Health, Bethesda, MD) software was used to analyze all of the images. A minimum of 10 images for each sample were quantified, and equal threshold values were applied for all of the samples. Mitochondrial fluorescence for OmpU and staurosporine-treated cells was normalized with respect to buffer-treated cells.
Detection of Cytochrome c Release by Flow Cytometry
Release of cytochrome c was detected using the protocol of Waterhouse and Trapani (41) with some modifications. Briefly, THP-1 monocytes were treated with OmpU (10 μg/ml) or buffer and incubated for 24 h. After incubation, cells were harvested and washed with PBS. Further, cells were incubated in the permeabilization buffer (1% FBS, 0.1% saponin, and 0.1% sodium azide in PBS) to selectively permeabilize the cellular membrane. Permeabilized cells were immediately fixed with 2% paraformaldehyde by incubating for 30 min at room temperature. Fixed cells were washed twice with ice-cold PBS and incubated in blocking buffer (3% BSA + 0.5% saponin in PBS) for 30 min at room temperature. After blocking, rabbit anti-human cytochrome c antibody (Sigma-Aldrich) was added at a dilution of 1:50 in the blocking buffer, and cells were incubated overnight at 4 °C. Further, cells were washed twice with PBS and incubated with FITC-conjugated anti-rabbit IgG (Sigma-Aldrich) antibody at a dilution of 1:100 in blocking buffer for 1 h at room temperature. Stained cells were washed twice with PBS (ice-cold), resuspended in 500 μl of blocking buffer, and analyzed by flow cytometry.
Determination of ATP
THP-1 monocytes were plated as described previously and treated with OmpU (10 μg/ml) or buffer for 24 h. Following incubation, cells were harvested and subjected to an ATP determination assay using the ATP bioluminescence assay kit HS II (Roche Diagnostics). This assay is based on the measurement of bioluminescence produced by the enzyme luciferase, which uses ATP from lysed cells to convert the substrate into bioluminescent product. This luminescence is then detected using a luminometer and is proportional to the amount of ATP present in the cells. The assay was performed as per the manufacturer's instructions, and data were presented as -fold change in the amount of ATP in OmpU-treated cells with respect to buffer.
Preparation of Mitochondrial Fraction
Enriched mitochondrial fraction from cultured cells was prepared using a mitochondria isolation kit (Sigma-Aldrich) according to the manufacturer's protocol. Briefly, THP-1 cells were treated with OmpU (10 μg/ml) and incubated for different time periods (0, 12, and 24 h). HEK 293 cells and Caco-2 cells were treated in a similar manner as THP-1 monocytes. After the respective incubations, cells were harvested and washed twice with ice-cold PBS. The mitochondrial fraction was prepared from cells using the detergent lysis method. Lysed cells were subjected to low speed centrifugation (600 × g) to eliminate debris, followed by high speed centrifugation (11,000 × g) to pellet down the mitochondria. The pellet so obtained was enriched in mitochondrial fraction, and the cytoplasmic fraction was present in the supernatant.
Preparation of Nuclear Fraction
THP-1 monocytes, HEK 293 cells, and Caco-2 cells were treated with OmpU (10 μg/ml) or buffer and incubated for 24 h. Cells were harvested and washed with ice-cold PBS. Washed cells were resuspended in 500 μl of buffer A (10 mm HEPES, 1.5 mm MgCl2, 10 mm KCl, 1.5 mm DTT, 0.05% Nonidet P-40) and incubated on ice for 5 min with intermittent vortexing and further subjected to centrifugation at 650 × g for 5 min. Pellet was resuspended in buffer B (5 mm HEPES, 1.5 mm MgCl2, 0.2 mm EDTA, 0.5 mm DTT, 26% glycerol) containing 300 mm NaCl and homogenized on ice, followed by centrifugation at 24,000 × g for 20 min at 4 °C. Supernatant obtained after centrifugation contained the nuclear fraction.
Detection of AIF Translocation from Mitochondria to Nucleus and Cytochrome c Release from mitochondria to Cytoplasm
Mitochondrial, nuclear, and cytoplasmic fractions were estimated for their protein concentrations using a Bradford (Sigma-Aldrich) assay. Equal amounts of protein were subjected to SDS-PAGE. Following separation, proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Merck Millipore, Darmstadt, Germany). Transferred membranes were blocked for 1 h at room temperature with 5% BSA in TBST (20 mm Tris, pH 7.6, 137 mm NaCl, 0.1% Tween 20). After blocking, membranes were incubated with rabbit anti-human AIF antibody (Sigma-Aldrich) or mouse anti-human cytochrome c (Sigma-Aldrich) diluted in TBST (1:1000) for 4 h at room temperature. Membranes were washed with TBST and incubated with anti-rabbit IgG or anti-mouse IgG coupled with horseradish peroxidase (1:5000; Sigma-Aldrich) for 1 h. TIM23, TOM22, or TOM20 was used as the loading control for mitochondrial samples and detected with mouse anti-human TIM23 or rabbit anti-human TOM22 or TOM20 antibody (Sigma-Aldrich). Lamin B1 detected by rabbit anti-human lamin B1 antibody (Santa Cruz Biotechnology, Inc.) was used as the loading control for nuclear lysates, and GAPDH detected by rabbit anti-human GAPDH antibody was used as the loading control for the cytoplasmic fraction.
Detection of OmpU in Mitochondria
THP-1 monocytes were treated with OmpU (10 μg/ml) and incubated for different time periods (15, 30, 60, 120, and 240 min). HEK 293 cells were treated in a similar manner and incubated for different time periods up to 6 h. Caco-2 cells were also treated with the same dose of OmpU and incubated for 2 h. After the respective incubations, cells were harvested, and mitochondrial lysates of each sample were prepared using the same method as mentioned previously. Centrifugation speeds were modified to 1000 × g (low speed centrifugation) and 3000 × g (high speed centrifugation) in order to obtain more purified mitochondrial fraction. Prepared lysates were analyzed for translocation of OmpU by Western blotting in a similar manner as mentioned above using primary antibody specific to V. cholerae OmpU (rabbit anti-OmpU) at a dilution of 1:1000, followed by secondary antibody, which was anti-rabbit IgG coupled with horseradish peroxidase (1:5000 dilution). TIM23 or TOM22 (mitochondrial markers; Sigma-Aldrich) was used as the loading control. Rabbit anti-human LAMP 1 (Abcam) and rabbit anti-human GAPDH (Santa Cruz Biotechnology) antibodies were used to check for the presence of lysosomes and cytoplasm, respectively, in the mitochondrial lysates. Mouse anti-human transferrin receptor (Invitrogen) and mouse anti-α-1 sodium potassium ATPase (Abcam) antibodies were used as plasma membrane markers in order to detect plasma membrane contamination in the mitochondrial preparation.
In order to detect the mitochondrial translocation of OmpU by flow cytometry, THP-1 monocytes were treated with OmpU (10 μg/ml) for 60 and 120 min or with buffer for 120 min. Following incubations, cells were harvested, and mitochondria were isolated as described previously. Isolated mitochondria were resuspended in 1× storage buffer provided in the mitochondria isolation kit (Sigma-Aldrich) and incubated with rabbit anti-OmpU at a dilution of 1:50 for 30 min at room temperature. Mitochondria were washed once with storage buffer by centrifugation at 17,500 × g for 5 min and incubated with FITC-tagged anti-rabbit IgG at a dilution of 1:100 at room temperature. Mitochondria were washed once with storage buffer, resuspended in 300 μl of storage buffer, and analyzed by flow cytometry using a BD AccuriTM C6 cytometer (BD Biosciences).
Detection of OmpU in Different Organelles by Cell Fractionation
THP-1 monocytes were plated and incubated with OmpU for 90 min. Following incubation, cells were harvested and subjected to a cell fractionation protocol for the preparation of mitochondrial, nuclear, and cytoplasmic + light membrane fractions. Mitochondrial fraction was prepared as described previously. For the preparation of nuclear fraction, cells were ruptured by homogenization in buffer A (1 m HEPES, 1 m MgCl2, 2.5 m KCl, 1 m DTT), following which they were centrifuged at 228 × g for 5 min at 4 °C to pellet down the nuclei along with other fragments. The supernatant was stored as the cytoplasmic + light membrane fraction from which mitochondria were removed by centrifuging at 3000 × g for 10 min. Nuclear pellet was further resuspended in 3 ml of buffer 1 (0.25 m sucrose, 10 mm MgCl2) and layered over buffer 2 (0.88 m sucrose, 0.5 mm MgCl2), followed by centrifugation at 2800 × g for 10 min at 4 °C. Pellet so obtained was resuspended in 1× radioimmune precipitation assay buffer and sonicated in ice. Lysate was centrifuged at 2800 × g for 10 min at 4 °C, and the supernatant was stored as nuclear fraction.
Detection of OmpU in Plasma Membrane
HEK 293 cells were plated and incubated with OmpU (10 μg/ml) for 15, 30, 60, and 120 min. Further, to obtain plasma membrane-enriched light membrane fraction, the supernatant containing cytoplasm + light membranes (described above) was ultracentrifuged at 100,000 × g for 1 h. Pellet so obtained was washed once with fractionation buffer, homogenized again, and finally collected as light membrane fraction. The final pellet was resuspended in lysis buffer. Equal amounts of all of the fractions were then analyzed for the presence of OmpU along with the specific markers, as described previously.
Detection of OmpU Translocation to Mitochondria from the Bacterial Cell
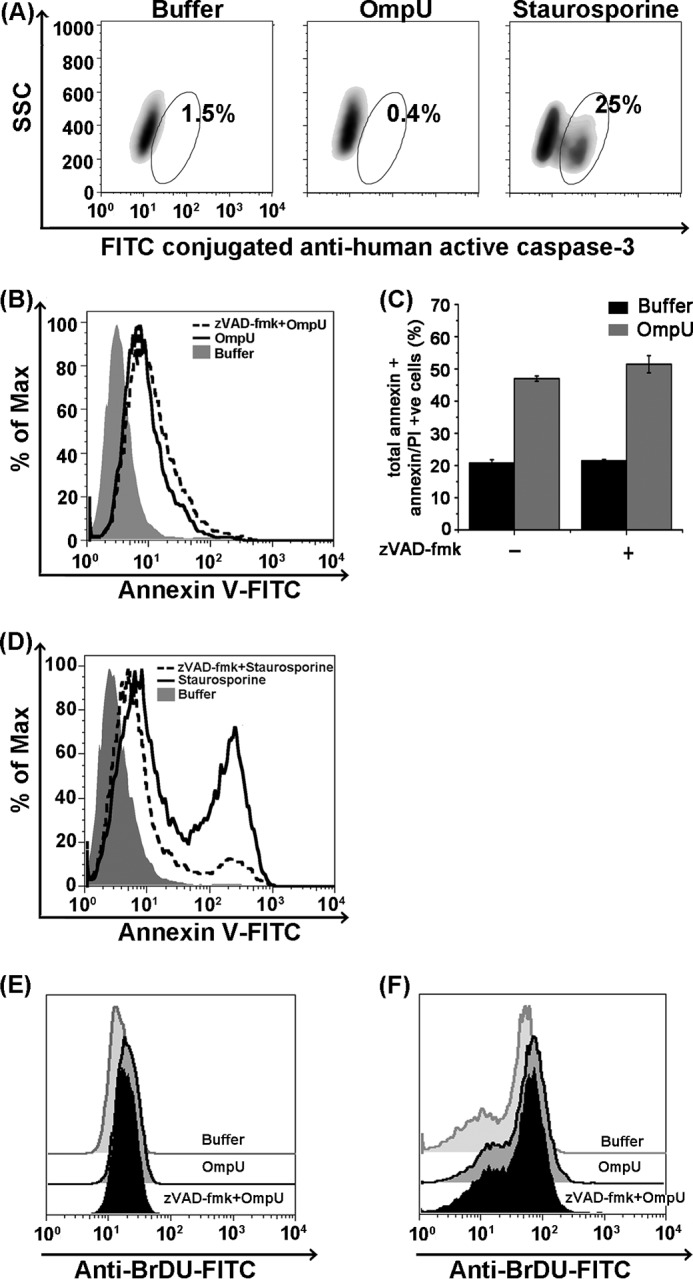
V. cholerae El Tor O1 strain (obtained from the Microbial Type Culture Collection and Gene Bank of the Institute of Microbial Technology (Chandigarh, India); MTCC code 3905) was cultured in brain heart infusion broth until A600 = 3. An aliquot of this culture was taken separately and subjected to incubation at 60 °C for 1 h for heat inactivation of the bacteria. Inactivation was confirmed by spreading the culture on a Luria agar plate and incubating at 37 °C for 16 h. Both live and heat-inactivated cultures were pelleted and washed three times with ice-cold PBS. THP-1 monocytes were treated with the bacteria (resuspended in sterile ice-cold PBS) at a multiplicity of infection of 10 and incubated at 37 °C for 15 min, 30 min, 1 h, and 2 h. Medium containing only bacterial cells, and THP-1 monocytes treated with PBS, were included as controls. All of the test samples and the controls were processed similarly. Following the respective incubations, THP-1 cells were harvested and washed three times with ice-cold PBS by centrifuging at 200 × g for 5 min. Mitochondria were isolated from all samples (as mentioned above). Streaking on agar plates from isolated mitochondria was done to detect the presence of any bacterial contaminants in the final lysates. Mitochondrial lysates were analyzed for the presence of OmpU by means of Western blotting as described previously. From the mitochondrial lysates obtained from 2-h treatments, another blot was performed for the detection of RNA polymerase β as the bacterial cytoplasmic marker and lipid A, a component of LPS, as the bacterial outer membrane marker in all of the samples. RNA polymerase β was detected using mouse anti-RNA polymerase β antibody (Abcam), whereas lipid A was detected using anti-lipid A antibody (Abcam). As a positive control for these markers, equal aliquots of live and heat-inactivated V. cholerae cultures (A600 = 3) were taken and pelleted. Pellets were dissolved in sample buffer (50 μl each) and heated at 100 °C for 15–20 min to completely lyse the bacterial cells. Following this, equal volumes of sample buffer containing lysed cells were analyzed in the same blot along with mitochondrial lysates for the respective bacterial markers.
Analysis of Direct Effect of OmpU on Isolated Mitochondria
Mitochondria were isolated from THP-1 monocytes using a mitochondria isolation kit (Sigma-Aldrich) as per the manufacturer's protocol. Isolated mitochondria were resuspended in storage buffer provided in the kit. Equal aliquots of isolated mitochondria were incubated with OmpU (5 μg/ml) or buffer for 30 min at 37 °C. Following incubation, mitochondria were stained with JC-1 dye and analyzed for loss of aggregates in the FL-2 channel by flow cytometry.
For detection of AIF and cytochrome c release, equal aliquots of isolated mitochondria were treated with OmpU (5 μg/ml) and incubated for different time periods (30 min, 1 h, and 2 h) or treated with buffer and incubated for 2 h. After the treatment, mitochondria were centrifuged at 17,500 × g for 5 min. Mitochondrial pellets were solubilized in sample buffer, and supernatant was collected separately for each sample. Supernatants and mitochondrial pellets were separated by SDS-PAGE and analyzed by Western blotting to visualize the release of AIF and cytochrome c. TIM23 was used as the mitochondrial loading control. For the inhibition study, freshly isolated mitochondria were preincubated with 1 μm cyclosporine A for 30 min, followed by treatment with OmpU for 90 min.
Statistical Analysis
Data are represented as the mean ± S.E. Statistical analysis was done using Student's two-sided t test.
Results
OmpU Induces Death of Target Cells
Different bacterial antigens can induce a multitude of host cellular responses, and cell death is one of them. To determine the cell viability, MTT is a very well established assay. Therefore, to probe whether any deterioration of cell health occurs in response to OmpU, THP-1 monocytes and HEK 293 cells were treated with different concentrations of OmpU or buffer, and following incubations, cells were analyzed for their viability using an MTT assay. The results showed that OmpU-treated cells undergo increasing loss of cell viability with increasing doses (Fig. 1, A and B). In response to 10 μg/ml protein treatment, around 18–28% and 16–26% loss in cell viability was observed after 24 h in THP-1 and HEK 293 cells with respect to buffer (Fig. 1, A and B). These observations led to the conclusion that OmpU affects target cell health and induces a loss in total cell viability.
FIGURE 1.

OmpU induces apoptosis-like features in THP-1 monocytes and HEK 293 cells. A and B, bar graphs showing OmpU-induced cell death measured as a loss of cell viability by an MTT assay in target cells. THP-1 monocytes (A) and HEK 293 (B) cells were exposed to different doses of OmpU for 24 h and subjected to an MTT assay to assess their cell viability. Bar graphs represent mean ± S.E. (error bars) of the percentage of cell viability calculated using values from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus buffer control for all of the doses. C, dot plots showing increase in total annexin V single positive + annexin V/PI double-positive population with increasing doses of OmpU treatment. THP-1 cells were treated with different doses of OmpU and incubated for 24 h. Cells were harvested and stained with annexin V-FITC and PI (dot plots are representative of three or more independent experiments). D, bar graphs showing an increase in the percentage of total annexin V single-positive + annexin V/PI double-positive cells with increasing doses of OmpU. Shown are the mean ± S.E. percentages of total annexin V single-positive + annexin V/PI double-positive cells for three or more independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus buffer control for all of the doses. E, histogram plot showing that LPS does not contribute to OmpU-mediated PCD. THP-1 monocytes were pretreated with or without polymyxin B, followed by treatment with 10 μg/ml OmpU or buffer for 24 h. Treated cells were stained with annexin V-FITC and PI and analyzed by flow cytometry. A histogram overlay shows a comparison of annexin V-positive plus annexin V/PI double-positive population between polymyxin B + OmpU- and OmpU-treated cells. F and G, dot plots showing that OmpU induced cell shrinking and increased cell granularity in OmpU- and buffer-treated THP-1 monocytes as analyzed by flow cytometry under the parameters of forward scatter (FSC) and side scatter (SSC).
V. cholerae OmpU Induces Morphological and Biochemical Changes That Suggest Programmed Cell Death
After we observed that OmpU induces cell death, we probed whether OmpU induced features that are characteristic of apoptosis. Apoptosis involves flipping of phosphatidylserine (PS) from the inner to the outer leaflet of the plasma membrane. To investigate whether V. cholerae OmpU induces PS flipping in target cells, THP-1 monocytes were treated with different doses of OmpU and incubated for 24 h. Cells were harvested and stained with annexin V-FITC and PI. Cells with increased PS exposure were detected as an annexin V-FITC-positive population, and late apoptotic/necrotic cells were detected as an annexin V-FITC and PI double-positive population. We observed that the total annexin V single-positive (Fig. 1C, bottom right quadrant) plus annexin V/PI double-positive population (top right quadrant) increased in a dose-dependent manner from 1.5 to 10 μg/ml, but a significant increase was observed at a concentration of 10 μg/ml only (38 ± 10.4% (S.D.)) compared with the buffer treatment (16 ± 4.9% (S.D.)). The result so obtained suggested that OmpU not only induces cell death but also induces certain initial changes like membrane blebbing that might lead to the detection of PS flipping in OmpU-treated cells (Fig. 1, C and D).
Endotoxin levels in different batches of purified protein were non-detectable (<0.06 endotoxin units by the limulus amebocyte lysate test), and in a separate experiment, cells were incubated with or without polymyxin B (5 μg/ml) for 30 min, followed by incubation with OmpU (10 μg/ml) for 24 h. No change was observed in the total annexin V-positive or annexin V/PI double-positive population in polymyxin B + OmpU-treated cells with respect to only OmpU-treated cells, suggesting that LPS plays no role in OmpU-mediated cell death (Fig. 1E).
In flow cytometric analysis, forward scatter gives the measure of cell size, and side scatter gives the measure of cell granularity. We observed that cells treated with OmpU showed a substantial decrease in forward scatter and corresponding increase in side scatter, implying that OmpU induces increased cell granularity along with shrinking of target cells (Fig. 1, F and G). However, the cells undergoing morphological changes appear higher than the cells undergoing biochemical changes. Therefore, all of the cells undergoing morphological changes may not be undergoing cell death at the indicated time point. All of these morphological and biochemical events induced by OmpU suggested that it could induce programmed cell death similar to apoptosis.
OmpU Induces DNA Fragmentation but Not DNA Laddering Typical of Apoptotic Cells
In classical apoptosis, the last step in the cascade is removal of the inhibitor from the CAD or caspase activated DNases. Nuclear DNA is cleaved by CAD, and this gives rise to several DNA fragments that, when resolved in an agarose gel, generate a distinct laddering pattern (42). Other types of cell death processes also generate DNA fragments that may be larger in size. These fragments may not properly resolve in the agarose gel and thus do not produce DNA ladder, as in the case of AIF-mediated cell death (8, 18).
To probe whether V. cholerae OmpU induces DNA fragmentation, THP-1 monocytes were treated with OmpU and incubated for different time periods. A TUNEL assay was performed, and a time-dependent increase in the percentage of cells with increased DNA fragmentation was observed upon OmpU treatment with respect to buffer control. Moreover, a significant increase was observed at 36 h as compared with the cells treated for 12 h (Fig. 2, A and C). To probe whether OmpU has the ability to induce DNA fragmentation in HEK 293 cells, cells were subjected to OmpU (10 μg/ml) treatment for 24 h. DNA fragmentation was observed in OmpU-treated cells with respect to buffer (Fig. 2B).
FIGURE 2.

OmpU induces DNA fragmentation atypical of apoptosis in target cells. A, histogram plots showing an increase in DNA fragmentation with increase in the incubation time after OmpU treatment as compared with the buffer treatment. THP-1 monocytes were treated with 10 μg/ml OmpU or buffer for the indicated time periods and subjected to a TUNEL assay. The increase in the FITC fluorescence of treated cells over buffer gives a measure of increased DNA fragmentation. Histogram overlays represent a comparison between OmpU- and buffer-treated cells in terms of their BrdU-FITC fluorescence (DNA fragmentation). Plots are representative of three independent experiments. B, histogram overlay showing increased DNA fragmentation by a TUNEL assay in HEK 293 cells treated with OmpU as compared with the buffer control. The histogram overlay represents a comparison between OmpU- and buffer-treated cells in terms of BrdU-FITC fluorescence and is representative of three independent experiments. C, bar graphs showing time-dependent increase in the percentage of THP-1 cells with increased DNA fragmentation upon OmpU treatment. Shown are the mean ± S.E. (error bars) percentages of BrdU-positive cells from three independent experiments (***, p < 0.001 for 24 h versus corresponding buffer control; **, p < 0.01 for 36 h versus 12 h). D, agarose gel showing an absence of DNA laddering pattern in OmpU-treated cells as compared with staurosporine, which is a known inducer of apoptosis. E, bar graph representation of OmpU-induced cytotoxicity in target cells as measured by LDH release. THP-1 monocytes were treated with the indicated doses of OmpU or buffer and subjected to an LDH release assay after 24 h. Bar graphs show the mean ± S.E. percentage of cell cytotoxicity calculated using values from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus buffer control for all of the doses.
Further, to check whether OmpU induced DNA laddering typical of apoptosis, THP-1 monocytes were treated with 10 μg/ml OmpU for different time periods. Staurosporine-treated cells were used as a positive control, and DNA from treated cells was checked by agarose gel electrophoresis. The resulting gel clearly showed a DNA laddering pattern for staurosporine, which is a known inducer of apoptosis. However, OmpU-treated cells did not show any DNA laddering pattern (Fig. 2D).
These results led to the conclusion that OmpU does induce DNA fragmentation, but it is not similar to classical apoptosis. This suggests a cell death mechanism that could be programmed but is different from apoptosis.
OmpU Induces LDH Release in Treated Cells
An LDH release assay is commonly used to determine the level of cytotoxicity, but according to some studies, it determines cytotoxicity of necrotic cells in the total cell population (43). Because the DNA fragmentation pattern in the case of OmpU-treated cells pointed toward a different kind of programmed cell death mechanism, we wanted to test whether OmpU-treated cells released LDH.
Supernatants from THP-1 cells treated with different doses of OmpU or buffer for 24 h were collected and subjected to an LDH release assay. Results so obtained showed a significant release of LDH, indicating lysis in OmpU-treated cells (average 33% at a concentration of 10 μg/ml) with respect to buffer (Fig. 2E). The above results led to the conclusion that OmpU-mediated PCD could have features of necrosis.
OmpU-induced Cell Death Does Not Involve Activation of Apoptosis-related Caspases
In the case of classical apoptosis, caspase-3 is the ultimate caspase activated by both extrinsic and intrinsic pathways. Therefore, we probed the activation of caspase-3 in OmpU-mediated cell death. Cells were treated with OmpU or buffer and incubated for different time periods, and cells treated with staurosporine were used as the positive control. We observed that OmpU did not induce any activation of caspase-3 at any of the time points. (Fig. 3A shows the plots for 24 h which are representative of all of the time points.)
FIGURE 3.
OmpU induces caspase-independent PCD in THP-1 monocytes and HEK 293 cells. A, density plots showing caspase-3 activation in staurosporine-treated cells serving as positive control but no caspase-3 activation in OmpU-treated cells. THP-1 monocytes were treated with 10 μg/ml OmpU or buffer for different time periods or with staurosporine for 4 h. Cells were harvested and stained with FITC-conjugated anti-active caspase-3 antibody. Increased FITC fluorescence (gated population) gives a measure of increased cleavage of pro-caspase-3 to the active caspase-3. Density plots for the 24 h time point serve as representative plots for all of the time points. Two independent experiments for each time point were performed. B, histogram overlay showing no inhibition in OmpU-mediated PS flipping (annexin V-positive cells) upon the use of total caspase inhibitor. THP-1 monocytes were preincubated with or without total caspase inhibitor Z-VAD-fmk followed by incubation with OmpU or buffer. Cells were harvested and stained with annexin V-FITC and PI. As mentioned under “Experimental Procedures,” quadrant gate was applied to all of the tests. Histogram overlay represents the population of cells in the bottom left quadrant plus bottom right quadrant. This is a representative plot of three independent experiments. C, bar graphs showing almost no change in percentage of total annexin V single-positive (bottom right quadrant) plus annexin V/PI double-positive (top right quadrant) cells upon OmpU treatment with/without caspase inhibitor. Quadrant gate was applied to all of the tests. The bar graph shows mean ± S.E. (error bars) for three independent experiments. D, histogram overlay showing a decrease in the percentage of annexin V positive population in Z-VAD-fmk + staurosporine-treated cells as compared with cells treated with staurosporine only. E and F, caspase inhibitor could not block DNA fragmentation induced by OmpU. THP-1 monocytes (E) or HEK 293 cells (F) were pretreated with Z-VAD-fmk, followed by treatment with OmpU or buffer, and incubated for 24 h. Cells were harvested and stained for the TUNEL assay. Offset histogram overlays represent a comparison between Z-VAD-fmk + OmpU-, OmpU only-, and buffer-treated cells in terms of BrdU-FITC fluorescence (DNA fragmentation). Each plot is representative of three independent experiments.
Further, we treated THP-1 cells with OmpU or staurosporine with or without total caspase inhibitor Z-VAD-fmk. We observed that inhibition of caspase activation did not affect OmpU-induced PS exposure (Fig. 3, B and C) and DNA fragmentation (Fig. 3E), whereas staurosporine-mediated apoptosis was mostly inhibited (Fig. 3D). Similarly, treatment with Z-VAD-fmk did not inhibit OmpU-mediated DNA fragmentation in HEK 293 cells, proving that OmpU mediates caspase-independent cell death in epithelial cells as well (Fig. 3F). Therefore, the above results indicated that OmpU-mediated programmed cell death is not apoptosis.
OmpU Induces MMPT in Target Cells
The absence of caspase activation and DNA laddering in OmpU-mediated PCD pointed toward a distinct pathway, and generally mitochondria are the central units for such processes (6, 44). In order to understand whether OmpU-mediated cell death mechanism involved MMPT, THP-1 monocytes were treated with different doses of OmpU. Treated cells were analyzed for changes in mitochondrial membrane potential (MMP), using JC-1 dye. Cells with intact MMP accumulate JC-1 dye as aggregates, but cells with disrupted mitochondrial membrane potential do not accumulate JC-1 in their mitochondrial membranes and release the dye into the cytoplasm, where the dye exists as monomers. We observed that with an increase in incubation time there was an increase in JC-1 monomers, suggesting a time-dependent increase in MMPT (Fig. 4, A and B). Similarly, in HEK 293 cells, OmpU-induced MMPT was analyzed using JC-1 dye by flow cytometry (Fig. 4C).
FIGURE 4.

OmpU induces MMPT in target cells. A, contour plots showing an increase in MMPT with increase in time of incubation after OmpU treatment. THP-1 monocytes were treated with 10 μg/ml OmpU or buffer and incubated for 4, 8, and 24 h. Cells were harvested and stained with JC-1 dye for detection of MMPT. Contour plots are representative of three independent experiments. B, bar graphs showing an increase in the percentage of JC-1 monomers in THP-1 cells with increase in time of incubation after OmpU treatment. Shown are the mean ± S.E. (error bars) percentages of cells with increased JC-1 monomers for three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus buffer control for all of the time points. C, histogram overlay showing more JC-1 monomers in OmpU-treated HEK 293 cells compared with buffer. HEK 293 cells were treated and incubated with 10 μg/ml OmpU or buffer, incubated for 24 h, and stained with JC-1 dye. The plot is representative of three independent experiments. D, microscopic analysis of loss of MMP in target HEK 293 cells. HEK 293 cells were plated and treated with 10 μg/ml OmpU or buffer for 24 h. Cells treated with staurosporine for 4 h were used as a positive control. Cells were stained using MitoTracker Red CMXros and analyzed by confocal microscopy. Images are representative of two independent experiments.
Another dye, MitoTracker Red CMXros, was used to visualize the loss of MMP in HEK 293 cells by confocal microscopy. CMXros is a potential sensitive dye that accumulates in mitochondria with intact MMP and becomes fluorescent. Therefore, accumulation of the dye and hence its fluorescence is proportional to MMP. In the event of loss of MMP, mitochondria are not able to retain the dye; therefore, relative fluorescence decreases. This loss of fluorescence is proportional to the extent of mitochondrial depolarization or MMPT.
HEK 293 cells were seeded on coverslips and treated with OmpU or buffer for 24 h. Cells treated with staurosporine for 4 h were taken as a positive control for MMPT. The resulting images clearly show a considerable loss of mitochondrial fluorescence corresponding to decreased MMP in OmpU-treated cells with respect to buffer control (Fig. 4D). The above data indicated that mitochondria could play a crucial role in OmpU-mediated PCD.
OmpU Induces Release of Cytochrome c from Host Cell Mitochondria
Cytochrome c as a member of the electron transport chain remains in the intact mitochondrial membrane, but MMPT usually leads to cytochrome c release from mitochondria. Once released, cytochrome c generally induces caspase activation, although some reports suggest that cytochrome c is released in the case of programmed necrosis as well (45). Therefore, cytochrome c release can be considered as a marker of MMPT and not necessarily a prerequisite of caspase-mediated intrinsic apoptotic cell death. Although there is no caspase activation in OmpU-mediated cell death, OmpU-mediated MMPT prompted us to check whether cytochrome c is released into the cytosol. THP-1 cells were treated with OmpU or buffer and incubated for 24 h. It was observed that a significant proportion of the total population of OmpU-treated cells (p < 0.001 for OmpU-treated versus buffer control) had lost cytochrome c from their mitochondria with respect to buffer-treated cells in a flow cytometry-based assay (Fig. 5A). The above results were confirmed by means of Western blotting in which a similar pattern of cytochrome c release from mitochondria was observed along with a simultaneous increase in the cytoplasm (Fig. 5B). Staurosporine was included as the positive control. These observations led to the conclusion that OmpU triggered major changes in mitochondria by disrupting mitochondrial membrane potential and inducing cytochrome c release into the cytoplasm.
FIGURE 5.

OmpU induces cytochrome c and AIF release from mitochondria along with translocation of AIF to nucleus. A, dot plots showing considerable loss of cytochrome c in OmpU-treated cells compared with buffer. THP-1 monocytes were treated with 10 μg/ml OmpU or buffer and incubated for 24 h. Treated cells were harvested and stained with anti-human cytochrome c followed by FITC-conjugated anti-rabbit IgG. Decreased FITC fluorescence (gated population) corresponds to the decrease in cytochrome c in the mitochondria. Dot plots are representative of three independent experiments. B, Western blot showing release of cytochrome c from mitochondria to cytoplasm. THP-1 monocytes were treated with 10 μg/ml OmpU for different incubation periods and incubated for 0, 12, and 24 h. Cells were harvested, and their mitochondrial and cytoplasmic fractions were prepared, which were then analyzed for the release of cytochrome c by means of Western blot analysis. Blots are representative of three independent experiments. C, bar graph showing a reduction in the amount of ATP in OmpU-treated cells with respect to buffer-treated cells. THP-1 cells were treated with OmpU or buffer for 24 h and subjected to an ATP determination assay. The bar graph shows the mean ± S.E. (error bars) -fold change in the amount of ATP in OmpU-treated cells with respect to buffer calculated from three independent experiments. D and E, Western blot showing release of AIF from the mitochondrial fraction and its translocation to the nuclear fraction in OmpU-treated cells compared with control. THP-1 monocytes (D) and HEK 293 cells (E) were incubated with 10 μg/ml OmpU for 0, 12, and 24 h for analysis of their mitochondrial fractions and with OmpU or buffer for 24 h for analysis of their nuclear fractions. Mitochondrial and nuclear fractions were isolated from treated cells and analyzed for the presence of AIF by Western blotting. The blots are representative of three or more independent experiments.
Because release of cytochrome c did not induce caspase activation in THP-1 monocytes, we probed the possibility that ATP depletion could hamper the association of cytochrome c with Apaf-1, thereby preventing the formation of the complex called apoptosome. This could prevent the activation of caspase cascade despite the release of cytochrome c. We observed a significant depletion (37–43% decrease with respect to buffer; Fig. 5C) in the amount of ATP in cells treated with OmpU as compared with buffer. Therefore, the loss of ATP could be a possible reason for the lack of caspase activation in OmpU-mediated PCD.
V. cholerae OmpU Induces Translocation of AIF from Host Cell Mitochondria to Its Nucleus
As a result of MMPT, AIF could be released from the mitochondrial intermembrane space to the cytosol. Upon translocation to nucleus from the cytosol, it can cause DNA fragmentation (18). In order to probe whether OmpU-mediated cell death involved AIF release from mitochondria and its translocation to the nucleus, THP-1 monocytes were treated with OmpU and incubated for 0, 12, or 24 h. We observed that release of AIF from mitochondria increased with increasing incubation periods (Fig. 5D). Because the AIF release from the mitochondria was maximal at 24 h, nuclear lysates were prepared from cells incubated with OmpU or buffer for 24 h. A considerable amount of AIF translocation was observed in the nucleus of OmpU-treated cells as compared with the buffer control (Fig. 5D). Similarly, a clear translocation of AIF from the mitochondria to the nucleus was observed in HEK 293 cells as well when treated with OmpU (Fig. 5E).
V. cholerae OmpU Is Translocated to Mitochondria of Targeted Host Cell
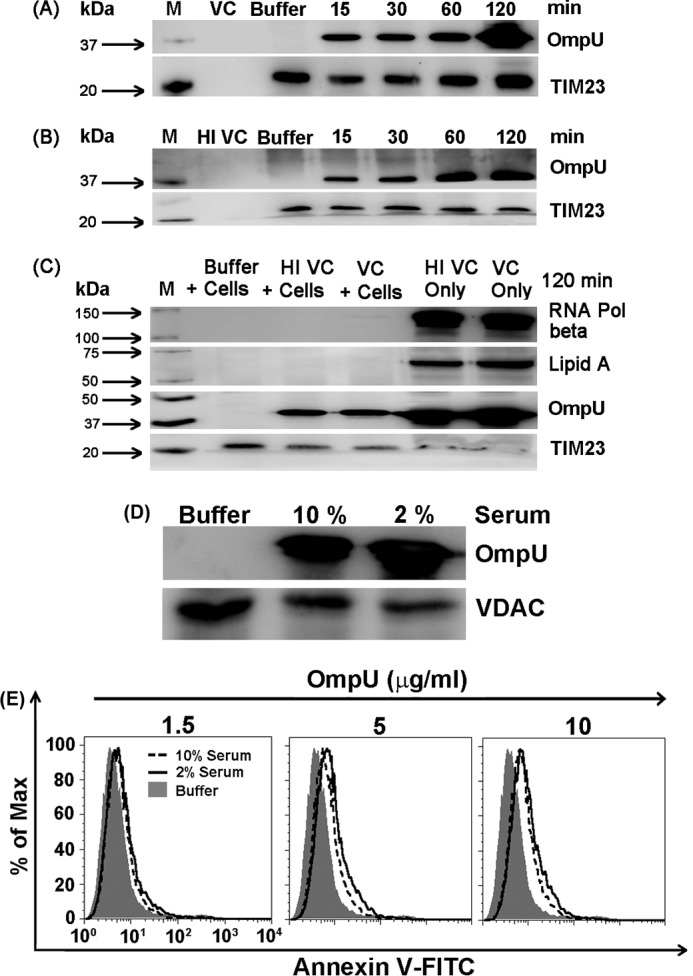
THP-1 cells and HEK 293 cells were treated with OmpU and incubated for different time periods. Mitochondrial fractions were prepared from treated cells and were analyzed for the presence of OmpU. TOM22 and TIM23 were used as the mitochondrial markers as well as the loading controls. GAPDH was used as the cytoplasmic marker, and LAMP 1 was used as the lysosomal marker. The resulting blots showed that mitochondrial preparation was almost pure with negligible contamination from cytoplasm or lysosomes. A clear translocation of OmpU to the mitochondria of treated cells was observed even at 15 min (Fig. 6A). A considerable amount of OmpU was observed in the mitochondria of THP-1 monocytes at the 1 and 2 h time points (Fig. 6, A and B) and at 2 and 4 h in HEK 293 cells (Fig. 6C).
FIGURE 6.

OmpU translocates to the mitochondria of target cells. A, Western blot result showing translocation of OmpU to the mitochondria of THP-1 monocytes. THP-1 monocytes were treated with 10 μg/ml OmpU for different incubation periods or buffer (for the maximum incubation period). After respective incubations, mitochondrial fractions were isolated and analyzed for the presence of OmpU by Western blotting using anti-OmpU antibody. TOM22 was used as a mitochondrial marker as well as a loading control. GAPDH and LAMP 1 were used as cytoplasmic and lysosomal markers. The blot is representative of three or more independent experiments. B, histogram showing mitochondrial translocation of OmpU in THP-1 monocytes. THP-1 monocytes were treated with 10 μg/ml OmpU (1 and 2 h) or buffer (2 h), and mitochondria were isolated. Isolated mitochondria were stained with anti-OmpU antibody followed by FITC-conjugated secondary antibody and analyzed by flow cytometry. The histogram overlay represents a comparison between mitochondria isolated from OmpU and buffer-treated cells in terms of FITC fluorescence that gives a measure of OmpU translocation. The overlay plot is representative of two independent experiments. C, Western blot result showing translocation of OmpU to the mitochondria of HEK 293 cells. Cells were plated and incubated with OmpU for different incubation periods (up to 6 h) or buffer (for 6 h) and analyzed for the presence of OmpU in their mitochondria as described for THP-1 monocytes. The blot is representative of three or more independent experiments. D, Western blot showing that OmpU predominantly localizes in the mitochondria of target cells in comparison with other organelles. THP-1 monocytes were treated with OmpU and incubated for 90 min. Cells were harvested, and mitochondrial, nuclear, and light membrane plus cytoplasmic fractions were prepared, which were analyzed for the presence of OmpU by means of Western blotting. Blots are representative of three independent experiments. E, Western blot showing a comparison of the kinetics of association of OmpU in plasma membrane with its association in mitochondria. HEK 293 cells were treated with 10 μg/ml OmpU for different incubation periods followed by preparation of mitochondrial fraction or plasma membrane enriched light membrane fraction. Resulting fractions were subjected to Western blotting using anti-OmpU antibody along with antibodies for plasma membrane and mitochondrial markers.
Furthermore, to find out whether OmpU localizes in the mitochondria only or other organelles as well, mitochondrial, nuclear, and light membrane + cytoplasmic fractions from OmpU-treated cells were prepared and subjected to Western blotting for the detection of OmpU in different fractions. The results showed that OmpU predominantly localizes in the mitochondria of treated cells as compared with other fractions (Fig. 6D).
We also checked the kinetics of association of OmpU in plasma membrane and compared it with its association in mitochondria. HEK 293 cells were treated with 10 μg/ml OmpU and incubated for 15 min, 30 min, 1 h, and 2 h. Purified mitochondrial fraction and light membrane fraction predominantly comprising plasma membrane were prepared from treated cells and analyzed for the presence of OmpU by means of Western blotting. The results showed that association of OmpU with plasma membrane at 15 min is much higher as compared with mitochondria. Moreover, with increasing incubation periods, the amount of OmpU in mitochondria increases, whereas in plasma membrane, it slightly declines at 30 min and becomes almost constant with the increase in incubation time until 2 h (Fig. 6E).
Therefore, we can conclude that OmpU associates with the plasma membrane and then begins to translocate to the mitochondria. Initially, its amount increases in mitochondria with increasing incubation periods, whereas in plasma membrane corresponding to the increase of OmpU in the mitochondrial fraction, there is a decrease in the amount of OmpU after 15 min.
Because purified OmpU translocated to mitochondria, we checked its translocation from V. cholerae. Live V. cholerae culture was prepared, and an aliquot of this culture was subjected to heat inactivation. Complete heat inactivation of the bacterial culture was confirmed by spreading live and inactivated cultures on Luria agar plates and incubating up to 16 h. Bacterial lawn was observed in live bacterial plates within 4 h. Heat-inactivated bacteria did not grow even after 16 h. THP-1 monocytes were infected with live and heat-inactivated cultures. Mitochondria were isolated from each sample, and mitochondrial lysates were analyzed for the presence of OmpU. In order to rule out the possibility of co-purification of the bacteria with mitochondria, lipid A of LPS as the outer membrane marker and RNA polymerase β as the bacterial cytoplasmic marker were probed in the mitochondrial lysates. OmpU translocation to the mitochondria was observed in cells cultured with live bacteria as well as heat-inactivated bacteria, whereas none of the bacterial cell markers appeared in the mitochondrial lysates (Fig. 7, A–C).
FIGURE 7.
OmpU is translocated to the mitochondria of target cells co-cultured with live and heat-inactivated V. cholerae. A, Western blots showing translocation of OmpU to the target cell mitochondria from live V. cholerae cells. THP-1 monocytes were infected with live V. cholerae (VC) at a multiplicity of infection of 10 and subjected to different incubation periods. PBS (Buffer)-treated cells and media containing only V. cholerae were used as controls. After respective incubations, mitochondrial fractions were isolated from all of the samples and analyzed for the presence of OmpU. The blots are representative of three or more independent experiments. B, Western blots showing translocation of OmpU to the host cell mitochondria from the heat-inactivated V. cholerae. THP-1 monocytes were infected with heat-inactivated V. cholerae (HI VC) at a multiplicity of infection of 10 for different incubation periods and analyzed for the presence of OmpU in a similar manner as mentioned above. The blots are representative of three independent experiments. C, control blot showing the absence of any bacterial component in purified mitochondria from co-culture of V. cholerae and mammalian cells. THP-1 monocytes were infected with live and heat-inactivated V. cholerae for 2 h. Mitochondrial lysates and bacterial cell lysates (live and heat-inactivated V. cholerae) were prepared and subjected to Western blot analysis for the detection of RNA polymerase beta (bacterial cytoplasmic marker) and lipid A, a component of LPS (bacterial outer membrane marker), along with OmpU and TIM23. D, an increase in serum concentration from 2 to 10% does not affect OmpU translocation to mitochondria. The Western blot shows mitochondrial translocation of OmpU to cells cultured in 2 and 10% serum-containing media. THP-1 monocytes were plated in 2 or 10% serum-containing medium, treated with 10 μg/ml OmpU, and incubated for 2 h. A mitochondrial fraction was isolated from all of the samples and analyzed for the presence of OmpU by Western blotting. Voltage-dependent anion channel (VDAC) was used as the mitochondrial marker and loading control. The blot is representative of two independent experiments. E, increase in serum concentration from 2 to 10% does not affect OmpU-induced PCD. A histogram overlay shows only the annexin V-positive population of OmpU-treated cells cultured in 2 and 10% serum-containing media. THP-1 monocytes cultured in 2 and 10% serum-containing media were treated with different doses of OmpU (1.5, 5, or 10 μg/ml) or buffer. Cells were harvested and stained with annexin V-FITC and PI. The histogram overlay represents a gated population comprising only annexin V-FITC single-positive cells. The plots are representative of three independent experiments.
Therefore, from our results, we clearly conclude that not only purified OmpU, but also OmpU from live or heat-inactivated V. cholerae, is translocated from the bacterial outer membrane to the mitochondria of host cells. Further, we observed that increasing the amount of serum in the culture medium had no effect on OmpU translocation to mitochondria and OmpU-induced PCD (Fig. 7, D and E).
OmpU Directly Induces Mitochondrial Changes That Could Lead to PCD in Target Cells
The observation that V. cholerae OmpU translocates to the mitochondria of treated cells made it important to check whether translocated OmpU was able to directly induce the mitochondrial changes, such as MMPT and release of AIF from the mitochondria of treated cells. Mitochondria were isolated from untreated THP-1 monocytes and were treated with OmpU or buffer. Treated mitochondria showed a considerable loss of MMP with respect to buffer, which was visualized as decreased JC-1 aggregates in isolated mitochondria (Fig. 8A). Furthermore, to analyze AIF and cytochrome c release, isolated mitochondria were incubated with OmpU for different time periods. A time-dependent release of AIF and cytochrome c from OmpU-treated mitochondria was observed, which started at 30 min and increased with time with respect to buffer (Fig. 8, B and C). At the same time, their amounts in the corresponding supernatants of OmpU-treated mitochondria increased with increasing incubation periods (Fig. 8, B and C).
FIGURE 8.

OmpU directly induces loss of mitochondrial membrane potential and AIF release from freshly isolated mitochondria. A, histogram overlay represents the increase in loss of mitochondrial membrane potential in OmpU-treated mitochondria compared with buffer treatment. Mitochondria were isolated from 3 × 107 THP-1 monocytes, and exactly equal aliquots of isolated mitochondria were incubated with 5 μg/ml OmpU or buffer for 30 min. Following incubation, mitochondria were stained with JC-1 dye. In the corresponding histogram overlay, the decrease in the red fluorescence of JC-1 aggregates gives a measure of decreased mitochondrial membrane potential in mitochondria. The plot is representative of three independent experiments. B and C, Western blots showing release of cytochrome c (B) and AIF (C) from the treated mitochondria and corresponding increase in the supernatant with an increase in incubation time after OmpU treatment. Equal aliquots of isolated mitochondria were treated with 5 μg/ml OmpU and incubated for different time periods or with buffer for 2 h. After the respective incubations, mitochondria were harvested, and supernatants were collected separately for each sample. Supernatants and mitochondria were analyzed for the presence of cytochrome c and AIF by Western blot analysis. The blots are representative of three or more independent experiments. D, Western blots showing inhibition of AIF release from isolated mitochondria by CsA. Freshly isolated mitochondria were pretreated with 1 μm CsA for 30 min, followed by treatment with 5 μg/ml OmpU for 90 min. Following incubation, samples were subjected to Western blotting as described above. Blots are representative of three independent experiments.
To check whether opening of the mitochondrial permeability transition pore is involved in OmpU-mediated MMPT, cyclosporine A (CsA) was used. CsA is a known inhibitor that prevents the opening of the mitochondrial permeability transition pore and hence MMPT. Freshly isolated mitochondria were pretreated with or without CsA, followed by treatment with OmpU (5 μg/ml) or buffer. Following treatment, mitochondria and corresponding supernatants were analyzed for the release of AIF by Western blotting. The resulting blots showed that CsA partially inhibited the release of AIF (Fig. 8D). This result further confirmed the role of mitochondrial permeability transition in the release of AIF from OmpU-treated isolated mitochondria. On the basis of all of the above results, we conclude that OmpU can directly induce changes in freshly isolated mitochondria that ultimately lead to PCD.
OmpU Induces Cell Death in Intestinal Epithelial Cells through the Same Mechanism as in Other Cell Lines
Because V. cholerae colonizes the small intestine, we probed the ability of OmpU to induce cell death in intestinal epithelial cells using the human colon carcinoma cell line Caco-2. Caco-2 cells treated with different doses of OmpU or buffer were subjected to an MTT assay and an LDH release assay, and the results showed that a significant percentage of OmpU-treated cells were induced to die even at a 5 μg/ml protein concentration (Fig. 9, A and B). Moreover, pretreatment with Z-VAD-fmk did not have any effect on OmpU-mediated cell death in Caco-2 cells, suggesting a caspase-independent mechanism as in other cell types (Fig. 9, C and D). A substantial translocation of OmpU to the mitochondria of Caco-2 cells was observed at 2 h (Fig. 9E). Finally, we checked the role of AIF and observed that OmpU induces AIF release from mitochondria of treated cells and its translocation to the nucleus (Fig. 9F). Therefore, we conclude that OmpU induces the same mechanism of caspase-independent PCD in intestinal epithelial cells as it induces in other cell lines, which involves translocation of OmpU to the mitochondria of target cells and translocation of AIF from the mitochondria to the nucleus of treated cells.
FIGURE 9.

OmpU induces caspase-independent cell death involving OmpU translocation to mitochondria in Caco-2 cells. A, bar graphs showing cell death induced by OmpU in Caco-2 cells measured as loss of cell viability by an MTT assay. Caco-2 cells were plated and treated with OmpU (5 and 10 μg/ml) or buffer for 24 h and subjected to an MTT assay. Bar graphs show the mean ± S.E. percentage of cell viability calculated using values from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus buffer control for all of the doses. B, bar graphs showing cell cytotoxicity measured by LDH release induced by OmpU in Caco-2 cells. Caco-2 cells were plated and treated as described for the MTT assay and subjected to an LDH release assay. Bar graphs show the mean ± S.E. (error bars) percentages of cell cytotoxicity calculated using values from three independent experiments. C and D, bar graphs showing that inhibition of apoptosis-related caspases does not inhibit OmpU-mediated cell death. Caco-2 cells were plated and pretreated with 20 μm Z-VAD-fmk, followed by treatment with OmpU (10 μg/ml) or buffer. Cells were subjected to either an MTT assay or an LDH release assay. Bar graphs show the mean ± S.E. percentage of cell viability (C) or percentage of cell cytotoxicity (D) calculated using values from three independent experiments. E, Western blot result showing translocation of OmpU in Caco-2 cells. Caco-2 cells were treated with 10 μg/ml OmpU for 2 h. The mitochondrial fraction was prepared and analyzed for the presence of different markers along with OmpU by Western blotting. F, Western blot result showing the release of AIF from the mitochondria of Caco-2 cells treated with 10 μg/ml OmpU for the indicated times and its translocation to the nucleus. Blots are representative of two independent experiments.
Discussion
Porins from Gram-negative bacteria are reported to be pro-apoptotic in two cases. In N. gonorrheae and P. aeruginosa, porin-mediated apoptosis contributes to the mode of pathogenesis (23, 24, 46). In the case of N. meningitides, porin protects the target cells from apoptosis (30). Therefore, it is evident that porins from different bacteria require separate evaluation regarding their effect on cell health and viability. OmpU, one of the major porins of V. cholerae, is considered to be crucial for bacterial pathogenesis and is a potential vaccine candidate. Therefore, OmpU was evaluated for its role in the induction of host cell death.
In the current study, we established that OmpU induces death of the target cells as analyzed by various assays like the MTT assay, the LDH release assay, and annexin V-FITC/PI staining (Figs. 1 (A–D), 2E, and 9 (A and B)). All of the results from different assays indicated that OmpU at 10 μg/ml induced a significant amount of cell death, although the percentage of dying cells varied slightly from assay to assay. The LDH release assay showed a higher percentage of dead cells as compared with the MTT assay or PI staining, but the difference can be attributed to the differences in assay conditions and different sensitivities of the assays (47). In addition, the LDH release assay depends only on the permeabilization of plasma membrane as opposed to PI staining, which requires nuclear membrane permeability as well. Therefore, it is possible that annexin V-positive cells could also contribute to the LDH release. It has been shown that early membrane blebbing, which can start even before PS flipping, can be associated with limited plasma membrane permeabilization. Further, it has been shown that LDH release can be blocked by inhibiting early membrane blebbing (48, 49). The appearance of a higher percentage in the LDH release assay could also be due to its higher sensitivity (47). Therefore, we assume that the percentage of non-viable cells, as indicated by the MTT assay, is close to the actual percentage of cell death in response to OmpU.
Results obtained on polymyxin B pretreatment of OmpU-treated THP-1 monocytes (Fig. 1E) and induction of cell death in HEK 293 cells (Figs. 1B and 2B) completely rule out the involvement of endotoxin and Toll-like receptors in OmpU-mediated cell death.
Moreover, we observed that OmpU also induces certain morphological and biochemical changes similar to that of apoptotic cell death (Fig. 1, F and G). In normal cell types, PS is asymmetrically distributed on the cell membrane and is predominantly present in the inner leaflet of the membrane lipid bilayer. In cells undergoing apoptosis, PS flipping takes place, and accordingly, its exposure increases in the outer leaflet of the plasma membrane (50). Moreover, cells undergoing apoptosis exhibit cell shrinking and increased cell granularity. Our data showed that OmpU induces the above mentioned changes that are commonly involved in apoptosis (Fig. 1). However, DNA fragmentation leading to DNA laddering in agarose gel is another major hallmark of apoptosis (2, 12). Although OmpU-mediated cell death showed DNA fragmentation, as evident in Fig. 2 (A–C), the fragments did not resolve in the agarose gel as a typical ladder as seen in apoptosis (Fig. 2D).
Caspases are the key enzymes involved in apoptosis (51). Among different caspases, caspase-3 is the main executioner enzyme involved in all caspase-dependent apoptotic pathways (16, 52). We observed that there was no induction of caspase-3 activation upon OmpU treatment (Fig. 3A). Moreover, there was no inhibition of OmpU-mediated PS flipping and DNA fragmentation with the use of total caspase inhibitor Z-VAD-fmk (Figs. 3 (B, D, and E) and 9 (C and D)). Results from the LDH release assay (Fig. 2E) and the absence of caspase activation imply that although the cell death mechanism induced by OmpU has certain characteristics of apoptosis as well as necrosis, it is different from both classical apoptosis and necrosis.
Further, we observed that though V. cholerae is a non-invasive bacterium, V. cholerae OmpU translocates to the mitochondria of target cells (Figs. 6, 7, and 9E). Mitochondria play a central role in different kinds of cell death processes (7, 53). Many factors, such as chemicals or pathogen-derived molecules, cause alterations in cellular stress responses, resulting in the dissipation of mitochondrial membrane potential (ΔΨm) (i.e. disruption of the proton gradient across the inner mitochondrial membrane). This leads to MMPT, which allows passage of small molecules, including apoptogenic factors, from the mitochondria to the cytosol (54, 55). We observed that OmpU is capable of inducing MMPT in both THP-1 (Fig. 4, A and B) and HEK 293 cells (Fig. 4, C and D).
Cytochrome c, a heme-containing protein complex, is a part of the electron transport chain present in the mitochondrial intermembrane space (56). It is known that cytochrome c is released into the cytoplasm following disruption of the mitochondrial membrane potential. Cytochrome c is instrumental in caspase-dependent apoptosis. However, some reports suggest that cytochrome c is released in other types of cell death processes, such as programmed necrosis, and may not always lead to caspase activation, and therefore, its release leaves the cells unaffected (45, 57, 58). Our data show that OmpU induces cytochrome c release from mitochondria (Fig. 5, A and B). However, it does not lead to caspase activation, which could probably be due to the loss of ATP in OmpU-treated cells (Fig. 5C). Cytochrome c binds to Apaf 1 in the presence of dATP which is required for apoptosome formation and is followed by caspase activation (59).
As a result of MMPT, AIF could also be released from mitochondria. AIF is present in the mitochondrial intermembrane space and is reported to have limited NADH or NADPH oxidase activity (60). AIF or apoptosis-inducing factor can induce cell death by chromatin condensation and DNA degradation upon translocation to the nucleus from mitochondria. In the absence of caspase activation, AIF plays a central role in the induction of caspase-independent PCD (8, 18, 61). OmpU-induced MMPT leads to the release of AIF from mitochondria (Figs. 5 (D and E) and 9F). Further, our study showed that the released AIF translocates to the nucleus (Figs. 5 (D and E) and 9F). Another very important observation during the course of our study was that when isolated mitochondria were treated directly with OmpU, they were induced to undergo MMPT along with AIF release (Fig. 8).
Furthermore, our results showed that CsA, which is a known inhibitor of MMPT, leads to partial inhibition of AIF release from freshly isolated mitochondria (Fig. 8E). This result confirms that OmpU directly interacts with isolated mitochondria inducing MMPT, which further results in AIF release. The above observations suggest that OmpU might itself act as a trigger inducing mitochondrial changes that lead to caspase-independent programmed cell death in target cells.
Therefore, we conclude that V. cholerae OmpU translocates to host cell mitochondria and induces MMPT, which leads to the release of cytochrome c and AIF but no caspase activation. AIF translocates to the nucleus and presumably induces large scale DNA fragmentation and cell death (Fig. 10).
FIGURE 10.
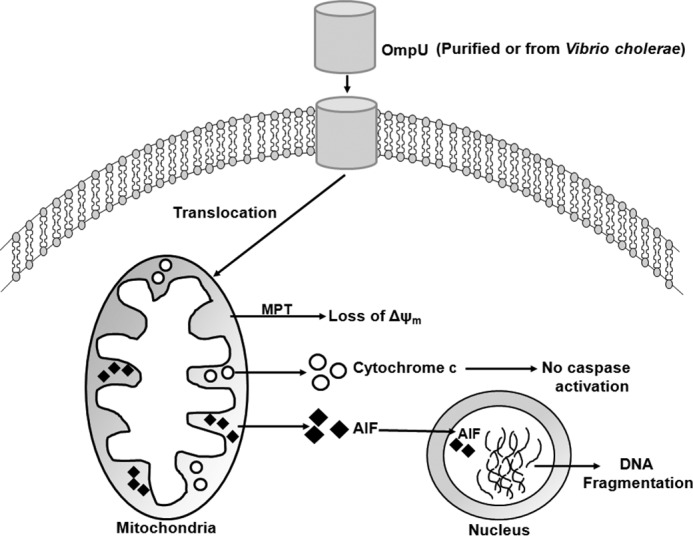
Schematic diagram depicting the pathway of OmpU-mediated apoptosis. Purified OmpU or OmpU from V. cholerae translocates across the cell membrane to the mitochondria. Translocated OmpU induces loss of mitochondrial membrane potential (ΔΨm), followed by release of cytochrome c and AIF into the cytoplasm. Released cytochrome c does not induce caspase activation. AIF translocates to the nucleus and induces DNA fragmentation and hence apoptosis-like programmed cell death.
Although PS exposure is a typical characteristic of cells undergoing apoptosis, there are reports that suggest that some level of PS exposure is also observed in cells undergoing AIF-mediated programmed cell death (8, 62, 63). As reported previously, released AIF can further act on other mitochondria, leading to an amplification of the process (18). Some reports have suggested that AIF-mediated PCD could be referred to as apoptosis-like PCD (8, 64, 65) because it includes biochemical and morphological changes similar to that of apoptosis. In the current study, our results evidently point toward this phenomenon in the case of OmpU.
PorB porin of N. gonorrhoeae is also reported to integrate into the mitochondria but induces caspase-mediated apoptosis (23, 46). Mitochondrial translocation of N. gonorrhoeae porin is blocked by serum. However, we observed that serum does not have any effect on V. cholerae OmpU translocation to the mitochondria (Fig. 7D). In the case of P. aeruginosa, no mitochondrial translocation of bacterial porin is reported (24).
Non-invasive Gram-negative bacteria mostly translocate their virulence factors inside the host cell through secretory vesicles (66) or via secretion pathways (67). In the present study, we showed that purified OmpU and OmpU from heat-inactivated bacteria that are probably incapable of producing vesicles can translocate to the host cell mitochondria. This observation suggests that from the vaccine strains, also OmpU can translocate into the host cell mitochondria and modulate cellular responses leading to death. To the best of our knowledge, this is the first report regarding porin translocation from a non-invasive bacterium to the host cell mitochondria and induction of apoptosis-like programmed cell death but not classical apoptosis.
Author Contributions
S. G. planned experiments, performed experiments, analyzed data, and wrote the paper; G. V. R. K. P. performed experiments; and A. M. planned experiments, analyzed data, and wrote the paper.
Acknowledgments
We acknowledge Dr. Samarjit Bhattacharyya (Indian Institute of Science Education and Research (IISER) Mohali) for providing anti-LAMP 1 antibody and helping with microscopy experiments, Dr. Mahak Sharma (IISER Mohali) for providing anti-transferrin antibody, and Dr. Sudip Mandal (IISER Mohali) for helping with the ATP determination assay.
This work was supported by Department of Biotechnology, India, Grant BT/PR1205/MED/29/318/2011 (to A. M.) and funding from the Indian Institute of Science Education and Research Mohali. The authors declare that they have no conflicts of interest with the contents of this article.
- PCD
- programmed cell death
- MMPT
- mitochondrial membrane permeability transition
- AIF
- apoptosis-inducing factor
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- LDH
- lactate dehydrogenase
- PI
- propidium iodide
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- PS
- phosphatidylserine
- MMP
- mitochondrial membrane potential
- CsA
- cyclosporine A
- LAMP
- lysosome-associated membrane protein.
References
- 1. Kerr J. F., Wyllie A. H., and Currie A. R. (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saraste A., and Pulkki K. (2000) Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 45, 528–537 [DOI] [PubMed] [Google Scholar]
- 3. Elmore S. (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peter M. E. (2011) Programmed cell death: apoptosis meets necrosis. Nature 471, 310–312 [DOI] [PubMed] [Google Scholar]
- 5. Chandra D., Choy G., Deng X., Bhatia B., Daniel P., and Tang D. G. (2004) Association of active caspase 8 with the mitochondrial membrane during apoptosis: potential roles in cleaving BAP31 and caspase 3 and mediating mitochondrion-endoplasmic reticulum cross talk in etoposide-induced cell death. Mol. Cell. Biol. 24, 6592–6607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bröker L. E., Kruyt F. A., and Giaccone G. (2005) Cell death independent of caspases: a review. Clin. Cancer Res. 11, 3155–3162 [DOI] [PubMed] [Google Scholar]
- 7. Kroemer G., and Martin S. J. (2005) Caspase-independent cell death. Nat. Med. 11, 725–730 [DOI] [PubMed] [Google Scholar]
- 8. Boujrad H., Gubkina O., Robert N., Krantic S., and Susin S. A. (2007) AIF-mediated programmed necrosis: a highly regulated way to die. Cell Cycle 6, 2612–2619 [DOI] [PubMed] [Google Scholar]
- 9. Pasparakis M., and Vandenabeele P. (2015) Necroptosis and its role in inflammation. Nature 517, 311–320 [DOI] [PubMed] [Google Scholar]
- 10. Volbracht C., Leist M., Kolb S. A., and Nicotera P. (2001) Apoptosis in caspase-inhibited neurons. Mol. Med. 7, 36–48 [PMC free article] [PubMed] [Google Scholar]
- 11. Weiss J. N., Korge P., Honda H. M., and Ping P. (2003) Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 93, 292–301 [DOI] [PubMed] [Google Scholar]
- 12. Ziegler U., and Groscurth P. (2004) Morphological features of cell death. News Physiol. Sci. 19, 124–128 [DOI] [PubMed] [Google Scholar]
- 13. Pawlowski J., and Kraft A. S. (2000) Bax-induced apoptotic cell death. Proc. Natl. Acad. Sci. U.S.A. 97, 529–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dewson G., and Kluck R. M. (2009) Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J. Cell Sci. 122, 2801–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Susin S. A., Lorenzo H. K., Zamzami N., Marzo I., Brenner C., Larochette N., Prévost M. C., Alzari P. M., and Kroemer G. (1999) Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med. 189, 381–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lakhani S. A., Masud A., Kuida K., Porter G. A. Jr., Booth C. J., Mehal W. Z., Inayat I., and Flavell R. A. (2006) Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311, 847–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi Y. (2001) A structural view of mitochondria-mediated apoptosis. Nat. Struct. Biol. 8, 394–401 [DOI] [PubMed] [Google Scholar]
- 18. Susin S. A., Lorenzo H. K., Zamzami N., Marzo I., Snow B. E., Brothers G. M., Mangion J., Jacotot E., Costantini P., Loeffler M., Larochette N., Goodlett D. R., Aebersold R., Siderovski D. P., Penninger J. M., and Kroemer G. (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441–446 [DOI] [PubMed] [Google Scholar]
- 19. Ashida H., Mimuro H., Ogawa M., Kobayashi T., Sanada T., Kim M., and Sasakawa C. (2011) Cell death and infection: a double-edged sword for host and pathogen survival. J. Cell Biol. 195, 931–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cohen J. (2002) The immunopathogenesis of sepsis. Nature 420, 885–891 [DOI] [PubMed] [Google Scholar]
- 21. Lin C. F., Chen C. L., Huang W. C., Cheng Y. L., Hsieh C. Y., Wang C. Y., and Hong M. Y. (2010) Different types of cell death induced by enterotoxins. Toxins 2, 2158–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kennedy C. L., Smith D. J., Lyras D., Chakravorty A., and Rood J. I. (2009) Programmed cellular necrosis mediated by the pore-forming α-toxin from Clostridium septicum. PLoS Pathog. 5, e1000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Müller A., Günther D., Düx F., Naumann M., Meyer T. F., and Rudel T. (1999) Neisserial porin (PorB) causes rapid calcium influx in target cells and induces apoptosis by the activation of cysteine proteases. EMBO J. 18, 339–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Buommino E., Morelli F., Metafora S., Rossano F., Perfetto B., Baroni A., and Tufano M. A. (1999) Porin from Pseudomonas aeruginosa induces apoptosis in an epithelial cell line derived from rat seminal vesicles. Infect. Immun. 67, 4794–4800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Achouak W., Heulin T., and Pagès J. M. (2001) Multiple facets of bacterial porins. FEMS Microbiol. Lett. 199, 1–7 [DOI] [PubMed] [Google Scholar]
- 26. Biswas T. (2000) Role of porin of Shigella dysenteriae type 1 in modulation of lipopolysaccharide mediated nitric oxide and interleukin-1 release by murine peritoneal macrophages. FEMS Immunol. Med. Microbiol. 29, 129–136 [DOI] [PubMed] [Google Scholar]
- 27. Alurkar V., and Kamat R. (1997) Immunomodulatory properties of porins of some members of the family Enterobacteriaceae. Infect. Immun. 65, 2382–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sakharwade S. C., Sharma P. K., and Mukhopadhaya A. (2013) Vibrio cholerae porin OmpU induces pro-inflammatory responses, but down-regulates LPS-mediated effects in RAW 264.7, THP-1 and human PBMCs. PLoS One 8, e76583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khan J., Sharma P. K., and Mukhopadhaya A. (2015) Vibrio cholerae porin OmpU mediates M1-polarization of macrophages/monocytes via TLR1/TLR2 activation. Immunobiology 220, 1199–1209 [DOI] [PubMed] [Google Scholar]
- 30. Massari P., Ho Y., and Wetzler L. M. (2000) Neisseria meningitidis porin PorB interacts with mitochondria and protects cells from apoptosis. Proc. Natl. Acad. Sci. U.S.A. 97, 9070–9075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chakrabarti S. R., Chaudhuri K., Sen K., and Das J. (1996) Porins of Vibrio cholerae: purification and characterization of OmpU. J. Bacteriol. 178, 524–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Skorupski K., and Taylor R. K. (1997) Control of the ToxR virulence regulon in Vibrio cholerae by environmental stimuli. Mol. Microbiol. 25, 1003–1009 [DOI] [PubMed] [Google Scholar]
- 33. Provenzano D., Lauriano C. M., and Klose K. E. (2001) Characterization of the role of the ToxR-modulated outer membrane porins OmpU and OmpT in Vibrio cholerae virulence. J. Bacteriol. 183, 3652–3662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crawford J. A., Kaper J. B., and DiRita V. J. (1998) Analysis of ToxR-dependent transcription activation of ompU, the gene encoding a major envelope protein in Vibrio cholerae. Mol. Microbiol. 29, 235–246 [DOI] [PubMed] [Google Scholar]
- 35. Mathur J., and Waldor M. K. (2004) The Vibrio cholerae ToxR-regulated porin OmpU confers resistance to antimicrobial peptides. Infect. Immun. 72, 3577–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakasone N., and Iwanaga M. (1998) Characterization of outer membrane protein OmpU of Vibrio cholerae O1. Infect. Immun. 66, 4726–4728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sperandio V., Girón J. A., Silveira W. D., and Kaper J. B. (1995) The OmpU outer membrane protein, a potential adherence factor of Vibrio cholerae. Infect. Immun. 63, 4433–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goel A. K., Jain M., Kumar P., and Jiang S. C. (2010) Molecular characterization of Vibrio cholerae outbreak strains with altered El Tor biotype from southern India. World J. Microbiol. Biotechnol. 26, 281–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Paauw A., Trip H., Niemcewicz M., Sellek R., Heng J. M., Mars-Groenendijk R. H., de Jong A. L., Majchrzykiewicz-Koehorst J. A., Olsen J. S., and Tsivtsivadze E. (2014) OmpU as a biomarker for rapid discrimination between toxigenic and epidemic Vibrio cholerae O1/O139 and non-epidemic Vibrio cholerae in a modified MALDI-TOF MS assay. BMC Microbiol. 14, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khan J., Gupta S., Chattopadhyay K., and Mukhopadhaya A. (2012) Refolding and functional assembly of the Vibrio cholerae porin OmpU recombinantly expressed in the cytoplasm of Escherichia coli. Protein Expr. Purif. 85, 204–210 [DOI] [PubMed] [Google Scholar]
- 41. Waterhouse N. J., and Trapani J. A. (2003) A new quantitative assay for cytochrome c release in apoptotic cells. Cell Death Differ. 10, 853–855 [DOI] [PubMed] [Google Scholar]
- 42. Wolf B. B., Schuler M., Echeverri F., and Green D. R. (1999) Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation factor-45/inhibitor of caspase-activated DNase inactivation. J. Biol. Chem. 274, 30651–30656 [DOI] [PubMed] [Google Scholar]
- 43. Chan F. K., Moriwaki K., and De Rosa M. J. (2013) Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 979, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Norberg E., Orrenius S., and Zhivotovsky B. (2010) Mitochondrial regulation of cell death: processing of apoptosis-inducing factor (AIF). Biochem. Biophys. Res. Commun. 396, 95–100 [DOI] [PubMed] [Google Scholar]
- 45. Li Y. Z., Li C. J., Pinto A. V., and Pardee A. B. (1999) Release of mitochondrial cytochrome c in both apoptosis and necrosis induced by β-lapachone in human carcinoma cells. Mol. Med. 5, 232–239 [PMC free article] [PubMed] [Google Scholar]
- 46. Müller A., Günther D., Brinkmann V., Hurwitz R., Meyer T. F., and Rudel T. (2000) Targeting of the pro-apoptotic VDAC-like porin (PorB) of Neisseria gonorrhoeae to mitochondria of infected cells. EMBO J. 19, 5332–5343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bopp S. K., and Lettieri T. (2008) Comparison of four different colorimetric and fluorometric cytotoxicity assays in a zebrafish liver cell line. BMC Pharmacol. 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Andrade R., Crisol L., Prado R., Boyano M. D., Arluzea J., and Aréchaga J. (2010) Plasma membrane and nuclear envelope integrity during the blebbing stage of apoptosis: a time-lapse study. Biol. Cell 102, 25–35 [DOI] [PubMed] [Google Scholar]
- 49. Wickman G. R., Julian L., Mardilovich K., Schumacher S., Munro J., Rath N., Zander S. A., Mleczak A., Sumpton D., Morrice N., Bienvenut W. V., and Olson M. F. (2013) Blebs produced by actin-myosin contraction during apoptosis release damage-associated molecular pattern proteins before secondary necrosis occurs. Cell Death Differ. 20, 1293–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mariño G., and Kroemer G. (2013) Mechanisms of apoptotic phosphatidylserine exposure. Cell Res. 23, 1247–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cohen G. M. (1997) Caspases: the executioners of apoptosis. Biochem. J. 326, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Porter A. G., and Jänicke R. U. (1999) Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 6, 99–104 [DOI] [PubMed] [Google Scholar]
- 53. Estaquier J., Vallette F., Vayssiere J. L., and Mignotte B. (2012) The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 942, 157–183 [DOI] [PubMed] [Google Scholar]
- 54. Ly J. D., Grubb D. R., and Lawen A. (2003) The mitochondrial membrane potential (ΔΨm) in apoptosis: an update. Apoptosis 8, 115–128 [DOI] [PubMed] [Google Scholar]
- 55. Vander Heiden M. G., Plas D. R., Rathmell J. C., Fox C. J., Harris M. H., and Thompson C. B. (2001) Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol. Cell. Biol. 21, 5899–5912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ow Y. P., Green D. R., Hao Z., and Mak T. W. (2008) Cytochrome c: functions beyond respiration. Nat. Rev. Mol. Cell Biol. 9, 532–542 [DOI] [PubMed] [Google Scholar]
- 57. Jemmerson R., LaPlante B., and Treeful A. (2002) Release of intact, monomeric cytochrome c from apoptotic and necrotic cells. Cell Death Differ. 9, 538–548 [DOI] [PubMed] [Google Scholar]
- 58. Von Ahsen O., Waterhouse N. J., Kuwana T., Newmeyer D. D., and Green D. R. (2000) The “harmless” release of cytochrome c. Cell Death Differ. 7, 1192–1199 [DOI] [PubMed] [Google Scholar]
- 59. Hu Y., Benedict M. A., Ding L., and Núñez G. (1999) Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J. 18, 3586–3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hangen E., Blomgren K., Bénit P., Kroemer G., and Modjtahedi N. (2010) Life with or without AIF. Trends Biochem. Sci. 35, 278–287 [DOI] [PubMed] [Google Scholar]
- 61. Susin S. A., Daugas E., Ravagnan L., Samejima K., Zamzami N., Loeffler M., Costantini P., Ferri K. F., Irinopoulou T., Prévost M. C., Brothers G., Mak T. W., Penninger J., Earnshaw W. C., and Kroemer G. (2000) Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 192, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cocco R. E., and Ucker D. S. (2001) Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol. Biol. Cell 12, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hirt U. A., Gantner F., and Leist M. (2000) Phagocytosis of nonapoptotic cells dying by caspase-independent mechanisms. J. Immunol. 164, 6520–6529 [DOI] [PubMed] [Google Scholar]
- 64. Cregan S. P., Fortin A., MacLaurin J. G., Callaghan S. M., Cecconi F., Yu S. W., Dawson T. M., Dawson V. L., Park D. S., Kroemer G., and Slack R. S. (2002) Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J. Cell Biol. 158, 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lorenzo H. K., and Susin S. A. (2004) Mitochondrial effectors in caspase-independent cell death. FEBS Lett. 557, 14–20 [DOI] [PubMed] [Google Scholar]
- 66. Ellis T. N., and Kuehn M. J. (2010) Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev. 74, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Coburn B., Sekirov I., and Finlay B. B. (2007) Type III secretion systems and disease. Clin. Microbiol. Rev. 20, 535–549 [DOI] [PMC free article] [PubMed] [Google Scholar]