

Background: Complement activation and oxidative damage contribute to retinal pigment epithelium (RPE) pathology in age-related macular degeneration (AMD).
Results: C5a induces interleukin-1β in human RPE cells and enables its release in response to lipofuscin phototoxicity.
Conclusion: C5a primes RPE cells for inflammasome activation by lipofuscin-mediated photooxidative damage.
Significance: This mechanism provides a new link between key factors of AMD pathogenesis.
Keywords: complement system, eye, immunology, inflammasome, interleukin 1 (IL-1), oxidative stress, retinal degeneration
Abstract
Complement activation, oxidative damage, and activation of the NLRP3 inflammasome have been implicated in retinal pigment epithelium (RPE) pathology in age-related macular degeneration (AMD). Following priming of RPE cells, the NLRP3 inflammasome can be activated by various stimuli such as lipofuscin-mediated photooxidative damage to lysosomal membranes. We investigated whether products of complement activation are capable of providing the priming signal for inflammasome activation in RPE cells. We found that incubation of primary human RPE cells and ARPE-19 cells with complement-competent human serum resulted in up-regulation of C5a receptor, but not C3a receptor. Furthermore, human serum induced expression of pro-IL-1β and enabled IL-1β secretion in response to lipofuscin phototoxicity, thus indicating inflammasome priming. Complement heat-inactivation, C5 depletion, and C5a receptor inhibition suppressed the priming effect of human serum whereas recombinant C5a likewise induced priming. Conditioned medium of inflammasome-activated RPE cells provided an additional priming effect that was mediated by the IL-1 receptor. These results identify complement activation product C5a as a priming signal for RPE cells that allows for subsequent inflammasome activation by stimuli such as lipofuscin-mediated photooxidative damage. This molecular pathway provides a functional link between key factors of AMD pathogenesis including lipofuscin accumulation, photooxidative damage, complement activation, and RPE degeneration and may provide novel therapeutic targets in this disease.
Introduction
Age-related macular degeneration (AMD)2 is the leading cause of blindness in all industrialized countries (1). For the majority of patients, in particular those affected by the intermediate stage and the atrophic late stage of the disease, there is currently no effective treatment available. Elucidating the still unresolved pathogenesis of this multifactorial, complex disease will help to identify potential targets for therapeutic intervention. The retinal pigment epithelium (RPE), a monolayer of post-mitotic support cells essential for photoreceptor function, is primarily affected by AMD. Oxidative/photooxidative damage to the RPE contributes to AMD, and antioxidative treatment has been demonstrated to slow disease progression in clinical trials (2). This damage is believed to be mediated at least in part by the photoreactive properties of lipofuscin and lipofuscin component A2E that accumulate in the macular RPE in large amounts over a lifetime (3, 4).
In addition, several lines of evidence indicate that processes of the innate immune system play a critical role in the pathogenesis of AMD. Activated components of the complement system such as C3a and C5a are detectable both locally in the sub-RPE space and systemically in plasma of AMD patients (5, 6). Genetic polymorphisms in several complement components and regulators such as CFH, C2, C3, and CFB are strongly associated with AMD (7). Another part of the innate immune system, the NLRP3 inflammasome, has recently been proposed to also contribute to AMD pathogenesis. Activation of the NLRP3 inflammasome in RPE cells was demonstrated in both atrophic and neovascular AMD (8, 9), and increased intravitreal and systemic levels of the inflammasome activation products IL-1β and IL-18 have been reported in AMD patients (10, 11). The inflammasome protein complex serves as an intracellular sensor for various signals of cell damage (12). Its activation results in the secretion of highly pro-inflammatory cytokines such as IL-1β and IL-18 and eventually in cell death by pyroptosis.
Activation of the NLRP3 inflammasome is a two-step process that requires an initial priming signal and a subsequent activation signal (12). The priming signal results in NF-κB-dependent transcriptional induction of NLRP3 and pro-IL-1β. The activation signal subsequently triggers assembly of NLRP3 and other protein components into the active inflammasome protein complex that results in caspase-1-mediated cleavage of pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18. Several substances have been suggested to provide the inflammasome activation signal in AMD including drusen components such as C1q (13) and amyloid-β (14), Alu RNA accumulation secondary to DICER1 deficiency (8), the lipofuscin component N-retinylidene-N-retinyl-ethanolamine (A2E) (15), and the lipid peroxidation product 4-hydroxynonenal (HNE) (16). We have recently suggested an additional mechanism by demonstrating that photooxidative damage to the RPE, enhanced by accumulated lipofuscin, can activate the NLRP3 inflammasome by inducing lysosomal membrane permeabilization and cytosolic leakage of lysosomal enzymes (17, 18).
In contrast to inflammasome activation, the mechanism of inflammasome priming in AMD has been little investigated so far. Interestingly, a recent study in patients with early or intermediate AMD demonstrated the CFH risk genotype to be associated with significantly increased plasma levels of the inflammasome-regulated cytokine IL-18, suggesting a role for activated complement components like C3a and C5a in inflammasome activation in AMD (19). Inflammasome priming by complement activation products has also been proposed in the context of other diseases such as atherosclerosis and gout (20, 21). In this study, we investigated the capacity of activated complement components to prime human RPE cells for inflammasome activation by lipofuscin-mediated photooxidative damage.
Experimental Procedures
Cell Culture and Treatments
Human fetal primary RPE (pRPE) cells (Clonetics H-RPE; Lonza, Cologne, Germany) were cultured in medium provided by the manufacturer (Clonetics RtEGM; Lonza) containing 2% heat-inactivated fetal bovine serum (FBS) and were used in experiments for a maximum of 6 cell culture passages. The human non-transformed RPE cell line ARPE-19 (CRL-2302; ATCC, Rockville, MD) was cultured as previously reported using medium containing 10% heat-inactivated FBS (22). For inflammasome priming, culture medium was exchanged by FBS-free medium supplemented with the indicated priming agents as described below.
For analysis of C5a receptor (C5aR) expression, cells were treated with 50 ng/ml C5a (R&D Systems, Wiesbaden, Germany) (23, 24). Cathepsin B inhibitor CA-074 (Merck/Calbiochem, Darmstadt, Germany) and cathepsin L inhibitor Z-FF-FMK (Merck/Calbiochem) were used at a concentration of 10 μm each for 1 h prior to and during irradiation treatment. For inhibition of caspase-1, we applied 10 μm of Z-YVAD-FMK (BioVision, Munich, Germany) 30 min prior to and during irradiation. Binding to C5aR was blocked using 0.5 μm of an inhibitory mouse monoclonal IgG antibody directed against human C5aR (clone S5/1; Biolegend, Fell, Germany). The drug anakinra (Kineret; Swedish Orphan Biovitrum, Langen, Germany) was used at a concentration of 100 ng/ml to inhibit the IL-1 receptor (IL1R).
Immunocytochemistry and Western Blot Analysis
For immunocytochemical detection of ZO-1 and C5aR, cells were stained with a rabbit polyclonal anti human ZO-1 antibody (Life Technologies, Darmstadt, Germany) and a mouse monoclonal anti human C5aR antibody (clone S5/1; Biolegend, Fell, Germany), respectively. Cells were fixed with 4% paraformaldehyde for immunocytochemistry. No cell permeabilization agent was applied prior to immunodetection of C5aR to limit this staining to cell surface proteins.
For Western blot analysis of C5aR and pro-IL-1β, we employed a mouse monoclonal anti human C5aR antibody (clone S5/1; Biolegend, Fell, Germany) and a goat polyclonal anti human IL-1β antibody (R&D Systems, Wiesbaden, Germany), respectively. Cells were lysed using RIPA buffer, and total protein content of cell lysates was quantified by Bradford assay (Sigma-Aldrich, Munich, Germany). Equal amounts of 50 μg of total protein per sample were separated by electrophoresis in 4–12% SDS-polyacrylamide gels (Lonza, Cologne, Germany) prior to transfer onto nitrocellulose membranes (Thermo Scientific) and subsequent immunodetection.
RT-PCR and Quantitative Real-time PCR
For conventional RT-PCR, isolation of total RNA from RPE cells and reverse transcription into cDNA was carried out with the Power SYBR Green Cells-to-Ct Kit (Life Technologies, Darmstadt, Germany) as recommended by the manufacturer. PCR was performed with 40 cycles using the KAPA2G Fast PCR Kit (PEQLAB Biotechnologie, Erlangen, Germany). The primers used for detection of C5aR, C3a receptor (C3aR), and C5a-like receptor 2 (C5L2) have been described (25). For human glyceraldehyde-3-phosphate dehydrogenase (GAPDH), we employed the sense primer 5′-CTCTGCTCCTCCTGTTCGAC-3′ and the antisense primer 5′-GCGCCCAATACGACCAAATC-3′. PCR products were run on 2% agarose gel with a 100 bp DNA ladder marker (Sigma-Aldrich). The negative control contained all PCR components but no cDNA template.
Quantitative real-time PCR (qPCR) was performed using again the Power SYBR Green Cells-to-Ct Kit (Life Technologies, Darmstadt, Germany) according to the manufacturer's protocol on a real-time PCR system (LightCycler 480 II; Roche, Basel, Switzerland) with the primers described above. The amount of target mRNA in test samples was normalized to GAPDH, and the comparative Ct method was used to evaluate gene expression.
Inflammasome Priming
For inflammasome priming, cells were treated with the indicated priming agents during the last 48 h of incubation with photoreceptor outer segments (POS). For priming with interleukins, cells were treated with 4 ng/ml recombinant human IL-1α (R&D Systems, Wiesbaden, Germany) (9) or 50 pg/ml recombinant human IL-1β (R&D Systems). For priming with normal human serum (NHS), full blood was drawn from a healthy donor into anticoagulant-free tubes. Blood samples were sedimented at room temperature for 30 min. Serum was separated by centrifugation (2000 × g, 5 min) and immediately stored at −80 °C. Heat inactivation of complement components was performed by incubating NHS in a water bath of 56 °C for 30 min. For inflammasome priming, NHS, heat-inactivated NHS, or C5-depleted human serum (Sigma-Aldrich) was added to FBS-free cell culture medium at a concentration of 25% each. In an additional treatment group, cell culture medium containing C5-deficient serum was re-supplemented with 50 ng/ml C5a (R&D Systems, Wiesbaden, Germany).
Inflammasome Activation
Following inflammasome priming, inflammasome activation by lipofuscin-mediated photooxidative damage was induced in RPE cells as previously described (17). Briefly, isolated porcine POS were covalently modified with the lipid peroxidation product HNE (5 mm) to stabilize them against lysosomal degradation (26). RPE cells were incubated with modified POS (concentration equivalent to 4 mg total POS protein per cm2 cell growth area) daily for 7 days, resulting in lipofuscinogenesis (22). During the last 48 h of POS treatment, cells were co-incubated with the respective priming agent as indicated. Subsequently, medium was changed, and cells were irradiated with blue light (peak wave length, 448 nm; irradiance, 0.8 milliwatt/cm2) for the indicated times of up to 6 h to induce photooxidative lysosomal membrane permeabilization and subsequent NLRP3 inflammasome activation (17). Irrespective of the duration of irradiation, medium was collected 6 h after the medium change in all treatment and control groups. Secretion of IL-1β secondary to inflammasome activation was measured by specific ELISA (BD OptEIA Human IL-1β ELISA Kit II; BD Biosciences, Heidelberg, Germany). To analyze loss of plasma membrane integrity secondary to inflammasome-mediated cell death, we assessed lactate dehydrogenase (LDH) release (Cytotoxicity Detection Kit; Roche, Mannheim, Germany).
Priming by Conditioned Medium
Conditioned medium was obtained from ARPE-19 cells following inflammasome activation by lipofuscin phototoxicity. This was induced by HNE-POS incubation, IL-1α priming, and blue light irradiation as described above. Before the start of irradiation, cells were washed and medium was changed. After 6 h of irradiation, conditioned medium was collected for use in priming experiments. In control experiments, inflammasome activation was achieved by treatment with l-leucyl-l-leucine methyl ester (Leu-Leu-OMe) (9). For this, ARPE-19 cells were primed by IL-1α, incubated with 1 mm Leu-Leu-OMe (Bachem, Bubendorf, Switzerland) for 1 h, washed, and incubated with new medium without Leu-Leu-OMe for another 2 h before conditioned medium was collected. For inflammasome priming by conditioned medium, treatment-naive cells were incubated with conditioned medium of HNE-POS/blue light-treated or Leu-Leu-OMe-treated cells for 48 h.
Statistical Analysis
Experiments were performed in duplicates (Figs. 3A, 3C, 4A, 4C, 6A, 6B) following the assay manufacturer's recommendation or in triplicates (Figs. 2, 3B, 3D, 4B, 4D). Results are presented as mean ± standard deviation. Statistical analysis was performed using paired (Fig. 2) or unpaired (Figs. 3, 4, 6) two-tailed Student's t tests (Microsoft Excel 2013; Microsoft, Redmond, WA). Differences were considered statistically significant at p < 0.05. In experiments with multiple group comparisons (Figs. 3, 4), significant differences were confirmed by additional analysis using one-way ANOVA with post-hoc analysis by Tukey's range test (GraphPad InStat 3.06, GraphPad Software, La Jolla, CA).
FIGURE 3.

NHS primes RPE cells for inflammasome activation by lipofuscin-mediated photooxidative damage. ARPE-19 cells (A, B) and pRPE cells (C, D) were incubated with unmodified POS (POS) or HNE-modified POS (HNE) to induce lipofuscinogenesis whereas control cells were incubated without POS (Co.). Lipofuscin-loaded cells were primed for inflammasome activation by treatment with IL-1α, complement-competent normal human serum (NHS), NHS and caspase-1 inhibitor Z-YVAD-FMK, or NHS and cathepsin B inhibitor CA-074. Subsequently, inflammasome activation was induced by cell irradiation with blue light for 3 or 6 h. Inflammasome activation under these experimental conditions was measured by means of inflammasome-regulated secretion of IL-1β (A, C) and inflammasome-induced cytotoxicity (LDH release) (B, D). Experiments were performed in duplicates (A, C) following the assay manufacturer's recommendation or in triplicates (B, D), and results are presented as mean ± S.D.
FIGURE 4.

Complement component C5a is the active priming agent in NHS. ARPE-19 cells (A, B) and pRPE cells (C, D) were examined. Treatment groups and labels are identical to Fig. 3. However, inflammasome priming in this experiment was performed by incubation with complement-competent normal human serum (NHS), heat-inactivated human serum (HI-NHS), NHS and a C5aR inhibitory antibody, C5-deficient NHS, or C5-deficient NHS and recombinant human C5a. Inflammasome activation was assessed be means of secretion of IL-1β (A, C) and cytotoxicity (B, D). Experiments were performed in duplicates (A, C) following the assay manufacturer's recommendation or in triplicates (B, D), and results are presented as mean ± S.D.
FIGURE 6.

Inflammasome priming in RPE cells is enhanced by a paracrine amplification loop. A, inflammasome activation was induced in IL-1α-primed ARPE-19 cells by HNE-POS/blue light treatment or incubation with Leu-Leu-OMe. Cells treated with HNE-POS but without blue light irradiation served as controls. Conditioned medium of these cells was collected. B, subsequently, new ARPE-19 cells were primed with conditioned medium of HNE-POS-treated control cells, conditioned medium of HNE-POS/blue light-treated cells conditioned medium of HNE-POS/blue light-treated cells and the IL1R inhibitory drug anakinra, conditioned medium of Leu-Leu-OMe-treated cells, or recombinant human IL-1β. Inflammasome activation was induced in all treatment groups by HNE-POS/blue light treatment and analyzed by means of IL-1β secretion. C, expression of pro-IL-1β protein (36 kDa) was assessed in ARPE-19 cells following priming with either conditioned medium of HNE-POS/blue light-treated cells or conditioned medium of HNE-POS/blue light-treated cells and co-incubation with the IL1R inhibitor anakinra, as well as in unprimed control cells. Experiments (A, B) were performed in duplicates following the assay manufacturer's recommendation, and results are presented as mean ± S.D.
FIGURE 2.

C5aR, but not C3aR, is up-regulated following incubation with activated complement. Using qPCR, we analyzed expression kinetics of (A) C5aR in ARPE-19 cells incubated with complement-competent NHS, (B) C5aR in ARPE-19 cells incubated with heat-inactivated NHS, (C) C3aR in ARPE-19 cells incubated with complement-competent NHS, (D) C5aR in ARPE-19 cells incubated with recombinant human C5a, and (E) C5aR in pRPE cells incubated with recombinant human C5a. Target mRNAs were normalized to GAPDH. Experiments were performed in triplicates, and results are presented as mean ± standard deviation. F, increased expression of C5aR protein (37 kDa) following priming with C5a was detected by (F) Western blot analysis and (G) immunocytochemistry in ARPE-19 cells.
Results
Anaphylatoxin Receptors C3aR, C5aR, and C5L2 Are Constitutively Expressed by Human RPE Cells
C3a and C5a represent the two dominant anaphylatoxins during complement activation. C3a binds to C3aR. C5a is a ligand for both C5aR and C5L2 with most biological effects being mediated by C5aR. Expression of C5aR and C3aR has been demonstrated in ARPE-19 cells (24, 25, 27). Expression of C5aR in RPE cells has also been detected by immunohistochemistry of human donor eyes where it was found to be localized predominantly on the basolateral cell side (28). We extended these previous studies by investigating the expression of C3aR, C5aR, and C5L2 in ARPE-19 and pRPE cells. Under the culture conditions employed in our experiments, both cell types exhibited characteristics of differentiated RPE cells including epithelial monolayer formation, hexagonal cell morphology, and intercellular ZO-1-positive tight junctions (Fig. 1A). In both RPE cell types, we detected constitutive expression of all three anaphylatoxin receptors (Fig. 1B).
FIGURE 1.
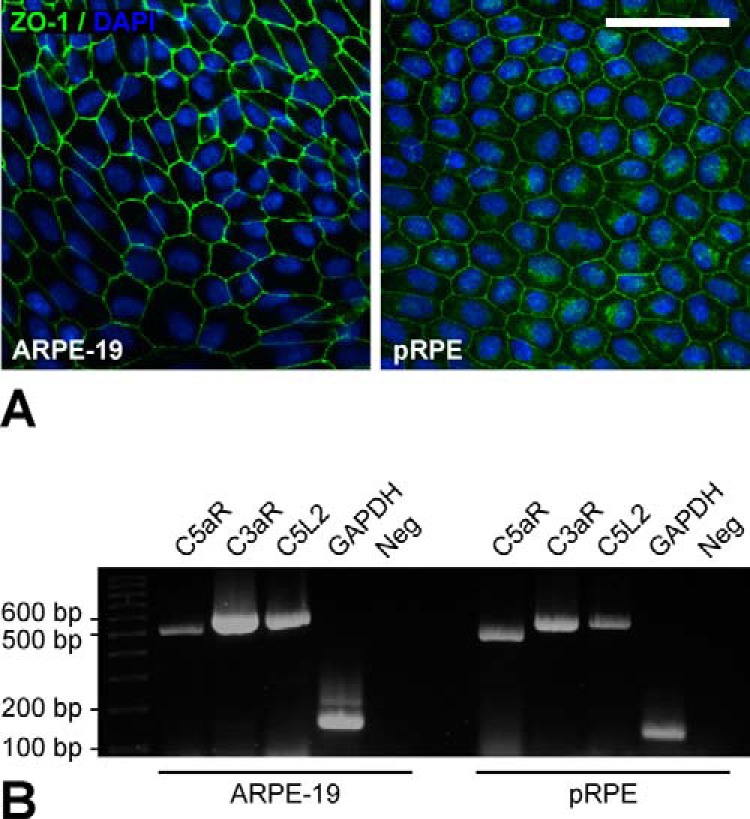
Anaphylatoxin receptors C5aR, C3aR, and C5L2 are constitutively expressed by human RPE cells. A, cells of the human RPE cell line ARPE-19 and primary human RPE cells (pRPE) were used for expression analysis. Both RPE cell types exhibited RPE-characteristic morphology in cell culture, including epithelial cell monolayer formation with polygonal cell shape and intercellular tight junctions as evident from immunostaining of ZO-1 (green). Nuclei were visualized by DAPI staining (blue). Scale bar, 50 μm. B, expression of C5aR (PCR product, 500 bp), C3aR (585 bp), and C5L2 (585 bp) in RPE cells was analyzed by RT-PCR. Expression of GAPDH (121 bp) was used as a positive control. PCR reaction mix without cDNA template served as negative control (Neg).
C5aR, but Not C3aR, Is Up-regulated following Incubation with Activated Complement
In normal human serum (NHS) in vitro, complement activation occurs rapidly when incubated at 37 °C, and thus complement-competent NHS in vitro is a rich source for complement activation products even without addition of complement activators such as zymosan (29). In contrast, heating of NHS to 56 °C for 30 min inactivates complement components and prevents complement activation but preserves the activity of other less heat-labile serum proteins. To assess the effects of activated complement components on anaphylatoxin receptors in human RPE cells, we measured expression of C5aR and C3aR in ARPE-19 cells and pRPE cells after incubation with complement-competent NHS and heat-inactivated NHS (HI-NHS) by qPCR analysis.
Studies investigating the time course of C5aR expression in ARPE-19 cells following stimulation with inflammatory cytokines reported a up-regulation with a maximum after 6 h for mRNA expression and after 24 h for cell surface protein expression (24). We likewise found that incubation of ARPE-19 cells with complement-competent NHS induced a significant up-regulation of C5aR expression (p = 0.007) with a peak 6-fold induction after 6 h (Fig. 2A). Heat inactivation of complement components completely prevented the effect of NHS on C5aR expression (Fig. 2B). In contrast to C5aR, expression of C3aR was not significantly affected by incubation with complement-competent NHS (Fig. 2C). Similar to NHS, recombinant C5a induced a maximum 6-fold up-regulation of C5aR expression in ARPE-19 cells (p = 0.011) after 6 h (Fig. 2D). This result was confirmed in pRPE cells, which likewise exhibited a significant up-regulation of C5aR epression (p = 0.0097) following incubation with C5a (Fig. 2E). Western blot analysis performed in ARPE-19 cells at different time points up to 24 h after beginning of an incubation with C5a for 6 h confirmed that the observed mRNA induction resulted in increased C5aR protein expression (Fig. 2F). Similarly, immunocytochemistry performed without cell permeabilization demonstrated increased C5aR cell surface staining in ARPE-19 cells 24 h following start of a C5a incubation for 6 h (Fig. 2G).
Our findings indicate that human RPE cells respond to incubation with activated complement components by up-regulation of C5aR expression whereas expression of C3aR is not affected. These results suggest C5aR as a mediator of complement effects on RPE cells and triggered us to further investigate C5a as a potential priming signal for the NLRP3 inflammasome in RPE cells.
NHS Primes RPE Cells for Inflammasome Activation by Lipofuscin-mediated Photooxidative Damage
We previously demonstrated that the in vitro model of lipofuscin-mediated photooxidative damage in RPE cells employed in this study results in activation of the NLRP3 inflammasome with activation of caspase-1 and subsequent release of IL-1β and IL-18 (17, 18). For our experiments, ARPE-19 cells and pRPE cells were incubated with unmodified POS or POS modified with the lipid peroxidation product HNE (HNE-POS) to induce intracellular accumulation of low and high levels of lipofuscin-like material, respectively. Subsequently, lipofuscin-loaded RPE cells were irradiated with blue light for up to 6 h. Inflammasome activation was assessed by means of inflammasome-regulated IL-1β secretion and inflammasome-induced pyroptotic cell death in both ARPE-19 cells (Fig. 3, A and B) and pRPE cells (Fig. 3, C and D).
Without prior inflammasome priming, no inflammasome activation was detectable in RPE cells following blue light irradiation. In contrast, in positive control cells treated with the priming agent IL-1α (9), blue light irradiation resulted in significant inflammasome activation with IL-1β secretion and pyroptosis. Inflammasome activation increased with light dose. Incubation with NHS exerted a strong priming effect similar to IL-1α. Inflammasome activation of NHS-primed cells was dependent on activity of the inflammasome component caspase-1 and the lysosomal protease cathepsin B, consistent with the previously reported mechanism of inflammasome activation by lysosomal enzyme leakage (17). There was good agreement between results in ARPE-19 cells and pRPE cells. The data demonstrate that complement-competent NHS contains a factor capable of providing the priming signal for subsequent inflammasome activation in RPE cells.
Complement Component C5a Is the Active Priming Agent in NHS
To identify the active priming agent in NHS, different complement components were inhibited during priming, and subsequent inflammasome activation by lipofuscin/blue light treatment was assessed again by means of IL-1β secretion and cell death in ARPE-19 cells (Fig. 4, A and B) and pRPE cells (Fig. 4, C and D). First, we inactivated all complement components in NHS by heating. We found that the priming effect of NHS was completely suppressed after heat inactivation, suggesting that heat-labile serum components such as complement components mediate inflammasome priming by NHS. To further delineate the responsible complement component in NHS, cells were treated with a C5aR inhibitor during priming with NHS with resulted in a significant reduction of the priming effect. Likewise, depletion of C5 prevented the priming effect of NHS. Supplementation with recombinant C5a resulted in complete restoration of the priming capacity of C5-depleted NHS.
To confirm that the observed IL-1β secretion is indeed a result of inflammasome priming we analyzed the induction of pro-IL-1β protein expression that represents a key element of inflammasome priming. In agreement with our results regarding IL-1β secretion, pro-IL-1β protein expression in ARPE-19 cells was strongly induced by incubation with IL-1α, NHS, or C5a as compared with control cells incubated with heat-inactivated NHS or C5-depleted NHS (Fig. 5). As previous studies demonstrated that ARPE-19 cells, unlike other cell types, constitutively express NLRP3 even in unprimed conditions and do not induce NLRP3 expression after priming (9) we did not include NLRP3 in this analysis. In summary, our results identify the complement component C5a as the active priming agent in NHS and demonstrate that C5a is capable of priming human RPE cells for inflammasome activation by lipofuscin-mediated photooxidative damage.
FIGURE 5.
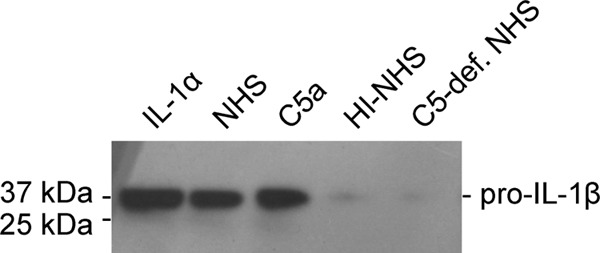
Priming by C5a induces expression of pro-IL-1β protein. Expression of pro-IL-1β protein (36 kDa) was assessed in ARPE-19 cells following priming with IL-1α, complement-competent normal human serum (NHS), recombinant human C5a, heat inactivated human serum (HI-NHS), or C5-deficient NHS.
Inflammasome Priming in RPE Cells Is Enhanced by a Paracrine Amplification Loop
Inflammasome priming involves induction of IL-1β expression, and IL-1β is known to induce its own expression by an auto-/paracrine amplification loop (30, 31). Therefore, we investigated whether inflammasome activation in RPE cells results in paracrine priming of the inflammasome in neighboring RPE cells. To test for this, we performed priming experiments with conditioned medium of cells following inflammasome activation. First, ARPE-19 cells were incubated with HNE-POS to induce lipofuscin accumulation, primed with IL-1α, and subsequently irradiated with blue light to trigger inflammasome activation as described above. As a positive control, inflammasome activation was induced in ARPE-19 cells by IL-1α priming and subsequent treatment with Leu-Leu-OMe (9). We confirmed that inflammasome activation resulted in a significant induction of IL-1β secretion in both treatment groups (Fig. 6A). Conditioned medium of lipofuscin/blue light-treated and Leu-Leu-OMe-treated RPE cells was collected for further priming experiments.
In a second step, new, untreated ARPE-19 cells were loaded with lipofuscin by HNE-POS treatment, incubated with the collected conditioned medium for 48 h, thoroughly washed to remove any IL-1β contained in the conditioned medium, and irradiated with blue light. Subsequently, inflammasome activation was assessed by measuring IL-1β secretion. In these experiments, conditioned medium derived from both lipofuscin/blue light-treated and Leu-Leu-OMe-treated cells exerted a strong priming effect that enabled inflammasome activation (Fig. 6B). However, when cells were co-incubated with the IL1R-inhibitory drug anakinra during priming with conditioned medium from lipofuscin/blue light-treated cells, the priming effect was significantly reduced (p = 0.020). This indicates that the priming effect of conditioned medium is mediated by an IL1R ligand such as IL-1β. Indeed, incubation of RPE cells with recombinant IL-1β alone instead of conditioned medium likewise resulted in a strong priming effect.
Additional analysis of inflammasome priming by means of pro-IL-1β protein expression produces results consistent with IL-1β secretion measurements (Fig. 6C). As described above, conditioned medium was collected from ARPE-19 cells following priming with IL-1α, treatment with HNE-POS, and irradiation with blue light. Incubation of new, untreated ARPE-19 cells with the conditioned medium resulted in marked induction of pro-IL-1β protein expression as compared with unprimed control cells incubated with unconditioned medium. The induction of pro-IL-1β was partially suppressed when cells were co-incubate with the IL1R inhibitor anakinra during priming. Together, these results indicate that during inflammasome activation in RPE cells in vitro, inflammasome-regulated cytokines such as IL-1β initiate a paracrine amplification loop of inflammasome priming that is amendable to intervention by IL1R-inhibitory drugs.
Discussion
Blue light irradiation of RPE cells in the presence of oxygen results in generation of reactive oxygen species in a lipofuscin-dependent manner (3) and subsequent permeabilization of lysosomal membranes by oxidative damage (32, 33). We have previously shown that lysosomal membrane permeabilization by lipofuscin-mediated photooxidative damage activates the NLRP3 inflammasome in primed RPE cells (17, 18). This mechanism may underlie the inflammasome activation observed in the RPE of AMD patients (8, 9) and may contribute to RPE pathology in this disease.
Activation of the NLRP3 inflammasome is a posttranscriptionally regulated event mediated by assembly of inflammasome components and subsequent proteolytic maturation of interleukin precursors. In most cells, however, inflammasome component NLRP3 and interleukin precursor pro-IL-1β are not expressed constitutively or only to low amounts. Therefore, inflammasome activation requires a prior priming signal to induce expression of these proteins. Most previous studies including our own investigated the mechanisms of inflammasome activation in RPE cells by utilizing well-established priming agents such as LPS and IL-1α (9, 15–18). However, the relevance of these substances as priming agents of the RPE in vivo in the context of AMD is unclear. We therefore investigate activated complement components as potential priming agents in RPE cells.
Chronic complement activation is associated with AMD, and activated complement components like C3a and C5a are deposited in the sub-RPE space in AMD (5). Thus, RPE cells are in constant, direct contact with these bioactive substances that, therefore, represent candidates for the inflammasome priming signal in AMD via anaphylatoxin receptors such as C5aR that is expressed on the basolateral side of the RPE (28). Indeed, AMD patients with the CFH risk genotype exhibit significantly increased systemic levels of the inflammasome-regulated cytokine IL-18 as compared with AMD patients without the CFH risk genotype, supporting a role for activated complement components in inflammasome activation in AMD (19). In other autoinflammatory diseases such as atherosclerosis and gout, inflammasome priming by complement activation products has likewise been proposed (20, 21). To elucidate the role of complement activation products in inflammasome activation in AMD, we studied the capacity of activated complement components to provide the priming signal in human RPE cells for subsequent NLRP3 inflammasome activation by lipofuscin-mediated photooxidative damage.
Our experiments were performed in the human RPE cell line ARPE-19 and primary fetal human RPE cells. In both cell types be detected constitutive expression of the anaphylatoxin receptors C5aR, C3aR, and C5L2. Employing inflammasome-regulated IL-1β secretion and inflammasome-mediated pyroptotic cell death as measures for inflammasome activation, we demonstrated distinct priming effects for activated complement in human serum as well as for recombinant C5a. Complement heat-inactivation, C5 depletion, and C5a receptor inhibition suppressed the priming effect of human serum, indicating that C5a represents the active priming agent in complement-activated human serum. Priming by C5a enabled subsequent inflammasome activation by lipofuscin-mediated photooxidative damage. Inflammasome activation was dependent on activity of caspase-1 and cathepsin B. Unlike the priming signal, complement-activated serum and C5a were found to be unable to provide the activation signal and, thus, to directly induce inflammasome activation in RPE cells which is consistent with previous reports (34).
Proteins covalently modified with the lipid peroxidation product carboxyethylpyrrole (CEP) have been reported to prime the NLRP3 inflammasome via TLR2 (13). However, subsequent reports have questioned the priming ability of CEP-adducted proteins and rather found it to potentiate inflammasome priming by other signals (35). In our experiments, we did not observe a priming effect of HNE-adducted POS in RPE cells. For example, inflammasome activation was not inducible in cells incubated with HNE-POS alone (Fig. 3, unprimed group) but only in cells co-incubated with the priming agent IL-1α (Fig. 3, IL-1α group). In similar experiments, we determined that POS modified by malondialdehyde (MDA) likewise did not induce inflammasome priming in RPE cells (data not shown). Thus, proteins modified by the lipid peroxidation products HNE and MDA do not seem to represent inflammasome priming signals for the RPE.
While immunological and inflammatory processes are usually believed to contribute to AMD pathogenesis in a detrimental way, the role of inflammasome activation in AMD is still controversial and may vary depending on disease stage and subtype. Inflammasome activation in retinal microglial cells and macrophages has been suggested to reduce choroidal neovascularization via IL-18 and thus to be protective in neovascular AMD (13, 36). In contrast, inflammasome activation in RPE cells has been reported to result in RPE degeneration which may contribute to the development of atrophic AMD (8, 37). While patients with neovascular AMD can be effectively treated with VEGF blocking drugs, no effective therapeutic options are currently available for atrophic AMD. Therefore, the unmed need for identification of potential pharmaceutical targets in atrophic AMD is of crucial importance for clinical ophthalmology.
Inhibitors of C5 and C5a as well as of complement components upstream of C5a generation such as CFD and C3 are currently evaluated in clinical studies in patients with AMD. In our study, inhibition of the C5a/C5aR axis reduced inflammasome activation by lipofuscin phototoxicity in RPE cells. This result supports the rational for therapeutic complement inhibition in atrophic AMD. Moreover, our results suggest the presence of a paracrine amplification loop of inflammasome priming in RPE cells via IL1R. Treatment by the IL1R-inhibitory drug anakinra significantly reduced inflammasome activation in our in vitro experiments, thus providing another potential treatment strategy. Finally, direct therapeutic interference with inflammasome activation has been demonstrated to be effective in vivo, for example using small molecules that provide specific inhibition of NLRP3 (38), and could be tested in future clinical trials for AMD.
In summary, our study identifies complement component C5a as a priming agent for the inflammasome in RPE cells that enables subsequent NLRP3 inflammasome activation by stimuli such as lipofuscin-mediated photooxidative damage. This molecular pathway links hallmark events of AMD pathogenesis including complement activation, lipofuscin accumulation, oxidative damage, and RPE degeneration and may provide novel treatment targets. Inflammasome-inhibiting therapeutics may serve as a potential future means of prevention and treatment of atrophic AMD.
Author Contributions
F. G. H. and T. U. K. coordinated the study and provided funding. C. B. and T. U. K. designed the experiments. C. B. performed the experiments. C. B. and T. U. K. analyzed the data and wrote the manuscript. F. G. H. revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Claudine Strack, BTA, for expert technical assistance.
The study was supported by German Research Foundation (DFG), Bonn, Germany, Grant KR 2863/7-1; Pro Retina Foundation, Bonn, Germany; University of Bonn, BONFOR and SciMed Programs, Bonn, Germany; and Dr. Eberhard and Hilde Rüdiger Foundation, Bonn, Germany (all to T. U. K.). The paper was presented at the 2014 annual meeting of the Association of Research in Vision and Ophthalmology (ARVO) in Orlando, FL (Brandstetter, C., Holz, F. G., Krohne, T.U. (2014) Complement Component C5a Primes the NLRP3 Inflammasome in Retinal Pigment Epithelial Cells. Invest. Ophthalmol. Vis. Sci. 55: E-Abstract 3444). Conflict of interest: CB, none. FGH, research grants: Acucela, Alcon, Allergan, Bayer, Carl Zeiss Meditec, Genentech, Heidelberg Engineering, Novartis, Optos; consultancy honoraria, lecture fees, travel grants: Acucela, Alcon, Allergan, Bayer, Genentech, Heidelberg Engineering, Novartis, Roche. TUK, research grants: Alcon, Novartis; consultancy honoraria, lecture fees, travel grants: Bayer, Heidelberg Engineering, Novartis.
- AMD
- age-related macular degeneration
- RPE
- retinal pigment epithelium
- HNE
- 4-hydroxynonenal
- qPCR
- quantitative real-time PCR
- C5aR
- C5a receptor
- A2E
- N-retinylidene-N-retinyl-ethanolamine
- LDH
- lactate dehydrogenase
- MDA
- malondialdehyde.
References
- 1. Resnikoff S., Pascolini D., Etya'ale D., Kocur I., Pararajasegaram R., Pokharel G. P., and Mariotti S. P. (2004) Global data on visual impairment in the year 2002. Bull. World Health Organ. 82, 844–851 [PMC free article] [PubMed] [Google Scholar]
- 2. Age-Related Eye Disease Study Research Group (2001) A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch. Ophthalmol. 119, 1417–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rózanowska M., Jarvis-Evans J., Korytowski W., Boulton M. E., Burke J. M., and Sarna T. (1995) Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J. Biol. Chem. 270, 18825–18830 [DOI] [PubMed] [Google Scholar]
- 4. Sparrow J. R., Nakanishi K., and Parish C. A. (2000) The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest. Ophthalmol. Vis. Sci. 41, 1981–1989 [PubMed] [Google Scholar]
- 5. Anderson D. H., Mullins R. F., Hageman G. S., and Johnson L. V. (2002) A role for local inflammation in the formation of drusen in the aging eye. Am. J. Ophthalmol. 134, 411–431 [DOI] [PubMed] [Google Scholar]
- 6. Scholl H. P. N., Charbel Issa P., Walier M., Janzer S., Pollok-Kopp B., Börncke F., Fritsche L. G., Chong N. V., Fimmers R., Wienker T., Holz F. G., Weber B. H. F., and Oppermann M. (2008) Systemic complement activation in age-related macular degeneration. PloS One 3, e2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lim L. S., Mitchell P., Seddon J. M., Holz F. G., and Wong T. Y. (2012) Age-related macular degeneration. Lancet 379, 1728–1738 [DOI] [PubMed] [Google Scholar]
- 8. Tarallo V., Hirano Y., Gelfand B. D., Dridi S., Kerur N., Kim Y., Cho W. G., Kaneko H., Fowler B. J., Bogdanovich S., Albuquerque R. J., Hauswirth W. W., Chiodo V. A., Kugel J. F., Goodrich J. A., Ponicsan S. L., Chaudhuri G., Murphy M. P., Dunaief J. L., Ambati B. K., Ogura Y., Yoo J. W., Lee D. K., Provost P., Hinton D. R., Nuñez G., Baffi J. Z., Kleinman M. E., and Ambati J. (2012) DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149, 847–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tseng W. A., Thein T., Kinnunen K., Lashkari K., Gregory M. S., D'Amore P. A., and Ksander B. R. (2013) NLRP3 Inflammasome Activation in Retinal Pigment Epithelial Cells by Lysosomal Destabilization: Implications for Age-Related Macular Degeneration. Invest. Ophthalmol. Vis. Sci. 54, 110–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao M., Bai Y., Xie W., Shi X., Li F., Yang F., Sun Y., Huang L., and Li X. (2015) Interleukin-1β Level Is Increased in Vitreous of Patients with Neovascular Age-Related Macular Degeneration (nAMD) and Polypoidal Choroidal Vasculopathy (PCV). PloS One. 10, e0125150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ijima R., Kaneko H., Ye F., Nagasaka Y., Takayama K., Kataoka K., Kachi S., Iwase T., and Terasaki H. (2014) Interleukin-18 induces retinal pigment epithelium degeneration in mice. Invest. Ophthalmol. Vis. Sci. 55, 6673–6678 [DOI] [PubMed] [Google Scholar]
- 12. Stutz A., Golenbock D. T., and Latz E. (2009) Inflammasomes: too big to miss. J. Clin. Invest. 119, 3502–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doyle S. L., Campbell M., Ozaki E., Salomon R. G., Mori A., Kenna P. F., Farrar G. J., Kiang A. S., Humphries M. M., Lavelle E. C., O'Neill L. A., Hollyfield J. G., and Humphries P. (2012) NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 18, 791–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu R. T., Wang A., To E., Gao J., Cao S., Cui J. Z., and Matsubara J. A. (2014) Vinpocetine inhibits amyloid-beta induced activation of NF-κB, NLRP3 inflammasome and cytokine production in retinal pigment epithelial cells. Exp. Eye Res. 127, 49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anderson O. A., Finkelstein A., and Shima D. T. (2013) A2E induces IL-1β production in retinal pigment epithelial cells via the NLRP3 inflammasome. PloS One 8, e67263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kauppinen A., Niskanen H., Suuronen T., Kinnunen K., Salminen A., and Kaarniranta K. (2012) Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells–implications for age-related macular degeneration (AMD). Immunol. Lett. 147, 29–33 [DOI] [PubMed] [Google Scholar]
- 17. Brandstetter C., Mohr L. K. M., Latz E., Holz F. G., and Krohne T. U. (2015) Light induces NLRP3 inflammasome activation in retinal pigment epithelial cells via lipofuscin-mediated photooxidative damage. J. Mol. Med. Berl. Ger. 93, 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mohr L. K. M., Hoffmann A. V., Brandstetter C., Holz F. G., and Krohne T. U. (2015) Effects of Inflammasome Activation on Secretion of Inflammatory Cytokines and Vascular Endothelial Growth Factor by Retinal Pigment Epithelial Cells. Invest. Ophthalmol. Vis. Sci. 56, 6404–6413 [DOI] [PubMed] [Google Scholar]
- 19. Cao S., Ko A., Partanen M., Pakzad-Vaezi K., Merkur A. B., Albiani D. A., Kirker A. W., Wang A., Cui J. Z., Forooghian F., and Matsubara J. A. (2013) Relationship between systemic cytokines and complement factor H Y402H polymorphism in patients with dry age-related macular degeneration. Am. J. Ophthalmol. 156, 1176–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Samstad E. O., Niyonzima N., Nymo S., Aune M. H., Ryan L., Bakke S. S., Lappegård K. T., Brekke O.-L., Lambris J. D., Damås J. K., Latz E., Mollnes T. E., and Espevik T. (2014) Cholesterol Crystals Induce Complement-Dependent Inflammasome Activation and Cytokine Release. J. Immunol. 192, 2837–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. An L.-L., Mehta P., Xu L., Turman S., Reimer T., Naiman B., Connor J., Sanjuan M., Kolbeck R., and Fung M. (2014) Complement C5a potentiates uric acid crystal-induced IL-1β production. Eur. J. Immunol. 44, 3669–3679 [DOI] [PubMed] [Google Scholar]
- 22. Krohne T. U., Stratmann N. K., Kopitz J., and Holz F. G. (2010) Effects of lipid peroxidation products on lipofuscinogenesis and autophagy in human retinal pigment epithelial cells. Exp. Eye Res. 90, 465–471 [DOI] [PubMed] [Google Scholar]
- 23. Nozaki M., Raisler B. J., Sakurai E., Sarma J. V., Barnum S. R., Lambris J. D., Chen Y., Zhang K., Ambati B. K., Baffi J. Z., and Ambati J. (2006) Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. U.S.A. 103, 2328–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu M., Liu B., Jawad S., Ling D., Casady M., Wei L., and Nussenblatt R. B. (2011) C5a contributes to intraocular inflammation by affecting retinal pigment epithelial cells and immune cells. Br. J. Ophthalmol. 95, 1738–1744 [DOI] [PubMed] [Google Scholar]
- 25. Cortright D. N., Meade R., Waters S. M., Chenard B. L., and Krause J. E. (2009) C5a, But Not C3a, Increases VEGF Secretion in ARPE-19 Human Retinal Pigment Epithelial Cells. Curr. Eye Res. 34, 57–61 [DOI] [PubMed] [Google Scholar]
- 26. Krohne T. U., Kaemmerer E., Holz F. G., and Kopitz J. (2010) Lipid peroxidation products reduce lysosomal protease activities in human retinal pigment epithelial cells via two different mechanisms of action. Exp. Eye Res. 90, 261–266 [DOI] [PubMed] [Google Scholar]
- 27. Fukuoka Y., and Medof E. M. (2001) C5a receptor-mediated production of IL-8 by the human retinal pigment epithelial cell line, ARPE-19. Curr. Eye Res. 23, 320–325 [DOI] [PubMed] [Google Scholar]
- 28. Skeie J. M., Fingert J. H., Russell S. R., Stone E. M., and Mullins R. F. (2010) Complement component C5a activates ICAM-1 expression on human choroidal endothelial cells. Invest. Ophthalmol. Vis. Sci. 51, 5336–5342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mollnes T. E., Garred P., and Bergseth G. (1988) Effect of time, temperature and anticoagulants on in vitro complement activation: consequences for collection and preservation of samples to be examined for complement activation. Clin. Exp. Immunol. 73, 484–488 [PMC free article] [PubMed] [Google Scholar]
- 30. Warner S. J., Auger K. R., and Libby P. (1987) Interleukin 1 induces interleukin 1. II. Recombinant human interleukin 1 induces interleukin 1 production by adult human vascular endothelial cells. J. Immunol. 139, 1911–1917 [PubMed] [Google Scholar]
- 31. Hornung V., and Latz E. (2010) Critical Functions of Priming and Lysosomal Damage for Nlrp3 Activation. Eur. J. Immunol. 40, 620–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davies S., Elliott M. H., Floor E., Truscott T. G., Zareba M., Sarna T., Shamsi F. A., and Boulton M. E. (2001) Photocytotoxicity of lipofuscin in human retinal pigment epithelial cells. Free Radic. Biol. Med. 31, 256–265 [DOI] [PubMed] [Google Scholar]
- 33. Schütt F., Davies S., Kopitz J., Holz F. G., and Boulton M. E. (2000) Photodamage to human RPE cells by A2-E, a retinoid component of lipofuscin. Invest. Ophthalmol. Vis. Sci. 41, 2303–2308 [PubMed] [Google Scholar]
- 34. Laudisi F., Spreafico R., Evrard M., Hughes T. R., Mandriani B., Kandasamy M., Morgan B. P., Sivasankar B., and Mortellaro A. (2013) Cutting Edge: The NLRP3 Inflammasome Links Complement-Mediated Inflammation and IL-1? Release. J. Immunol. Author Choice. 191, 1006–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saeed A. M., Duffort S., Ivanov D., Wang H., Laird J. M., Salomon R. G., Cruz-Guilloty F., and Perez V. L. (2014) The oxidative stress product carboxyethylpyrrole potentiates TLR2/TLR1 inflammatory signaling in macrophages. PloS One 9, e106421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Doyle S. L., Ozaki E., Brennan K., Humphries M. M., Mulfaul K., Keaney J., Kenna P. F., Maminishkis A., Kiang A.-S., Saunders S. P., Hams E., Lavelle E. C., Gardiner C., Fallon P. G., Adamson P., Humphries P., and Campbell M. (2014) IL-18 attenuates experimental choroidal neovascularization as a potential therapy for wet age-related macular degeneration. Sci. Transl. Med. 6, 230ra44. [DOI] [PubMed] [Google Scholar]
- 37. Kim Y., Tarallo V., Kerur N., Yasuma T., Gelfand B. D., Bastos-Carvalho A., Hirano Y., Yasuma R., Mizutani T., Fowler B. J., Li S., Kaneko H., Bogdanovich S., Ambati B. K., Hinton D. R., Hauswirth W. W., Hakem R., Wright C., and Ambati J. (2014) DICER1/Alu RNA dysmetabolism induces Caspase-8-mediated cell death in age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 111, 16082–16087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Coll R. C., Robertson A. A. B., Chae J. J., Higgins S. C., Muñoz-Planillo R., Inserra M. C., Vetter I., Dungan L. S., Monks B. G., Stutz A., Croker D. E., Butler M. S., Haneklaus M., Sutton C. E., Núñez G., Latz E., Kastner D. L., Mills K. H. G., Masters S. L., Schroder K., Cooper M. A., and O'Neill L. A. J. (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]