

 Doxorubicin and lipid-loading (steatosis) interact synergistically in Huh7 hepatoma cells. This results in enhanced cytotoxicity and pro-inflammatory ROS accumulation.
Doxorubicin and lipid-loading (steatosis) interact synergistically in Huh7 hepatoma cells. This results in enhanced cytotoxicity and pro-inflammatory ROS accumulation.
Abstract
There has been a dramatic increase in the number of clinically obese individuals in the last twenty years. This has resulted in an increasingly common scenario where obese individuals are treated for other diseases, including cancer. Here, we examine interactions between lipid-induced steatosis and doxorubicin treatment in the human hepatoma cell line Huh7. The response of cells to either doxorubicin, lipid-loading or a combination were examined at the global level by DNA microarray, and for specific endpoints of cytotoxicity, lipid-loading, reactive oxygen species, anti-oxidant response systems, and apoptosis. Both doxorubicin and lipid-loading caused a significant accumulation of lipid within Huh7 cells, with the combination resulting in an additive accumulation. In contrast, cytotoxicity was synergistic for the combination compared to the individual components, suggesting an enhanced sensitivity of lipid-loaded cells to the acute hepatotoxic effects of doxorubicin. We demonstrate that a synergistic increase in reactive oxygen species and deregulation of protective anti-oxidant systems, most notably metallothionein expression, underlies this effect. Transcriptome analysis confirms synergistic changes at the global level, and is consistent with enhanced pro-inflammatory signalling in steatotic cells challenged with doxorubicin. Such effects are consistent with a potentiation of progression along the fatty liver disease spectrum. This suggests that treatment of obese individuals with doxorubicin may increase the risk of both acute (i.e. hepatotoxicity) and chronic (i.e. progress of fatty liver disease) adverse effects. This work highlights the need for more study in the growing therapeutic area to develop risk mitigation strategies.
Introduction
The incidence of breast cancer in women has remained stubbornly high, affecting approximately one in eight women in the Western world during their lifetimes.1 In contrast, the incidence of obesity and its related morbidities has rapidly increased over the past twenty years.2 One consequence of this is the increased probability of treating obese individuals for breast cancer, especially given the positive correlation between obesity and breast cancer in post-menopausal women.3 An important co-morbidity associated with obesity is the spectrum of fatty liver diseases, ranging from simple steatosis, through steatohepatitis to cirrhosis and/or hepatocarcinogenesis. The molecular underpinnings of each of these conditions is still not fully elucidated, nor the rationale for progression through the disease spectrum fully understood.2 However, what is clear is that the liver undergoes a number of metabolic changes as it progresses through this spectrum, initially as an adaptation to excess lipid, and then as a result of the development of pathology. These changes alter the liver's ability to both maintain body homeostasis and to efficiently handle therapeutic agents.4 An important question is how the impact of breast cancer treatment with standard therapeutics alters in individuals with fatty liver disease. Such impacts could include an enhanced adverse effect profile over both acute (e.g. increased cytotoxicity against non-malignant tissues) or chronic (e.g. increased progression through the fatty liver disease spectrum) time periods. Given the increasing success of chemotherapy, producing an ever-increasing pool of cancer survivors who must live with the potential long-term consequences of their chemotherapy, it is important to understand these chronic effects.5,6
Doxorubicin (Dox) is a member of the quinone-containing anthracycline antibiotics, and due to its wide distribution throughout the body it is effective in the treatment of a number of human cancers, including breast cancer.7–9 Due to the non-targeted nature of doxorubicin's mode of action, adverse side effects are diverse, most notably cardiotoxicity and to a lesser extent hepatotoxicity.10,11 At present, the published literature regarding interactions between doxorubicin and fatty acids are conflicting. For example, both adverse12,13 and protective14,15 interactions between doxorubicin and omega-3 fatty acids have been reported. In addition, Magnolia seed extract, which is rich in linoleate, oleate and palmitate has been reported to ameliorate doxorubicin cardiomyocyte toxicity in vitro.16 Finally, epidemiological studies suggest that doxorubicin may promote progression through the fatty liver disease spectrum.17,18 This contradictory evidence between studies suggests a complex interaction that may be context (e.g. cell type/species/concentration) dependent, and thus requires further study.
In this work we confirm that both free fatty acids and doxorubicin cause lipid-loading (steatotic phenotype) in human hepatoma cells, and that their combination results in an additive effect on intracellular lipid accumulation. In contrast, cytotoxicity is synergistic, as is the increase in reactive oxygen species. Such alterations, we believe, are consistent with enhanced sensitivity of obese individuals to the acute adverse effects of doxorubicin, more specifically hepatotoxicity. In addition, the observed synergistic increase in oxidative stress and pro-inflammatory signalling is likely to potentiate transition along the fatty liver disease spectrum, creating an increased health burden for those who survive their cancer treatment.
Materials and methods
Materials
Doxorubicin, fatty acid free BSA, oleate, palmitate and staurosporine were all purchased from Sigma Aldrich (Poole, UK). Primary antibodies were purchased from Santa Cruz Biotechnology (TX, USA) for Metallothionein (FL-61), while secondary antibodies and Cell Tag 700 Stain were purchased from Li-Cor Biosciences (Cambridge, UK).
Cell culture
The human hepatoma cell line Huh719 was a kind gift from Steve Hood (GlaxoSmithKline, Ware, UK) and were cultured in Dulbecco's Modified Eagle Medium with 2 mM l-glutamine, 4.5 g L–1 glucose, 100 units per mL1 each penicillin and streptomycin, and 10% foetal bovine serum (FBS), at 37 °C and 5% CO2. Huh7 cells were seeded at 1 × 104 cell per cm2 into appropriate vessels 24 h prior to treatment. For lipid-loading of cells, palmitate and oleate were first dissolved in DMSO and conjugated to fatty acid free BSA at the required concentration as described by Wang et al. 20 Cells were exposed to the BSA-conjugated lipid mixture for 24 h prior to subsequent experiments to allow lipid-loading to occur.
Viability assay
Cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl tetrazolium bromide (MTT) assay, as previously described.21 Briefly, Huh7 cells were exposed to lipid and/or doxorubicin for the required time; during the final 180 min of incubation, 0.5 mg mL–1 MTT was added. At the end of the incubation period, the resultant formazan salt was dissolved in DMSO and its absorbance measured at 540 nm. Results are expressed as a percentage of vehicle control; each data point represents the mean of a minimum of three independent experiments of 6 wells per experiment, with error bars representing the standard error of the mean (SEM). Combination index was calculated using Compusyn.22
Intracellular lipid analysis
Intracellular lipid content was determined fluorometrically based on staining with Nile Red. Following exposure of Huh7 cells to lipid and/or doxorubicin, cells were processed by two methods. For microscopy, cells were washed and then fixed with 3.7% para-formaldehyde for 10 minutes. Next, 1 μM Nile Red was added and cells incubated at room temperature for 15 min in the dark. Images were acquired using a Nikon Eclipse TS100 inverted fluorescence microscope (λ ex 510–560 nm, λ em 590 nm). For quantitation of intracellular lipid, cells were recovered from the flasks, stained with 1 μM Nile Red as before and lipid content quantified using a Molecular Devices SpectraMax Gemini XS fluorescence spectrophotometer (CA, US) (λ ex 485 nm, λ em 535 nm). These values were exported to Excel, background fluorescence (medium only) removed, and then normalised for cellular protein. Lipid content was expressed as relative fluorescence units per mg protein.
Intracellular reactive oxygen species analysis
Intracellular ROS levels were assessed using the ROS-Glo™ H2O2 assay from Promega (Southampton, UK) following the manufacturer's instructions. Briefly, Huh7 cells were exposed to lipid and/or doxorubicin as described previously; during the final 6 hours of incubation, 25 μM H2O2 substrate solution was added to the cells. At the end of incubation time, ROS-Glo Detection solution was added, incubated for 20 minutes and then luminescence was measured using a BMG LABTECH FLUOstar Omega plate reader (Ortenberg, Germany).
Caspase activity assay
Caspase 3/7 activity was measured using the Caspase-Glo 3/7assay kit (Promega, UK) as previously reported.23 Briefly, Huh7 cells were exposed to lipid and/or doxorubicin as required. Next, Caspase-Glo reagent was added, mixed, and incubated at room temperature for one hour. Luminescence was measured using a BMG LABTECH FLUOstar Omega plate reader (Ortenberg, Germany).
Transcriptomic analysis
Huh7 cells (naïve or pre-loaded with 300 μM FFA mixture for 24 hours) were exposed to doxorubicin for 4 or 12 hours, and then total RNA was extracted from cells using the RNeasy Plus Mini Kit (QIAGEN-UK) according to manufacturer's instructions. RNA samples were sent to the Central Biotechnology Services (Cardiff University, UK), for quality control, synthesis of biotin-labeled cRNA, and hybridisation against Illumina Human-HT12 (Illumina, Inc., Hayward, CA) chips. Washed chips were scanned using a Bead Station 500× (Illumina) and the signal intensities quantified with BeadStudio (Illumina). Analysis of array output files was performed within the Bioconductor R suite:24 data pre-processing was performed using the beadarray and illuminaHumanv4.db packages;25 differential gene expression analysis was undertaken using the limma package.26 Microarray data files are available through ArrayExpress (; https://www.ebi.ac.uk/arrayexpress/), accession number E-MTAB-3523. Functional clustering analysis was undertaken using the DAVID suite to identify statistically over-represented Gene Ontology (GO) terms.27 Network interactions were visualised using hive plots generated by the R package HiveR.
Protein analysis
Cells were exposed to lipid and/or doxorubicin as required. At the end of incubation period, cells were fixed in 3.7% para-formaldehyde and permeabilised with 0.1% Triton X-100. Non-specific binding was blocked with Odyssey Blocking buffer for 1.5 h at room temperature with gentle shaking. Primary antibodies (1 : 100) were incubated overnight at 4 °C with shaking, followed by IRDye 800 CW secondary anti rabbit antibody (1 : 600) and 0.2 μM Cell Tag 700 Stain for 1 h at room temperature with gentle shaking. Quantification was undertaken with an Odyssey Imaging System (LI-COR Biosciences) at 800 nm for IRDye 800 CW and 700 nm for Cell Tag 700 Stain.
Statistical analysis
Statistical analysis was undertaken using GraphPad Prism v6.01 (GraphPad Software Inc, La Jolla, USA). Datasets were compared through either a one-way ANOVA with Tukey's multiple comparison test or a two-way ANOVA with Sidak's mulitple comparison test, as appropriate. The level of statistical significance was set a priori at p < 0.05.
Results and discussion
Doxorubicin and FFA-mediated lipid-loading elicit a synergistic increase in cytotoxicity
To examine the potential for a steatotic phenotype to alter the acute toxic liability associated with doxorubicin the Huh7 human hepatoma cell line was employed. These cells have previously been demonstrated to respond to a range of chemical agents, and possess a transcriptomic and metabolomic profile similar to in vivo hepatocytes.28 The impact of both the C16 saturated fatty acid palmitate, and the C18 : 1 unsaturated fatty acid oleate on Huh7 cell viability was examined. These fatty acids were chosen as they represent the commonest saturated and unsaturated fatty acids found in Western diets, respectively.29 Oleate was non-toxic at all concentrations tested (maximum 1 mM), while palmitate caused a concentration dependent reduction in cell viability that reached statistical significance at 150 μM (Fig. 1a). As these fatty acids are rarely found in the diet in isolation, the impact of a free fatty acid (FFA) mixture was examined. As expected, toxicity of this FFA mixture was predominantly driven by the toxicity of palmitate, with all mixtures containing greater than 200 μM palmitate causing significant toxicity (data not shown). An FFA mixture of 300 μM (2 : 1 v/v oleate : palmitate) caused no significant toxicity (Fig. 1a), and is consistent with clinical data in the Western population where no-fasting serum levels of palmitate and oleate are reported as approximately 140 μM and 90 μM, respectively.30 This FFA mixture was used in all subsequent studies.
Fig. 1. Synergistic cytotoxicity between doxorubicin and lipid-loading in Huh7 cells. (A) Huh7 cells were exposed to varying concentrations of fatty acids or a 300 μM FFA mixture (2 : 1 v/v oleate : palimitate) for 24 h and then cell viability measured by MTT assay. Huh7 cells, either naïve (black bars) or pre-loaded with 300 μM FFA mixture for 24 h (open bars), were exposed to varying concentrations of fatty acids, or doxorubicin for 24 h, and then cell viability (B) or (C) caspase 3/7 activity was measured. A combination index was calculated to examine potential interactions between the FFA mixture and doxorubicin. Each data point represents the mean of three independent experiments. Error bars represent the standard error of the mean (SEM). * = p < 0.05, ** = p < 0.01, *** = p < 0.001 compared to vehicle control or the indicated comparison.

Exposure of cells to doxorubicin caused a concentration-dependent toxicity, with an IC50 value of 17 ± 4.6 μM in naïve hepatocytes, consistent with previous observations (Fig. 1b; ref. 31). Cells that have been lipid-loaded with the 300 μM FFA mixture for 24 h prior to doxorubicin exposure showed an enhanced sensitivity, with an IC50 value of 3.6 ± 5.4 μM (Fig. 1b). This result is not specific to the FFA mixture, as pre-loading with 1 mM oleate also caused a significant increase in sensitivity, producing an IC50 value of 6.8 ± 4.5 μM (data not shown).
In both cases, the observed toxicity from the combination was synergistic compared to the individual treatments, with maximal combination indexes of 0.2 and 0.1 for doxorubicin : FFA mixture and doxorubicin : oleate, respectively (Fig. 1b).
Previous data demonstrates that doxorubicin can induce apoptosis in many cell lines, including Huh7 cells,32 and we thus examined the activity of executioner caspases in our system. As can be seen in Fig. 1c, both lipid loading and doxorubicin elicited a statistically significant increase in caspase-3/7 activity. However, the combination of doxorubicin treatment of 300 μM FFA lipid-loaded cells demonstrated a clear synergy when compared to the individual treatments alone.
This suggests that steatotic hepatocytes are significantly more sensitive to the cytotoxic effects of doxorubicin than naïve hepatocytes, with lipid-loading acting to prime the cells against any subsequent perturbations.
Both lipid loading and doxorubicin have been reported to induce intracellular lipid accumulation (steatosis) in a number of experimental systems.33–36 To confirm that this was the case in our model system, the extent of lipid accumulation was also assessed under these conditions. Treatment of cells with 300 μM FFA or 3.6 μM doxorubicin resulted in significant intracellular lipid accumulation (Fig. 2a), which was confirmed to be in the form of lipid droplets (Fig. 2b). Lipid accumulation was approximately 3.4-fold and 4.7-fold above control for 300 μM FFA and 3.6 μM doxorubicin, respectively. Treatment of 300 μM FFA lipid-loaded hepatocytes with 3.6 μM doxorubicin resulted in an approximate 10-fold increase in lipid levels (Fig. 2), consistent with an additive effect.
Fig. 2. Additive lipid-accumulation between doxorubicin and lipid-loading in Huh7 cells. Huh7 cells, either naïve or pre-loaded with 300 μM FFA mixture (2 : 1 v/v oleate : palimitate) for 24 h, were exposed to 3.6 μM doxorubicin for 24 h. Intracellular lipid accumulation was detected by Nile Red and (A) quantified or (B) visualised by fluorescence microscopy. For lipid quantitation, each data point represents the mean of three independent experiments. Error bars represent the standard error of the mean (SEM). * = p < 0.05.

Current literature suggests that doxorubicin-mediated toxicity may be either ameliorated or potentiated by fatty acid (pre)treatment.12–14,16 These data are sourced from a number of cell backgrounds (e.g. renal, breast, cardiac), species (e.g. human, rat, mouse) and protocols (including in vitro versus in vivo measurement). As such, this contradictory data may suggest that the interaction between doxorubicin and fatty acids is context-dependent, meaning that it is important to study responses in human liver models to best predict the response in this organ in vivo.
The data presented here supports an adverse, synergistic interaction between lipid-loading and doxorubicin in human hepatocytes. This interaction could be the explained via either a direct mechanistic interaction between doxorubicin and FFAs, or an indirect action whereby, for example, lipid-loading alters the intracellular concentration of doxorubicin. Robust measurement of intracellular doxorubicin and lipid concentrations would be required to definitively answer this question, but we do note that doxorubicin has been demonstrated to inhibit palmitate uptake into cardiomyocytes.37 However, as we observe both additive and synergistic effects this would support an interaction as opposed to a simple shift along the concentration-response curve.
Transcriptomic analysis of the differential response of naïve and lipid loaded hepatocytes to doxorubicin treatment
Having demonstrated that lipid-loaded hepatocytes were more sensitive to the acute cytotoxic effects of doxorubicin, we next undertook a transcriptomic analysis to examine the molecular mechanisms that may underlie this effect. In addition, such a study may help to predict genotype-phenotype relationships that underpin the chronic response to doxorubicin of steatotic hepatocytes.38,39 Huh7 cells (naïve or pre-loaded with 300 μM FFA mixture for 24 h) were exposed to doxorubicin for 4 hours or 12 hours and then their transcriptomes analysed. Two concentrations of doxorubicin were examined: 3.6 μM and 0.1 μM, representing the previously determined IC50 value and a non-steatotic level. In addition, these concentrations are consistent with the clinical scenario: peak plasma concentrations are typically 5–15 μM, with free doxorubicin concentrations in the range 1–4 μM.40 Raw data files are deposited within ArrayExpress (accesssion ID: E-MTAB-3523), and differentially expressed genes (DEGs) provided as ESI.†
Lipid-loading with the 300 μM FFA mixture caused a significant transcriptome change at the early and late time points, eliciting 577 and 440 statistically significant alterations, respectively (adjusted p-value <0.01). As expected, exposure of naïve Huh7 cells to 0.1 μM doxorubicin elicited only minor transcriptome changes, consistent with its minimal impact on cellular phenotype. In contrast, 3.6 μM doxorubicin caused 578 and 535 significant transcript alterations following 4 and 12 hours exposure, respectively. Analysis of the transcriptomic data revealed lipid loading and doxorubicin exposures caused shared regulation of 35 and 152 genes following 4 h and 12 h treatment, respectively. When compared to the total number of genes altered, this represents a 45% overlap, which suggests a highly conserved phenotypic core between these two distinct treatments.
When the transcriptome perturbations elicited by the two treatments were compared, 401/578 (69%, 95% CI 64–74%) and 416/535 (78%, 95% CI 72–83%) changes were found to be shared following 4 h and 12 h treatment, respectively. This suggests a highly conserved core response to these two, quite different, perturbations.
Hive plots allow an unbiased analysis of the relationships within a network.41 As nodes are mapped on, and positioned within, an axis based upon structural properties, hive plots demonstrate network characteristics in a reproducible and unbiased manner. Fig. 3a provides hive plots for each condition; axes represent differentially expressed genes (DEGs; grey nodes), while DEGs common between treatments are linked by arcs.
Fig. 3. Transcriptomic analysis of Huh7 cells exposed to doxorubicin and/or lipids reveals marked commonality in response. Huh7 cells, either naïve or pre-loaded with 300 μM FFA mixture (2 : 1 v/v oleate : palimitate) for 24 h, were exposed to 3.6 μM doxorubicin for 4 h or 12 h, and then transcriptomes analysed by Illumina bead array. Differentially expressed genes (DEGs) are presented as Hive plots: each axis represents and individual comparison, with DEGs presented as grey nodes, with position along the axis determined by fold-change; where DEGs are common between two axes, they are connected by an arc. DEGs were determined from three independent treatments for each condition, with an adjusted p-value <0.05 deemed significant.

For 0.1 μM doxorubicin treatment (Fig. 3, top row), only lipid-loading produced significant alterations in transcript levels. For 3.6 μM doxorubicin treatment (Fig. 3, bottom row), both doxorubicin treatment and lipid-loading produced DEGs at both time points, with many of these perturbations being shared (red arcs). As the position of a DEG node on an axis is determined by its expression level, it is possible to see that the level of differential expression caused by both treatments is similar, with connecting arcs being concentric in nature.
We note that at the early time point of 4 h, the combination treatment results in no DEGs, suggesting that the effect of the two treatments effectively cancel out each other.
In contrast, following 12 h exposure to both 300 μM FFA and doxorubicin we observe many DEGs, the majority of which are common with doxorubicin or lipid-loading alone (green and blue arcs, respectively). While, DEG fold changes were consistent between the two treatments when given alone, combination treatment results in significant alterations in the fold change of many DEGs, as indicated by the non-concentric nature of the connecting arcs (blue and green). The synergistic nature of the transcript changes are also supported by the maximal fold changes observed, as indicated by the length of the axis: maximal fold change observed for 300 μM FFA or 3.6 μM doxorubicin alone are 6.0-fold and 6.2-fold, respectively, while this increases to 14.9-fold for the combination treatment.
To explore the biological consequences of these transcriptome alterations we used the DAVID tool to examine for over-representation of gene ontology terms within the DEGs. Biological process terms associated with lipid metabolism, insulin signalling, proliferation, inflammation and drug metabolism were identified following both 300 μM FFA and doxorubicin treatment, and with combination treatment. Such over-represented terms are consistent with the previously reported biological effects for these perturbations.42 For example, after twelve hours exposure to the combination treatment the expression of genes involved in lipid homeostasis (e.g. acetyl CoA carboxylase, microsomal triglyceride transfer protein, and sterol O-acyltransferease 2) were all significantly down-regulated. In contrast, the expression of genes associated with lipid biosynthesis (e.g. HMG-CoA2, fatty acid desaturase 1/2, fatty acid synthase) were all up-regulated. Such data are consistent with a shift toward an increasing lipid load within the cells, with lipid biosynthesis increasing at a greater rate than lipid metabolism. In addition, a cellular response to ROS-mediated damage can be seen through the modulation of expression of genes associated with inflammation (e.g. alpha2 macroglubulin, interleukin 8, leukotriene A4 hydroxylase, and Factor 11 receptor), and, as highlighted in Fig. 4, the metallothionein rescue response system (e.g. metallothionein proteins, HAMP).
Fig. 4. Synergistic oxidative stress between doxorubicin and lipid-loading in Huh7 cells. Huh7 cells, either naïve or pre-loaded with 300 μM FFA mixture (2 : 1 v/v oleate : palimitate) for 24 h, were exposed to the indicated concentration of doxorubicin for various times and then various parameters measured: (A) ROS, (B) transcripts associated with oxidative stress response, and (C) metallothionein protein levels. Each data point represents the mean of three independent experiments. Error bars represent the standard error of the mean (SEM) * = p < 0.05, ** = p < 0.01, *** = p < 0.001.

Enhanced acute toxicity of doxorubicin in lipid-loaded hepatocytes is mediated through increased oxidative stress
Both doxorubicin and lipid-loading have been previously associated with increased reactive oxygen species production in various cell types, including liver cells.43 It is therefore logical to hypothesise that oxidative stress is the mechanistic underpinning for the observed synergistic toxicity between these two treatments. To examine this hypothesis, we measured the level of ROS in cells treated with lipids or doxorubicin alone, or in combination. As shown in Fig. 4a both doxorubicin and lipid-loading alone were capable of inducing a significant increase in ROS within Huh7 cells in a time-dependent manner, with peak ROS levels observed following 24 h of treatment. Importantly, the combination treatment produced a significantly enhanced ROS level at all the time points examined, greater than that expected through simple effect additivity. We have confirmed this synergistic interaction using an alternate reporter dye, 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA), and present this data as ESI Fig. 1.†
The induction of intracellular ROS would be expected to lead to a number of biological responses, with the aim of returning the cell to homeostasis: consistent with this, transcriptome analysis revealed an over-representation of GO-terms associated with oxidative stress responses. To further explore these changes, the individual transcript levels for all oxidative stress response DEGs are presented in Fig. 4b. The main DEGs identified were members of the metallothionein family (MTs), glutathione peroxidase (GPX) and uncoupling protein 2 (UCP2), but the behaviours were significantly different. For GPX and UCP2, 3.6 μM doxorubicin or 300 μM FFA alone produced an up-regulation at 4 hours, which reversed to a down-regulation at 12 hours. Up-regulation of GPX has been previously reported the HepG2 hepatoma cell line44 and in TR9-7 cells following doxorubicin exposure.45 In contrast, members of the metallothionein protein family, which are a complementary protective system in the maintenance of ROS homeostasis within the cell46,47, show the opposite behaviour: they are down-regulated at 4 hours and up-regulated by 12 hours by the individual treatments. One hypothesise is that activation of metallothionein proteins may act to protect cells from ROS in the absence of glutathione peroxidase, and hence the two systems demonstrate an inverse relationship.
We note with interest that the combination of doxorubicin treatment in previously lipid-loaded cells has an inverse expression pattern when compared to both treatments alone. Transcripts for MT are up-regulated at 4 h and down-regulated by twelve hours, with GPX and UCP2 showing the opposite pattern. We confirmed this differential regulation of MT by doxorubicin in naïve and lipid-loaded cells levels through in-cell westerns (Fig. 4c).
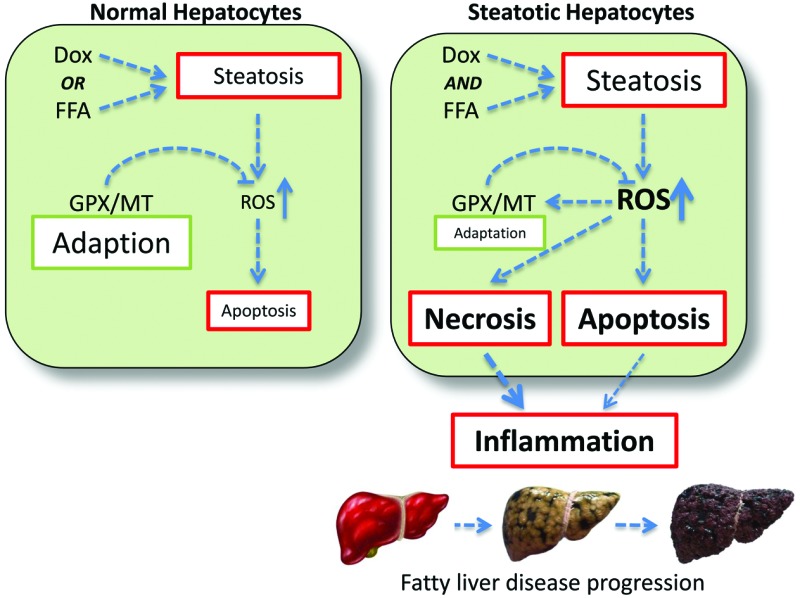
One possible explanation for the observed data is the complementary nature of the GPX and MT oxidant defence systems. As depicted in Fig. 5, the response to a single perturbation (i.e. doxorubicin or FFA) is an increase in intracellular ROS, which in turn activates the GPX/MT defence mechanism. This adaptation allows return to homeostasis with minimal cell loss, which will be via programmed cell death. However, in conditions of multiple perturbations (i.e. doxorubicin and FFA-induced steatosis), the level of intracellular ROS accumulation are much higher, overwhelming the oxidative stress response system and leading to increased levels of cytotoxicity.
Fig. 5. Synergistic interactions between doxorubicin and lipid-loading in hepatocytes. In naïve hepatocytes (left panel), exposure to either doxorubicin or free fatty acids (FFA) results in steatosis and oxidative stress. GPX/MT-mediated adaptation acts to reduce ROS levels, minimising the necessity for cell death through apoptosis. In steatotic hepatocytes (right panel), the combination of doxorubicin and FFA exposure leads to additive lipid accumulation, and synergistic intracellular ROS levels. This overwhelms the GPX/MT adaptive system, resulting in increased cell death, release of pro-inflammatory signals and potentiation through the fatty liver disease spectrum.

In addition, as both necrotic and apoptotic cell death have been associated with pro-inflammatory signalling in hepatocytes,48,49 this will potentiate an inflammatory response, leading to progression through the fatty liver disease spectrum.2 Such a proposed mechanism is consist with the epidemiological data supporting a positive association between doxorubicin treatment and progression along the fatty liver disease spectrum.17,18
Conclusions
Within Western society, obesity is now recognised as being a health problem of epidemic proportions. Not only does this raise serious issues with regard to the direct treatment of obesity and its related morbidities, but it also means that many therapeutics are now being used to treat an obese population. Given that the majority of clinical trials specifically exempt obese individuals, this means that many therapeutics are being prescribed to patient populations in which they have not been extensively tested. As such, it is important to consider both the acute and chronic implications of such a treatment scenario, and in particular in disease conditions such as cancer where aggressive treatment regimens have led to improved survivorship. We provide more data to confirm the observation that obese individuals may be more sensitive to the acute adverse effects of drugs. As depicted in Fig. 5, our data is consistent with a scenario where exposure alone to doxorubicin or lipid-loading elicits steatosis plus accompanying oxidative stress. In this case, the cell is able to mount an effective response and adapt to this steatotic phenotype through enhanced expression of anti-oxidant defence mechanisms. However, when the treatments are combined the additive effect on intracellular lipid accumulation produces a synergistic increase in oxidative stress, which we believe overwhelms the cell. The adaptive response is muted, leading to higher levels of cell death, including necrosis, resulting in an inflammatory response in the local tissue (Fig. 5). Importantly, this supports the notion that therapeutic agents such as doxorubicin may synergise with pre-existing cellular steatosis to generate a pro-oxidative, pro-inflammatory environment. Given that inflammatory infiltration is a key determinant in progression from simple steatosis to the more complex (and less benign) steatohepatitis,2 our data suggests that treatment of obese patients with doxorubicin may act to progress individuals through the fatty liver disease spectrum toward more complex phenotypes with significant health and wellbeing implications. This area requires significantly more research to understand how such a paradigm translates to the clinic, and to investigate treatment strategies to minimize such a possibility.
Acknowledgments
SA was funded by Kingdom of Saudi Arabia Ministry of Education/King Abdulaziz University; VL was funded by the U.K. Biotechnology and Biological Sciences Research Council (BB/K501694/1).
Footnotes
References
- Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D. M., Forman D., Bray F. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- Fisher C. P., Kierzek A. M., Plant N. J., Moore J. B. World J. Gastroenterol. 2014;20:15070–15078. doi: 10.3748/wjg.v20.i41.15070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves G. K., Pirie K., Beral V., Green J., Spencer E., Bull D., C. Million Women Study BMJ. 2007;335:1134–1139. doi: 10.1136/bmj.39367.495995.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrell M. D., Cherrington N. J. Drug Metab. Rev. 2011;43:317–334. doi: 10.3109/03602532.2011.577781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilici A., Ozguroglu M., Mihmanh I., Turna H., Adaletli I. Med. Oncol. 2007;24:367–371. doi: 10.1007/s12032-007-0034-8. [DOI] [PubMed] [Google Scholar]
- Griggs J. J., Sorbero M. E. S., Lyman G. H. Arch. Intern. Med. 2005;165:1267–1273. doi: 10.1001/archinte.165.11.1267. [DOI] [PubMed] [Google Scholar]
- Young R. C., Ozols R. F., Myers C. E. N. Engl. J. Med. 1981;305:139–153. doi: 10.1056/NEJM198107163050305. [DOI] [PubMed] [Google Scholar]
- Rouzier R., Perou C. M., Symmans W. F., Ibrahim N., Cristofanilli M., Anderson K., Hess K. R., Stec J., Ayers M., Wagner P., Morandi P., Fan C., Rabiul I., Ross J. S., Hortobagyi G. N., Pusztai L. Clin. Cancer Res. 2005;11:5678–5685. doi: 10.1158/1078-0432.CCR-04-2421. [DOI] [PubMed] [Google Scholar]
- Chan K. K., Cohen J. L., Gross J. F., Himmelstein K. J., Bateman J. R., Tsulee Y., Marlis A. S. Cancer Treat. Rep. 1978;62:1161–1171. [PubMed] [Google Scholar]
- Singal P. K., Iliskovic N. N. Engl. J. Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- Kalender Y., Yel M., Kalender S. Toxicology. 2005;209:39–45. doi: 10.1016/j.tox.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Fahrmann J. F., Hardman W. E. Lipids Health Dis. 2013:12. doi: 10.1186/1476-511X-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibet S., Goupille C., Bougnoux P., Steghens J.-P., Gore J., Maheo K. Free Radical Biol. Med. 2008;44:1483–1491. doi: 10.1016/j.freeradbiomed.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Hsu H.-C., Chen C.-Y., Chen M.-F. J. Biomed. Sci. 2014:21. doi: 10.1186/s12929-014-0101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Hou C.-C., Cheng C.-F., Chiu T.-H., Hsu Y.-H., Sue Y.-M., Chen T.-H., Hou H.-H., Chao Y.-C., Cheng T.-H., Chen C.-H. Mol. Pharmacol. 2007;72:1238–1245. doi: 10.1124/mol.107.037523. [DOI] [PubMed] [Google Scholar]
- Park K.-H., Kim S.-Y., Gul R., Kim B.-J., Jang K. Y., Chung H.-T., Sohn D.-H. Biol. Pharm. Bull. 2008;31:809–815. doi: 10.1248/bpb.31.809. [DOI] [PubMed] [Google Scholar]
- Vauthey J. N., Pawlik T. M., Ribero D., Wu T. T., Zorzi D., Hoff P. M., Xiong H. Q., Eng C., Lauwers G. Y., Mino-Kenudson M., Risio M., Muratore A., Capussotti L., Curley S. A., Abdalla E. K. J. Clin. Oncol. 2006;24:2065–2072. doi: 10.1200/JCO.2005.05.3074. [DOI] [PubMed] [Google Scholar]
- Peppercorn P. D., Reznek R. H., Wilson P., Slevin M. L., Gupta R. K. Br. J. Cancer. 1998;77:2008–2011. doi: 10.1038/bjc.1998.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabayashi H., Taketa K., Miyano K., Yamane T., Sato J. Cancer Res. 1982;42:3858–3863. [PubMed] [Google Scholar]
- Wang X. L., Zhang L., Youker K., Zhang M.-X., Wang J., LeMaire S. A., Coselli J. S., Shen Y. H. Diabetes. 2006;55:2301–2310. doi: 10.2337/db05-1574. [DOI] [PubMed] [Google Scholar]
- Gee R. H., Spinks J. N., Malia J. M., Johnston J. D., Plant N. J., Plant K. E. Toxicology. 2015;329:40–48. doi: 10.1016/j.tox.2015.01.005. [DOI] [PubMed] [Google Scholar]
- Chou T.-C. Pharmacol. Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- Plant K. E., Anderson E., Simecek N., Brown R., Forster S., Spinks J., Toms N., Gibson G. G., Lyon J., Plant N. Toxicol. Appl. Pharmacol. 2009;235:124–134. doi: 10.1016/j.taap.2008.10.019. [DOI] [PubMed] [Google Scholar]
- Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y. C., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A. J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J. Y. H., Zhang J. H. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning M. J., Smith M. L., Ritchie M. E., Tavare S. Bioinformatics. 2007;23:2183–2184. doi: 10.1093/bioinformatics/btm311. [DOI] [PubMed] [Google Scholar]
- Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G., Sherman D. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A. Genome Biol. 2003;4:R60. [PubMed] [Google Scholar]
- Lin J., Schyschka L., Muhl-Benninghaus R., Neumann J., Hao L., Nussler N., Dooley S., Liu L., Stockle U., Nussler A. K., Ehnert S. Arch. Toxicol. 2012;86:87–95. doi: 10.1007/s00204-011-0733-y. [DOI] [PubMed] [Google Scholar]
- Ervin R. B., Wright J. D., Wang C.-Y., Kennedy-Stephenson J. Adv. Data. 2004:1–6. [PubMed] [Google Scholar]
- Mitropoulos K. A., Armitage J. M., Collins R., Meade T. W., Reeves B. E. A., Wallendszus K. R., Wilson S. S., Lawson A., Peto R. Eur. Heart J. 1997;18:235–241. doi: 10.1093/oxfordjournals.eurheartj.a015225. [DOI] [PubMed] [Google Scholar]
- Castaneda F., Kinne R. K. H. J. Cancer Res. Clin. Oncol. 1999;125:1–8. doi: 10.1007/s004320050235. [DOI] [PubMed] [Google Scholar]
- Eom Y. W., Kim M. A., Park S. S., Goo M. J., Kwon H. J., Sohn S., Kim W. H., Yoon G., Choi K. S. Oncogene. 2005;24:4765–4777. doi: 10.1038/sj.onc.1208627. [DOI] [PubMed] [Google Scholar]
- Feldstein A. E., Canbay A., Guicciardi M. E., Higuchi H., Bronk S. F., Gores G. J. J. Hepatol. 2003;39:978–983. doi: 10.1016/s0168-8278(03)00460-4. [DOI] [PubMed] [Google Scholar]
- Gomez-Lechon M. J., Donato M. T., Martinez-Romero A., Jimenez N., Castell J. V., O'Connor J.-E. Chem.-Biol. Interact. 2007;165:106–116. doi: 10.1016/j.cbi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Arunachalam S., Pichiah P. B. T., Achiraman S. FEBS Lett. 2013;587:105–110. doi: 10.1016/j.febslet.2012.11.019. [DOI] [PubMed] [Google Scholar]
- King P. D., Perry M. C. Oncologist. 2001;6:162–176. doi: 10.1634/theoncologist.6-2-162. [DOI] [PubMed] [Google Scholar]
- Beanlands R. S. B., Shaikh N. A., Wen W. H., Dawood F., Ugnat A. M., McLaughlin P. R., Carere R., Liu P. P. J. Mol. Cell. Cardiol. 1994;26:109–119. doi: 10.1006/jmcc.1994.1012. [DOI] [PubMed] [Google Scholar]
- Plant N. J. Toxicol. Res. 2015;4:9–22. doi: 10.1039/c5tx00173k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C. P., Plant N. J., Moore J. B., Kierzek A. M. Bioinformatics. 2013;29:3181–3190. doi: 10.1093/bioinformatics/btt552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Zheng H., Tang M., Ryu Y.-C., Wang X. Am. J. Physiol.: Heart Circ. Physiol. 2008;295:H2541–H2550. doi: 10.1152/ajpheart.01052.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M., Birol I., Jones S. J. M., Marra M. A. Brief. Bioinform. 2012;13:627–644. doi: 10.1093/bib/bbr069. [DOI] [PubMed] [Google Scholar]
- De Gottardi A., Vinciguerra M., Sgroi A., Moukil M., Ravier-Dall'Antonia F., Pazienza V., Pugnale P., Foti M., Hadengue A. Lab. Invest. 2007;87:792–806. doi: 10.1038/labinvest.3700590. [DOI] [PubMed] [Google Scholar]
- Rogalska A., Koceva-Chyla A., Jozwiak Z. Chem.-Biol. Interact. 2008;176:58–70. doi: 10.1016/j.cbi.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Gong P. F., Cederbaum A. I., Nieto N. Free Radical Biol. Med. 2004;36:307–318. doi: 10.1016/j.freeradbiomed.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Hussain S. P., Amstad P., He P. J., Robles A., Lupold S., Kaneko I., Ichimiya M., Sengupta S., Mechanic L., Okamura S., Hofseth L. J., Moake M., Nagashima M., Forrester K. S., Harris C. C. Cancer Res. 2004;64:2350–2356. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- Kondoh M., Inoue Y., Atagi S., Futakawa N., Higashimoto M., Sato M. Life Sci. 2001;69:2137–2146. doi: 10.1016/s0024-3205(01)01294-2. [DOI] [PubMed] [Google Scholar]
- Zalewska M., Trefon J., Milnerowicz H. Proteomics. 2014;14:1343–1356. doi: 10.1002/pmic.201300496. [DOI] [PubMed] [Google Scholar]
- Faouzi S., Burckhardt B. E., Hanson J. C., Campe C. B., Schrum L. W., Rippe R. A., Maher J. J. J. Biol. Chem. 2001;276:49077–49082. doi: 10.1074/jbc.M109791200. [DOI] [PubMed] [Google Scholar]
- Rock K. L. and Kono H., in Annual Review of Pathology-Mechanisms of Disease, 2008, vol. 3, pp. 99–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.