

Abstract
Excessive intramyocellular triglycerides (muscle lipids) are associated with reduced contractile function, insulin resistance, and Type 2 diabetes, but what governs lipid accumulation in muscle is unclear. Here we report a role of Lkb1 in regulating lipid metabolism in muscle stem cells and their descendent mature muscles. We used MyodCre and Lkb1flox/flox mice to specifically delete Lkb1 in myogenic cells including stem and differentiated cells, and examined the lipid accumulation and gene expression of myoblasts cultured from muscle stem cells (satellite cells). Genetic deletion of Lkb1 in myogenic progenitors led to elevated expression of lipogenic genes and ectopic lipid accumulation in proliferating myoblasts. Interestingly, the Lkb1-deficient myoblasts differentiated into adipocyte-like cells upon adipogenic induction. However, these adipocyte-like cells maintained myogenic gene expression with reduced ability to form myotubes efficiently. Activation of AMPK by AICAR prevented ectopic lipid formation in the Lkb1-null myoblasts. Notably, Lkb1-deficient muscles accumulated excessive lipids in vivo in response to high-fat diet feeding. These results demonstrate that Lkb1 acts through AMPK to limit lipid deposition in muscle stem cells and their derivative mature muscles, and point to the possibility of controlling muscle lipid content using AMPK activating drugs.
Worldwide emerging epidemics of obesity and its associated Type 2 diabetes (T2D), hypertension, cardiovascular diseases, and cancer urge an imperative need for understanding mechanisms underlying fat deposition. Accumulating evidence supports that obesity is caused by excess lipid deposition not only in adipose tissues, but also in non-adipose tissues such as skeletal muscles (van Herpen and Schrauwen-Hinderling, 2008). The skeletal muscle comprises about 40% of the body mass of adults and plays an important role in regulating whole body glucose metabolism and lipid utilization (Zierath and Hawley, 2004). Excessive lipid accumulation in skeletal muscle (called intramyocellular triglyceride, IMTG) is associated with the development of insulin resistance and T2D (Corcoran et al., 2007). Therefore, understanding the molecular regulation of muscle lipid biogenesis and metabolism may lead to promising strategies to treat obesity and diabetes.
Skeletal muscle is made up of multinucleated muscle cells called myofibers, which can be broadly classified as type I and type II (IIa, IIx, and IIb) fibers. Type I and IIa fibers appear red and contain numerous mitochondria, empowering them with a high oxidative capacity and the ability to efficiently utilize lipids (triglycerides) as an energy source (Zierath and Hawley, 2004). By contrast, Type IIx and IIb fibers appear white and have less lipid deposition due to their glycolytic metabolism (Dyck et al., 1997; He et al., 2001). During development, myofibers are formed by fusion of mononuclear myocytes differentiated from satellite cells, a population of myogenic stem cells. Satellite cells are also responsible for the growth, maintenance, and repair of postnatal skeletal muscle (Collins et al., 2005; Montarras et al., 2005; Kuang et al., 2007). Previous studies have demonstrated that satellite cells and their descendant myoblasts are plastic and capable of transdifferentiation into non-myogenic cells such as adipocytes (Seale et al., 2008; Kajimura et al., 2009; Yin et al., 2013), osteoblasts (Rauch et al., 2002), and other types of cells (Brack et al., 2007). Several genes or pathways, including PRMD16, C/EBPβ, MicroRNA-133, and Wnt signaling, have been implicated in the myoblast to adipocyte transition (Ross et al., 2000; Vertino et al., 2005; Seale et al., 2008; Kajimura et al., 2009; Yin et al., 2013). Furthermore, deposition of intramyocellular lipids is affected by age, obesity, and exercise (Taylor-Jones et al., 2002; Franklin and Kanaley, 2009). However, molecular mechanisms governing lipid metabolism in muscle stem cells are still elusive.
Serine/threonine protein kinase 11 (Stk11), commonly known as liver kinase B1 (Lkb1), is reported as a master regulator of many cellular processes including energy metabolism (Nakada et al., 2010), cellular polarity (Amin et al., 2009; Granot et al., 2009), cell adhesion (Zagorska et al., 2010), and cell death (Karuman et al., 2001). Skeletal muscle-specific deletion of Lkb1 mediated by MCK-Cre or HSA-Cre leads to alterations of glucose uptake, insulin sensitivity, and lipid and fatty acid oxidation (Koh et al., 2005; Sakamoto et al., 2005; Koh et al., 2006; Thomson et al., 2007; Jeppesen et al., 2013). However, the metabolic phenotype of the muscle-specific Lkb1 knockout mice appeared to be paradoxical: whereas the Lkb1-null mice had improved insulin sensitivity and glucose homeostasis, they also had defective fatty acid oxidation and reduced contraction-mediated glucose uptake (Koh et al., 2005; Sakamoto et al., 2005; Koh et al., 2006; Thomson et al., 2007; Jeppesen et al., 2013). Although these previous studies have led to the realization that Lkb1 is an important regulator of glucose and lipid metabolism in adult muscle, how these metabolic alterations observed in the postmitotic myofibers develop is unknown. Particularly, the function of Lkb1 in myogenic progenitor cells, especially its role in lipid metabolism of muscle stem cells is completely unknown. Here, we address this question by using the MyodCre mouse line to specifically delete Lkb1 in satellite cells and their descendent myoblasts and adult muscles. Our results demonstrate that Lkb1 acts through AMPK to limit ectopic lipid accumulation in muscle stem cells and mature muscles.
Materials and Methods
Animals
All procedures involving mice were guided by Purdue University Animal Care and Use Committee. Mice were housed in the animal facility with free access to standard rodent chow and water. The mouse strains were derived from Jackson Laboratory (Bar Harbor, ME) under these stock numbers: MyoDCre (#014140), Lkb1flox/flox (#014143).
Primary myoblast isolation and culture
Primary myoblasts were isolated as previously described (Shan et al., 2013a). Briefly, the hind limb skeletal muscles from the WT and MyoD-Lkb1 mice were collected, minced, and digested. The digestions were stopped with F-10 Ham’s medium containing 20% fetal bovine serum (FBS; HyClone, Logan, UT) and centrifuged at 450 × g for 5 min. Then the cells were seeded on collagen-coated dishes and cultured in growth medium containing F-10 Ham’s medium, with 20% FBS, 4ng/ml basic fibroblast growth factor, and 1% penicillin–streptomycin at 37 °C with 5% CO2. The medium was changed every 2 days.
Myogenic and adipogenic differentiation
For myogenic differentiation, the WT and MyoD-Lkb1 myoblasts were induced with horse serum induction medium contains DMEM and 2% horse serum for 3 days. For adipogenic differentiation, the WT and MyoD-Lkb1 myoblasts were induced with adipogenic induction medium contains DMEM, 10% FBS, 2.85 μM insulin, 0.3 μM dexamethasone (DEXA), and 0.63 mM 3-isobutyl-methylxanthine (IBMX) for 4 days and then induced in differentiation medium contains DMEM, 200 nM insulin, and 10 nM T3 for 2 days. After differentiation, the cells were used for staining and extracting total RNA and protein.
Total RNA extraction, cDNA synthesis and real-time PCR
Total RNA extraction, cDNA synthesis and real-time PCR were performed as described (Shan et al., 2013a,b). Briefly, total RNA was extracted from cells and muscle tissues using Trizol Reagent according to the manufacturer’s instructions. The purity and concentration of total RNA were measured by a spectrophotometer (Nanodrop 3000, Thermo Fisher) at 260 nm and 280 nm. Ratios of absorption (260/280 nm) of all samples were between 1.8 and 2.0. Then 5 μg of total RNA was reversed transcribed using random primers and MMLV reverse transcriptase. Real-time PCR was carried out with a Roche Lightcycler 480 PCR System using SYBR Green Master Mix and DD gene-specific primers. The 2−ΔΔCT method was used to analyze the relative changes in gene expression normalized against 18S rRNA as internal control.
Protein extraction and Western blot analysis
Protein extraction and Western blot were conducted as previously described (Shan et al., 2013a). Briefly, total protein was isolated from cells or tissues using RIPA buffer contains 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS. Protein concentrations were determined using Pierce BCA Protein Assay Reagent (Pierce Biotechnology, Rockford, IL). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE), transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore Corporation), blocking in 5% fat-free milk for 1 h at RT, then incubated with first antibodies in 5% milk overnight at 4 °C. Acetyl-CoA carboxylase (ACC) and phosphor-ACC (pACC) antibodies are from cell signaling, FABP4 antibody is from Abcam, all the other antibodies (Lkb1, C/EBPα, phosphor-AMPK (pAMPK), AMPK, and GAPDH) were from Santa Cruz Biotechnology (Santa Cruz). Secondary antibodies (anti-rabbit IgG or anti-mouse IgG, Jackson ImmunoResearch) were diluted 8,000-fold. Immunodetection was performed using enhanced chemiluminescence (ECL) Western blotting substrate (Pierce Biotechnology) and detected with a Gel Logic 2200 imaging system (Carestream, Rochester, NY).
Oil Red O (ORO) staining
Cultured cells and muscle sections were washed with PBS and fixed with 10% formaldehyde for 15 min at room temperature. Then the cells were stained using the ORO working solutions containing 6 ml ORO stock solution (5 g/L in isopropanol) and 4 ml ddH2O for 30 min. After staining, the cells and muscle sections were washed with 60% isopropanol and pictured.
Immunostaining and image acquisition
Immunostaining was performed as previously describe (Shan et al., 2013b). Briefly, cells were fixed with 4% PFA and blocked with blocking buffer containing 5% goat serum, 2% BSA, 0.2% triton X-100, and 0.1% sodium azide in PBS for 1 h. Then the samples were incubated with primary antibodies diluted in blocking buffer overnight at 4 °C. After washing with PBS, the samples were incubated with secondary antibodies and DAPI for 45 min at room temperature. Fluorescent images were captured using Leica DM 6000B fluorescent microscope.
Data Analysis
All experimental data are presented as means ± SEM. Comparisons were made by unpaired two-tailed Student’s t-tests or one-way ANOVA, as appropriate. Effects were considered significant at P <0.05.
Results
Lkb1 expression in muscle is inversely correlated with adipogenic gene expression
Slow myofibers accumulate more IMTG than fast myofibers (Dyck et al., 1997; He et al., 2001). To establish whether Lkb1 expression is correlated to IMTG content, we investigated the relative expression of Lkb1 in soleus (Sol) and extensor digitorum longus (EDL) muscles, known to be enriched with slow-intermediate (Type I/IIa) and fast (Type IIx/IIb) myofibers, respectively. Consistent with previous reports, we found that the expression of adipogenic genes including Adipoq and Lep was higher in Sol muscles compared to EDL muscles (Fig. S1A). In addition, higher expression levels of lipid metabolism related genes, including Lpl, Fasn, Pnpla2, Lipe, Pparg, Cebpa, and Fabp4 were also found in Sol muscles (Fig. S1A–B). Conversely, the protein and the mRNA levels of Lkb1 were lower in Sol muscles than those in EDL muscles (Fig. S1B–C). Similar inverse correlations between adipogenic gene and Lkb1 expression were detected in EDL and Sol muscle-derived myoblasts when induced to undergo adipogenic differentiation (Fig. S1D–E).
The inverse correlation between Lkb1 and adipogenic gene expression suggests that Lkb1 may regulate lipid metabolism in muscle progenitors and mature muscles.
Deletion of Lkb1 enhances lipid accumulation in cultured muscle progenitor cells and their derivative myotubes
To directly address if Lkb1 regulate muscle lipid metabolism, we established a myogenic lineage specific Lkb1 knockout mouse model by crossing MyoDCre and Lkb1flox/flox mice. In the MyodCre/Lkb1flox/flox (henceforth MyoD-Lkb1) offspring, essentially all embryonic myogenic progenitors, their descendent muscles and postnatal satellite cells should have the Lkb1 gene deleted. With this tool we examined how deletion of Lkb1 affects lipid accumulation in muscle progenitor cells by isolating primary myoblasts from WT (Lkblflox/flox) and Lkb1 conditional knockout (MyoD-Lkb1 cKO) mice. Using Oil Red O (ORO) staining as an indicator of lipid content, we found that the MyoD-Lkb1 myoblasts had more abundant ORO signal than did the WT myoblasts (Fig. 1A). Immunostaining with muscle progenitor cell-specific Pax7 indicated that the ORO abundant cells expressed Pax7 and therefore were bona fide myoblasts (Fig. 1B). Realtime PCR and Western blot analyses confirmed that the reduced expression of Lkb1 in the MyoD-Lkb1 myoblasts was associated with increased expression of Fabp4 and Adipoq (Fig. 1C–E), classical markers of adipocytes and lipid metabolism. Interestingly, Lkb1 deletion also leads to elevated expression of muscle progenitor cell markers Pax7, Myf5, and MyoD, but reduced expression of Myog, a classical myogenic differentiation marker (Fig. 1F). These data indicate that deletion of Lkb1 enhances lipid accumulation and expression of lipid related genes in proliferating myoblasts.
Fig. 1.
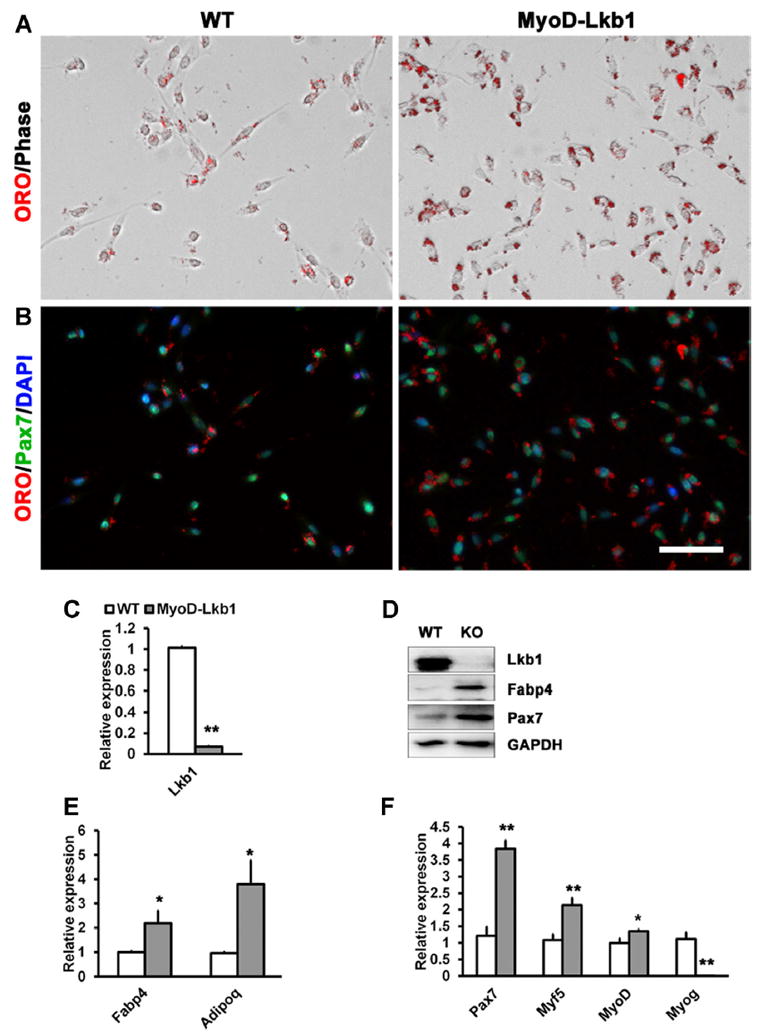
Deletion of Lkb1 enhances lipid accumulation in myoblast. (A) Oil Red O (ORO) staining of the WT and MyoD-Lkb1 myoblast. (B) Myoblasts were stained with ORO and Pax7 antibody to label lipid accumulation and satellite cells. Nuclei were counterstained with DAPI. (C) mRNA levels of Lkb1 in WT and MyoD-Lkb1 myoblasts. (D) Protein levels of Lkb1, Fabp4 and Pax7 in WT and MyoD-Lkb1 myoblasts. (E, F) Expression of adipogenic (E) and myogenic (F) related genes in WT and MyoD-Lkb1 myoblasts. Error bars represent SEM, n =5. “*” P <0.05, “**” P <0.01. Scale bars: 50 μm.
As the MyoD-Lkb1 cKO myoblasts have altered expression of adipogenic and myogenic genes, we next examined their myogenic and adipogenic differentiation potentials. When induced to undergo myogenic differentiation by serum withdrawal, WT myoblasts readily formed multinucleated myotubes that express sarcomeric myosin heavy chain (MHC) (Fig. S2A–B). By contrast, MyoD-Lkb1 myoblasts formed only few and small MHC+ myotubes (Fig. S2A–B). Western blot and Realtime PCR confirmed that compared to WT myotubes, MyoD-Lkb1 myotubes expressed lower levels of differentiation and fusion marker genes, including Des, Cav3, Myh3, Myh4, and Myh8, but higher levels of undifferentiated progenitor cell marker Pax7 (Fig. S2C–D). These results demonstrate that deletion of Lkb1 inhibits myogenic differentiation.
We further examined the adipogenic differentiation of the MyoD-Lkb1 cKO myoblasts after induced with adipogenic medium containing insulin, dexamethasone, and T3. Under this condition, WT myoblasts differentiated into multinuclear myotubes, but the Lkb1-null myoblasts formed few myotubes that were very small in size (Fig. 2A). Instead, Lkb1 deleted myoblasts differentiated into adipocyte-like cells with a number of lipid droplets (Fig. 2A). ORO and MHC antibody (MF20) staining revealed robust lipid accumulation (ORO+) but few myotubes (MHC+) in Lkb1-deficient myoblasts, compared to strong MHC but weak ORO signals in the WT myoblast culture (Fig. 2B). Consistent with morphological alteration, Lkb1-null cells had lower expression levels of myogenic differentiation marker genes, including Myog, Myf6, Des, Myh3, Myh8 (Fig. 2C–D), but higher expression levels of adipogenic genes such as Fabp4, Adipoq, and Lep (Fig. 2E). Interestingly, the expression of classical brown adipose specific genes, Prdm16, Cidea, Pgc1a, and Ppara, were reduced in the MyoD-Lkb1 cKO cultures (Fig. 2F), suggesting that the adipocyte-like cells differentiated from the Lkb1-deleted myoblasts are more similar to white adipocytes. These results together demonstrate that Lkb1 deletion promotes adipogenic differentiation of myoblasts at the expense of myogenic differentiation.
Fig. 2.
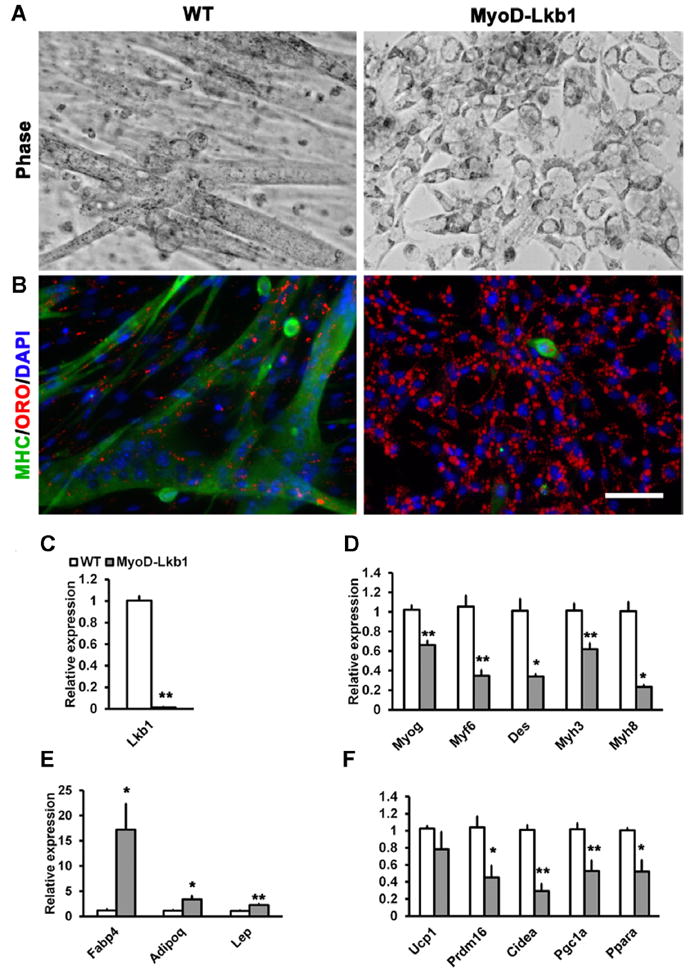
Lkb1 deletion promotes adipogenic differentiation. (A) Representative images of WT and MyoD-Lkb1 myoblasts after adipogenic differentiation. (B) WT and MyoD-Lkb1cells were stained with Oil Red O and MHC antibody to label lipid accumulation and myotubes. Nuclei were counterstained with DAPI. (C–F) The expression of (C) Lkb1, (D) Myogenic-related genes, (E) adipogenic-related genes and (F) brown marker genes in WT and MyoD-Lkb1 myoblasts after adipogenic differentiation. Error bars represent SEM, n =5–7. “*” P <0.05, “**” P <0.01. Scale bars: 50 μm.
Satellite cells are plastic and can transdifferentiate into adipocytes (Ross et al., 2000; Vertino et al., 2005; Seale et al., 2008; Kajimura et al., 2009; Yin et al., 2013). In addition to the gain of adipogenic gene expression, a hallmark of transdifferentiation is the loss of myogenic gene expression. To address if lipid accumulation in the adipocyte-like cells is due to a myoblast to adipocyte transdifferentiation, we examined myogenic gene expression in the lipid-filled cells. The adipocyte-like cells differentiated from Lkb1-deficient myoblasts still expressed high levels of Pax7, Myf5, and MyoD (Fig. S3), indicating that ectopic lipid accumulation in Lkb1-null myoblasts is not due to a cell identity switch to adipocytes.
Deletion of Lkb1 leads to ectopic lipid deposition in vivo after feeding with HFD
To investigate the physiological relevance of the observed lipid accumulation in cultured Lkb1-null myoblasts, we examined muscle lipid content of the MyoD-Lkb1 mice. We fed mice with high-fat diet (HFD) to mimic the eutrophic condition in culture medium to promote lipid uptake and accumulation. Prior to HFD feeding, the WT and MyoD-Lkb1 mice expressed similar levels of adipogenic genes in the Sol muscles (Fig. S4A) and accumulated little lipid in myofibers (Data not shown). After HFD feeding, however, higher levels of adipogenic genes (Adipoq, Lep, Cebpa, Srebp-1c, and Fasn) were detected in the MyoD-Lkb1 cKO mice than in their WT littermates (Fig. S4B). The elevated expression of adipogenic genes was associated with readily detectable lipid deposition in the Myod-Lkb1 myofibers revealed by ORO staining (Fig. S4C). These results indicate that alterations in lipid metabolism observed in the Lkb1-null progenitors have led to metabolic consequences in the adult muscle.
Lkb1 deletion alters the expression of lipid metabolism related genes in myoblasts
Lipid accumulation in cells is a dynamic process involving fatty acid transport and uptake, and lipid synthesis and utilization. In muscle cells, lipid metabolism is regulated by enzymes involved in lipogenesis, lipolysis, fatty acid synthase, and oxidation (Li et al., 2011; Lee et al., 2014). We examined whether Lkb1 deletion alters the expression of genes involved in these processes. MyoD-Lkb1 cKO myoblasts expressed elevated levels of Cebpa, Fasn, Acaca, and Dgat1 (Fig. 3A–B), key factors mediating triglyceride synthesis (formation of lipid droplets). In addition, genes related to fatty acid transport and mobilization, Fabp4 and Lpl, were also upregulated in the MyoD-Lkb1 cKO myoblasts (Fig. 3A–B). On the other hand, rate-limiting enzymes mediating lipolysis (Atgl/Pnpla2, hydrolyzes glycerides) and fatty acid oxidation (Cpt1b) were downregulated in MyoD-Lkb1 cKO myoblasts (Fig. 3C). Also downregulated was pACC (Fig. 3D), which blocks the activity of ACC, a key enzyme that bidirectionally inhibits beta-oxidative but promotes lipogenesis. Interestingly, expression of Hsl/Lipe, which hydrolyzes DG to form MG was increased in the MyoD-Lkb1 cKO myoblasts (Fig. 3C). Similar gene expression patterns were observed in the MyoD-Lkb1 cKO myoblasts after they were induced to undergo adipogenic differentiation (Fig. 3E–F). Together, the combined upregulation of lipid synthesis and transport genes, and downregulation of lipolysis and fatty acid oxidation-related genes underscore the enhanced lipid accumulation in the MyoD-Lkb1 cKO myoblasts.
Fig. 3.

Lkb1 absence affects the expression of lipid metabolism related genes. (A, B) The expression of lipogenesis and fatty acid synthase related genes and proteins in WT and MyoD-Lkb1 myoblasts before adipogenic differentiation. (C–D) The expression of lipolysis and oxidation related genes/proteins in WT and MyoD-Lkb1 myoblasts before adipogenic differentiation. (E–F) The expression of lipid metabolism related genes in WT and MyoD-Lkb1 myoblasts after adipogenic differentiation. Error bars represent SEM, n =5–7. “*” P <0.05, “**” P <0.01.
Lkb1 affects lipid metabolism through AMPK pathway
To understand the molecular mechanisms through which Lkb1 regulates lipid metabolism in muscle stem cells, we focused on the canonical AMPK pathway downstream of Lkb1 (Lizcano et al., 2004). As expected, we found that the levels of pAMPK (T172) were significantly attenuated in cultured myoblasts of MyoD-Lkb1 compared to WT mice (Fig. 4A, B). The mRNA level of Prkaa2 was also significantly lower in Lkb1-deleted myoblasts (Fig. 4C). Consistent with lower levels of Lkb1 expression in SOL compared to EDL muscles (Fig. S1), lower levels of Prkaa2 were found in SOL muscles (Fig. 4D).
Fig. 4.

AMPK signal pathway affects lipid metabolism related genes expression in myoblasts. (A–C) Deletion of Lkb1 decreased protein levels of phosphorylated AMPK (A, B) and mRNA levels of Prkaa2 (C). (D) Low expression of Prkaa2 in SOL muscle. (E, F) AMPK activator AICAR treatment significantly increased pAMPK level and reduced the protein levels of C/EBPa in WT primary myoblast. (G) Activation of AMPK by AICAR decreased the mRNA expression of lipid metabolism related genes in WT primary myoblast. Error bars represent SEM, n =5–7. “*” P <0.05, “**” P <0.01.
We further examined if activation of AMPK can conversely inhibit lipid deposition in myoblasts. We used an established AMPK activator AICAR to activate AMPK, and AICAR treatment significantly increased pAMPK level but decreased C/EBPα expression (Fig. 4E and F). In addition, the expression of a panel of adipogenic genes including Adipoq, Cebpa, Lpl, Fasn, and Acaca were significantly decreased, while the expression of fatty acid oxidation-related gene Cpt1b was significantly increased by AICAR treatment (Fig. 4G). Furthermore, AICAR treatment reduced lipid accumulation in WT myoblasts, as manifested by reduced ORO staining signal (Fig. 5A). These results demonstrate that activation of AMPK inhibits lipid accumulation in WT myoblasts.
Fig. 5.

Deletion of Lkb1 enhances lipid accumulation in myoblasts through AMPK pathway. (A) AICAR treatment decreased lipid accumulation in WT and MyoD-Lkb1 myoblasts. (B, C) AICAR treatment increased the level of pAMPK in WT and MyoD-Lkb1 myoblast. n =3. (D, E) AICAR treatment decreased the expression of Fabp4 (D), but upregulated the level of phosphor-ACC (pACC) (E). (F) AICAR treatment abolished the effect of Lkb1 deletion on the expression of the adipogenic and fatty acid synthesis related genes. n =5–7. Error bars represent SEM, “*” P <0.05, “**” P <0.01. Scale bars: 50 μm.
Finally, we examined if activation of AMPK can rescue the ectopic lipid deposition observed in the MyoD-Lkb1 cKO myoblasts. AICAR treatment indeed reduced ORO signal in the MyoD-Lkb1 cKO myoblasts, to a similar level observed in vehicle treated WT myoblasts (Fig. 5A). At the molecular level, AICAR treatment significantly decreased the expression of Fabp4, but upregulated the levels of pAMPK and pACC (Fig. 5B–E). Importantly, AICAR treatment nearly abolished the effect of Lkb1 deletion on the expression of the adipogenic and fatty acid synthesis-related genes (Fig. 5F). Collectively, these data indicate that Lkb1 limits lipid accumulation in myoblasts through activating the AMPK pathway.
Discussion
We identified in this study a role of Lkb1 in lipid metabolism of muscle stem cells. We used the MyodCre to drive muscle lineage specific deletion of Lkb1. Previous studies have demonstrate the MyodCre marks all embryonic progenitors that give rise to muscle satellite cells and mature muscles (Kanisicak et al., 2009), therefore the MyoD-Lkb1 cKO mouse model provides key information regarding the role of Lkb1 in muscle progenitors. We provided evidence that deletion of Lkb1 enhanced lipid accumulation and the expression of adipogenic marker genes in myoblasts. We further elucidated the downstream signaling through which Lkb1 regulates the lipid metabolism in muscle stem cells. Our study provides critical understanding of the development of metabolic phenotypes reported in mature muscle specific Lkb1 knockout mice (Koh et al., 2005; Sakamoto et al., 2005).
Lipid accumulation is regulated by the expression of several genes related to the fatty acids uptake (Lpl), lipogenesis (Acc, Fasn), triglycerides synthesis (Dgat1), mitochondrial lipid/fatty acid oxidation (Cpt1b), and lipolysis (Atgl/Pnpla2, Hsl/Lipe, and Mgl) (Li et al., 2011; Lee et al., 2014). Atgl deficiency mice have severe defects in TG hydrolysis, leading to TG accumulation in muscle (Haemmerle et al., 2006; Nunes et al., 2012), while overexpression of Atgl decreased TG content in myotubes (Badin et al., 2011). Muscle-specific overexpression of Lpl (Voshol et al., 2001; Tamilarasan et al., 2012) or Dgat1 (Roorda et al., 2005; Liu et al., 2007; Liu et al., 2011) also results in increased accumulation of TG in skeletal muscles. By contrast, deletion of Lpl (Wang and Eckel, 2009; Wang et al., 2009) or Dgat1 (Liu et al., 2011) in skeletal muscle reduces TG accumulation. Cpt1 overexpression similarly reduces lipid storage in mice (Bruce et al., 2007). Acetyl-CoA carboxylase (ACC) catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, and phosphorylation of ACC reduces its enzyme activity. We found that Lkb1 KO myoblasts have reduced level of pACC, which should lead to increased levels of malonyl-CoA. Malonyl-CoA not only accelerates fatty acid synthesis, but also inhibits the activity of Cpt1. A reduction of pACC would further inhibit Cpt1 activity and decrease the uptake of fatty acyl-CoA into the mitochondria for subsequent oxidation. Our gene expression analysis results are consistent with these previous reports and demonstrate that Lkb1 acts as negative regulator of lipogenic genes, and its ablation contributes to lipid accumulation in muscle cells.
Hsl is the rate-limiting enzyme for the cellular catabolism of DG and Hsl deficiency causes the accumulation of DG in skeletal muscle and adipose tissue (Haemmerle et al., 2002). The interesting finding that Lkb1 deletion increases Hsl expression may be due to a negative feedback regulation in response to reduced DG and MG levels. In this scenario, reduced Atgl expression not only led to accumulation of TG, but also reduced levels downstream products (DG and MG). The reduced DG and MG in turn act as negative feedback to increase Hsl expression.
Lkb1 can phosphorylate a number of substrates including AMPK (Lizcano et al., 2004), a central metabolic sensor of lipid and glucose metabolism in muscle and adipose tissue (Hardie et al., 2003; Koh et al., 2006). We found that the levels of pAMPK (T172) were reduced in MyoD-Lkb1 myoblasts. AMPK can directly phosphorylate several substrates critically involved in lipid metabolism, including ACC, Atgl, Hsl, and SREBP1 (Watt et al., 2006; Koh et al., 2008; Ahmadian et al., 2011; Mihaylova and Shaw, 2011). Previous studies have suggested that the phosphorylation of AMPK in mammalian cells induced by AICAR treatment might be mediated through Lkb1-dependent/independent manner (Rattan et al., 2005; Sakamoto et al., 2005; Sun et al., 2007). Here, we found that AICAR treatment upregulated pAMPK and pACC in WT and MyoD-Lkb1 myoblast. Importantly, AICAR abolished the effects of Lkb1 deletion on lipid accumulation in myoblast. These data indicate that Lkb1 affects lipid accumulation in myoblast through AMPK-dependent pathways. In conclusion, our data provide novel insights into the role of Lkb1 in lipid metabolism of skeletal muscle stem cells. These results demonstrate that targeting Lkb1 and its downstream signaling represents an effective approach to regulate lipid metabolism in myocytes and treat obesity and diabetes.
An interesting question remains to be addressed is if ectopic lipid accumulation is a cause or a consequence of the impaired myogenic differentiation. An analogical question exists in the adult muscle. Ectopic lipid accumulation is associated with impaired muscle function in muscle wasting diseases (Grounds et al., 2014). Under such conditions muscle lipid accumulation is considered as a consequence of loss of muscle integrity and function. However, lipid accumulation has also been reported to lead to myopathy (Saini-Chohan et al., 2012). The observation that under adipogenic culture conditions wild-type satellite cells accumulate lipids while maintaining myogenic gene expression (Starkey et al., 2011) suggests that lipid accumulation in the Lkb1-null myoblasts is not due to secondary effect of impaired myogenic differentiation. Thus, Lkb1 deletion might have directly accelerated ectopic lipid accumulation in myoblasts independent of its differentiation status. This is supported by our observation that Lkb1-null myoblasts accumulate lipids prior to induction of differentiation.
Supplementary Material
Acknowledgments
The project was partially supported by funding from NIH (R01AR060652) and an incentive grant from Purdue University Office of Vice President for Research (OVPR) to SK. We thank Jun Wu for mouse colony maintenance.
Contract grant sponsor: NIH;
Contract grant number: R01AR060652.
Abbreviations
- Acc
acetyl-CoA carboxylase
- AMPK
AMP-activated protein kinase
- Atgl
adipose triglyceride lipase
- BAT
brown adipose tissue
- Cebpa
CCAAT/enhancer-binding protein alpha
- Cpt1
carnitine palmitoyltransferase-1
- DAPI
4’,6-diamidino-2-phenylindole
- DEXA
dexamethasone
- DG
diacylglycerol (diglyceride)
- Dgat1
diacylglycerol acyltransferase 1
- EDL
extensor digitorum longus
- Fabp4
fatty acid-binding protein 4
- Fas
fatty acid synthase
- FBS
fetal bovine serum
- Hsl
hormone-sensitive lipase
- IBMX
3-isobutyl-methylxanthine
- Lkb1
Liver kinase B1
- Lpl
lipoprotein lipase
- Pax7
paired-box transcription factor 7
- Pparg
Peroxisome proliferator-activated receptor γ
- Sol
soleus
- Srebp-1c
sterol regulatory element-binding Protein 1c
Footnotes
Conflict of interest: The authors declare no conflict of interests.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
Literature Cited
- Ahmadian M, Abbott MJ, Tang TY, Hudak CSS, Kim Y, Bruss M, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, Wang YH, Duncan RE, Kang C, Sul HS. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011;13:739–748. doi: 10.1016/j.cmet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin N, Khan A, St Johnston D, Tomlinson I, Martin S, Brenman J, McNeill H. LKB1 regulates polarity remodeling and adherens junction formation in the Drosophila eye. Proc Natl Acad Sci USA. 2009;106:8941–8946. doi: 10.1073/pnas.0812469106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badin PM, Louche K, Mairal A, Liebisch G, Schmitz G, Rustan AC, Smith SR, Langin D, Moro C. Altered skeletal muscle lipase expression and activity contribute to insulin resistance in humans. Diabetes. 2011;60:1734–1742. doi: 10.2337/db10-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- Bruce CR, Brolin C, Turner N, Cleasby ME, van der Leij FR, Cooney GJ, Kraegen EW. Overexpression of carnitine palmitoyltransferase I in skeletal muscle in vivo increases fatty acid oxidation and reduces triacylglycerol esterification. Am J Physiol-Endoc M. 2007;292:E1231–E1237. doi: 10.1152/ajpendo.00561.2006. [DOI] [PubMed] [Google Scholar]
- Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA, Morgan JE. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Corcoran MP, Lamon-Fava S, Fielding RA. Skeletal muscle lipid deposition and insulin resistance: Effect of dietary fatty acids and exercise. Am J Clin Nutr. 2007;85:662–677. doi: 10.1093/ajcn/85.3.662. [DOI] [PubMed] [Google Scholar]
- Dyck DJ, Peters SJ, Glatz J, Gorski J, Keizer H, Kiens B, Liu S, Richter EA, Spriet LL, van der Vusse GJ, Bonen A. Functional differences in lipid metabolism in resting skeletal muscle of various fiber types. Am J Physiol. 1997;3:E340–E351. doi: 10.1152/ajpendo.1997.272.3.E340. [DOI] [PubMed] [Google Scholar]
- Franklin RM, Kanaley JA. Intramyocellular lipids: Effect of age, obesity, and exercise. Phys Sportsmed. 2009;37:20–26. doi: 10.3810/psm.2009.04.1679. [DOI] [PubMed] [Google Scholar]
- Granot Z, Swisa A, Magenheim J, Stolovich-Rain M, Fujimoto W, Manduchi E, Miki T, Lennerz JK, Stoeckert CJ, Jr, Meyuhas O, Seino S, Permutt MA, Piwnica-Worms H, Bardeesy N, Dor Y. LKB1 regulates pancreatic beta cell size, polarity, and function. Cell Metab. 2009;10:296–308. doi: 10.1016/j.cmet.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grounds MD, Terrill JR, Radley-Crabb HG, Robertson T, Papadimitriou J, Spuler S, Shavlakadze T. Lipid accumulation in dysferlin-deficient muscles. Am J Pathol. 2014;184:1668–1676. doi: 10.1016/j.ajpath.2014.02.005. [DOI] [PubMed] [Google Scholar]
- Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefier G, Zechner R. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- Haemmerle G, Zimmermann R, Hayn M, Theussl C, Waeg G, Wagner E, Sattler W, Magin TM, Wagner EF, Zechner R. Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J Biol Chem. 2002;277:4806–4815. doi: 10.1074/jbc.M110355200. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- He J, Watkins S, Kelley DE. Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes. 2001;50:817–823. doi: 10.2337/diabetes.50.4.817. [DOI] [PubMed] [Google Scholar]
- Jeppesen J, Maarbjerg SJ, Jordy AB, Fritzen AM, Pehmoller C, Sylow L, Serup AK, Jessen N, Thorsen K, Prats C, Qvortrup K, Dyck JRB, Hunter RW, Sakamoto K, Thomson DM, Schjerling P, Wojtaszewski JFP, Richter EA, Kiens B. LKB1 regulates lipid oxidation during exercise independently of AMPK. Diabetes. 2013;62:1490–1499. doi: 10.2337/db12-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP, Spiegelman BM. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009;460:1154–1158. doi: 10.1038/nature08262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanisicak O, Mendez JJ, Yamamoto S, Yamamoto M, Goldhamer DJ. Progenitors of skeletal muscle satellite cells express the muscle determination gene. MyoD Dev Biol. 2009;332:131–141. doi: 10.1016/j.ydbio.2009.05.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuman P, Gozani O, Odze RD, Zhou XC, Zhu H, Shaw R, Brien TP, Bozzuto CD, Ooi D, Cantley LC, Yuan JY. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell. 2001;7:1307–1319. doi: 10.1016/s1097-2765(01)00258-1. [DOI] [PubMed] [Google Scholar]
- Koh HJ, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, Li YF, Liew CW, Ho RC, Hirshman MF, Kulkarni RN, Kahn CR, Goodyear LJ. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves, glucose homeostasis, and decreases TRB3. Mol Cell Biol. 2006;26:8217–8227. doi: 10.1128/MCB.00979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh HJ, Brandauer J, Goodyear LJ. LKB1 and AMPK and the regulation of skeletal muscle metabolism. Curr Opin Clin Nutr Metab Care. 2008;11:227–232. doi: 10.1097/MCO.0b013e3282fb7b76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh HJ, Hirshman MF, Fujii N, Arnolds DE, Rogers MJ, Mukai N, Jessen N, Ho RC, Goodyear LJ. Skeletal muscle-specific knockout of LKB1 causes AICAR resistance but improves glucose tolerance. Diabetes. 2005;54:A384–A384. [Google Scholar]
- Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Pramyothin P, Karastergiou K, Fried SK. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim Biophys Acta. 2014;1842:473–481. doi: 10.1016/j.bbadis.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Paran C, Wolins NE, Horowitz JF. High muscle lipid content in obesity is not due to enhanced activation of key triglyceride esterification enzymes or the suppression of lipolytic proteins. Am J Physiol Endocrinol Metab. 2011;300:E699–E707. doi: 10.1152/ajpendo.00316.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yu SQ, Khan RS, Ables GP, Bharadwaj KG, Hu YY, Huggins LA, Eriksson JW, Buckett LK, Turnbull AV, Ginsberg HN, Blaner WS, Huang LS, Goldberg IJ. DGAT1 deficiency decreases PPAR expression and does not lead to lipotoxicity in cardiac and skeletal muscle. J Lipid Res. 2011;52:732–744. doi: 10.1194/jlr.M011395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang YY, Chen N, Shi XJ, Tsang B, Yu YH. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J Clin Invest. 2007;117:1679–1689. doi: 10.1172/JCI30565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309:2064–2067. doi: 10.1126/science.1114758. [DOI] [PubMed] [Google Scholar]
- Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–U669. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes PM, van de Weijer T, Veltien A, Arnts H, Hesselink MKC, Glatz JFC, Schrauwen P, Tack CJ, Heerschap A. Increased intramyocellular lipids but unaltered in vivo mitochondrial oxidative phosphorylation in skeletal muscle of adipose triglyceride lipase-deficient mice. Am J Physiol-Endoc M. 2012;303:E71–E81. doi: 10.1152/ajpendo.00597.2011. [DOI] [PubMed] [Google Scholar]
- Rattan R, Giri S, Singh AK, Singh I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J Biol Chem. 2005;280:39582–39593. doi: 10.1074/jbc.M507443200. [DOI] [PubMed] [Google Scholar]
- Rauch C, Brunet AC, Deleule J, Farge E. C2C12 myoblast/osteoblast transdifferentiation steps enhanced by epigenetic inhibition of BMP2 endocytosis. Am J Physiol Cell Physiol. 2002;283:C235–C243. doi: 10.1152/ajpcell.00234.2001. [DOI] [PubMed] [Google Scholar]
- Roorda BD, Hesselink MKC, Schaart G, Moonen-Kornips E, Martinez-Martinez P, Losen M, De Baets MH, Mensink RP, Schrauwen P. DGAT1 overexpression in muscle by in vivo DNA electroporation increases intramyocellular lipid content. J Lipid Res. 2005;46:230–236. doi: 10.1194/jlr.M400416-JLR200. [DOI] [PubMed] [Google Scholar]
- Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, MacDougald OA. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- Saini-Chohan HK, Mitchell RW, Vaz FM, Zelinski T, Hatch GM. Delineating the role of alterations in lipid metabolism to the pathogenesis of inherited skeletal and cardiac muscle disorders: Thematic Review Series: Genetics of Human Lipid Diseases. J Lipid Res. 2012;53:4–27. doi: 10.1194/jlr.R012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie, Ashworth D, Alessi A. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. Embo J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, Tempst P, Rudnicki MA, Beier DR, Spiegelman BM. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan TZ, Liang XR, Bi PP, Kuang SH. Myostatin knockout drives browning of white adipose tissue through activating the AMPK-PGC1 alpha-Fndc5 pathway in muscle. Faseb J. 2013;27:1981–1989. doi: 10.1096/fj.12-225755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan TZ, Liu WY, Kuang SH. Fatty acid binding protein 4 expression marks a population of adipocyte progenitors in white and brown adipose tissues. Faseb J. 2013;27:277–287. doi: 10.1096/fj.12-211516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkey JD, Yamamoto M, Yamamoto S, Goldhamer DJ. Skeletal muscle satellite cells are committed to myogenesis and do not spontaneously adopt nonmyogenic fates. J Histochem Cytochem. 2011;59:33–46. doi: 10.1369/jhc.2010.956995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Connors KE, Yang DQ. AICAR induces phosphorylation of AMPK in an ATM-dependent, LKB1-independent manner. Mol Cell Biochem. 2007;306:239–245. doi: 10.1007/s11010-007-9575-6. [DOI] [PubMed] [Google Scholar]
- Tamilarasan KP, Temmel H, Das SK, Al Zoughbi, Schauer W, Vesely S, Hoefier PW. Skeletal muscle damage and impaired regeneration due to LPL-mediated lipotoxicity. Cell Death & Disease. 2012;3:e354. doi: 10.1038/cddis.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Jones JM, McGehee RE, Rando TA, Lecka-Czernik B, Lipschitz DA, Peterson CA. Activation of an adipogenic program in adult myoblasts with age. Mech Ageing Dev. 2002;123:649–661. doi: 10.1016/s0047-6374(01)00411-0. [DOI] [PubMed] [Google Scholar]
- Thomson DM, Brown JD, Fillmore N, Condon BM, Kim HJ, Barrow JR, Winder WW. LKB1 and the regulation of malonyl-CoA and fatty acid oxidation in muscle. Am J Physiol Endocrinol Metab. 2007;293:E1572–E1579. doi: 10.1152/ajpendo.00371.2007. [DOI] [PubMed] [Google Scholar]
- van Herpen NA, Schrauwen-Hinderling VB. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav. 2008:94231–241. doi: 10.1016/j.physbeh.2007.11.049. [DOI] [PubMed] [Google Scholar]
- Vertino AM, Taylor-Jones JM, Longo KA, Bearden ED, Lane TF, McGehee RE, Jr, MacDougald OA, Peterson CA. Wnt10b deficiency promotes coexpression of myogenic and adipogenic programs in myoblasts. Mol Biol Cell. 2005;16:2039–2048. doi: 10.1091/mbc.E04-08-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voshol PJ, Jong MC, Dahlmans VE, Kratky D, Levak-Frank S, Zechner R, Romijn JA, Havekes LM. In muscle-specific lipoprotein lipase-overexpressing mice, muscle triglyceride content is increased without inhibition of insulin-stimulated whole-body and muscle-specific glucose uptake. Diabetes. 2001;50:2585–2590. doi: 10.2337/diabetes.50.11.2585. [DOI] [PubMed] [Google Scholar]
- Wang H, Eckel RH. Lipoprotein lipase: From gene to obesity. Am J Physiol-Endoc M. 2009;297:E271–E288. doi: 10.1152/ajpendo.90920.2008. [DOI] [PubMed] [Google Scholar]
- Wang H, Knaub LA, Jensen DR, Jung DY, Hong EG, Ko HJ, Coates AM, Goldberg IJ, de la Houssaye BA, Janssen RC, McCurdy CE, Rahman SM, Choi CS, Shulman GI, Kim JK, Friedman JE, Eckel RH. Skeletal muscle-specific deletion of lipoprotein lipase enhances insulin signaling in skeletal muscle but causes insulin resistance in liver and other tissues. Diabetes. 2009;58:116–124. doi: 10.2337/db07-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt MJ, Holmes AG, Pinnamaneni SK, Garnham AP, Steinberg GR, Kemp BE, Febbraio MA. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am J Physiol-Endoc M. 2006;290:E500–E508. doi: 10.1152/ajpendo.00361.2005. [DOI] [PubMed] [Google Scholar]
- Yin H, Pasut A, Soleimani VD, Bentzinger CF, Antoun G, Thorn S, Seale P, Fernando P, van IJcken W, Grosveld F, Dekemp RA, Boushel R, Harper ME, Rudnicki MA. MicroRNA-133 controls brown adipose determination in skeletal muscle satellite cells by targeting Prdm16. Cell Metab. 2013;17:210–224. doi: 10.1016/j.cmet.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagorska A, Deak M, Campbell DG, Banerjee S, Hirano M, Aizawa S, Prescott AR, Alessi DR. New roles for the LKB1-NUAK pathway in controlling myosin phosphatase complexes and cell adhesion. Sci Signal. 2010;3(115) doi: 10.1126/scisignal.2000616. [DOI] [PubMed] [Google Scholar]
- Zierath JR, Hawley JA. Skeletal muscle fiber type: Influence on contractile and metabolic properties. PLoS Biol. 2004;2:e348. doi: 10.1371/journal.pbio.0020348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.