

Abstract
Rv2466c is a key oxidoreductase that mediates the reductive activation of TP053, a thienopyrimidine derivative that kills replicating and non-replicating Mycobacterium tuberculosis, but whose mode of action remains enigmatic. Rv2466c is a homodimer in which each subunit displays a modular architecture comprising a canonical thioredoxin-fold with a Cys19-Pro20-Trp21-Cys22 motif, and an insertion consisting of a four α-helical bundle and a short α-helical hairpin. Strong evidence is provided for dramatic conformational changes during the Rv2466c redox cycle, which are essential for TP053 activity. Strikingly, a new crystal structure of the reduced form of Rv2466c revealed the binding of a C-terminal extension in α-helical conformation to a pocket next to the active site cysteine pair at the interface between the thioredoxin domain and the helical insertion domain. The ab initio low-resolution envelopes obtained from small angle x-ray scattering showed that the fully reduced form of Rv2466c adopts a “closed” compact conformation in solution, similar to that observed in the crystal structure. In contrast, the oxidized form of Rv2466c displays an “open” conformation, where tertiary structural changes in the α-helical subdomain suffice to account for the observed conformational transitions. Altogether our structural, biochemical, and biophysical data strongly support a model in which the formation of the catalytic disulfide bond upon TP053 reduction triggers local structural changes that open the substrate binding site of Rv2466c allowing the release of the activated, reduced form of TP053. Our studies suggest that similar structural changes might have a functional role in other members of the thioredoxin-fold superfamily.
Keywords: conformational change, enzyme, Mycobacterium tuberculosis, oxidation-reduction (redox), small-angle x-ray scattering (SAXS), thioredoxin, x-ray crystallography
Introduction
Among infectious human diseases, tuberculosis (TB)3 is the second greatest killer worldwide due to a single infectious agent, and remains a major challenge to human health care. In 2013, there were about 9 million new cases and 1.5 million deaths from TB, with an estimated one-third of the human population carrying a latent infection (1). First-line treatment for drug-susceptible TB requires the administration of a combination of four drugs during a period of 6 months: isoniazid, rifampicin, ethambutol, and pyrazinamide. Lengthy treatment regimens, unpleasant side effects, and patient noncompliance have provided conditions for the generation of multidrug-resistant and extensively drug-resistant cases of TB (2). Thus, the discovery and development of novel anti-TB drugs with bactericidal mechanisms different from those of currently available agents has become an urgent need. In that context, bedaquiline, a diarylquinoline that inhibits the c subunit of ATP synthase from Mycobacterium tuberculosis, has been recently approved by the Food and Drug Administration (FDA) for the treatment of multidrug-resistant TB in adults (3–5). Moreover, several candidate molecules are currently in preclinical studies, phase II and III clinical trials (6, 7). However, up to date, only a few drugs are capable of effectively killing non-replicating M. tuberculosis, thus allowing TB reactivation (8). Interestingly, we have recently discovered a new series of thienopyrimidine (TP) compounds that kill both replicating and non-replicating bacilli (Fig. 1) (9). Structure-activity relationship analysis demonstrated that a NO2 group at position C6 is essential for anti-TB activity as any alternative substitution, including NH2, carbonitrile, carboxylate, or carboxamide, resulted in an inactive TP compound. In addition, the most active compound TP053, displayed a hydrophobic phenyl and N-methyl groups at positions 2 and 4, respectively, accounting for a minimum inhibitory concentration value of 0.125 μg/ml against M. tuberculosis strain H37Rv.
FIGURE 1.
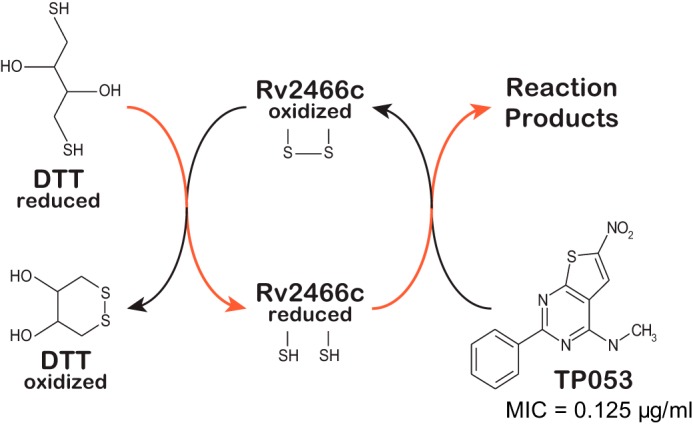
Rv2466c mediates the reduction of TP053 to kill M. tuberculosis. Model of the catalytic cycle of Rv2466c. Reduced Rv2466c reacts with TP053 to oxidized Rv2466c and reduced reaction products. Oxidized Rv2466c is then recycled to its reduced form by reduced DTT via disulfide exchange. The proposed electron flow during redox exchange is indicated in red. MIC, minimum inhibitory concentration.
To elucidate the mechanism of action of these TP derivatives, spontaneous resistant mutants of M. tuberculosis H37Rv to TP053 were isolated (9). This strategy had proven useful to study antibiotic resistance in M. tuberculosis and other human bacterial pathogens (10, 11). Whole-genome sequencing and bioinformatics analysis revealed a non-synonymous g83c mutation in the rv2466c gene, resulting in replacement of Trp28 by a serine residue. Interestingly, the overexpression of wild-type Rv2466c increased the sensitivity of M. tuberculosis wild-type and resistant mutant strains to TP053, indicating that TP053 is a prodrug activated by Rv2466c, and the W28S mutation impairs the function of Rv2466c. Rv2466c is a soluble protein localized in the reducing environment of the cytosol, whose expression is transcriptionally regulated during the oxidative stress response (12). Several lines of experimental evidence strongly support the notion that Rv2466c is a thioredoxin-like reductase that transfers reducing equivalents from intracellular thiols to substrates. Initial velocities recorded at different TP053 concentrations in the presence of the reductant 1 mm dithiothreitol (DTT) and catalytic amounts of Rv2466c were consistent with Michaelis-Menten kinetics (kcat = 0.0403 s−1; Km = 6.489 μm) indicating that Rv2466c catalyzes the reduction of TP053 by DTT. This conclusion was supported by the observation that Rv2466c in its oxidized state and in the absence of DTT was unable to metabolize TP053 (9). The rate-limiting step in the catalytic cycle of Rv2466c, in the presence of 1 mm DTT, is the reduction of TP053 by reduced Rv2466c, and not the reduction of oxidized Rv2466c by DTT. The experimental data support a model in which the oxidized form of Rv2466c accepts electrons from a thiol reductant (DTT in the in vitro experiments), whereas the reduced form of Rv2466c, directly or indirectly, transfers the electrons to TP053 (Fig. 1). In combination with structure-activity relationship studies, it is suggested that the nitro group is being reduced to generate toxic anion radicals and/or a variety of other highly reactive compounds including nitroso and hydroxylamine derivatives (9), as these products are extremely toxic, readily damage unknown cellular targets including proteins, membrane lipids, and DNA and likely leading to M. tuberculosis death (9).
The crystal structure of reduced Rv2466c has been solved recently, revealing a unique homodimer in which a β-strand is swapped between the thioredoxin domains of each subunit, and an α-helical subdomain inserted into each thioredoxin domain (9). A large, mostly hydrophobic groove harbors a redox active-site motif CPWC, typical of the thioredoxin superfamily of oxidoreductases (13–16). Using a combination of x-ray crystallography, small angle x-ray scattering, limited proteolysis, circular dichroism, site-directed mutagenesis and activity measurements, we show that Rv2466c undergoes significant conformational changes upon formation of its oxidized state, providing unprecedented insight into the molecular mechanism of TP053 activation.
Experimental Procedures
Methods
Recombinant Rv2466c wild-type and mutants were produced in Escherichia coli and purified to apparent homogeneity as previously described (9). Rv2466c-C19S, Rv2466c-C22S, and Rv2466c-H99A, mutants were synthetized by GenScript using the pET29a-Rv2466c construct as template. Activity measurements were performed as previously described (9).
Rv2466c-CT-His Expression and Purification
The Rv2466c gene was amplified by standard PCR using oligonucleotide primers Rv2466c_CT_His_NdeI_TEV_Fwd (5′-GGAATTCCATATGCTCGAGAAGGCCCCCCAGAAGTCTGTCGCCG-3′) and Rv2466c_CT_His_HindIII_TEV_Rev (5′-CCCAAGCTTGTCGAACTGAGGCGGCTCGGTGCG-3′), Phusion DNAPolymerase (New England Biolabs), and genomic DNA from M. tuberculosis H37Rv as DNA template. The PCR fragment was digested with NdeI and HindIII and ligated to the expression vector pET23b (Novagen) generating pET23b-Rv2466c-CT-His. The recombinant Rv2466c-CT-His (220 residues) has an additional peptide of 13 amino acids (KLAAALEHHHHHH) at the C terminus that includes a histidine tag (Fig. 3). E. coli BL21(DE3)pLysS cells transformed with pET23b-Rv2466c-CT-His were grown in 3000 ml of 2× YT medium supplemented with 100 μg/ml of carbenicillin and 34 μg/ml of chloramphenicol at 37 °C. When the culture reached an A600 value of 0.8, the Rv2466c-CT-His expression was induced by adding 0.5 mm isopropyl β-d-thiogalactopyranoside (MIP). After ∼16 h at 18 °C, cells were harvested and resuspended in 40 ml of 20 mm imidazol, 50 mm Tris-HCl, pH 7.5, 500 mm NaCl (solution A), containing protease inhibitors (Complete EDTA-free, Roche). Cells were disrupted by sonication (five cycles of 1 min) and the suspension was centrifuged for 20 min at 10,000 × g. Supernatant was applied to a HisTrap chelating column (1 ml, GE HealthCare) equilibrated with solution A. The column was washed with solution A until no absorbance at 280 nm was detected. Elution was performed with a linear gradient of 20–500 mm imidazole in 40 ml of solution A at 1 ml/min. Fractions containing Rv2466c-CT-His were pooled and loaded into a HiLoad 16/60 Superdex 200 column (GE HealthCare) equilibrated in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl (solution B). Aliquots of 12 μl were mixed with 3 μl of 250 mm Tris-HCl, pH 6.8, 10% SDS, 50% glycerol, 500 mm DTT, and 0.01% bromphenol blue at the indicated times. Samples were boiled for 3 min and run onto a NuPAGE® 4–12% gel (Invitrogen). Protein bands were visualized by staining the gel with SimplyBlueTM SafeStain (Invitrogen). The resulting preparation displayed a single protein band. Purified Rv2466c-CT-His was concentrated to 10–50 mg ml−1 in 10 mm Tris-HCl, pH 7.5, and stored at −80 °C.
FIGURE 3.

Limited proteolysis experiments show significant conformational differences between the redox forms of Rv2466c. A, SDS-PAGE showing the trypsin cleavage profile for Rv2466cOX, Rv2466cRED, and Rv2466cRED in the presence of TP053. B, the peptide bonds that are cleaved specifically by trypsin are shown with arrows. The N-terminal sequences of selected proteolytic fragments are underlined in the amino acid sequence and labeled in dark green in the schematic representation of Rv2466c.
Rv2466c-CT-His Crystallization and Data Collection
Rv2466c-CT-His was crystallized in the presence of the 13-residue long C-terminal tag containing 6 histidine residues (Fig. 3). Two crystal structures were obtained, referred thereafter as Rv2466c-HT1 and Rv2466c-HT2. Rv2466c-HT1 was obtained by mixing 0.25 μl of Rv2466c-CT-His at 10 mg/ml in 20 mm Tris-HCl, pH 7.5, with 0.25 μl of a solution containing 100 mm sodium acetate, pH 5.5, 0.3 m sodium acetate, 25% (w/v) PEG MME 2,000. Crystals grew in 7–8 days as rhomboid prisms to maximum dimensions of 140 × 80 × 80 μm and cryo-cooled in liquid nitrogen using 100 mm sodium acetate, pH 5.5, 0.3 m sodium acetate, 10% (w/v) PEG MME 2,000, and 25% (v/v) ethylene glycol. X-ray diffraction data were collected at beamline ID29 (European Sychrotron Radiation Facility, Grenoble, France) and processed with XDS program (17) to a maximum resolution of 1.70 Å. Rv2466c-HT1 crystallized in space group P21 with two independent molecules in the asymmetric unit (Table 1). The second structure, Rv2466c-HT2, was obtained by mixing 0.25 μl of Rv2466c at 10.0 mg/ml with 0.25 μl of 0.1 m Tris-HCl, pH 8.5, 0.1 m magnesium chloride, and 17% (w/v) PEG 20,000. Crystals grew in 1–2 days as rhomboid prisms to maximum dimensions of 100 × 50 × 50 μm. Prior to data collection the crystals were cryo-cooled in liquid nitrogen by using 0.1 m Tris-HCl, pH 8.5, 0.1 m magnesium chloride, and 10% (w/v) PEG 20,000 and 20% ethylene glycol as cryo-protectant solution. X-ray diffraction data were collected at beamline I04 (Diamond Light Source, United Kingdom) and processed with XDS program (17). Crystals diffracted to a maximum resolution of 1.51 Å. Rv2466c-HT2 crystallized in P21 with two independent molecule in the asymmetric unit (Table 1).
TABLE 1.
X-ray crystallography data
| Rv2466c-His | |
|---|---|
| Beamline | I04 (DLS) |
| Wavelength (Å) | 0.953700 |
| Resolution range (Å) | 40.26—1.507 (1.561—1.507) |
| Space group | P 21 |
| Unit cell | 40.649 46.814 104.308 90 97.97 90 |
| Total reflections | 249,842 (19424) |
| Unique reflections | 60,540 (5200) |
| Multiplicity | 4.1 (3.7) |
| Completeness (%) | 98.29 (85.07) |
| Mean I/σ(I) | 10.47 (2.11) |
| Wilson B-factor | 11.86 |
| Rsym | 0.09497 (0.6503) |
| R-factor | 0.1314 (0.1958) |
| R-free | 0.1762 (0.2530) |
| Number of non-H atoms | 3861 |
| Macromolecules | 3415 |
| Ligands | 1 |
| Water | 426 |
| Protein residues | 419 |
| R.m.s. (bonds) | 0.009 |
| R.m.s. (angles) | 1.15 |
| Ramach. favored (%) | 100 |
| Ramach. outliers (%) | 0 |
| Clashscore | 3.00 |
| Average B-factor | 19.40 |
| Macromolecules | 17.70 |
| Ligands | 38.30 |
| Solvent | 32.00 |
Rv2466c-CT-His Structure Determination and Refinement
Structure determination of Rv2466c-CT-His was carried out by molecular replacement using Phaser (18) and the PHENIX suite (19), and our previous Rv2466c crystal structure (9; PDB 4NXI) as search model. The built model was further extended by Buccaneer (20) and the CCP4 suite (21). The final manual building was carried out with Coot (22) and refined with phenix.refine (23). The structures were validated by MolProbity(24). Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession code 4ZIL.
Preparation of Oxidized Rv2466c
Oxidation of Rv2466c was accomplished with 5,5′-dithiobis(2-nitrobenzoic acid) (Sigma) in 100 mm NaH2PO4-NaOH, pH 8.0. To prepare oxidized Rv2466c (Rv2466cOX), 0.5 mg of protein at 1 mg/ml was mixed with 5 times molar excess of 5,5′-dithiobis(2-nitrobenzoic acid) followed by purification with illustra NAP-5 columns (GE Healthcare). To test whether Rv2466c was fully oxidized, we repeated the incubation of Rv2466cOX with 5,5′-dithiobis(2-nitrobenzoic acid). We could no longer detect the release of 5-thio-2-nitrobenzoic acid via an absorbance increase at 412 nm, confirming the absence of free thiol groups.
Limited Proteolysis of Rv2466c
Recombinant purified Rv2466cRED (25 μg) was incubated with 0.25 μg of trypsin (Sigma) in 100 μl of 20 mm Tris-HCl, pH 7.5, in the presence of 1 mm DTT and 1 mm CaCl2 for 0–90 min at 37 °C. 12-μl aliquots were mixed with 6 μl of 250 mm Tris-HCl, pH 6.8, 10% SDS, 50% glycerol, 500 mm DTT, and 0.01% bromphenol blue at the indicated times. Samples were boiled for 3 min and run in a NuPAGE®12% gel (Invitrogen). Protein bands were visualized by staining the gel with SimplyBlueTM SafeStain (Invitrogen). The proteolysis of recombinant Rv2466cOX was essentially performed following the same protocol as for Rv2466cRED but using 0.05 μg of trypsin, and in the absence of DTT. The proteolysis of Rv2466cRED in the presence of TP053 (110 μm) was carried out by using 0.05 μg of trypsin, and in the absence of DTT.
N-terminal Sequence Analyses
Samples were run in a NuPAGE®12% gel (Invitrogen). The gel was washed for 10 min with Milli-Q-purified water twice and then with NuPAGE transfer buffer (Invitrogen) during 15 min at room temperature. Proteolytic fragments were electrotransferred to a PVDF membrane using the iBlot dry blotting system (Invitrogen) during 7 min. The PVDF membrane was washed for 5 min with Milli-Q-purified water three times and bands were then stained with a solution containing 0.1 g/liter of Coomassie Brilliant Blue G-250, 40% methanol, and 1% glacial acetic acid. N-terminal sequence analysis was performed using an Applied Biosystems 494 procise cLC protein sequencer, in the Functional Genomics Center Zurich at the ETH Zurich, Switzerland.
Circular Dichroism Spectra
Far- and near-UV circular dichroism spectra were recorded on a JASCO J-715 CD spectropolarimeter at protein concentrations of 0.1 mg/ml (Far-UV CD spectra) and 0.2 mg/ml (Near-UV CD spectra) in 5 mm NaH2PO4-NaOH, pH 7.0, at 20 °C. Spectra of the reduced proteins were recorded in the same buffer containing 1 mm DTT.
Thermal Unfolding
Thermal unfolding transitions were recorded on a Jasco J-715 spectropolarimeter equipped with a computer controlled water bath. Thermal unfolding was recorded at 222 nm in 5 mm NaH2PO4-NaOH at pH 7.0 between 20 and 90 °C with a heating rate of 1 °C/min. Transitions were tentatively fitted according to Equation 1 to obtain the apparent melting temperature (Tm; 25),
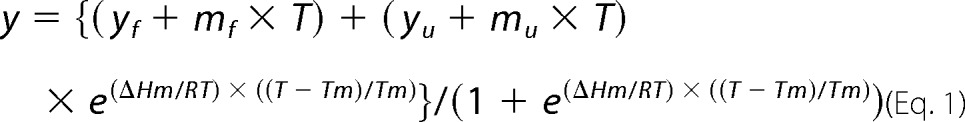 |
where y represents the observed CD signal at 222 nm, yf and yu are the y axis intercepts, and mf and mu the slopes of the pre- and post-transition baselines, respectively, T is the temperature in K, Tm is the melting temperature, and ΔHm is the enthalpy change of unfolding at Tm. Curve fitting was performed with Prism GraphPad.
Small Angle X-ray Scattering Measurements
Synchrotron x-ray diffraction data for recombinant purified WT Rv2466c and mutants were collected on the P12 beamline of the European Molecular Biology Laboratory (storage ring Petra-3, DESY, Hamburg) on a pixel Pilatus 2M detector. Scattering patterns were measured with a 1-s exposure time for protein samples at a minimum of three different protein concentrations ranging from 1 to 10 mg/ml. Samples were incubated with 2 mm DTT or 2 mm ascorbic acid in the case of WT Rv2466cOX before data collection. To check for radiation damage, 20 50-ms exposures were compared; no radiation damage was observed. Using the sample-to-detector distance of 3.1 m, the range of momentum transfer values is 0.008 < s < 0.47 Å−1 (s = 4π sin(θ)/λ where 2θ is the scattering angle and λ = 1.5 Å is the x-ray wavelength). Data were processed using standard procedures and extrapolated to infinite dilution by the program package PRIMUS (26). The forward scattering (I(0)) was evaluated using the Guinier approximation (27) assuming the intensity is represented as I(s) = I(0)exp(−(qRg)2/3) for a very small range of momentum transfer values (s < 1.3/Rg). The maximum dimensions (Dmax), the interatomic distance distribution functions (P(r)), and the radii of gyration (Rg) were computed using GNOM (28). The molecular mass of the protein was evaluated by comparison of the forward scattering with that from a reference solution of bovine serum albumin.
Ab Initio Shape Determination
The low-resolution structures of WT Rv2466c and mutants were calculated ab initio by using DAMMIF (29). Structure clustering and averaging were carried out with DAMAVER and DAMCLUST (30). The results and statistics are summarized in Table 2. For each data set, pairwise alignment of structures was carried out, and the normalized spatial discrepancy (NSD) was calculated (31). The clustering process groups structures with lower NSD. In general, NSD values close to 1 indicate that the two structures are similar; structures with NSD larger than 2 are taken as outliers and removed. Average NSD values within clusters were between 0.6 and 1, indicating a very good structural agreement for each cluster. Furthermore, average NSD values for structures of different clusters are between 0.8 and 1.1 also indicating similarity between the clusters.
TABLE 2.
SAXS data and refinement parameters
| Data collection parameters | Rv2466c reduced | Rv2466c C19S | Rv2466c C22S | Rv2466c oxidized | Rv2466c H99A |
|---|---|---|---|---|---|
| Instrument | Beamline P12 (Petra-3) | Beamline P12 (Petra-3) | Beamline P12 (Petra-3) | Beamline P12 (Petra-3) | Beamline P12 (Petra-3) |
| Wavelength (Å) | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 |
| S range (Å−1)a | 0.007–0.370 | 0.008–0.322 | 0.014–0.307 | 0.009–0.370 | 0.015–0.316 |
| Exposure time (s) | 1 | 1 | 1 | 1 | 1 |
| Concentration range (mg ml−1) | 1–10 | 1–10 | 1–10 | 1–10 | 1–10 |
| Temperature (K) | 283 | 283 | 283 | 283 | 283 |
| Structural parametersb | |||||
| I(0) (cm−1) (from P(r)) | 37.43(1) | 35.99(1) | 37.72(1) | 38.52(1) | 37.60(1) |
| Rg (Å) (from P(r)) | 25.7(1) | 25.4(1) | 25.7(1) | 27.2(1) | 26.4(1) |
| I(0) (cm−1) (from Guinier) | 37.65(7) | 35.91(2) | 37.62(5) | 38.5(1) | 37.57(3) |
| RR (Å) (from Guinier) | 24.2(7) | 25.0(7) | 25.5(7) | 27(1) | 26.2(8) |
| Dmax (Å) | 81.0 | 81.3 | 84.9 | 90.0 | 89.5 |
| Porod volume estimate (Å3) | 79,400 | 73,400 | 73,800 | 76,700 | 76,800 |
| Dry volume calculated from sequence (Å3) | 27,900 | 27,900 | 27,900 | 27,900 | 27,900 |
| Ab initio modeling (χ2 value) | 0.973(1) | 1.179(7) | 0.890(3) | 0.747(1) | 0.965(1) |
| Average NSD between clusters | 0.91(2) | 0.8(1) | 0.86c | 0.9(2) | 0.89c |
| Average NSD within clusters | 0.6(1) | 0.6(1) | 0.58(1) | 0.59(8) | 0.61(8) |
| Molecular mass determinationd | |||||
| Molecular mass (from I(0)) (kDa) | 45(3) | 43(3) | 45(3) | 46(3) | 45(3) |
| Calculated monomeric from sequence (kDa) | 23.1 | 23.1 | 23.1 | 23.1 | 23.1 |
| Software employed | |||||
| Primary data reduction | SASFLOW | SASFLOW | SASFLOW | SASFLOW | SASFLOW |
| Data processing | PRIMUS | PRIMUS | PRIMUS | PRIMUS | PRIMUS |
| Ab initio analysis | DAMMIF | DAMMIF | DAMMIF | DAMMIF | DAMMIF |
| Validation and averaging | DAMAVER | DAMAVER | DAMAVER | DAMAVER | DAMAVER |
| Rigid-body modeling | SASREF | SASREF | SASREF | SASREF | SASREF |
| Computation of model intensities | CRYSOL | CRYSOL | CRYSOL | CRYSOL | CRYSOL |
a S-range used for calculation of P(r) function and ab initio modeling.
b Values in parentheses are estimated errors approximated to the last decimal place.
c Two clusters have been identified, thus no standard deviation reported.
d Molecular mass estimated by calculation of I(0) and comparison against bovine serum albumin.
Fitting the Crystal Structure to Small Angle X-ray Scattering (SAXS) Envelopes
The crystal structure of Rv2466c (PDB code 4NXI) was fitted to the SAXS envelopes using a semiautomatic approach. First, ab initio bead models obtained by DAMMIF were converted into surface maps using Chimera molmap command (32), followed by fitting of the crystal structure into the maps using Chimera fit-in-map module (32). To reconcile the SAXS envelope of Rv2466c in its oxidized state and mutant H99A with the available crystal structure, the latter was subdivided into domains and refined multiple times against the SAXS data using SASREF (33). This program performs rigid body modeling using subunits with known atomic structures against solution scattering data. For the analysis the crystal structure was subdivided in 5 fragments. Fragment 1 corresponds to residues A.8-A.82, A.130-A.205, B.8-B.82, and B.130-B.205; fragment 2 contains residues A.83-A.118, fragment 3 contains residues B.83-B.118, fragment 4 contains residues A.119-A.129, and fragment 5 contain residues B.119-B.129. During SASREF refinement, several constrains were applied: (i) position of fragment 1 was fixed, (ii) at the intersection of fragments, the distance between two neighboring Cα atoms in different fragments was fixed to 3.8 Å. The SASREF refinement was carried out without symmetry constrains, as the flexibility of the open form was likely to break the 2-fold axis. Moreover, ab initio DAMMIF reconstructions indicated the absence of internal symmetry for Rv2466cOX and H99A in solution. The hybrid SASREF models were fitted into the reconstructed SAXS envelope using the Chimera fit-in-map module (32).
Structural Transition between the Compact and Extended Conformational States of Rv2466c
The video (supplemental Video S1) shows a swinging back-and-forth movement of the helical bundle. To calculate the morph trajectory, we first used Modeler (34) to construct ab initio all missing side chains in the crystallographic structures of the Rv2466c reduced state (PDB code 4NXI). The hybrid SASREF model of the Rv2466c oxidized state was calculated following the methodology described under “Fitting the Crystal Structure to SAXS Envelopes.” After superposition of their thioredoxin domains, we used Chimera (UCSF) to generate coarse conformational intermediates by interpolating the atomic positions. The interpolation was based on the Corkscrew method, as implemented in Chimera (32). We show 171 linear interpolation steps calculated between the reduced and oxidized Rv2466c forms. For each interpolation step, the hydrogen-bonding network was calculated and displayed. The conformational intermediates of only one monomer in the dimer are shown for clarity (chain A).
Structural Analysis and Alignment
Structural analysis and graphics for publications were performed with Chimera (32).
Results and Discussion
Rv2466c Displays a Modular Architecture with Structural Variability at an α-Helical Subdomain
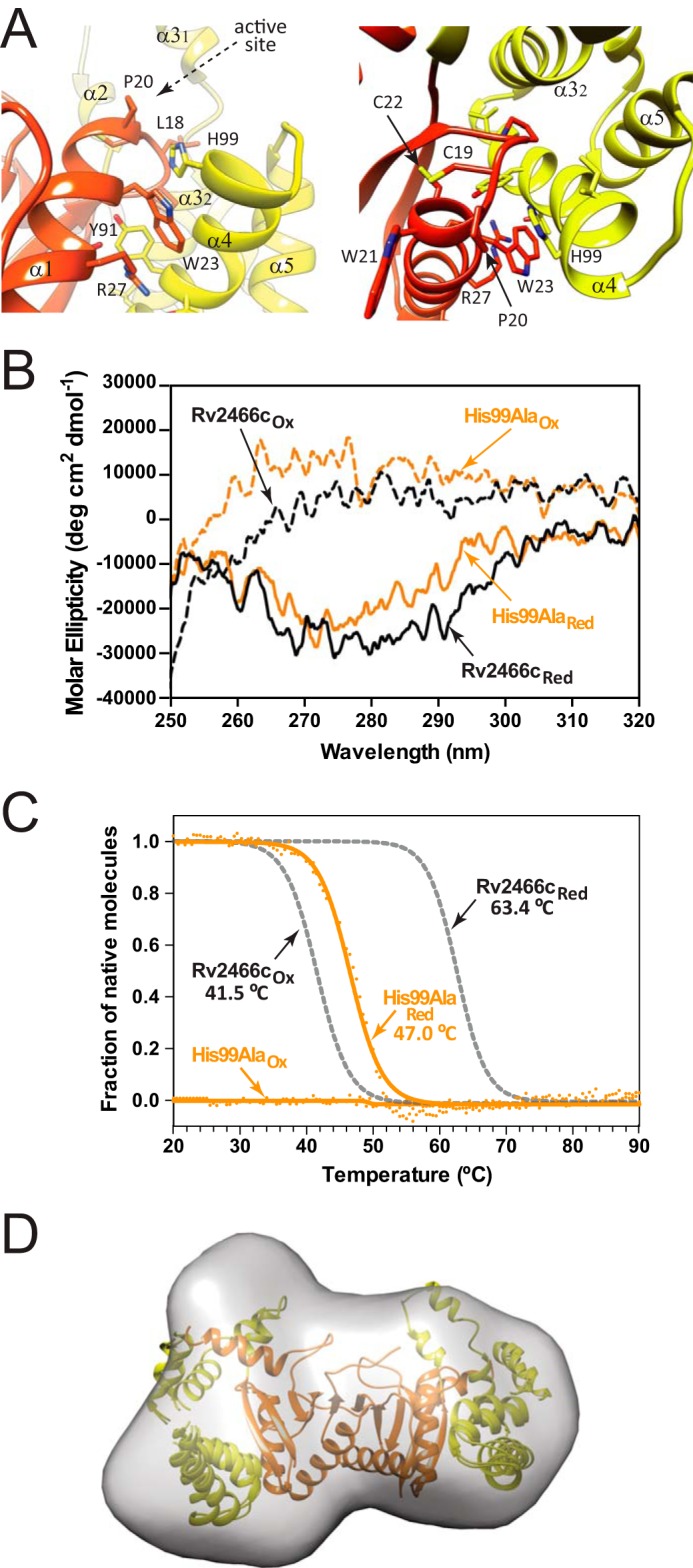
The crystal structure of Rv2466c in its reduced state revealed a modular architecture comprising a canonical thioredoxin-fold (residues Met1 to Val44 and Asp133 to Ser189), an insertion consisting of an antiparallel four α-helical bundle (residues Trp69 to Tyr132; α3″-α4-α5-α6) and a short α-helical hairpin (residues Met45 to Ala68; α2-α3′; Fig. 2A)(9, 14). Importantly, the thioredoxin domain and the α-helical bundle communicate with each other through an extensive interface of 1380 Å2. This interface comprises residues Phe15, Asp16, Pro17, and Leu18 (β1-α1 loop), Trp23, Arg27, and Leu30 (α1), Phe42, and Val44 (β2; from the thioredoxin-fold domain); and Val72 and Ile76 (α3), Leu87, Asp88, Tyr91, Thr92, Asn96, Ile98, His99, and Asn100 (α4; located in the α-helical subdomain; 9). The last 17 residues of the protein (Tyr190 to Asp207) drives protein dimerization via a β-strand (β5) swapping mechanism (Fig. 2A) (9). The Cys19-Pro20-Trp21-Cys22 active-site motif is located on top of α1 in the thioredoxin-fold, and in close proximity to the α2-α3′ hairpin and the four-helix bundle of the insertion domain. It is worth noting that the lateral chain of Trp28 makes hydrophobic interactions with three secondary structural elements of the core of the thioredoxin domain of Rv2466c. The mutation of W28S predicted structural defects in the thioredoxin-fold, likely including the destabilization of the dimerization core and the Cys19-Pro20-Trp21-Cys22 motif, impairing the activity of Rv2466c against TP053. Structural superposition of the two monomers of the protein dimer suggested that the helical subdomain might display structural flexibility (9).
FIGURE 2.

Crystal structure of reduced Rv2466c-CT-His. A, schematic representation showing the structure of Rv2466c-CT-His. Monomer A (Rv2466c) is shown in orange (thioredoxin domain) and yellow (α-helical subdomain), whereas monomer B (Rv2466c′) is shown in cyan. The C-terminal peptide of 13 amino acids (KLAAALEHHHHHH) is shown in blue. B, schematic representation showing the catalytic motif (Cys19-Pro20-Trp21-Cys22) in close proximity to the α-helical subdomain, comprising a four α-helical bundle (α3″–α4–α5–α6) and a short α-helical hairpin (α2-α3′). The C terminus peptide of 13 amino acids binds to the active site and folds into an α-helix. C, close view of the active site of Rv2466c showing the molecular interactions with the peptide. D, conformational changes triggered by binding of the C-terminal peptide segment. Structural superposition of two selected monomers from the reduced Rv2466c (9) and Rv2466c-CT-His constructs, with the α9 in blue, suggesting that the α2–α3′ hairpin region displays structural variability. E, schematic representation showing the comparison between the crystal structure of Rv2466c-CT-His and the structural homologue from M. leprae (PDB code 4WKW). The canonical thioredoxin folds are shown in orange. The α-helical subdomains of Rv2466c-CT-His and the M. leprae homologue are shown in yellow and green, respectively. F, schematic representation showing the comparison of all monomers from the crystal structures of the Rv2466c (9), Rv2466c-CT-His, and the structural homologue from M. leprae (PDB code 4WKW).
Here we determined the crystal structure of a second construct of Rv2466c (Rv2466c-CT-His; 220 residues) in its reduced form (Rv2466cRED), containing an additional peptide of 13 amino acids at the C terminus that includes a hexahistidine tag (208KLAAALEHHHHHH220), at 1.51-Å resolution (Fig. 2 and Table 1). The close inspection of the electron density maps revealed that the C-terminal peptide extension binds to the active site pocket, being in direct contact with the Cys19-Pro20-Trp21-Cys22 motif (Fig. 2, B and C). Strikingly, the peptide adopts an α-helix conformation (α9; Fig. 2C). The protein-peptide interaction promotes several conformational changes in Rv2466c, including the reorientation by about 20 degrees of the α2-α3′ hairpin, which opens the groove next to the active thiol pair that accommodates the peptide (r.m.s. deviation for residue range 45–68 is 2.8–3.0 Å; Fig. 2D). A short kink (residues 64 to 67) located between α3′ and α3″ helices adopts an α-helical conformation allowing these two helices to merge into an elongated α3 helix of about 38 Å in length (r.m.s. deviation for residue range 59–82 is 1.9 Å; Fig. 2D) (35). In addition, the connecting loop α7-β3, located at the central hydrophobic groove becomes partially disordered in both monomers.
More recently, the crystal structure of the reduced, unliganded form of a Rv2466c homologue from Mycobacterium leprae has been solved at 2.3-Å resolution (PDB code 4WKW; 80.6% amino acid sequence identity; Z-score of 13.0; r.m.s. deviation value of 1.26 for 177 aligned residues for monomer B, and Z-score of 12.3; r.m.s. deviation value of 1.35 for 175 aligned residues for monomer A). Structural comparison of Rv2466c-CT-His with the M. leprae homologue revealed that (i) the Cys19-Pro20-Trp21-Cys22 active-site motif and (ii) the residues located at the interface between the thioredoxin-fold and the α-helical subdomain are strictly conserved in both proteins. However, very important differences were observed in the conformation of the α-helical subdomain in both monomers of the protein dimer, indicating structural variability and a tendency of C2 symmetry breakage (Fig. 2, E and F). Interestingly, the α-helical hairpin was partially disordered in monomer A, as observed in the crystal structure of the untagged Rv2466c (9). Taken together, the experimental data suggest that the artificial C-terminal tag in Rv2466c binds next to the active-site disulfide in a manner that mimics the structure of a complex between Rv2466c and a putative peptide substrate and that the substrate is accommodated via a conformational change in the helical bundle as a result of the intrinsic flexibility within the helical bundle. In addition, the structural data suggest that the interface between the thioredoxin domain and the α-helical subdomain might play an important role during substrate recognition and catalysis.
Conformation of Oxidized and Reduced Rv2466c
To investigate the effect of the redox state on the conformation of Rv2466c, we first performed limited proteolysis experiments (36, 37). As depicted in Fig. 3A, when incubated with trypsin, oxidized Rv2466c (Rv2466cOX) was rapidly degraded. However, similar proteolysis experiments carried out with Rv2466cRED suggest, in contrast, a conformational rearrangement relative to Rv2466cOX, as the protein was markedly protected from digestion by trypsin even after 90 min incubation (Fig. 3A). Edman sequencing of the two predominant small species of 15 and 8 kDa revealed the sequence EGMAR for both fragments, located in the kink between α3′ and α3″ helices (Fig. 3B). Interestingly, trypsin digestion of Rv2466cRED in the presence of TP053 with the protease gave rise to two additional fragments of 13 and 9 kDa. The 13-kDa band proved to be a mixture of 4 fragments with the N-terminal sequences 63EGMAR67, 126AATSD130 (α6), 138SHHAG142 (α7), and 139KSHHA143 (α7), whereas the 8-kDa fragment showed the sequence 126AATSD130, located in the last α-helix of the bundle. Moreover, whereas TP053 becomes reduced, oxidized Rv2466c was formed (70% of Rv2466cOX after 150 min incubation with TP053), adopting a similar proteolytic pattern to that observed for Rv2466cOX.
Second, the overall shapes of Rv2466cOX and Rv2466cRED in solution were obtained by SAXS, a powerful technique capable of providing structural information on flexible and dynamic proteins in solution (38, 39) (Table 2; see Fig. 4A for experimental SAXS data and DAMMIF-calculated fits from corresponding overall solution structures). The interatomic distance distribution functions (P(r)) computed for Rv2466cOX and Rv2466cRED are shown in Fig. 4A. The molecular mass determined from the scattering data confirmed that the protein is a homodimer in solution in agreement with size exclusion chromatography and dynamic light scattering (9). The radius of gyration (Rg) values obtained for Rv2466cOX (27.2(1) Å) and Rv2466cRED (25.7(1) Å) revealed a reduction in Rg (ΔRg) of about −1.5 Å, indicating that Rv2466cOX is in an extended conformation compared with the reduced state.
FIGURE 4.

Conformation of oxidized and reduced Rv2466c, as characterized by SAXS. A, upper panel: scattering curves of Rv2466cRED and Rv2466cOX. Lower panel: P(r) function distributions of Rv2466cRED and Rv2466cOX. B and C, low resolution models of Rv2466cRED and Rv2466cOX in solution. Average low resolution structure of Rv2466cRED (B) and Rv2466cOX (C) with the high resolution crystal structure of Rv2466cRED (Protein Data Bank code 4NXI) fitted by rigid body docking, respectively.
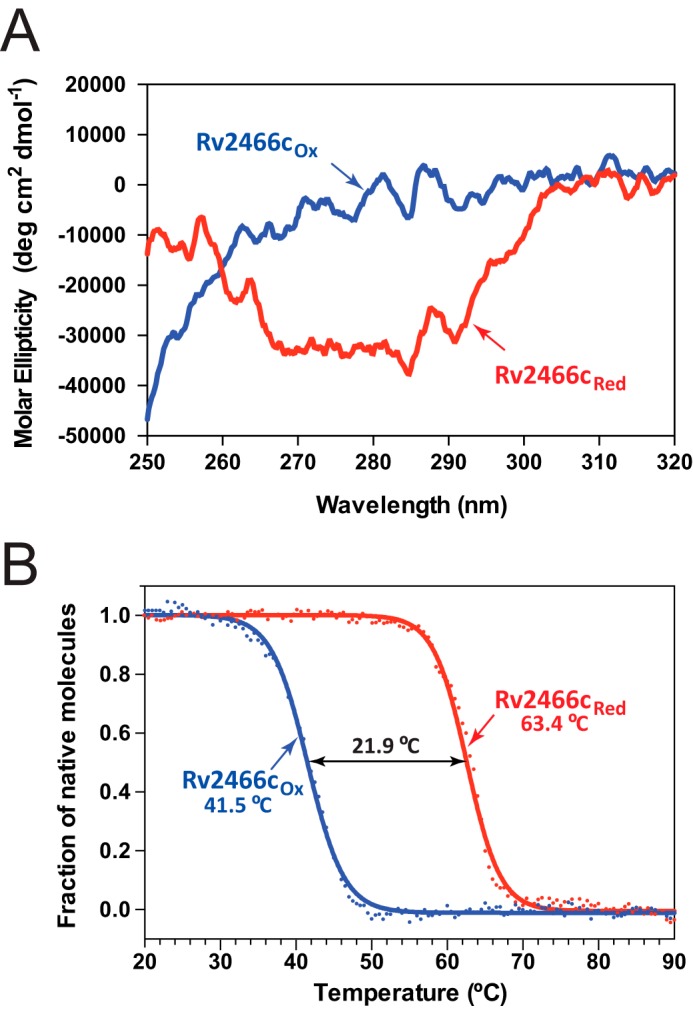
The ab initio low-resolution envelopes of Rv2466cOX and Rv2466cRED were reconstructed using SAXS data (Fig. 4; Table 2). The crystal structure (PDB 4NXI) readily fits into the ab initio low-resolution envelope of Rv2466cRED (Fig. 4B). It is worth noting that even though symmetry constrains were not applied in the ab initio reconstruction, the low-resolution envelopes of Rv2466cRED clearly show internal symmetry, in agreement with its homodimeric nature. In contrast, the ab initio shape of Rv2466cOX appears asymmetric and significantly differs from the crystal structure of Rv2466cRED (Fig. 4C) pointing to major differences between the redox states. The structural differences are most remarkable in the region of the α-helical domains; in the case of Rv2466cRED both apical lobes display a similar compact conformation, in which the crystallographic conformation readily fits. To reconcile the structure of Rv2466cRED observed in the crystal and the oxidized state suggested by SAXS we performed a refinement as implemented in the program SASREF (see “Experimental Procedures” for details). The method employed consists in allowing a certain degree of flexibility within α-helical domains, whereas fixing the thioredoxin domain. Using this refinement strategy, it was possible to construct a hybrid model agreeing with the solution SAXS envelope and fitting the experimental SAXS data from Rv2466cRED with discrepancy χ2 around 1.0. These results indicate that tertiary structural changes in the α-helical subdomain are sufficient to account for the observed variability (Fig. 4C). To further explore this hypothesis, we investigated the secondary and tertiary structure changes of Rv2466c under reducing and oxidizing conditions by near-UV circular dichroism (CD). The near-UV CD spectra revealed clear differences in the local environment of the aromatic side chains in Rv2466cOX and Rv2466cRED (Fig. 5A). In addition, thermal unfolding followed by the far-UV CD signal at 222 nm indicated dramatic differences in protein stability between the two states (Fig. 5B). Specifically, the apparent melting temperatures (Tm) of Rv2466cOX and Rv2466cRED were 41.5 and 63.4 °C, indicating that the reduced state is about 22 °C more stable than the oxidized form. In summary, CD analysis indicates that the formation of the catalytic disulfide bond in Rv2466cOX leads to large tertiary structure changes and a less stable conformation. These results are in good agreement with the predicted opening of the α-helix subdomain of the protein in oxidative conditions, as observed by SAXS.
FIGURE 5.
Tertiary structure differences between the oxidized and reduced forms of Rv2466c analyzed by CD spectroscopy. A, near-UV CD spectra for Rv2466cRED and Rv2466cOX. B, thermal unfolding transitions of Rv2466cRED and Rv2466cOX monitored by the change in CD signal at 222 nm. The Tm values are given in the figures and represent the apparent melting temperature. The transition was tentatively fitted according to a two-state equilibrium (solid lines) and normalized.
The Redox State of the CPWC Motif Regulates Rv2466c Conformation and Activity
Rv2466c displays a catalytic disulfide located in the Cys19-Pro20-Trp21-Cys22 motif. In Rv2466cRED, the N-terminal cysteine (Cys19) is solvent-exposed, whereas the C-terminal cysteine (Cys22) is buried, as observed in other members of the thiol-disulfide oxidoreductase superfamily (Fig. 6A) (40). Interestingly, Rv2466cRED adopts a “closed” conformation and is able to metabolize TP053, whereas Rv2466cOX adopts an “open” conformation, and is unable to activate TP053. To gain further insight into the relevance of the redox state on the conformational changes triggered in Rv2466c and its ability to metabolize TP053, we studied the single-cysteine variants C19S and C22S. As depicted in Fig. 6B, the C19S mutant proved to be completely inactive, whereas the C22S mutant displayed only 6% residual activity. This is consistent with the fact that Cys19 is the active-site cysteine responsible for the initial nucleophilic attack during the reduction of TP053 (13, 40). Interestingly, the near-UV CD spectra of the C19S and C22S mutants recorded under oxidized conditions have similar signatures compared with that of Rv2466cRED state, with no signs of major changes in the tertiary structure (Fig. 6C). These results correlate very well with thermal unfolding experiments, with the C19S and C22S mutants showing melting temperatures of 56 and 55 °C, respectively, closer to that observed for Rv2466cRED (Table 2; Fig. 6D). Furthermore, SAXS analysis of both mutants indicate that they adopt a similar structural arrangement as observed for the reduced form of Rv2466c. Specifically, the reduced and oxidized forms of C19S and C22S mutants displayed a similar radius of gyration than Rv2466cRED (Rg (C19S) = 25.4(1); Rg (C22S) = 25.7(1); Rg (WT) = 25.7(1)). In addition, the ab initio envelope reconstructions (χ2 value C19S = 1.179(7) and χ2 value C22S = 0.890(3)) of both mutants fitted with the atomic coordinates of the crystal structure of Rv2466c (Fig. 6E). Thus, we postulate that the transfer of electrons from Rv2466c to TP053 alters the conformation of the protein, from a closed, more stable reduced conformation to an open, less stable oxidized state.
FIGURE 6.

The redox state of the CPWC motif regulates Rv2466c conformation and activity. A, localization of Cys19 and Cys22 in the active site of Rv2466c (top of α1 helix). B, activity measurements of Rv2466c WT and Rv2466c variants against TP053 (9). C, near-UV CD spectra of the C19S and C22S variants. D, thermal unfolding of the C19S and C22S variants as monitored by CD spectroscopy. E, low resolution models of C19S and C22S mutants in solution. Average low resolution structure of C19S and C22S mutants with the high resolution crystal structure of Rv2466cRED (Protein Data Bank code 4NXI) fitted by rigid body docking, respectively.
The Open to Closed Motion Is Mediated by Conformational Changes at the Interface between the Thioredoxin Domain and the α-Helical Subdomain
The α-helical subdomain of Rv2466c comprises an antiparallel four-helix bundle (α3″-α4-α5-α6) and a short α-helical hairpin (α2-α3′). The α3″, α4, α5, and α6 helices display an amphipathic character with short connecting loops, and a topology that diverges from a canonical four α-helical bundle. This deviation is accounted by steric restrictions and intramolecular interactions imposed by the neighboring thioredoxin-fold. Specifically, the structural elements β1, β2, and α1 located in the thioredoxin-fold seem to be important to maintain the stability of the α-helical bundle (Fig. 7A). The lateral chain of Tyr91 (α4) establishes stacking interactions with the lateral chains of Phe15 (β1), Trp23 (α1), and Phe42 (β2). In addition, the phenolic hydroxyl group of Tyr91 is hydrogen bonded with the lateral chain of Ser26 (α1) and with the main chain carbonyl group of the catalytic Cys22 (α1). Furthermore, the connecting loop β1-α1 inserts into the hydrophobic core of the four-α-helix bundle, promoting multiple hydrophobic interactions, and including residues Pro17 and Leu18 (loop β1-α1), Pro71 and Val72 (α3″), Met94 and Ile98 (α4), and Leu106 (α5; Fig. 7A). The α4 is further stabilized by contacts with the N-terminal region of α1. Specifically, the lateral chain of His99 (α4) makes a hydrogen bond with the main chain amino group of Pro17, and stacks with the lateral chain of Trp23 (α1), which in turn is also in close contact with the main chain atoms of Gly95 and Asn96. Finally, the guanidinium group of Arg27 hydrogen bonds with the lateral chain hydroxyl group of Thr92.
FIGURE 7.
Conformational changes at the interface between the thioredoxin domain and the α-helical subdomain. A, two close views of the thioredoxin domain-α-helical subdomain interface, and the active site motif Cys19-Pro20-Trp21-Cys22. B, near-UV CD spectra of the oxidized and reduced H99A variant. C, normalized thermal unfolding transition of the oxidized and reduced H99A variant monitored by CD. D, low resolution model of the reduced H99A variant in solution deduced from SAXS data compared with the high-resolution crystal structure of Rv2466cRED (PDB code 4NXI).
To further advance on the understanding of the molecular mechanism of TP053 reduction by Rv2466c, we investigated the role of His99, located at the interface between the thioredoxin domain and the α-helical subdomain. As depicted in Fig. 6B, when His99 was replaced by alanine, the mutant undergoes a dramatic decrease on its ability to metabolize TP053, retaining only 18% of activity. The near-UV CD spectra of the H99A mutant recorded under reduced and oxidized conditions revealed differences in the tertiary structure with the Rv2466cRED state (Fig. 7B). Thermal unfolding followed by CD indicate that the reduced H99A variant has a stability similar to that of the oxidized mimics WT (Fig. 7C). In accordance with the above CD spectra, experimental SAXS data comparison of H99A and WT oxidized state are similar (DATCMP-calculated (ref) χ2 value of 1.35), and the Rg value obtained for H99A (26.4(1) Å) is comparable with that observed for the oxidized form of Rv2466cOX (27.2(1) Å), indicating that the mutant is in a more open conformation than Rv2466cRED (DATCMP-calculated χ2 value of 1.52; Table 2). In that sense, the comparison of the crystal structure of Rv2466cRED with the solution structures obtained by SAXS for H99A revealed differences in the conformation of the protein as visualized for Rv2466cOX (Fig. 7D). Thus, structural changes in the interdomain interface deregulate the transition from an open to a closed state, impairing the capacity of Rv2466c to metabolize TP053.
A Model of TP053 Activation by Rv2466c
Taken together, the experimental data support a model in which local structural changes in the thioredoxin-fold that result from Cys19-Cys22 disulfide bond formation are sufficient to alter the fine equilibrium of the α-helical subdomain, triggering its conformational arrangement from a closed to an open state (supplemental Video S1; see “Experimental Procedures” for details). Specifically, once TP053 binds to Rv2466cRED, it becomes reduced and Rv2466cOX is formed. The formation of the catalytic disulfide bond triggers a conformational transition from the closed to the open conformation that in turn allows the release of the reaction product.
The thioredoxin-fold is an ubiquitous protein folding motif in all organisms and found in enzymes catalyzing thiol-disulfide exchange reactions, including protein disulfide reduction (thioredoxin; 41), disulfide formation (disulfide oxidoreductase DsbA; 42, 43), disulfide isomerization (protein-disulfide isomerase; 44), and glutathionylation (glutathione S-transferase, GST; 45). Interestingly, the functional diversity in some thioredoxin family members seems to be due to some extent by the presence of an α-helical insertion between the canonical second β-strand and the second α-helix (corresponding to residues Met45 to Tyr132 in Rv2466c) of the thioredoxin-fold. For example, GSTs are a family of enzymes that primarily catalyze nucleophilic addition of the thiol of glutathione (GSH) to a variety of hydrophobic electrophiles in the cellular detoxification of cytotoxic and genotoxic compounds (46). Human κ class GST, the closest homologue with known structure to Rv2466c (Dali Z score 16.4, sequence ID 14%), contains a large α-helical region (Ser42 to Pro184) that also undergoes conformational changes upon substrate binding (Fig. 8A). Thus, flexibility of the α-helical subdomain allows substrate-dependent conformational changes that lead to assemble the GSH and acceptor binding sites. Strikingly, the region of the α-helical subdomain in GST that is most involved in substrate binding (Ser42-Ser96) corresponds to the region in the α-helical subdomain of Rv2466c that participates in recognition of the C-terminal tag (Met45-Trp69), suggesting a conserved mechanism of substrate binding between both members of the thioredoxin family. DsbA is another family member that contains an inserted α-helical bundle (Val61 to Pro151 in E. coli DsbA). The crystal structures of the DsbB-DsbA complex revealed how DsbA recognize DsbB, an E. coli membrane protein that maintains DsbA in its oxidized active form (47, 48). Interestingly, the hydrophobic groove of DsbA interacting with DsbB contains two loops within the α-helical bundle that show conformational variability upon substrate binding (Fig. 8B; loop 1, residues Val61-Gly65; loop 2, residues Leu147-Pro151). Importantly, the equivalent regions to loops 1 and 2 in Rv2466c and GST are involved in peptide/substrate binding. Thus, these similarities suggest a conserved mechanism of substrate recognition and product release.
FIGURE 8.

Structural location of the α-helical subdomain in GST and DsbA. A, schematic representation of human κ class glutathione transferase (hGSTk). The transfer of GST reduced glutathione (GSH) to a variety of hydrophobic electrophiles for cellular detoxification. In this instance, the apo state of hGSTk (PDB code 3RPP) and when bound to a reaction product, S-hexylglutathione (GTX; PDB code 3RPN) is shown. B, close-up view of overlapping hGSTk structures where structural variability of the helical bundle can be observed; most evident is the variability within Ser42-Ser96 residues, which contains a region that structures upon substrate binding. C, schematic representation of disulfide bond oxidoreductases DsbA (orange-yellow) and DsbB (gray). DsbA is a periplasmic dithiol oxidase responsible for protein disulfide formation, DsbB is an integral membrane protein catalyzing the oxidation of DsbA by ubiquinone. Three crystal structures are overlapped, corresponding to the E. coli C33A DsbA mutant in complex with DsbB (PDB code 2HI7), oxidized DsbA (PDB code 1DSB), and C33A DsbA mutant (PDB code 1TI1). D. The canonical α1 of the thioredoxin-fold undergoes large conformational changes depending on its redox state. The helical bundle (yellow) shows significant structural variability between structures.
Supporting this notion, the NMR structures of oxidized and reduced human and E. coli thioredoxin also revealed subtle structural differences in the active site and the surrounding regions triggered by a change in redox state (49, 50). The structural variability and available biochemical data (51), prompted to postulate that thioredoxin uses a chaperone-like mechanism of conformational changes to bind a diverse group of proteins and fast thiol-disulfide exchange chemistry in a hydrophobic environment to promote high rates of disulfide reduction (13). It is worth noting that helical bundles have been observed to undergo important functional conformational changes, including hemagglutinin (HA) protein and apolipophorin III (52–59). The interdomain analysis indicates that the stability of the α-helix bundle is very much dependent on the thioredoxin domain, in particular (i) changes in the conformation the χ1 rotational angle of Cys22 and (ii) changes in α1, which share multiple contacts with α4, could suffice to destabilize the α-helix bundle close conformation, allowing the opening into a less stable state. We postulate Rv2466c exploits the subtle structural changes in the thioredoxin-fold during disulfide bond formation to trigger a conformational change from a closed to an open state.
Concluding Remarks
The discovery of TP053 and the mechanism that Rv2466c employs to activate it represents a step forward toward the development of novel antibiotics to fight TB. Rv2466c is a member of the thiol-disulfide oxidoreductase superfamily that is responsible for the reduction of TP053, killing replicating and non-replicating M. tuberculosis (9). Rv2466c utilizes a chaperone-like mechanism of conformational changes to recognize TP053 and redox exchange chemistry in a hydrophobic environment to promote compound reduction. Rv2466c is able to undergo drastic conformational changes involving partial unfolding of an α-helical subdomain, comprising an α-helix bundle and a short α-helical hairpin. Importantly, the conformational state of this α-helix subdomain is synchronized with the redox state of the thioredoxin domain. When the Cys19-Cys22 disulfide bond is present, Rv2466c adopts an open inactive conformation, whereas upon reduction it returns to its active closed conformation. Local conformational changes mainly in the CXXC active site motif of the thioredoxin domain, alter the surface contact between the two domains, and trigger the open conformation, facilitating product release. Our studies contribute to the understanding of the molecular mechanisms of redox reactions in bacteria, suggesting that similar changes may also have a functional role in other members of the thioredoxin-fold superfamily.
Author Contributions
M. E. G. and D. A.-J. conceived the project. D. A.-J., N. C., M. T., E. M., S. U., and E. D. performed the biochemical, biophysical, and structural studies. D. A.-J., L. R. C., M. R. P., R. M., V. M., G. R., D. I. S., R. G., and M. E. G. analyzed the results. D. A.-J., D. I. S., R. G., and M. E. G. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We gratefully acknowledge Pedro Arrasate (Unit of Biophysics, CSIC,UPV/EHU, Spain), Dr. E. Ogando and Dr. T. Mercero (Scientific Computing Service UPV/EHU, Spain) for technical assistance. We thank the Diamond Light Source (DLS, Oxfordshire, UK), the European Molecular Biology Laboratory (EMBL, Hamburg, Germany), Positron-Electron Tandem Ring Accelerator (PETRA), and Deutsches Elektronen-Synchrotron (DESY) rings, and the French National Synchrotron Facility (SOLEIL, St. Aubin, France) staffs for the onsite assistance. We also acknowledge all members of the Structural Glycobiology Group (Spain) for valuable scientific discussions.
This work was supported by European Commission Contract HEALTH-F3-2011-260872, the Spanish Ministry of Economy and Competitiveness Contract BIO2013-49022-C2-2-R, and the Basque Government (to M. E. G.), the European Commission FP7 Programme under BioStruct-X Grant 283570, Diamond Light Source Proposals mx8302/10130, and SOLEIL Proposal 20140843. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Video S1.
The atomic coordinates and structure factors (code 4ZIL) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TB
- tuberculosis
- TP
- thienopyrimidine
- NSD
- normalized spatial discrepancy
- SAXS
- small angle x-ray scattering
- PDB
- Protein Data Bank
- r.m.s.
- root mean square.
References
- 1. World Health Organization (2014) Global Tuberculosis Report 2013, www.who.int/tb/publications/global_report/en
- 2. Zumla A., Raviglione M., Hafner R., and von Reyn C. F. (2013) Tuberculosis. N. Engl. J. Med. 368, 745–755 [DOI] [PubMed] [Google Scholar]
- 3. Andries K., Verhasselt P., Guillemont J., Göhlmann H. W., Neefs J. M., Winkler H., Van Gestel J., Timmerman P., Zhu M., Lee E., Williams P., de Chaffoy D., Huitric E., Hoffner S., Cambau E., Truffot-Pernot C., Lounis N., and Jarlier V. (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227 [DOI] [PubMed] [Google Scholar]
- 4. Koul A., Dendouga N., Vergauwen K., Molenberghs B., Vranckx L., Willebrords R., Ristic Z., Lill H., Dorange I., Guillemont J., Bald D., and Andries K. (2007) Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 3, 323–324 [DOI] [PubMed] [Google Scholar]
- 5. Cohen J. (2013) Infectious disease: approval of novel TB drug celebrated with restraint. Science 339, 130. [DOI] [PubMed] [Google Scholar]
- 6. Zumla A., Nahid P., and Cole S. T. (2013) Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discovery 12, 388–404 [DOI] [PubMed] [Google Scholar]
- 7. Jackson M., McNeil M. R., and Brennan P. J. (2013) Progress in targeting cell envelope biogenesis in Mycobacterium tuberculosis. Future Microbiol. 8, 855–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lechartier B., Rybniker J., Zumla A., and Cole S. T. (2014) Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol. Med. 6, 158–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Albesa-Jové D., Chiarelli L. R., Makarov V., Pasca M. R., Urresti S., Mori G., Salina E., Vocat A., Comino N., Mohorko E., Ryabova S., Pfieiffer B., Lopes Ribeiro A. L., Rodrigo-Unzueta A., Tersa M., Zanoni G., Buroni S., Altmann K. H., Hartkoorn R. C., Glockshuber R., Cole S. T., Riccardi G., and Guerin M. E. (2014) Rv2466c mediates the activation of TP053 to kill replicating and non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 9, 1567–1575 [DOI] [PubMed] [Google Scholar]
- 10. Hartkoorn R. C., Sala C., Neres J., Pojer F., Magnet S., Mukherjee R., Uplekar S., Boy-Röttger S., Altmann K. H., and Cole S. T. (2012) Towards a new tuberculosis drug: pyridomycin-nature's isoniazid. EMBO Mol. Med. 4, 1032–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neres J., Hartkoorn R. C., Chiarelli L. R., Gadupudi R., Pasca M. R., Mori G., Venturelli A., Savina S., Makarov V., Kolly G. S., Molteni E., Binda C., Dhar N., Ferrari S., Brodin P., Delorme V., Landry V., de Jesus Lopes Ribeiro A. L., Farina D., Saxena P., Pojer F., Carta A., Luciani R., Porta A., Zanoni G., De Rossi E., Costi M. P., Riccardi G., and Cole S. T. (2015) 2-Carboxyquinoxalines Kill Mycobacterium tuberculosis through noncovalent inhibition of DprE1. ACS Chem. Biol. 10, 705–714 [DOI] [PubMed] [Google Scholar]
- 12. Raman S., Song T., Puyang X., Bardarov S., Jacobs W. R. Jr., and Husson R. N. (2001) The alternative σ factor SigH regulates major components of oxidative and heat stress responses in Mycobacterium tuberculosis. J. Bacteriol. 183, 6119–6125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holmgren A. (1995) Thioredoxin structure and mechanism: conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure 3, 239–243 [DOI] [PubMed] [Google Scholar]
- 14. Martin J. L. (1995) Thioredoxin: a fold for all reasons. Structure 3, 245–250 [DOI] [PubMed] [Google Scholar]
- 15. Qi Y., and Grishin N. V. (2005) Structural classification of thioredoxin-like fold proteins. Proteins 58, 376–388 [DOI] [PubMed] [Google Scholar]
- 16. Hanschmann E. M., Godoy J. R., Berndt C., Hudemann C., and Lillig C. H. (2013) Thioredoxins, glutaredoxins, and peroxiredoxins-molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 19, 1539–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 21. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., and Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sotomayor-Pérez A. C., Subrini O., Hessel A., Ladant D., and Chenal A. (2013) Molecular crowding stabilizes both the intrinsically disordered calcium-free state and the folded calcium-bound state of a repeat in toxin (RTX) protein. J. Am. Chem. Soc. 135, 11929–11934 [DOI] [PubMed] [Google Scholar]
- 26. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 27. Guinier A. (1939) La diffraction des rayon's X aux tres petits angles: application a l'etude de phenoménes ultramicroscopies. Ann. Phys. 12, 161–237 [Google Scholar]
- 28. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 29. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kozin M. B., and Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 32. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 33. Petoukhov M. V., and Svergun D. I. (2005) Global rigid body modelling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martí-Renom M. A., Stuart A. C., Fiser A., Sánchez R., Melo F., and Sali A. (2000) Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 [DOI] [PubMed] [Google Scholar]
- 35. Görner K., Holtorf E., Waak J., Pham T. T., Vogt-Weisenhorn D. M., Wurst W., Haass C., and Kahle P. J. (2007) Structural determinants of the C-terminal helix-kink-helix motif essential for protein stability and survival promoting activity of DJ-1. J. Biol. Chem. 282, 13680–13691 [DOI] [PubMed] [Google Scholar]
- 36. Tian G., Kober F. X., Lewandrowski U., Sickmann A., Lennarz W. J., and Schindelin H. (2008) The catalytic activity of protein-disulfide isomerase requires a conformationally flexible molecule. J. Biol. Chem. 283, 33630–33640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mazon H., Gábor K., Leys D., Heck A. J., van der Oost J., and van den Heuvel R. H. (2007) Transcriptional activation by CprK1 is regulated by protein structural changes induced by effector binding and redox state. J. Biol. Chem. 282, 11281–11290 [DOI] [PubMed] [Google Scholar]
- 38. Giganti D., Alegre-Cebollada J., Urresti S., Albesa-Jové D., Rodrigo-Unzueta A., Comino N., Kachala M., López-Fernández S., Svergun D. I., Fernández J. M., and Guerin M. E. (2013) Conformational plasticity of the essential membrane-associated mannosyltransferase PimA from mycobacteria. J. Biol. Chem. 288, 29797–29808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blanchet C. E., and Svergun D. I. (2013) Small-angle x-ray scattering on biological macromolecules and nanocomposites in solution. Annu. Rev. Phys. Chem. 64, 37–54 [DOI] [PubMed] [Google Scholar]
- 40. Mössner E., Huber-Wunderlich M., and Glockshuber R. (1998) Characterization of Escherichia coli thioredoxin variants mimicking the active-sites of other thiol/disulfide oxidoreductases. Protein Sci. 7, 1233–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mössner E., Huber-Wunderlich M., Rietsch A., Beckwith J., Glockshuber R., and Aslund F. (1999) Importance of redox potential for the in vivo function of the cytoplasmic disulfide reductant thioredoxin from Escherichia coli. J. Biol. Chem. 274, 25254–25259 [DOI] [PubMed] [Google Scholar]
- 42. Inaba K., and Ito K. (2008) Structure and mechanisms of the DsbB-DsbA disulfide bond generation machine. Biochim. Biophys. Acta 1783, 520–529 [DOI] [PubMed] [Google Scholar]
- 43. Wunderlich M., Jaenicke R., and Glockshuber R. (1993) The redox properties of protein disulfide isomerase (DsbA) of Escherichia coli result from a tense conformation of its oxidized form. J. Mol. Biol. 233, 559–566 [DOI] [PubMed] [Google Scholar]
- 44. Ali Khan H., and Mutus B. (2014) Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front. Chem. 2, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dirr H., Reinemer P., and Huber R. (1994) X-ray crystal structures of cytosolic glutathione S-transferases: implications for protein architecture, substrate recognition and catalytic function. Eur. J. Biochem. 220, 645–661 [DOI] [PubMed] [Google Scholar]
- 46. Wu B., and Dong D. (2012) Human cytosolic glutathione transferases: structure, function, and drug discovery. Trends Pharmacol. Sci. 33, 656–668 [DOI] [PubMed] [Google Scholar]
- 47. Inaba K., Murakami S., Suzuki M., Nakagawa A., Yamashita E., Okada K., and Ito K. (2006) Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell 127, 789–801 [DOI] [PubMed] [Google Scholar]
- 48. Malojcić G., Owen R. L., Grimshaw J. P., and Glockshuber R. (2008) Preparation and structure of the charge-transfer intermediate of the transmembrane redox catalyst DsbB. FEBS Lett. 582, 3301–3307 [DOI] [PubMed] [Google Scholar]
- 49. Qin J., Clore G. M., and Gronenborn A. M. (1994) The high-resolution three-dimensional solution structures of the oxidized and reduced states of human thioredoxin. Structure 2, 503–522 [DOI] [PubMed] [Google Scholar]
- 50. Jeng M. F., Campbell A. P., Begley T., Holmgren A., Case D. A., Wright P. E., and Dyson H. J. (1994) High-resolution solution structures of oxidized and reduced Escherichia coli thioredoxin. Structure 2, 853–868 [DOI] [PubMed] [Google Scholar]
- 51. Singh R., and Whitesides G. M. (1990) Comparisons of rate constants for thiolate-disulfide interchange in water and in polar aprotic solvents using dynamic proton NMR line shape analysis. J. Am. Chem. Soc. 112, 1190–1197 [Google Scholar]
- 52. Bullough P. A., Hughson F. M., Skehel J. J., and Wiley D. C. (1994) Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371, 37–43 [DOI] [PubMed] [Google Scholar]
- 53. Carr C. M., and Kim P. S. (1993) A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 73, 823–832 [DOI] [PubMed] [Google Scholar]
- 54. Carr C. M., and Kim P. S. (1994) Flu virus invasion: halfway there. Science 266, 234–236 [DOI] [PubMed] [Google Scholar]
- 55. Carr C. M., Chaudhry C., and Kim P. S. (1997) Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. U.S.A. 94, 14306–14313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Han X., Bushweller J. H., Cafiso D. S., and Tamm L. K. (2001) Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 8, 715–720 [DOI] [PubMed] [Google Scholar]
- 57. Fan D., Zheng Y., Yang D., and Wang J. (2003) NMR solution structure and dynamics of an exchangeable apolipoprotein, Locusta migratoria apolipophorin III. J. Biol. Chem. 278, 21212–21220 [DOI] [PubMed] [Google Scholar]
- 58. Narayanaswami V., Kiss R. S., and Weers P. M. (2010) The helix bundle: a reversible lipid binding motif. Comp. Biochem. Physiol. A Mol. Intergr. Physiol. 155, 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang J., Sykes B. D., Ryan R. O. (2002) Structural basis for the conformational adaptability of apolipophorin III, a helix-bundle exchangeable apolipoprotein. Proc. Natl. Acad. Sci. U.S.A. 99, 1188–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.