

Background: HPLC enables quantification of bacterial cell-wall composition, yet systematic studies across strains, species, and chemical perturbations are lacking.
Results: UPLC coupled to computational modeling enables submicroliter injection volumes, and was applied to systematic analysis of several Gram-negative species.
Conclusion: Composition is largely decoupled from morphology, although large interspecies differences were evident.
Significance: UPLC and automated analysis accelerate discovery regarding peptidoglycan and physiology.
Keywords: cell wall, cytoskeleton, high-performance liquid chromatography (HPLC), morphogenesis, peptidoglycan, muropeptide
Abstract
The bacterial cell wall is a network of glycan strands cross-linked by short peptides (peptidoglycan); it is responsible for the mechanical integrity of the cell and shape determination. Liquid chromatography can be used to measure the abundance of the muropeptide subunits composing the cell wall. Characteristics such as the degree of cross-linking and average glycan strand length are known to vary across species. However, a systematic comparison among strains of a given species has yet to be undertaken, making it difficult to assess the origins of variability in peptidoglycan composition. We present a protocol for muropeptide analysis using ultra performance liquid chromatography (UPLC) and demonstrate that UPLC achieves resolution comparable with that of HPLC while requiring orders of magnitude less injection volume and a fraction of the elution time. We also developed a software platform to automate the identification and quantification of chromatographic peaks, which we demonstrate has improved accuracy relative to other software. This combined experimental and computational methodology revealed that peptidoglycan composition was approximately maintained across strains from three Gram-negative species despite taxonomical and morphological differences. Peptidoglycan composition and density were maintained after we systematically altered cell size in Escherichia coli using the antibiotic A22, indicating that cell shape is largely decoupled from the biochemistry of peptidoglycan synthesis. High-throughput, sensitive UPLC combined with our automated software for chromatographic analysis will accelerate the discovery of peptidoglycan composition and the molecular mechanisms of cell wall structure determination.
Introduction
Plant, fungal, and algal cells as well as most bacteria have a cell wall surrounding the cytoplasmic membrane that defines the shape of the cell (2) and provides mechanical resistance to expansion due to the osmotic pressure from within the cell (3). In bacteria, the cell wall is an important antibiotic target (4), with treatment often disrupting the integrity of the cell wall and eventually leading to cell lysis (5). The cell wall plays an important role in pathogenesis, in part due to the uncommon stereochemistry of the three d-isomers of amino acids that defend against most proteases that otherwise would degrade the cell wall (1, 6). Furthermore, many surface proteins anchored to the cell wall are involved in pathogenic processes such as host-cell invasion and immune system interactions (7). Cell morphology has been linked to many important behaviors, and cell size often varies with fitness (8–11). Thus, quantitative comparisons of the biochemical makeup of the cell wall of different species and strains as well as in the presence of chemical or environmental perturbations are critical for our global understanding of microbial physiology.
The bacterial cell wall (also known as the murein sacculus) is a single macromolecular polymer network composed of peptidoglycan, which consists of long strands of glycans that are bound together through cross-links between short strings of peptides (1). The disaccharide subunits composing the glycan strands contain one N-acetyl-glucosamine (GlcNAc) and one N-acetylmuramic acid (MurNAc), to which a peptide stem with five amino acids is appended through a d-lactic acid linkage (1). The disaccharide subunit and the peptide stem are collectively referred to as a muropeptide. Muropeptide precursors are synthesized in the cytoplasm, flipped across the inner membrane, and incorporated into an existing glycan strand by penicillin-binding proteins (12). In the rod-shaped bacterium Escherichia coli, mutations or changes to the expression levels of essential penicillin-binding proteins can result in subtle changes to rod dimensions (13) or cell rounding (13, 14). In rod-shaped bacteria, cell-shape determination and maintenance involve the spatiotemporal regulation of cell wall growth, which requires the actin homolog MreB (15). MreB forms polymers attached to the inner surface of the cytoplasmic membrane (15), and its localization dynamics guides the pattern of cell wall synthesis (16, 17). In E. coli, when MreB is depleted or inhibited by the small molecule A22, cells become round and eventually lyse (18, 19). Several point mutations in mreB have been shown to alter cell width (19), and some of these mutations confer an environment-dependent fitness advantage that scales with cell size (20). Thus, cell shape and size emerge from a complex system of interactions among MreB, the cell wall synthesis machinery, and peptidoglycan composition.
High performance liquid chromatography (HPLC) has been used for over 50 years for the separation, identification, and quantification of biomolecules (21). HPLC techniques have been tailored to analyze microbial biomolecules associated with cell wall growth, including muropeptides, cell wall digestion products, glycan strands, stem peptides, and d-amino acid incorporation (1, 22). For soluble muropeptides isolated from bacterial cell walls, typical HPLC runs require 1–2 h and 100–200 μl of sample injected to resolve individual peaks (1). Typical HPLC preparations result in ∼0.5 mg of muropeptides, which produces roughly 1 absorbance unit for the highest peaks (M4 or D44, Fig. 1A, supplemental Table S1). Muropeptide identities can be determined either by collecting the eluted volume from an individual peak and subjecting it to mass spectrometry (MS) (23), or by comparison to previous studies based on the elution time of the peak. Muropeptides fall into general groups of monomers, dimers, and trimers (tetramers are less common) that can be further classified based on the attached peptide stem(s) and/or the presence of an anhydro group on the disaccharide (1, 24). The abundance of each peak is usually estimated by integrating the absorbance between the two time points around the peak elution time at which the chromatogram reaches the baseline absorbance (1). The fraction of muropeptides that are cross-linked can be calculated from a ratio based on the overall abundance of monomers and multimers (25), and the average glycan strand length can be determined from the fraction of anhydro muropeptides, which designate the ends of glycan strands (1, 25, 26). In addition to the quantification of peptidoglycan composition across a variety of species, mutants, and conditions, HPLC muropeptide analyses have also been used to identify and characterize antibiotics and bacteriocins based on their effects on peptidoglycan (27–29).
FIGURE 1.

Muropeptide analysis using UPLC and Chromanalysis is high-throughput and highly sensitive. A, chromatograms resulting from injection of 0.2 and 4 μl of the same E. coli MG1655 sample yield quantitatively similar peak abundances. Muropeptide labels: M = monomer, D = dimer, T = trimer; (2, 3, 4, 5) indicate the number of amino acid stem peptides; modifications: G = glycine replacing l-alanine, L = two additional amino acids from Pronase E cleavage, D = 3,3-diaminopimelic acid (DAP)-DAP cross-bridge, N = terminating anhydro-muropeptide. B, subtraction of a water blank from the chromatogram produces a baseline with essentially zero absorbance, improving the accuracy of peak abundance quantification. C, schematic of Gaussian fitting by Chromanalysis, with blue circles denoting the centers and amplitudes of the fitted Gaussians. A skewed peak such as the M3L peak shown is well fit with a sum of two Gaussians. The M3L chemical structure is shown on the right. D, mean muropeptide species quantifications from seven E. coli MG1655 peptidoglycan samples is more accurate using Chromanalysis (blue) compared with the commercial software Empower (yellow). Error bars represent 1 S.D. E, with UPLC, all muropeptide moieties exhibit a linear relationship between injection volume and area under the peak for an E. coli MG1655 sample. Curves for monomers, dimers, and trimers are colored in red, green, and blue, respectively. Each curve was normalized so that the best-fit line with a y intercept of 0 had an area of 1 for an injection volume of 10 μl. Curve thickness is proportional to the relative abundance of the peak in a 10-μl injection. Inset, zoom of region with submicroliter volumes.
Although many studies have been conducted with HPLC, several important technical and biological questions have not been addressed. Due to the large injection volumes required for resolving low-abundance peaks with HPLC, a sample can only be used for at most a few chromatographic analyses. Moreover, for cells at low optical density, which have low peptidoglycan density per cell, or in which peptidoglycan recovery is challenging (such as from some Gram-positive species), a single preparation may not produce enough peptidoglycan for HPLC analysis; one such case is E. coli L-forms, a cell wall-deficient state that has been reported to have a few percent of the peptidoglycan levels of normal, rod-shaped E. coli cells (30, 31). Furthermore, although some model organisms such as E. coli have been the subject of numerous HPLC studies (1) (and others such as Pseudomonas aeruginosa have been the subject of surprisingly few), a systematic comparison among different strains of a given organism has yet to be undertaken; this lack may be due in part to the long preparation and run times required for HPLC. Full exploitation of the power of liquid chromatography for peptidoglycan analysis will require a highly sensitive, high-throughput, reproducible technique that is supported by quantitative analysis tools that allow systematic extraction and comparison of muropeptide abundances across sample volumes and conditions.
Ultra performance liquid chromatography (UPLC)4 addresses many of these challenges; the higher pressures relative to HPLC increase throughput by decreasing run time and potentially improve resolution. Here, we present a UPLC protocol for muropeptide analyses and report its application to three Gram-negative model organisms (E. coli, Vibrio cholerae, and P. aeruginosa) to compare peptidoglycan variability both across species and across common laboratory strains, over a range of morphologies and cell sizes. We demonstrate the importance of empirical determination of the baseline for accurate quantification of peak abundances, and we present a user-friendly, flexible software package for modeling the chromatogram as a sum of Gaussian absorbance peaks that allows for identification and quantification of moeity abundances. We also show that clinical isolates of P. aeruginosa have overall peptidoglycan content that is similar to that of non-pathogenic laboratory strains. Finally, we use UPLC to demonstrate that the cell widening caused by treatment with sublethal doses of A22 is not coupled to changes in either the abundances of any muropeptide species or peptidoglycan density.
Experimental Procedures
Ultra Performance Liquid Chromatography
Peptidoglycan samples were prepared from bacterial cultures as previously described (22). Briefly, bacterial cell walls were isolated using a combination of ultracentrifugation and digestion with Pronase E and muramidase. These enzymes do not perturb the cross-links or the anhydro muropeptides that are used to compute average glycan strand length (1). Soluble muropeptide volumes from 0.1 to 10 μl were injected onto a Waters Acquity UPLC H-Class system equipped with an Acquity UPLC BEH C18 1.7-μm column, PDA detector, and fraction collector. Absorbance was detected at 205 nm and separation of muropeptides was achieved using 50 mm sodium phosphate, pH 4.35, + 0.4% (v/v) sodium azide for solvent A, and 75 mm sodium phosphate, pH 4.95, + 15% (v/v) methanol for solvent B. Peaks were identified based on retention time, which has been extensively established using amino acid analysis, enzymatic digestion, Edman degradation, radioactive labeling, dansylation, paper chromatography, reduction, and altering temperature, pH, and ionic strength (25, 26, 32–40). Flow was set to 125 μl/min with a linear gradient over 50 min to complete elution of all muropeptides within 25 min. Blank runs were performed by injecting 10 μl of water and separating using the above conditions. Molar fractions of oligomers (dimers, trimers, and tetramers) are determined from the area under the peaks in the chromatogram, and the cross-linking percentage is calculated according to: % cross-linking = % molar fraction dimers + 2× (% molar fraction trimers) + 3× (% molar fraction tetramers). The multipliers in the formula above are used to account for the number of cross-links per oligomer; for instance, a trimer contains two cross-links and therefore is multiplied by 2. Average glycan strand length is calculated according to: average glycan strand length = 100 muropeptides/sum of (% molar fraction of all anhydro peaks).
Quantitation of Muropeptides using Chromanalysis
Chromanalysis is a Matlab-based software package designed to fit a chromatogram with a set of Gaussians. For chromatograms with an associated blank, the blank was first subtracted (Fig. 1B), setting the baseline to zero absorbance. A user-defined percentage can be removed from the beginning and end of the chromatogram to ignore peaks from salts in the buffers. Peaks and valleys were then identified using the “findpeaks” function. For chromatograms without an associated blank, the baseline was defined by connecting the valleys for which there was not an absorbance increase of >0.01 relative to the previous valley. The baseline was then iteratively refined by removing local maxima and replacing them with an interpolated value. The baseline was further refined by iteratively removing the points on either side of a valley, and the result was subtracted from the chromatogram.
For each of the 50 largest peaks, 50 time points on either side of the maximum were fit to a Gaussian to determine the center and amplitude of the peak. Any Gaussian fits with a center time shifted by >2% or an amplitude that was either negative or shifted by >10% relative to the peak maximum had their amplitude set to zero so that they could be re-fit at a later point in the algorithm. For pairs of peaks whose centers were closer than 1.5 times the sum of their widths, the corresponding region of the chromatogram was re-fit by a sum of two Gaussians, and the new fit was evaluated relative to the old fit by computing the sum of squared residuals.
Next, the resulting Gaussian fits were subtracted from the original chromatogram to identify smaller peaks that may have been masked by other larger, nearby peaks. These new peaks were again fit to Gaussians with the same filtering as in the previous step. The sum of the final set of Gaussians was usually indistinguishable from the initial chromatogram.
For peak identification, the chromatogram was compared with a standard for the given moieties over the windows of retention times in which the peaks are expected to elute. Standards can be either edited or newly defined in the Chromanalysis graphical user interface. The chromatogram peaks are aligned to the standard using a dynamic programming algorithm to account for differences in gaps between peaks. The user can then optimize the labeling by manually altering the retention times. Descriptions of other functionalities, such as the output of chromatogram analysis into various file formats, comparison of chromatograms, and an extensive tutorial video and manual for the use of the Chromanalysis interface, can be found in the supplemental materials.
Imaging and Analysis of Cell Morphology
Cells were grown overnight in lysogeny broth (41) at 37 °C. They were then diluted 1:100 in fresh medium and imaged when cultures reached early exponential phase (optical density at 600 nm = 0.3). Cells were placed on 1% agarose pads made from lysogeny broth and imaged within minutes. Phase-contrast images were acquired with a Nikon Ti-E inverted microscope using a 100× (NA 1.40) oil immersion objective and a Neo sCMOS camera (Andor Technology). The microscope was outfitted with an active-control environmental chamber for temperature regulation (HaisonTech). Images were acquired using μManager version 1.4 (42).
Custom Matlab (MathWorks) image-processing code was used to segment cells and to identify cell outlines from phase-contrast microscopy images (16). A local coordinate system was generated for each cell outline using a method adapted from MicrobeTracker (43). Cell widths were calculated by averaging the distances between contour points perpendicular to the cell midline, excluding contour points within the poles and sites of septation. Cell length was calculated as the length of the midline from pole to pole. Measurements of average cell width across a population were highly reproducible, and we previously validated our measurements using cell outlines extracted from cells stained with the membrane dye FM4-64 (20). All populations were imaged in an unsynchronized state, and cell lengths follow the distribution expected from a rod-shaped organism undergoing binary fission.
Protein Quantification and A22 Treatment
The protein content of each peptidoglycan sample was measured using the Bio-Rad DC Protein Assay. Sublethal treatment with A22 was performed as follows. An overnight culture of REL606 was diluted 1:100 into 50 ml of fresh LB medium and grown to early exponential phase (∼2 h, A600 = 0.3) before a 1:50 dilution into 250 ml of pre-warmed LB supplemented with a range of A22 concentrations. Cultures were grown to mid-exponential phase (A600 ∼ 0.7) before harvesting for extraction of muropeptides and image analysis for determination of cell morphology.
Statistical Analysis
The non-parametric Kruskal-Wallis test was employed for analysis of differences in mean characteristics between strains. An important feature of this test is its insensitivity to differences in sample size. For comparison of peptidoglycan measurements between species, pairwise t tests assuming equal variance were performed. In all cases, determination of significance accounted for multiple hypothesis testing with a Bonferroni correction.
Results
UPLC Accurately Quantifies Muropeptide Abundance from Extremely Low Volumes
To test the resolution of UPLC for muropeptide analysis, we first sought to identify elution conditions that maximize resolution and speed compared with HPLC. We prepared and digested sacculi from E. coli MG1655 cells using a protocol modified from standard HPLC analyses (“Experimental Procedures”) (22). Across various combinations of column temperature, gradient curve and length, and flow rate, we achieved a balance between optimal resolution and shorter analysis time with a column temperature of 55 °C, a linear gradient over 50 min, and a flow rate of 125 μl/min. These parameters enabled separation of all known E. coli muropeptide moieties in under 25 min (Fig. 1A), compared with the 2-h analysis times typically required for HPLC. In addition to this shorter time scale, UPLC achieved resolutions with 4-μl injections (Fig. 1A) that were comparable with those of the 200-μl injections typically required for HPLC (data not shown).
For UPLC muropeptide separations, we used buffers that were identical to those used for HPLC. Methanol was used in the elution buffer to elute muropeptides off the column based on hydrophobicity. Sodium azide was used in the equilibration buffer to compensate for methanol absorption of UV light at the monitoring wavelength of 205 nm. Despite this corrective measure, a flat baseline is often not achieved in muropeptide chromatographic analyses (44–50), which introduces error into the quantification of peak abundances because the integrated area is usually determined based on either the point at which the peaks reach the baseline or the valley between two peaks for non-baseline-resolved moieties. To directly measure the baseline, we ran a water blank before and after each sample injection on the UPLC, although injection of solvent A (see “Experimental Procedures”) can be used for a blank as well. We subtracted the preceding water blank chromatogram from the muropeptide chromatogram. This technique led to a flat baseline at zero UV absorbance from which peak abundances could be quantified (Fig. 1B).
To automate the detection and quantification of peaks in the chromatogram, we developed a Matlab-based software package (Chromanalysis) that identifies peaks through Gaussian fitting, aligns the peak elution times to a standard chromatogram to assign identities, and quantifies the area under each Gaussian based on its amplitude and width (Fig. 1C, supplemental Table S1, and ”Experimental Procedures“). Most moieties were well fit by a single Gaussian, and peaks with skew were usually fit by two, or at most three, Gaussians, perhaps indicating the presence of two moieties with similar elution times that create overlapping peaks. In cases where multiple Gaussians are fit in apparent single absorbance maxima in the chromatogram, our quantification method assigns the combined weight within user-defined intervals to a given moiety. Importantly, the quantification of a given peak is usually not affected by the presence of other nearby peaks; that is, when peaks overlap, Chromanalysis can nevertheless determine the abundances of the two peaks through the Gaussian-fitting process, despite the fact that neither peak may drop all the way to the baseline. Chromanalysis takes as input raw text files of chromatogram output from UPLC or HPLC sample runs, and allows the user to specify the type of baseline subtraction method, the subinterval of the run to be analyzed, and the standard for peak labeling. Peak identities can also be manually reassigned; furthermore, the software can be run in batches for rapid analysis. By comparison with the commercial software Empower, Chromanalysis provides more precise accurate quantification of replicate E. coli MG1655 peptidoglycan samples (Fig. 1D). Chromanalysis is open-source and is available as a Bitbucket repository, along with an instruction manual and tutorial video (supplemental materials).
Given that we were able to achieve peak heights and resolution from 4-μl injections on UPLC that were similar to those from 200-μl injections on HPLC, we next sought to test the limits of this increased detection sensitivity. We injected volumes of muropeptide samples in 1-μl increments from 1 to 10 μl, and in 0.1-μl increments from 0.1 to 0.5 μl. Even with an injection volume as small as 0.2 μl, the muropeptide profile mirrored that of a 4-μl UPLC injection (Fig. 1A), with quantitatively similar molar fractions of virtually all moieties. Moreover, the total integrated area of most muropeptide peaks increased linearly with injection volume (Fig. 1E), with linear fits having an R2 value >0.91, indicating that equivalent results to a 10-μl injection can be achieved with ∼50-fold less volume. A few moieties exhibited some deviation from a linear relationship between injection volume and peak area, particularly at volumes <1 μl (Fig. 1E, inset); these muropeptide moieties are consistently hard to accurately quantify in both large and small injections, and may consist of low-abundance anhydro moieties that are subject to more variable integration. Chromanalysis is well suited to the challenge of accurately quantifying low-abundance muropeptides, by robustly measuring relative peak abundances across different sample baselines and signal-to-noise ratios (Fig. 1E). The abundance of some moieties in injection volumes <1 μl was so small that they did not absorb enough UV to result in a chromatographic peak (Fig. 1E, inset), indicating that injection volumes of 1–5 μl are optimal to ensure accurate quantitation. Based on the estimate of 1 mg of peptidoglycan in a sample preparation, a 1-μl injection would be equivalent to <10 μg of peptidoglycan. Nonetheless, submicroliter volumes can be used for more abundant moieties, demonstrating the sensitivity advantage of UPLC over HPLC.
Robust Muropeptide Composition Across Common Laboratory Strains of Gram-negative Bacteria
By far, the majority of HPLC muropeptide analyses have focused on E. coli strains in different growth conditions (13, 19, 25, 26, 31, 51–54). A variety of laboratory strains have been used, with key quantities such as cross-linking varying over a range from 25% (25, 52) to 37% (31). We define cross-linking as the percentage of the number of cross-links per muropeptide, with one for the number of cross-links per dimer and two for each trimer. It is unclear whether this variability results from differences in genotype, growth medium, temperature, or sample preparation. To address the differences in peptidoglycan content across common laboratory strains of several model organisms, we prepared muropeptide samples from different wild-type strains of E. coli, V. cholerae, and P. aeruginosa. Cells from four E. coli K12 strains (MG1655, NCM3722, BW25113, MC1000), uropathogenic E. coli (UPEC), and E. coli B (REL606) were grown to an optical density of 0.3, and then imaged on agar pads to ascertain any differences in cell morphology (“Experimental Procedures”). From phase-contrast images, we measured the average length and width of hundreds to thousands of single cells from each strain using custom image analysis software (Fig. 2A, “Experimental Procedures”). Cells were measured at similar times after exit from stationary phase. The average widths of the six E. coli strains ranged from 0.8 to 1.04 μm, whereas their average lengths ranged from 3.36 to 5.22 μm. Nevertheless, UPLC analysis of these E. coli strains revealed that cross-linking and average glycan strand length were not significantly different between strains (Kruskal-Wallis test: cross-linking, p = 0.44; strand length, p = 0.52), with sample-to-sample variability typical of HPLC muropeptide analyses (5–7%) (Fig. 2B).
FIGURE 2.

Muropeptide composition is maintained across strains of E. coli and V. cholerae but varies substantially between the two species. A and C, cell size varies across six E. coli strains (A) and five V. cholerae strains (C). Average width and length calculated from phase-contrast images (“Experimental Procedures,” n = 743–2457 cells for E. coli, n = 1191–5448 cells for V. cholerae). Shown are the cells from each population with the smallest sum of squared deviation in length and width from the average. B, all E. coli strains have similar cross-linking percentages and average glycan strand lengths. n denotes the number of samples contributing to each data point. D, V. cholerae strains have similar cross-linking and glycan strand lengths across different strains, but have less cross-linking and shorter glycan strand lengths than E. coli. n denotes the number of samples contributing to each data point. Inset: smoothed curvature across the cell outlines aggregated for each population. The left peak is shifted to slightly positive values in all strains, particularly CA401, M010, and 0139, representing the overall curvature of the cell body. The right peak is due to the cell poles.
The Gram-negative bacterium V. cholerae has curved rod morphology, and HPLC analyses of strain N16961 previously detected shorter glycan strands than in E. coli (26, 55, 56). We performed a similar survey of morphology and peptidoglycan content from strains CA401, MO10, MZ02, MAK757, and 0139, which cover the El Tor (MO10, MAK757) and Classical (CA401) biogroups, and the O139 (MO10), O1 (CA401, MAK757), and non-O1, non-O139 (MZO2) serotypes. Single-cell quantification of morphology showed that average cell widths ranged from 0.9 to 1.13 μm, and average lengths ranged from 2.66 to 4.1 μm (Fig. 2C). Interestingly, the strains also displayed different levels of curvature along the cell outline (Fig. 2D, inset). Although it has been suggested that curved cells could be produced by varying the spatial pattern of cell wall cross-linking (57), our measurements indicated no significant differences in either cross-linking percentages or average glycan strand lengths across V. cholerae strains (Kruskal-Wallis test: cross-linking, p = 0.07; strand length, p = 0.33) (Fig. 2D). This observation agrees with previous HPLC analysis of the curved organism Caulobacter crescentus, which showed that cross-linking is unaffected in straight ΔcreS mutants (58). Together, our data suggest that in controlled conditions, there is little variability among strains of the model organisms E. coli and V. cholerae, but significant differences between the two species with respect to peptidoglycan cross-linking (average across strains and two-sided t test: E. coli, 31 ± 0.3% (standard error, S.E.); V. cholerae, 26 ± 0.4% (S.E.), p < 0.001) and glycan strand length (average across strains and two-sided t test: E. coli, 27 ± 1 (S.E.); V. cholerae, 12 ± 0.6 (S.E.), p < 0.001).
Clinical and Laboratory P. aeruginosa Strains Display Similar Peptidoglycan Content
P. aeruginosa is a Gram-negative, opportunistic human pathogen and is associated with infections in cystic fibrosis, burns, and immunocompromised patients (59–64). Some strains are resistant to many antibiotics, and therefore P. aeruginosa is of significant clinical interest (61–63, 65–67). The peptidoglycan of P. aeruginosa is of the A1γ chemotype, the same as E. coli (65, 67–69), and is characterized by a 4-3 cross-link involving the stem peptide diaminopimelic acid (24). However, a quantitative analysis of the chemical composition of P. aeruginosa peptidoglycan has not been undertaken, particularly in relationship to pathogenicity.
Because P. aeruginosa can cause chronic infections, we hypothesized that muropeptide abundances in clinical isolates of P. aeruginosa might differ compared with laboratory strains to evade immune system recognition. To determine this variability across lab strains, we analyzed three common wild-type strains (PA01, PA14, and PAK) and three clinical isolates (SMC715, SMC726, and SMC738) from eye samples (70). Morphological analysis revealed some variability in cell size (Fig. 3A), as with E. coli (Fig. 2A) and V. cholerae (Fig. 2C), but overall cell shape was qualitatively similar across non-pathogenic and pathogenic strains (Fig. 3A). Similar to E. coli and V. cholerae, cross-linking percentages and average glycan strand lengths were not significantly different across all strains (Kruskal-Wallis test: cross-linking, p = 0.07; strand length, p = 0.07) (Fig. 3B). As expected from a previous study (65), P. aeruginosa strains exhibited a small yet significant increase in cross-linking relative to E. coli (average across strains and two-sided t test: P. aeruginosa, 33 ± 0.2%, E. coli, 31 ± 0.3%, p < 0.001), and average glycan strand length (17 ± 0.4) (Fig. 3B) was intermediate between those of E. coli (27 ± 1) and V. cholera (12 ± 0.6) (Fig. 2, B and D). Differences in glycan strand length between species were highly significant (species compared and two-sided t test: P. aeruginosa versus E. coli, p < 0.001; P. aeruginosa versus V. cholerae, p < 0.001). There was less variability in glycan strand lengths among P. aeruginosa strains (Fig. 3B), suggesting that this quantity may be more tightly regulated in P. aeruginosa and V. cholerae than in E. coli. Our UPLC muropeptide analyses of clinical isolates revealed two peaks specific to these P. aeruginosa clinical isolates that appeared at elution times between 8 and 10 min, on either side of M3L; these peaks were at low abundance and hence had negligible effects on our estimates of cross-linking and glycan strand length.
FIGURE 3.
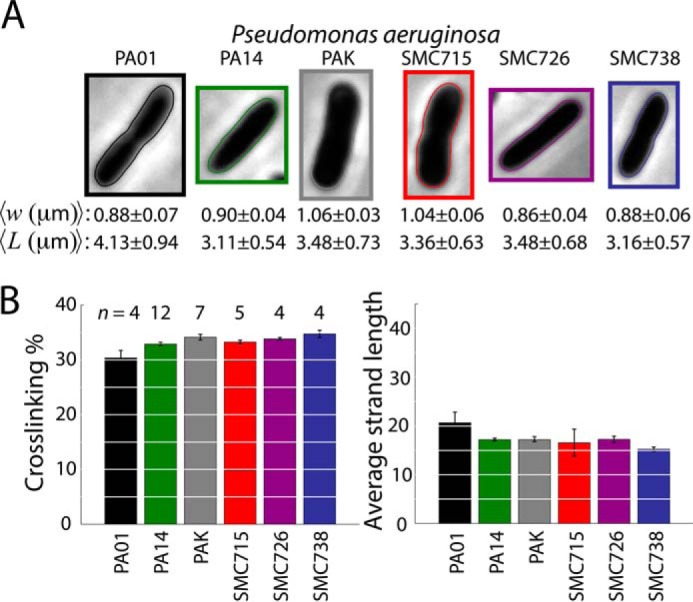
Clinical isolates of P. aeruginosa exhibit similar peptidoglycan composition to laboratory strains. A, laboratory strains and clinical isolates varyin cell size; average width and length were calculated from phase-contrast images (n = 810–13065 cells). Shown are the cells from each population with the smallest sum of squared deviation in length and width from the average. B, cross-linking percentage and average glycan strand lengths are similar across strains. Glycan strand length is intermediate between V. cholerae and E. coli. n denotes the number of samples contributing to each data point.
Chemically Induced Cell Size Increase Is Independent of Changes in Peptidoglycan Composition
We observed that peptidoglycan composition was reasonably constant across strains of E. coli, V. cholerae, and P. aeruginosa with a range of cell sizes, indicating that cell wall synthesis maintains its biochemical output despite changes in metabolism associated with variations in cell size in these organisms. However, it was not clear whether muropeptide abundance would be maintained for a given genotype when cell size changed. To systematically vary cell width (and hence surface area and volume), we employed the small molecule A22, which causes depolymerization of the MreB cytoskeleton (14, 16, 18). Although MreB is not directly responsible for any of the enzymatic steps in cell wall synthesis, we previously showed that sublethal doses of A22 increase the steady-state width of E. coli cells (71). Thus, although high concentrations of A22 have been reported to halt cell wall synthesis (54), sublethal doses clearly allow the cell to maintain at least some wall growth. We exposed E. coli REL606 cells to A22 concentrations between 0 and 2.5 μg/ml (just below the minimum inhibitory concentration) for 2.5 h during exponential growth, measured cell morphology, and then performed UPLC muropeptide analysis. As expected, the width w increased approximately linearly as a function of the A22 concentration c (Fig. 4A), with a best linear fit of,
where c is measured in μg/ml. In our UPLC muropeptide analyses, we found that the percentage of mole fractions of virtually all peaks were maintained across A22 concentrations (Fig. 4B), indicating that changes in cell size need not be coupled to changes in the biochemical composition of the cell wall. To determine whether peptidoglycan density changed as a function of A22 concentration, we measured the total amount of protein in each sample, which we expected to scale with the cell volume,
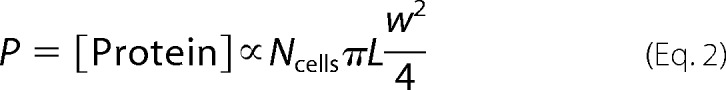 |
where Ncells is the number of cells in the sample and L is the average cell length. In contrast, the amount of peptidoglycan, which we estimated by the total integrated absorbance of the baseline-corrected chromatogram, should scale with the surface area,
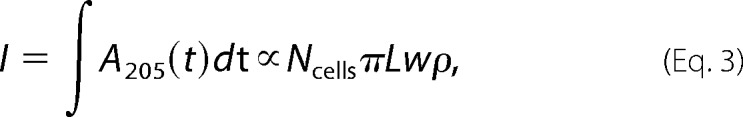 |
where ρ is the peptidoglycan density. Thus, if ρ is unaffected by sublethal A22 treatment, then the ratio of I and P should scale as 1/w. When we fit our measured ratios across different values of c to the function 1/(1 + αc), where α represents the constant of proportionality in the decrease of I/P with c, we obtained the value α = 0.23. This agrees well with our width measurements described by Equation 1, which would predict a value of 0.22/0.83 = 0.26 (Fig. 4C). Therefore, our data indicate that both peptidoglycan composition and density are unaffected by sublethal doses of A22.
FIGURE 4.

Sublethal treatment with the antibiotic A22 causes cell width to increase without altering peptidoglycan composition or density. A, cell width reaches a new steady-state value after 2.5 h of A22 treatment at sublethal concentrations, and increases approximately linearly as a function of A22 concentration. Average width and length were calculated from phase-contrast images (n = 195–549 cells). Shown are the cells from each population with the smallest sum of squared deviation in length and width from the average. B, relative abundances of each muropeptide moiety remain essentially unchanged under sublethal A22 treatment. C, the ratio of integrated absorbance of the baseline-subtracted chromatogram to the concentration of protein decreases with increasing A22 concentration (c). A fit to the function A/(1 + Bc) (blue dashed line) exhibits a trend similar to that of the inverse of the average cell width ⟨w⟩ (black dotted line), indicating that the ratio is proportional to the surface area-to-volume ratio, as expected for cylinders with constant peptidoglycan density. The standard errors of the estimates of A and B are 0.055 and 0.063, respectively. n denotes the number of samples contributing to each data point.
Discussion
Muropeptide chemical analyses using HPLC have provided a wealth of information that has guided studies of bacterial cell wall structure and function. The advent of UPLC with more robust submicron particles that can withstand higher pressures has the potential to extend the capabilities of liquid chromatography. Here, we have presented UPLC peptidoglycan analyses across a broad range of bacterial strains, species, and chemical treatments, thereby establishing a standard of quantitative data to which future analyses can be related.
The benefits of UPLC include higher throughput, with a reduction in run time from 2 h in HPLC to 25 min in UPLC (Fig. 1A) that allows maximum turnover, and a reduction in sample injection volume from 200 μl in HPLC to <1 μl in UPLC (Fig. 1E), which allows maximum efficiency from a single sample. The optimal UPLC separation of muropeptides involved running at a column temperature of 55 °C, a linear gradient over 50 min, and a flow rate of 125 μl/min. Smaller injection volumes enable flexibility of analysis. A standard experimental procedure for producing a sample of soluble muropeptides yields ∼200 μl of sample, enough for 1–2 HPLC injections. By injecting only a few microliters at a time on UPLC, orders of magnitude of more injections can be realized from each preparation without sacrificing accuracy. This allows multiple repeat injections for troubleshooting the separation of peaks while saving the sample for further muropeptide purification and subsequent analysis by MS, both of which greatly enhance the productive output and precision from the multiday sample preparation. Moreover, the reduction in run time to a quarter of the length of an HPLC run not only allows faster sample analysis, it also enables a leap in throughput through the simultaneous loading of 96 samples for continuous operation of the UPLC instrument. To realize the benefits of this increased throughput, we have also developed Matlab-based software to automate muropeptide quantification and peak identification. This software empowers all researchers, even those with limited familiarity with chromatographic techniques and analysis, to quickly and accurately quantify common chemical properties of cell wall samples. We are distributing this software open-source to facilitate HPLC/UPLC studies by the wider community.
To date, the vast majority of muropeptide HPLC analyses have been carried out on E. coli. To systematically define the variability in peptidoglycan composition across samples, strains, and species, we focused on laboratory and pathogenic strains of model Gram-negative species. The higher throughput of UPLC facilitated an extensive comparison with 115, 19, and 36 analyses of E. coli, V. cholerae, and P. aeruginosa strains, respectively. Within species, our analyses revealed a high degree of conservation in the global peptidoglycan properties of cross-linking percentage and average glycan strand length, despite significant variability in cell size (Figs. 2 and 3). These measurements suggest that peptidoglycan composition is likely maintained across closely related strains, although growth conditions (25, 26, 51) or mutations (51) may have a significant impact. We also found that sublethal concentrations of A22 increased cell width without changing peptidoglycan composition or density (Fig. 4); this result presents an interesting comparison with previous studies showing that high, lethal doses of A22 halt peptidoglycan synthesis (54). Nonetheless, peptidoglycan properties (particularly glycan strand length) varied across species, likely illustrating differences in the enzymatic kinetics of the synthesis machinery. The approximate maintenance of cross-linking percentage across species may reflect a conservation of the biophysical constraints on the cell wall imposed by the 4-3 cross-bridge linkage common to Gram-negative, rod-shaped bacteria.
Our results suggest that the spatiotemporal dynamics of the synthesis machinery may be more important for determining cell size and shape than the biochemical output of this machinery. Our UPLC analyses confirmed that cell wall synthesis is robust and tightly regulated. Future, more challenging analyses of peptidoglycan may benefit from the improved sensitivity of UPLC, particularly in the case of Gram-positive bacteria, which can have more highly cross-linked walls. Organisms with a broader range of muropeptide species, such as tetramers, will also be amenable to UPLC. Taken together, these investigations of a wide range of species with different growth patterns, ecological niches, and interactions with other organisms will yield insight into the general principles governing morphogenesis.
Author Contributions
S. M. D., M. A. D. P, and K. C. H. conceived and coordinated the study. S. M. D., A. M., and R. D. M. performed the experiments. S. M. D., C. T., and A. M. wrote the Chromanalysis software. All authors participated in the writing of the paper.
Acknowledgments
We thank the Campbell Laboratory of Ophthalmic Microbiology at UPMC for generously providing P. aeruginosa clinical isolates, Brian Hammer for V. cholerae strains, Shripa Patel and Dick Winant of the Stanford Protein and Nucleic Acids facility for help with mass spectrometry, and Thad Hughes for helpful discussions.
This work was supported by a Stanford Interdisciplinary Graduate Fellowship (to C. T.), a National Science Foundation Graduate Research Fellowship (to A. M.), the Laboratory for Infection Medicine Sweden, the Knut and Alice Wallenberg Foundation, the Swedish Research Council (to F. C.), a Bio-X Senior Postdoctoral Fellowship (to R. D. M.), and National Institutes of Health Director's New Innovator Award DP2OD006466 and National Science Foundation CAREER Award MCB-1149328 (to K. C. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table S1 and supplemental data.
- UPLC
- ultra performance liquid chromatography.
References
- 1. Desmarais S. M., De Pedro M. A., Cava F., and Huang K. C. (2013) Peptidoglycan at its peaks: how chromatographic analyses can reveal bacterial cell wall structure and assembly. Mol. Microbiol. 89, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barsanti L., Passarelli V., Evangelista V., Frassanito A. M., and Gualtieri P. (2011) Chemistry, physico-chemistry and applications linked to biological activities of beta-glucans. Nat. Prod. Rep. 28, 457–466 [DOI] [PubMed] [Google Scholar]
- 3. Koch A. L. (2000) The exoskeleton of bacterial cells (the sacculus): still a highly attractive target for antibacterial agents that will last for a long time. Crit. Rev. Microbiol. 26, 1–35 [DOI] [PubMed] [Google Scholar]
- 4. Bush K. (2012) Antimicrobial agents targeting bacterial cell walls and cell membranes. Rev. Sci. Tech. 31, 43–56 [DOI] [PubMed] [Google Scholar]
- 5. Yount N. Y., and Yeaman M. R. (2013) Peptide antimicrobials: cell wall as a bacterial target. Ann. N.Y. Acad. Sci. 1277, 127–138 [DOI] [PubMed] [Google Scholar]
- 6. Garimella R., Halye J. L., Harrison W., Klebba P. E., and Rice C. V. (2009) Conformation of the phosphate d-alanine zwitterion in bacterial teichoic acid from nuclear magnetic resonance spectroscopy. Biochemistry 48, 9242–9249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bruck S., Personnic N., Prevost M. C., Cossart P., and Bierne H. (2011) Regulated shift from helical to polar localization of Listeria monocytogenes cell wall-anchored proteins. J. Bacteriol. 193, 4425–4437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lenski R. E., and Travisano M. (1994) Dynamics of adaptation and diversification: a 10,000-generation experiment with bacterial populations. Proc. Natl. Acad. Sci. U.S.A. 91, 6808–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schaechter M., Maaloe O., and Kjeldgaard N. O. (1958) Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J. Gen. Microbiol. 19, 592–606 [DOI] [PubMed] [Google Scholar]
- 10. Young K. D. (2006) The selective value of bacterial shape. Microbiol. Mol. Biol. Rev. 70, 660–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Young K. D. (2010) Bacterial shape: two-dimensional questions and possibilities. Annu. Rev. Microbiol. 64, 223–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Typas A., Banzhaf M., Gross C. A., and Vollmer W. (2012) From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 10, 123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Philippe N., Pelosi L., Lenski R. E., and Schneider D. (2009) Evolution of penicillin-binding protein 2 concentration and cell shape during a long-term experiment with Escherichia coli. J. Bacteriol. 191, 909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee T. K., Tropini C., Hsin J., Desmarais S. M., Ursell T. S., Gong E., Gitai Z., Monds R. D., and Huang K. C. (2014) A dynamically assembled cell wall synthesis machinery buffers cell growth. Proc. Natl. Acad. Sci. U.S.A. 111, 4554–4559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gitai Z., Dye N., and Shapiro L. (2004) An actin-like gene can determine cell polarity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 101, 8643–8648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ursell T. S., Nguyen J., Monds R. D., Colavin A., Billings G., Ouzounov N., Gitai Z., Shaevitz J. W., and Huang K. C. (2014) Rod-like bacterial shape is maintained by feedback between cell curvature and cytoskeletal localization. Proc. Natl. Acad. Sci. U.S.A. 111, E1025–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Teeffelen S., Wang S., Furchtgott L., Huang K. C., Wingreen N. S., Shaevitz J. W., and Gitai Z. (2011) The bacterial actin MreB rotates, and rotation depends on cell-wall assembly. Proc. Natl. Acad. Sci. U.S.A. 108, 15822–15827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gitai Z., Dye N. A., Reisenauer A., Wachi M., and Shapiro L. (2005) MreB actin-mediated segregation of a specific region of a bacterial chromosome. Cell 120, 329–341 [DOI] [PubMed] [Google Scholar]
- 19. Kruse T., Bork-Jensen J., and Gerdes K. (2005) The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane-bound complex. Mol. Microbiol. 55, 78–89 [DOI] [PubMed] [Google Scholar]
- 20. Monds R. D., Lee T. K., Colavin A., Ursell T., Quan S., Cooper T. F., and Huang K. C. (2014) Systematic perturbation of cytoskeletal function reveals a linear scaling relationship between cell geometry and fitness. Cell Rep. 9, 1528–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Touchstone J. (1993) History of chromatography. J. Liquid Chromatogr. 16, 1647–1665 [Google Scholar]
- 22. Desmarais S. M., Cava F., de Pedro M. A., and Huang K. C. (2014) Isolation and preparation of bacterial cell walls for compositional analysis by ultra performance liquid chromatography. J. Vis. Exp. 83, e51183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wieser A., Schneider L., Jung J., and Schubert S. (2012) MALDI-TOF MS in microbiological diagnostics-identification of microorganisms and beyond (mini review). Appl. Microbiol. Biotechnol. 93, 965–974 [DOI] [PubMed] [Google Scholar]
- 24. Vollmer W., Blanot D., and de Pedro M. A. (2008) Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167 [DOI] [PubMed] [Google Scholar]
- 25. Glauner B. (1988) Separation and quantification of muropeptides with high-performance liquid chromatography. Anal. Biochem. 172, 451–464 [DOI] [PubMed] [Google Scholar]
- 26. Glauner B., Höltje J. V., and Schwarz U. (1988) The Composition of the murein of Escherichia coli. J. Biol. Chem. 263, 10088–10095 [PubMed] [Google Scholar]
- 27. Schäberle T. F., Vollmer W., Frasch H. J., Hüttel S., Kulik A., Röttgen M., von Thaler A. K., Wohlleben W., and Stegmann E. (2011) Self-resistance and cell wall composition in the glycopeptide producer Amycolatopsis balhimycina. Antimicrob. Agents Chemother. 55, 4283–4289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barreteau H., Bouhss A., Fourgeaud M., Mainardi J. L., Touzé T., Gérard F., Blanot D., Arthur M., and Mengin-Lecreulx D. (2009) Human- and plant-pathogenic Pseudomonas species produce bacteriocins exhibiting colicin M-like hydrolase activity towards peptidoglycan precursors. J. Bacteriol. 191, 3657–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vollmer W., Pilsl H., Hantke K., Höltje J. V., and Braun V. (1997) Pesticin displays muramidase activity. J. Bacteriol. 179, 1580–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Billings G., Ouzounov N., Ursell T., Desmarais S. M., Shaevitz J., Gitai Z., and Huang K. C. (2014) De novo morphogenesis in L-forms via geometric control of cell growth. Mol. Microbiol. 93, 883–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Joseleau-Petit D., Liébart J. C., Ayala J. A., and D'Ari R. (2007) Unstable Escherichia coli L forms revisited: growth requires peptidoglycan synthesis. J. Bacteriol. 189, 6512–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boylen C. W., and Ensign J. C. (1968) Ratio of teichoic acid and peptidoglycan in cell walls of Bacillus subtilis following spire germination and during vegetative growth. J. Bacteriol. 96, 421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de la Rosa E. J., de Pedro M. A., and Vázquez D. (1985) Penicillin binding proteins: role in initiation of murein synthesis in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 82, 5632–5635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hakenbeck R., Holtje J. V., and Labischinski H. (eds) (1983) The Target of Penicillin: The Murein Sacculus of Bacterial Cell Walls : Architecture and Growth: Proceedings, Walter De Gruyter Inc., Berlin, Germany [Google Scholar]
- 35. Kandler O., Schleifer K. H., and Dandl R. (1968) Differentiation of Streptococcus faecalis Andrewes and Horder and Streptococcus faecium Orla-Jensen based on the amino acid composition of their murein. J. Bacteriol. 96, 1935–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sutow A. B., and Welker N. E. (1967) Chemical composition of the cell walls of Bacillus stearothermophilus. J. Bacteriol. 93, 1452–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang W. S., and Lundgren D. G. (1968) Peptidoglycan of a chemolithotrophic bacterium, Ferrobacillus ferrooxidans. J. Bacteriol. 95, 1851–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weidel W., Frank H., and Martin H. H. (1960) The rigid layer of the cell wall of Escherichia coli strain B. J. Gen. Microbiol. 22, 158–166 [DOI] [PubMed] [Google Scholar]
- 39. Goodell E. W., and Schwarz U. (1977) Enzymes synthesizing and hydrolyzing murein in Escherichia coli: topographical distribution over the cell envelope. Eur. J. Biochem. 81, 205–210 [DOI] [PubMed] [Google Scholar]
- 40. Markiewicz Z., Glauner B., and Schwarz U. (1983) Murein structure and lack of dd- and ld-carboxypeptidase activities in Caulobacter crescentus. J. Bacteriol. 156, 649–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bertani G. (2004) Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186, 595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Edelstein A., Amodaj N., Hoover K., Vale R., and Stuurman N. (2010) Computer control of microscopes using μManager. Curr. Protoc. Mol. Biol. 14, Unit 14 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sliusarenko O., Heinritz J., Emonet T., and Jacobs-Wagner C. (2011) High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol. Microbiol. 80, 612–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu Z., Pan D. D., Guo Y., and Zeng X. (2013) Structure and anti-inflammatory capacity of peptidoglycan from Lactobacillus acidophilus in RAW-264.7 cells. Carbohydr. Polym. 96, 466–473 [DOI] [PubMed] [Google Scholar]
- 45. Koyama N., Tokura Y., Münch D., Sahl H. G., Schneider T., Shibagaki Y., Ikeda H., and Tomoda H. (2012) The nonantibiotic small molecule cyslabdan enhances the potency of β-lactams against MRSA by inhibiting pentaglycine interpeptide bridge synthesis. PloS One 7, e48981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang Z., Unrine J., Nonaka K., and Van Lanen S. G. (2012) Fe(II)-dependent, uridine-5′-monophosphate α-ketoglutarate dioxygenases in the synthesis of 5′-modified nucleosides. Methods Enzymol. 516, 153–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ronholm J., Wang L., Hayashi I., Sugai M., Zhang Z., Cao X., and Lin M. (2012) The Listeria monocytogenes serotype 4b autolysin IspC has N-acetylglucosaminidase activity. Glycobiology 22, 1311–1320 [DOI] [PubMed] [Google Scholar]
- 48. Garcia-Bustos J., and Tomasz A. (1990) A biological price of antibiotic resistance: major changes in the peptidoglycan structure of penicillin-resistant pneumococci. Proc. Natl. Acad. Sci. U.S.A. 87, 5415–5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Filipe S. R., and Tomasz A. (2000) Inhibition of the expression of penicillin resistance in Streptococcus pneumoniae by inactivation of cell wall muropeptide branching genes. Proc. Natl. Acad. Sci. U.S.A. 97, 4891–4896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Garcia-Bustos J. F., Chait B. T., and Tomasz A. (1988) Altered peptidoglycan structure in a pneumococcal transformant resistant to penicillin. J. Bacteriol. 170, 2143–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Evans K. L., Kannan S., Li G., de Pedro M. A., and Young K. D. (2013) Eliminating a set of four penicillin binding proteins triggers the Rcs phosphorelay and Cpx stress responses in Escherichia coli. J. Bacteriol. 195, 4415–4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kitano K., Tuomanen E., and Tomasz A. (1986) Transglycosylase and endopeptidase participate in the degradation of murein during autolysis of Escherichia coli. J. Bacteriol. 167, 759–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Obermann W., and Höltje J. V. (1994) Alterations of murein structure and of penicillin-binding proteins in minicells from Escherichia coli. Microbiology 140, 79–87 [DOI] [PubMed] [Google Scholar]
- 54. Uehara T., and Park J. T. (2008) Growth of Escherichia coli: significance of peptidoglycan degradation during elongation and septation. J. Bacteriol. 190, 3914–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lam H., Oh D. C., Cava F., Takacs C. N., Clardy J., de Pedro M. A., and Waldor M. K. (2009) d-Amino acids govern stationary phase cell wall remodeling in bacteria. Science 325, 1552–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Möll A., Dörr T., Alvarez L., Chao M. C., Davis B. M., Cava F., and Waldor M. K. (2014) Cell separation in Vibrio cholerae is mediated by a single amidase whose action is modulated by two nonredundant activators. J. Bacteriol. 196, 3937–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang K. C., Mukhopadhyay R., Wen B., Gitai Z., and Wingreen N. S. (2008) Cell shape and cell-wall organization in Gram-negative bacteria. Proc. Natl. Acad. Sci. U.S.A. 105, 19282–19287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cabeen M. T., Charbon G., Vollmer W., Born P., Ausmees N., Weibel D. B., and Jacobs-Wagner C. (2009) Bacterial cell curvature through mechanical control of cell growth. EMBO J. 28, 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Blackburn N. T., and Clarke A. J. (2002) Characterization of soluble and membrane-bound family 3 lytic transglycosylases from Pseudomonas aeruginosa. Biochemistry 41, 1001–1013 [DOI] [PubMed] [Google Scholar]
- 60. Scheurwater E. M., Pfeffer J. M., and Clarke A. J. (2007) Production and purification of the bacterial autolysin N-acetylmuramoyl-l-alanine amidase B from Pseudomonas aeruginosa. Protein Expr. Purif. 56, 128–137 [DOI] [PubMed] [Google Scholar]
- 61. Remans K., Vercammen K., Bodilis J., and Cornelis P. (2010) Genome-wide analysis and literature-based survey of lipoproteins in Pseudomonas aeruginosa. Microbiology 156, 2597–2607 [DOI] [PubMed] [Google Scholar]
- 62. Duchêne M., Barron C., Schweizer A., von Specht B. U., and Domdey H. (1989) Pseudomonas aeruginosa outer membrane lipoprotein I gene: molecular cloning, sequence, and expression in Escherichia coli. J. Bacteriol. 171, 4130–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Skurnik D., Roux D., Cattoir V., Danilchanka O., Lu X., Yoder-Himes D. R., Han K., Guillard T., Jiang D., Gaultier C., Guerin F., Aschard H., Leclercq R., Mekalanos J. J., Lory S., and Pier G. B. (2013) Enhanced in vivo fitness of carbapenem-resistant oprD mutants of Pseudomonas aeruginosa revealed through high-throughput sequencing. Proc. Natl. Acad. Sci. U.S.A. 110, 20747–20752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Czechowska K., McKeithen-Mead S., Al Moussawi K., and Kazmierczak B. I. (2014) Cheating by type 3 secretion system-negative Pseudomonas aeruginosa during pulmonary infection. Proc. Natl. Acad. Sci. U.S.A. 111, 7801–7806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Legaree B. A., Daniels K., Weadge J. T., Cockburn D., and Clarke A. J. (2007) Function of penicillin-binding protein 2 in viability and morphology of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 59, 411–424 [DOI] [PubMed] [Google Scholar]
- 66. Wolter D. J., and Lister P. D. (2013) Mechanisms of β-lactam resistance among Pseudomonas aeruginosa. Curr. Pharm. Des. 19, 209–222 [PubMed] [Google Scholar]
- 67. Mirelman D., Nuchamowitz Y., and Rubinstein E. (1981) Insensitivity of peptidoglycan biosynthetic reactions to β-lactam antibiotics in a clinical isolate of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 19, 687–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Heilmann H. D. (1972) On the peptidoglycan of the cell walls of Pseudomonas aeruginosa. Eur. J. Biochem. 31, 456–463 [DOI] [PubMed] [Google Scholar]
- 69. Heilmann H. D. (1974) On the peptidoglycan of the cell walls of Pseudomonas aeruginosa: structure of the peptide side chains. Eur. J. Biochem. 43, 35–38 [DOI] [PubMed] [Google Scholar]
- 70. Zegans M. E., Wozniak D., Griffin E., Toutain-Kidd C. M., Hammond J. H., Garfoot A., and Lam J. S. (2012) Pseudomonas aeruginosa exopolysaccharide Psl promotes resistance to the biofilm inhibitor polysorbate 80. Antimicrob. Agents Chemother. 56, 4112–4122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tropini C., Lee T. K., Hsin J., Desmarais S. M., Ursell T., Monds R. D., and Huang K. C. (2014) Principles of bacterial cell-size determination revealed by cell-wall synthesis perturbations. Cell Rep. 9, 1520–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]