

Abstract
Extended hydrogen bond networks are a common structural motif of enzymes. A recent analysis proposed quantum delocalization of protons as a feature present in the hydrogen bond network spanning a triad of tyrosines (Y16, Y32, and Y57) in the active site of ketosteroid isomerase (KSI), contributing to its unusual acidity and large isotope shift. In this study, we utilized amber suppression to substitute each tyrosine residue with 3-chlorotyrosine to test the delocalization model and the proton affinity balance in the triad. X-ray crystal structures of each variant demonstrated that the structure, notably the O–O distances within the triad, was unaffected by 3-chlorotyrosine substitutions. The changes in the cluster’s acidity and the acidity’s isotope dependence in these variants were assessed via UV–vis spectroscopy and the proton sharing pattern among individual residues with 13C nuclear magnetic resonance. Our data show pKa detuning at each triad residue alters the proton delocalization behavior in the H-bond network. The extra stabilization energy necessary for the unusual acidity mainly comes from the strong interactions between Y57 and Y16. This is further enabled by Y32, which maintains the right geometry and matched proton affinity in the triad. This study provides a rich picture of the energetics of the hydrogen bond network in enzymes for further model refinement.
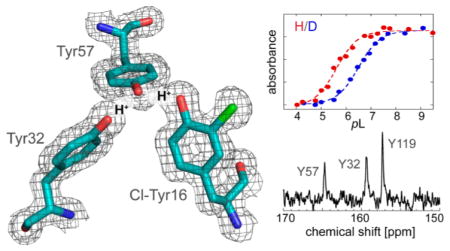
Extended hydrogen bond (H-bond) networks are common features in biological systems, including the active sites of many enzymes. In earlier work mapping the active site electrostatic environment of the enzyme ketosteroid isomerase from Pseudomonas putida (pKSI) by vibrational Stark spectroscopy, it was discovered unexpectedly that the low pKa of the oxyanion hole of the D40N mutant (which simulates the intermediate state) was associated with titration of a cluster of tyrosine residues.1 This was established by observing characteristic absorption spectral changes associated with the difference between neutral and ionized tyrosine and detailed 13C nuclear magnetic resonance (NMR) studies exploiting the sensitivity of the chemical shift of the carbon adjacent to the tyrosine hydroxyl group (Cζ) to the protonation state of tyrosine.1 A key feature of the tyrosine cluster, known from several high-resolution X-ray structures of KSI, is the short O–O distances among their hydroxyl groups.2 The pKa of the tyrosine cluster, involving residues 16, 32, and 57 in the active site (Figure 1), was found to be 6.3 in marked contrast to the pKa of tyrosine itself in aqueous solution, which is around 10.1. In addition to this large shift in pKa, a further unusually large H–D pKa isotope effect was observed. Path-integral molecular dynamics simulations were conducted on this system, leading to a model in which proton delocalization (a nuclear quantum effect) plays a key role to explain both the exceptional stabilization of the negative charge on the ionized tyrosine cluster and this large H–D equilibrium isotope effect.3 The simulations showed that protons delocalize because the zero-point energies of their O–H bonds are comparable to the potential energy barriers for proton transfer.
Figure 1.
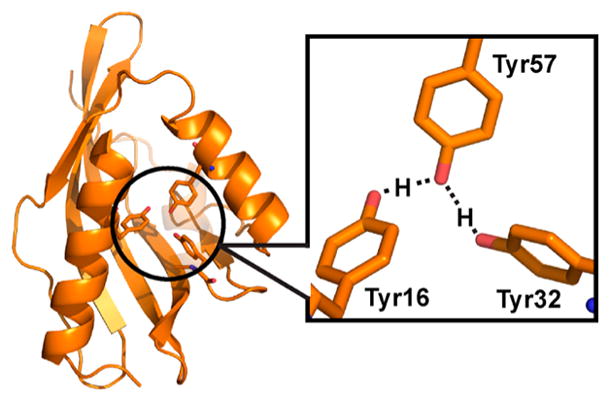
Proton delocalization in the H-bonding network of the intermediate state of KSI.3 In the intermediate state of apo-KSI, protons delocalize among the three tyrosine residues in the active site, leading to an unusual acidity and a large H–D pKa isotope effect of the tyrosine cluster.
An important element that determines the proton delocalization equilibrium in the H-bond network of KSI is the relative proton affinities of the tyrosines in the cluster, which are generally thought to be well-matched. In this study, we are interested in learning how sensitive the collective properties of the cluster are to proton affinity detuning at each site, and whether the interactions between different tyrosine residues contribute equally to proton delocalization in the H-bond network. We do so by selectively modifying the proton affinity of each tyrosine site in turn by using amber suppression to incorporate the unnatural amino acid 3-chlorotyrosine (Cl-Y, where Cl is ortho to the hydroxyl group) selectively at positions 16, 32, and 57, denoted KSI-Cl-Y16, KSI-Cl-Y32, and KSI-Cl-Y57, respectively. Because the solution pKa of Cl-Y is 8.3, its incorporation substantially “detunes” the intrinsic proton affinity balance compared with an all-tyrosine cluster without destroying the H-bond network in the cluster. Depending on the contribution of each tyrosine site in the cluster to proton delocalization, we expect quite different effects on both the cluster’s overall pKa and the H–D isotope effect on the pKa, and this can be used to further refine the model that describes the H-bond network.
MATERIALS AND METHODS
Synthesis of [13Cζ]-3-Chlorotyrosine
To a 25 mL round-bottom flask containing a suspension of [13Cζ]-L-tyrosine (0.25 g, 1.4 mmol, Cambridge Isotope Laboratories; the ζ-carbon is adjacent to the hydroxyl group) in 2 mL of glacial acetic acid was added dropwise SO2Cl2 (152 μL, 1.9 mmol). Within 5 min, a white slurry was formed. The slurry was stirred for 30 min at 20 °C, then filtered, and washed three times with 1 mL of glacial acetic acid.4 The recovered solid was dried under vacuum overnight (220 mg, 70%). The purity of the product was assessed by 1H NMR [300 MHz, D2O (Figure S1)] and 13C NMR [500 MHz, D2O (Figure S2); this figure also shows the large chemical shift difference, 9.4 ppm, between the protonated and deprotonated forms of [13Cζ]-3-chlorotyrosine].
Mutagenesis, Expression, and Purification of KSI-Cl-Y Variants
QuikChange site-directed mutagenesis (Stratagene) was used to introduce the amber codon TAG at position 16, 32, or 57 in the pKSI gene cloned into the pKK223-3 expression vector.5,6 A His tag was also introduced following a thrombin cleavage site at the C-terminus of the protein to facilitate a milder purification procedure, avoiding the unfolding–refolding step used to elute KSI from a deoxycholate affinity resin in earlier work (Table S1).5,6
To express KSI-Cl-Y variants, pEVOL-ClYRS was cotransformed with the mutated pKK223-3 plasmid into BL21(DE3) as described previously in ref 7 (we thank Professor Wang from the Institute of Biophysics, Chinese Academy of Sciences, for providing the pEVOL-ClYRS plasmid that contains the genes for the suppressor tRNA and an evolved tyrosyl tRNA synthetase from Methanococcus jannaschii).7,8 A single colony was grown overnight at 37 °C in 5 mL of LB medium supplemented with 0.6 mM 3-chlorotyrosine (or [13Cζ]-3-chlorotyrosine). The overnight culture was diluted into 1 L of LB, incubated at 37 °C, and induced with 1 mM isopropyl β-D-1-thiogalactopyranoside and 0.02% L-arabinose at an OD600 of 0.7–0.8. After induction, the cells were grown for an additional 12 h at 23 °C before being harvested. Following lysis by homogenization, the protein was isolated from the soluble fraction by Ni-NTA affinity chromatography followed by His tag cleavage, which leaves four additional amino acids at the C-terminal end, LVPR, compared with pKSI constructs used before. The cleaved protein was further purified by anion exchange chromatography (HiTrap Q HP 5 mL, GE Healthcare) in 20 mM Tris (pH 8) using a salt gradient from 10 to 220 mM NaCl. Unexpectedly, two populations of the KSI variant were separated by careful anion exchange chromatography but were found to have identical chemical compositions (Figure S3 and further discussion in the Supporting Information). Unless otherwise noted, all measurements discussed in the main text were taken on the protein population from the larger first elution peak, which was found to have properties consistent with earlier constructs, as discussed below. To prepare proteins for crystallization, the samples were taken through an additional purification step via size-exclusion chromatography (Superdex 75 10/300 GL, GE Healthcare).
For [13Cζ]tyrosine labeling of KSI-Cl-Y variants, protein expression was conducted in M9 minimal medium, augmented with 1 mM MgSO4, 0.2% glucose, MEM Vitamin Solution (GIBCO), 100 μg/mL 13Cζ-labeled tyrosine (Cambridge Isotope Laboratories), 130 μg/mL 3-chlorotyrosine (Sigma-Aldrich), and the remaining 19 naturally occurring L-amino acids (25 μg/mL), using previously published methods.1
pKa Values of D40N-Cl-Y Variants
A series of buffers were prepared with a pL (where L is H or D) between 4 and 10 as described previously.3 Samples for titration were prepared by combining 60 μL of 150–200 μM protein (in pure L2O), 150 μL of buffer, and 390 μL of L2O.
UV–visible spectra were recorded for each D40N-Cl-Y variant as a function of pL using a PerkinElmer Lambda 25 spectrophotometer with a 1 cm path length quartz cell. Difference absorbance spectra were obtained by subtraction of the absorbance spectra at a low pL of 4.0. For each D40N-Cl-Y variant, the peak position of the difference absorbance spectrum between high pL (9.0) and low pL (4.0) was determined and compared to the peak positions of the difference spectra of tyrosine (Y) (293 nm) and Cl-Y (300 nm) to determine the identity of the primary residue being titrated across the pL series. As will be discussed below, the degree of energy stabilization at the primary site of ionization dominated the measured pKa and pKa isotope effect. The change in extinction coefficient at 300 nm (6 nm red-shifted from the peak position of KSI-Cl-Y16 and KSI-Cl-Y32’s difference spectra, where Y57 is the primary residue being titrated) or 306 nm (6 nm red-shifted from the peak position of KSI-Cl-Y57’s difference spectrum, where Cl-Y57 is titrated) as a function of pL was then fit to a titration curve to determine the apparent pKa, employing a previous established method;3,9 the spectra were baseline-corrected to a value of 0 at 320 or 326 nm, respectively (see Results).
KSI-Cl-Y Kinetics
The isomerization of 5(10)-estrene-3,17-dione by KSI at 20 °C in 40 mM potassium phosphate (pH 7.2) was monitored as previously described.10 A concentrated substrate solution was prepared in dimethyl sulfoxide (DMSO) and added to the buffered enzyme sample to initiate the reaction (2% DMSO in the final solution). The values of kcat, KM, and kcat/KM were determined by fitting the initial rates of product formation as a function of initial substrate concentration to the Michaelis–Menten equation. For each KSI variant, three independent experiments with enzyme concentrations of 10, 20, and 50 nM were performed.
X-ray Crystallography
Crystals of all three D40N-Cl-Y variants were obtained using hanging drop vapor diffusion by mixing 1 μL of 20 mg/mL KSI variant with 1 μL of a reservoir solution [1.1–1.4 M ammonium sulfate, 40 mM potassium phosphate (pH 7.2), and 3–6% 2-propanol]. Blade- or cubic-shaped crystals appeared after incubation for 3 days at 20 °C. Cryoprotection was achieved by directly soaking crystals in 2.0 M sucrose prior to flash-freezing in liquid nitrogen. Single-crystal diffraction data for each D40N-Cl-Y variant were collected at Stanford Synchrotron Radiation Laboratory beamlines BL7-1 and BL11-1 and Advanced Light Source (Lawrence Berkeley National Laboratory) beamline BL502. Data were integrated and scaled using mosflm11 and CCP4,12 respectively. Data collection and refinement statistics are summarized in Table S2. An initial protein model for each structure was obtained by molecular replacement with Phaser13 using the coordinates from the previously published structure of KSI-D40N with bound 3-fluoro-4-nitrophenol [Protein Data Bank (PDB) entry 3VGN]. The protein model is chosen to minimize the effect of modeling bias in the structural comparisons (especially the active site residues) between D40N-Cl-Y and the wild type (see Results). Simulated annealing refinement was conducted using a maximum-likelihood amplitude-based target function as implemented in Phenix.14 Further refinement was conducted with Phenix, interspersed with manual model building in Coot.15 All structural figures were prepared using PyMOL.16
13C NMR Spectroscopy of D40N-Cl-Y
One-dimensional 13C NMR spectra were recorded on a Varian UNITYINOVA NMR spectrometer (500 MHz) running VNMR version 6.1C and equipped with a 5 mm PFG switchable probe operating at ambient temperature. Samples contained 0.3–0.5 mM KSI, 40 mM potassium phosphate (pH 7.2), 1 mM EDTA, 2 mM DTT, and 5% D2O as the lock solvent. Spectra were acquired in a 5 mm Shigemi symmetrical microtube for approximately 15000 scans and processed using a 10 Hz line broadening function.1 Peak positions are reported as the chemical shift value corresponding to the position of maximal intensity for each peak. The 13C NMR spectra of the ionized and neutral form of 3-chlorotyrosine were recorded in D2O at pD 13 and 4, respectively (Figure S2). All 13C chemical shifts were referenced to an external standard of sodium 3-trimethylsilylpropionate-2,2,3,3-d4 (0 ppm) under identical solvent and temperature conditions.
Three samples were prepared and measured: D40N-Cl-Y16 bearing 13Cζ-labeled Cl-Y16 and unlabeled tyrosines (Y32,57,119), D40N-Cl-Y16 bearing unlabeled Cl-Y16 and 13Cζ-labeled tyrosines at the other three sites, and D40N-Cl-Y57 bearing [13Cζ]Cl-Y57 and unlabeled tyrosines (Y16,32,119). For D40N-Cl-Y16, initial measurements were performed on the mixture of two populations from the anion exchange column. The samples were then further purified in an effort to elucidate the chemical shifts corresponding to the first population. The fractional ionization of each residue in the tyrosine triad was calculated using a method similar to that described previously.17 The chemical shifts of the neutral and ionized amino acids (Y or Cl-Y) in aqueous solution were used as the reference states for fractional ionization calculations.1
RESULTS
Preparation and Kinetic Characterization of KSI-Cl-Y Variants
KSI variants bearing Cl-Y at residue 16, 32, or 57 were expressed and purified and found to have the expected masses by ESI-MS. Tandem MS was further performed on the first population of the KSI variants to verify the incorporation of Cl-Y (Figure S4). The expression yield of KSI-Cl-Y variants is 3–5 mg/L, ~5 times lower than that of wild-type KSI (20–25 mg/L), a decrease typical for amber suppression observed in other systems.18 The enzymatic activities of the three KSI-Cl-Y variants were characterized and compared with that of wild-type (WT) KSI (Table 1). The proteins retained most of their activity upon incorporation of Cl-Y, with a modest 1.5–5-fold decrease in kcat; kcat/KM decreased by 8–10-fold for KSI-Cl-Y57 and KSI–Cl-Y16 and remained the same as that of the WT19 for KSI-Cl-Y32. The relatively small but systematic changes observed in these conservative mutants suggest structural features very similar to those of the wild type; nonetheless, they provide ideal tests for electric field/catalytic proficiency correlations mapped by vibrational Stark spectroscopy,20 and this will be discussed in a future publication (Y. Wu and S. G. Boxer, unpublished data).
Table 1.
Effects of Chlorotyrosine Substitution on KSI-Catalyzed Isomerization of 5(10)-Estrene-3,17-dione
| kcat (s−1) | KM (μM) | kcat/KM (×104 M−1 s−1) | kcat ratio (mutant/WT) | ΔΔG‡a (kcal mol−1) | kcat/KM ratio (mutant/WT) | |
|---|---|---|---|---|---|---|
| WTb | 9.0 ± 0.2 | 16 ±2 | 54 ±7 | 1 | 0 | 1 |
| KSI-Cl-Y57 | 1.8 ± 0.1 | 25 ± 2 | 7.2 ± 0.7 | 0.20 | 0.94 | 0.13 |
| KSI-Cl-Y32 | 5.1 ± 0.1 | 11 ±2 | 46 ± 8 | 0.57 | 0.34 | 0.85 |
| KSI-Cl-Y16 | 1.9 ± 0.1 | 44 ± 3 | 4.3 ± 0.4 | 0.21 | 0.90 | 0.080 |
ΔΔG‡ = ΔG‡ (mutant) − ΔG‡(WT). ΔG‡ = RT(29.5 – ln kcat). T = 293 K, and R is the gas constant.
Values from ref 19. The addition of the LVPR C-terminus was found not to affect the catalytic rates.
X-ray Crystal Structures of D40N-Cl-Y Variants
We determined X-ray crystal structures for the D40N variants bearing Cl-Y at positions 16, 32, and 57 (PDB entries 5D82, 5D83, and 5D81, respectively); the key active site residues in the tyrosine triad are shown in Figure 2, and the data collection statistics can be found in Table S2. The overall structures obtained for these variants were quite similar to those determined previously for P. putida KSI bound to equilenin (PDB entry 1OH0),2 with average root-mean-square deviation (rmsd) values of 0.24, 0.24, and 0.22 Å for the main-chain atoms of apo D40N-Cl-Y16, apo D40N-Cl-Y32, and apo D40N-Cl-Y57, respectively. Similar results were obtained for comparison with the apo WT structure (PDB entry 3VSY); however, the resolution of that structure (1.5 Å) is worse than that of PDB entry 1OH0 (1.1 Å), so the latter was used for our comparisons. In some cases, coordinate changes of up to 1–1.5 Å are observed in regions distant from the active site, for instance, at the α-helices around residue 35. Focusing on the active site residues, especially the O–O distances of the tyrosine triad, we found superposition of Cl-Y-containing KSI with WT KSI structures revealed conformational differences on the same scale as the 0.1–0.2 Å estimated coordinate error for the X-ray structures given their resolutions (Figure 2; see also Figure S5). Because small distances can affect the H-bond interactions in which we are interested, we also used an alternative way to estimate the errors in the O–O distances by refining the distances using Refmac5. The resulting O–O distances differ on average from those refined by Phenix by 0.06, 0.10, and 0.06 Å for apo D40N-Cl-Y16, apo D40N-Cl-Y32, and apo D40N-Cl-Y57, respectively. Therefore, within the coordinate uncertainty at the resolution of the X-ray structures, the incorporation of Cl-Y preserves the relative positioning of the residues participating in the H-bonding network. The highly conservative nature of these mutants, which can be achieved only through unnatural amino acid incorporation, allows us to study subtle aspects of the hydrogen bond network, as well as the catalytic role of active site electrostatics, in a systematic manner.
Figure 2.
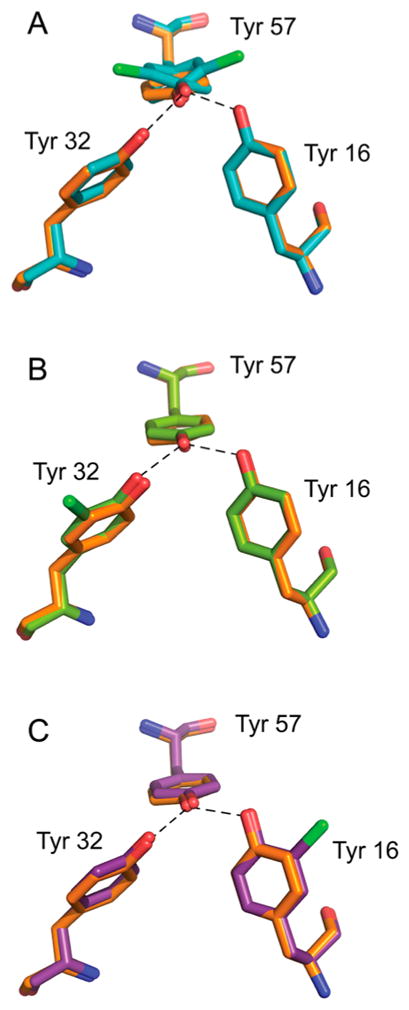
Structures of the tyrosine clusters in apo D40N-Cl-Y variants, superimposed onto a high-resolution structure of D40N, colored orange. (A) Superposition of 1.4 Å resolution D40N-Cl-Y57 with D40N. (B) Superposition of 1.7 Å resolution D40N-Cl-Y32 (chain b) with D40N. (C) Superposition of 1.4 Å resolution D40N-Cl-Y16 (chain a) with D40N. Oxygen, nitrogen, and chlorine are colored red, blue, and green, respectively.
For D40N-Cl-Y57, two orientations of Cl-Y57 with occupancies of 59 and 41% are observed. Compared to Y57 in the high-resolution WT KSI structure, the plane of the benzene ring is rotated approximately 20° toward either direction in the two conformations of D40N-Cl-Y57 (Figure 2). The O–O distances between Cl-Y57 and Y32 and Y16 are 2.73 and 2.42 Å, respectively, for the major conformation and 2.72 and 2.48 Å, respectively, for the minor conformation (Table 2). Two ordered water molecules within 3 Å of the tyrosine residues are observed in the oxyanion hole (Figure S5A).
Table 2.
O–O Distances of the Tyrosine Triad in Each Mutant
| chain | O57–O32 (Å) | O57–O16 (Å) | |
|---|---|---|---|
| D40N | a | 2.7(1) | 2.5(1) |
| b | 2.6(9) | 2.5(3) | |
| D40N-Cl-Y57 | 59% | 2.7(3) | 2.4(2) |
| 41% | 2.7(2) | 2.4(8) | |
| D40N-Cl-Y32 | a | 2.5(0) | 2.6(1) |
| b | 2.6(9) | 2.4(5) | |
| D40N-Cl-Y16 | a | 2.6(6) | 2.4(1) |
| b | 2.5(9) | 2.4(9) |
For both D40N-Cl-Y32 and D40N-Cl-Y16, two asymmetric units are observed in one unit cell. The two chains of each mutant differ by an overall rmsd value of ~0.19 Å from each other and have similar O–O distances among the three triad residues (Table 2). The two mutants share more similarities in their active site geometry: Cl-Y32 and Cl-Y16 both adopted a single orientation. Their benzene ring is coplanar to their tyrosine counterparts in KSI (PDB entry 1OH0), and the chlorine is pointing away from the H-bond core of the tyrosine triad. The only difference is that two ordered water molecules are found within 3 Å of the triad residues for D40N-Cl-Y16, while only one was observed for D40N-Cl-Y32 (Figure S5B, C).
pKa Values and Isotope Effect of D40N-Cl-Y Variants
We measured the pKa and the pKa isotope effect of the active site of each D40N-Cl-Y variant using UV absorbance spectroscopy (Figure 3). All measurements were taken in the D40N background as this is the pKSI mutant in which the highly perturbed pKa of Tyr57 was originally observed.1 The D40N variant used here differs from that in previous studies1,3 in that it has four extra amino acids at the C-terminus (LVPR) because of the purification procedure. This variant’s tyrosine cluster has a pKa of 5.8, which differs from that of the previously studied construct (pKa 6.3) by 0.5 pKa unit (Figure S6A). Despite this unexpected difference, the large and unusual isotope effect on the pKa (Figure S6A) matches that found earlier,3 and the 13C NMR spectrum of [13Cζ]tyrosine-labeled protein (Figure S6B) is likewise identical to that observed previously.1 Combined with the high degree of structural similarity described above (it appears that the active site, in particular the H-bonded Tyr cluster, is unchanged), we suggest that the extra four amino acids at the C-terminus, especially the positively charged arginine, are interacting with the water in a way that results in this difference in absolute pKa. The C-terminus of KSI ends at the side of the enzyme that has a large opening for water passage. The extra LVPR residues increase the length of the unstructured C-terminus from ~6 to 10 residues. Interestingly, we observed a sharp 13C NMR peak at 159.6 ppm in unlabeled (natural abundance) KSI (Figure S7) that was absent in the previous construct without the LVPR C-terminus. 1,17 It is likely that this peak originates from the fast rotating arginine side chain at the end of the extended C-terminus (the peak position of Arg’s ζ-carbon in water is at 159.67 ppm21).
Figure 3.
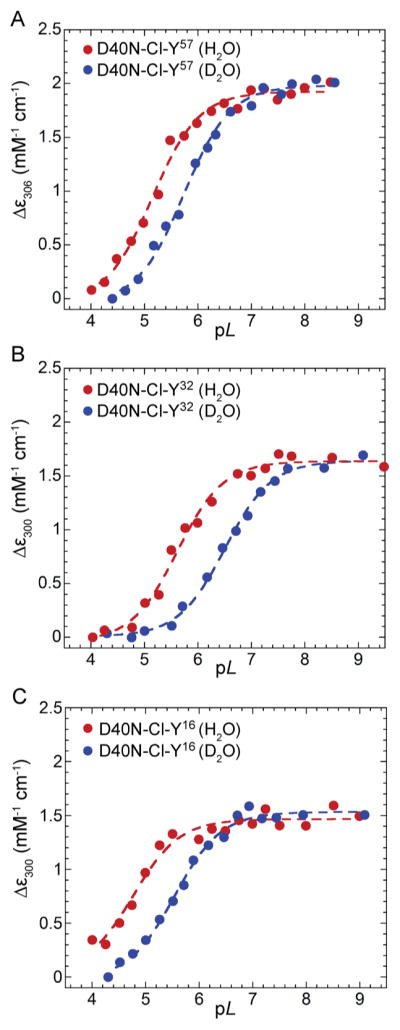
UV–vis titration curves of D40N-Cl-Y variants used to obtain the pKa and isotope effect (ΔpKa). (A) Titration curves of D40N-Cl-Y57 in buffered H2O (red) and D2O (blue). Fractional ionization of the phenolic moiety is monitored by measuring the change in absorption at 306 nm. (B) Titration curves of D40N-Cl-Y32. Fractional ionization is monitored by measuring the change in absorption at 300 nm. (C) Titration curves of D40N-Cl-Y16. Fractional ionization is monitored by measuring the change in absorption at 300 nm. All data sets were fit to sigmoidal curves to yield pKas and ΔpKas.
The intrinsic pKas of tyrosine (10.1) and 3-chlorotyrosine (8.2) differ by 1.87 pKa units (Figure S8),7 and their H–D isotope effects are both close to 0.5 pKa unit. The high pH–low pH difference spectra for Tyr and 3-Cl-Tyr (Figure S9A) show that the peak of the difference spectrum red shifts by 7 nm in 3-Cl-Tyr (from 293 to 300 nm), which provides a basis for evaluating the primary site of titration in the three D40N-Cl-Y variants. We discuss each D40N-Cl-Y variant separately.
D40N-Cl-Y57
In the case of D40N-Cl-Y57 (Figure S9B), a broad peak centered around 300 nm was observed in the difference spectra with extinction coefficient differences similar to that of Cl-Y in solution, identifying Cl-Y57 as the primary site being titrated across the pL series.9 The apparent pKa of D40N-Cl-Y57 was determined to be 5.1, 0.7 pKa unit lower than that of D40N (Figure 3A and Table 3). Upon H/D substitution, the pKa of D40N-Cl-Y57 increased by 0.61 pKa unit, representing a pKa isotope effect that is 0.26 pKa unit smaller than that of D40N (Table 3).
Table 3.
pKa Values and pKa Isotope Effects of D40N-Cl-Y Variants
| D40N | D40N-Cl-Y57 | D40N-Cl-Y32 | D40N-Cl-Y16 | |
|---|---|---|---|---|
| pKa (H) | 5.78 ± 0.11 | 5.15 ± 0.08 | 5.66 ± 0.08 | 4.86 ± 0.13 |
| pKa (D) | 6.65 ± 0.09 | 5.76 ± 0.07 | 6.53 ± 0.08 | 5.56 ± 0.04 |
| pKa shift relative to D40Na | – | −0.65 (+1.22b) | −0.10 | −0.90 |
| H–D pKa isotope effect (ΔpKa) | 0.87 ± 0.14 | 0.61 ± 0.10 | 0.87 ± 0.11 | 0.70 ± 0.14 |
| shift in H–D pKa isotope effect relative to D40Na | – | −0.26 | ~0 | −0.17 |
| primary site of titration | Y57 | Cl-Y57 | Y57 | Y57 |
The pKa and isotope effect of D40N are used as the reference states for comparison.
The partial contribution to the pKa shift from the loss of proton delocalization (see the text).
D40N-Cl-Y32
The high pH–low pH absorbance difference spectrum of D40N-Cl-Y32 overlapped well with that of tyrosine, with a peak centered around 295 nm (Figure S9C). The apparent pKa was determined to be 5.7, 0.1 pKa unit lower than that of D40N (Figure 3B and Table 3). Upon H/D substitution, the pKa of D40N-Cl-Y32 increased by 0.87 pKa unit, quite similar to that of D40N (Table 3).
D40N-Cl-Y16
The high pH–low pH absorbance difference spectrum of D40N-Cl-Y16 (Figure S9D) overlapped well with that of tyrosine, similar to that of D40N-Cl-Y32, which indicates the ionization of one tyrosine (mostly Y57) upon pL titration of the mutant. The apparent pKa of D40N-Cl-Y16 is 4.9, 0.9 pKa unit lower than that of D40N (Figure 3C and Table 3). Upon H/D substitution, the pKa of D40N-Cl-Y16 increased by 0.70 pKa unit, a pKa isotope effect slightly smaller than that of D40N (Table 3). Results for all variants are summarized in Table 3.
13C NMR of D40N-Cl-Y Variants
To further dissect the proton delocalization pattern into the roles of individual residues in the triad, we employed site-specific 13C isotope labeling to look at the fractional ionization of each tyrosine residue. The assignment of peaks to each residue is based on the similarity of their chemical shifts to those of D40N,17 as Cl-Y incorporation is expected to have only local effects confined to the tyrosine cluster (i.e., residue 57, the primary site of ionization, would still have the most downfield chemical shift). For D40N-Cl-Y57, one peak at 163.06 ppm was observed for 13Cζ-labeled Cl-Y57 (Figure 4C). For D40N-Cl-Y16, because only very small amounts of these isotopically modified and Cl-Y-modified proteins were made, we initially combined the two populations from ion exchange chromatography. In the spectra with both populations, the peak corresponding to 13Cζ-Y119, a residue at the dimer interface far from the active site, has the same chemical shift as D40N [157.3 ppm (Figure 4A)]. Two peaks at 157.3 and 158.0 ppm were observed for 13Cζ-labeled Cl-Y16 (Figure 4B), and two peaks at 158.7 and 159.3 ppm were observed for 13Cζ-Y32 (Figure 4A). For 13Cζ-Y57, two peaks at 164.2 and 164.8 ppm were observed (Figure 4A). Measurements on small samples of the separated population showed a single peak for each of the residues in the tyrosine triad (Figure 4A and and Figure 4B, bottom). By comparison, the peaks at 158.0, 158.7, and 164.2 ppm are assigned to Cl-Y16, Y32, and Y57, respectively, of the first population. The small peak at 159.6 ppm (Figure 4B, top) is likely from the same arginine side chain at the end of the C-terminus as that in the unlabeled KSI (Figure S7). The signal-to-noise ratios of the spectra in these cases were sufficient to distinguish this relatively sharp peak from the background signals.
Figure 4.

13C NMR spectra of D40N-Cl-Y variants. In panels A and B, the top spectra correspond to the protein samples containing both populations and the bottom spectra to the separated population (second population in panel A and first population in panel B). The spectrum in panel C corresponds to the first population. For the spectra of the individual population, the presence of the small peak that corresponds to the other population is a consequence of nonideal separation, rather than slow interconversion (Figure S10 and discussion in the Supporting Information). The peak assignment to each residue is determined by comparison with D40N. The values of the chemical shifts are discussed in the text and compiled in Table S3. (A) Spectra of D40N-Cl-Y16 with 13Cζ-labeled Tyr at positions 32, 57, and 119 and unlabeled Cl-Y16. The chemical shifts from the first population are derived by comparing the two spectra and are used for calculations of fractional ionizations (see the text). (B) Spectra of D40N-Cl-Y16 with 13Cζ-labeled Cl-Y16 and the other tyrosines not labeled. The chemical shifts from the first population are shown in the bottom spectrum. (C) Spectrum of D40N-Cl-Y57 with 13Cζ-labeled Cl-Y57 and the other tyrosines not labeled. The observed chemical shift is very close to that of ionized 3-chlorotyrosine (163.1 ppm).
DISCUSSION
pKa Values and Isotope Effects of D40N-Cl-Y Variants
Proton delocalization within the tyrosine triads of the three KSI-Cl-Y mutants is assessed from the shift in their overall pKa values and the difference in the pKa isotope effect upon H/D substitution compared to that of D40N. The earlier modeling and analysis of the origin of the abnormally low pKa of Y57 in D40N (the pKa of the primary site associated with the titration) demonstrated that the proton affinities of the three tyrosines in the triad are well-matched to allow strong interactions, and that the phenolic protons formally belonging to Y16 and Y32 on the sides of the cluster could delocalize onto Y57 at the center to stabilize its negative charge (Figure 1).3 Using this as a starting point, we can make some simple predictions for the effect of Cl-Y incorporation at each site and then compare them with the experimental results.
Addition of chlorine lowers the pKa of tyrosine by 1.8 pKa units from a pure inductive effect of the Cl substituent (Figure S8). We focus on two questions: how sensitive are the collective properties of the cluster to proton affinity detuning at each site, and do the interactions between different tyrosine residues contribute equally to proton delocalization? We will use the language developed in ref 3 and the simplified model developed by McKenzie22,23 to describe this qualitatively. McKenzie’s model uses a Morse potential at each site to describe the diabatic states that are then coupled by the H-bond interactions to give adiabatic potential curves. This is illustrated in Figure S11. The coupling strength depends sensitively on the O–O distance between the H-bond donor and the acceptor. Because the O–O distances among the hydroxyl groups of the triad residues are conserved within the uncertainty of the coordinates of the X-ray structures upon Cl-Y incorporation (Table 2), for each H-bond pair, we will discuss the changes in the cluster’s overall pKa in light of the site proton affinities, even though effects arising from subtle changes in geometry cannot be strictly ruled out. This allows us to directly attribute any change in proton delocalization between a Cl-Y variant and D40N to the proton affinity detuning of a single residue. While all three residues should be considered together, within the framework of this two-site model, we treat the H-bond network such that the interaction between Y57 and Y32 or between Y57 and Y16 stabilizes the system independently. For instance, when residue 16 is modified, because the positions of Y57 and Y32’s diabatic potentials are unperturbed, the energetic benefit from their interaction is not significantly altered from that of the wild type. We then define the in situ proton affinity of Y57 as referring to its adiabatic potential after coupling with Y32 (Figure S11A, dashed line), which is lower than the diabatic potential well of Y57 alone, and compare it with the proton affinity of Cl-Y16 to assess the additional stabilization effect from this second pairwise H-bonding interaction.
As shown in Table 3, the tyrosine triads of D40N-Cl-Y mutants exhibit noticeable changes up to 1.2 pKa units in their acidities from that of D40N, suggesting that proton affinity detuning effectively alters the proton delocalization behaviors in the H-bond network that provide the extra stabilization energy. Meanwhile, the H–D isotope effects (ΔpKa) of the mutants appear to respond less sensitively to the perturbation; the variation among the mutants and D40N is smaller compared to the variation in their overall pKa values. However, isotope effects larger than that observed for tyrosine or chlorotyrosine in solution (0.46 ± 0.08) are still present in all mutants, indicating that the tyrosine triad in these mutants preserves some features of the nuclear quantum effect observed in D40N.
In the case of D40N-Cl-Y57, the large intrinsic change in the pKa of Cl-Y57 should produce a mismatch in intrinsic proton affinity with the other two sites in the cluster. As a consequence, we expect a net loss of proton delocalization to stabilize the ionized residue 57, counterbalancing the fact that the residue, by itself, is more acidic. Indeed, the observed pKa shift of D40N-Cl-Y57 relative to that of D40N (0.65 pKa unit) exemplifies the outcome of these two competing effects: the reduction in the level of proton delocalization ostensibly decreased the cluster’s acidity by 1.1 pKa units (1.8 – 0.65), corresponding to 1.5 kcal mol−1 of lost stabilization energy at 298 K due to proton affinity mismatch. Moreover, the H–D pKa isotope effect of D40N-Cl-Y57 is 0.26 pKa unit smaller than that of D40N and in fact is quite similar to that of an isolated chlorotyrosine in solution, consistent with our prediction of a smaller degree of proton delocalization in this variant.
In the case of D40N-Cl-Y32, addition of chlorine lowers the intrinsic proton affinity of residue 32, making it more comparable to the in situ proton affinity of Y57 (the adiabatic potential after coupling with Y16). We might predict a larger degree of proton sharing between the two to stabilize the ionized tyrosine 57, leading to an effective pKa even lower than that of D40N. However, a minimal change in the pKa of D40N-Cl-Y32 from that of D40N (0.1 pKa unit) was observed along with indistinguishable isotope shifts, indicating that there is virtually no effect on the proton delocalization equilibrium when the intrinsic proton affinity of site 32 is modulated by 1.8 pKa units. On the other hand, it was previously found that if the tyrosine at position 32 is substituted with phenylalanine, the pKa of the cluster returns to that of a normal tyrosine pKa of >9.9 On this basis, we posit that the interaction between Y32 and Y57 is more similar to a normal H-bond, which favors the ionization via electrostatic stabilization (i.e., the dipole moment along Y32’s O–H bond stabilizes a negative charge on the oxygen atom of Y57). This description is consistent with the X-ray structures, in which the Y32 O atom is consistently found to be more distant from Y57 than Y16. The longer O–O distance leads to a weaker coupling between the two sites; thus, the energetics of the proton in the adiabatic potential is not significantly lowered. These data suggest that the nuclear quantum effect is smaller for this pairwise interaction, which is consistent with the insensitivity of the tyrosine cluster’s pKa and H–D pKa isotope effect on the precise affinity matching of Y32 to the other members of the triad. As the degree of proton delocalization across the H-bond in the KSI tyrosine triad strongly depends on the separation between the proton donor and acceptor oxygen atoms,3 it is not unreasonable to view the role of Y32 as a local modulator: it finely tunes the proton affinity of Y57 relative to that of Y16 and facilitates their closer approximation in the active site, which the overall protein architecture alone cannot enforce, so that the latter two can afford a larger degree of proton delocalization that is responsible for the unusual acidity of the cluster.
The proposed difference in the role of Y32 and Y16 in proton delocalization in the triad is further tested by the data we presented for D40N-Cl-Y16. D40N-Cl-Y16 is expected to have a degree of proton delocalization between Y57 and Cl-Y16 larger than that of D40N and a pKa lower than that of D40N, because the addition of chlorine brings the intrinsic proton affinity of site 16 closer to the in situ proton affinity of Y57 [the adiabatic potential after coupling with Y32 (Figure S11A)]. Indeed, a large overall pKa shift of 0.9 pKa unit in the active site upon substitution of Y16 with Cl-Y is observed (Figure 3C and Table 3); this is a striking observation considering that site 16 is not even the primary site of ionization based on the UV–vis difference spectrum. Moreover, the cluster is rendered even more acidic than when the primary site of ionization (Y57) is chlorinated. This provides compelling evidence that the additional stabilization energy of 1.2 kcal mol−1 is afforded by an increased capacity for the phenolic proton on site 16 to delocalize onto its ionized neighbor. Note that in all three mutants, the measured pKa reflects the stabilization of the negative charge at site 57 (Figure S9). In D40N-Cl-Y57, chlorine addition directly affects the stabilization of the negative charge on Cl-Y57. However, in D40N-Cl-Y16 and D40N-Cl-Y32, the inductive effect of chlorine addition on Cl-Y16 or Cl-Y32 is propagated to Y57 via the coupling of the two sites’ potentials, which changes the proton delocalization equilibrium and leads to the downward shift of the effective pKa. The contrast between the large pKa shift of 0.9 pKa unit upon substitution of Y16 with Cl-Y and the small shift upon replacement of Y32 is in line with the proposal described above: compared to a normal hydrogen bond, the interaction between Y57 and Y16 is stronger and more sensitive to the relative proton affinity of the two partners. Their interaction stabilizes the ionized active site by allowing proton delocalization between the two residues and is finely tuned via electrostatic interaction with residue 32.
This larger degree of proton delocalization in the active site of D40N-Cl-Y16 suggests that it should possess the largest H–D pKa isotope effect; however, the measured isotope shift is instead 0.17 pKa unit smaller than that of D40N, which appears to be inconsistent with what is expected for its effect on the pKa (Figure 3C and Table 3). We consider two possible explanations for this observation. First, in the UV–vis measurements, the spectra were fitted to a single titration curve to yield the pKa of the primary site of ionization in the cluster (site 57). Because a larger degree of proton delocalization between Y57 and Cl-Y16 was observed in D40N-Cl-Y16, it is possible that site 16 also exhibits a noticeable amount of ionization (Figure S9D). In this case, the stabilization energy gained from the larger degree of proton delocalization in the cluster could affect the properties of both residues in the pL range of titration and may only be fully accounted for when the isotope effects of both residues are considered. However, because of the overlap of the basis UV spectra of Cl-Y and Y, it is difficult to deconvolute the spectrum to assess the ionization of Y57 and Cl-Y16 separately at each pL. As we chose to fit the data at the peak position of ionized tyrosine, it is possible that we measured the isotope effect of Y57 that reflects only the partial effect of the proton delocalization in the cluster. Second, an alternative explanation for the apparent small isotope effect is that the energy barrier to H (D) transfer is so low in this system that it is described by a single-well potential. In this scenario, delocalization occurs through a classical mechanism, and quantum effects such as zero-point energy and tunneling are no longer necessary for the protons to delocalize. Indeed, in such a situation, one would expect a smaller pKa isotope effect (which reports on quantum effects).3 Interpretation of this curious pKa isotope effect of D40N-Cl-Y16 is further discussed along with the 13C NMR data in the next section.
Fractional Ionization of Tyrosine Residues Using 13C NMR
The changes in the proton delocalization equilibrium in D40N-Cl-Y16 and D40N-Cl-Y57 were clearly reflected from their altered pKas and isotope effects relative to those of the WT. To gain insight into how the ionization is shared among the three residues in the variants’ triads, we determined the fractional ionizations of each residue (Figure 4 and Table S3). The 13C chemical shifts of the ζ-carbons of tyrosine and 3-chlorotyrosine are sensitive to the ionization state of the adjacent hydroxyl group, with a dynamic range of approximately 10 ppm from neutral to fully ionized (Figure S2).1,24 Moreover, the proton transfers within the triad are much faster than the NMR time scale, so the observed peak position reflects the population-weighted average of all ionization states present for a given residue.17 These two observations form the basis for the interpretation of fractional ionization: a larger degree of proton delocalization in the active site of a given variant would cause the fractional ionization of individual residues to be more similar and vice versa. This is an approximate, but residue-specific, method for learning about charge localization.
The chemical shift of 13Cζ-labeled Cl-Y57 in D40N-Cl-Y57 is virtually identical to that of the ionized Cl-Y (Figure 4C), suggesting the (exclusive) localization of negative charge on residue 57. This result agrees well with the proton delocalization model and the conclusion from pKa titration that addition of chlorine at site 57 causes a larger mismatch of the intrinsic proton affinity among the three residues, largely diminishing the extent of proton delocalization in the active site of the variant.
Unlike D40N-Cl-Y57, the residues in the active site triad of D40N-Cl-Y16 all exhibit 13C chemical shifts between that of the ionized and neutral amino acids, indicating significant sharing of the negative charge among the three. The fractional ionization of each residue is calculated for the first population (Table S3 with details of the calculations). Y57 remains as the major ionized residue in the triad bearing the negative charge >60% of the time, suggesting an appreciable height of the energy barriers for proton transfer among the three residues. When compared with D40N, it shows more extensive sharing between the triad residues: the level of fractional ionization of residue 16 increased by ~6% (27%), while the level of fractional ionization of residue 57 decreased by ~5% (62%). The results indicate an increased degree of proton delocalization in the active site of D40N-Cl-Y16 from that of the wild type, in line with the lower effective pKa observed. A more quantitative analysis and comprehensive explanation for the one peculiar isotope effect await a more rigorous model following the methods described in ref 3, and this will be presented elsewhere (L. Wang, S. D. Fried, and T. E. Markland, unpublished data).
CONCLUDING REMARKS
In summary, we have observed a consistent correlation among proton affinity matching, extent of proton delocalization, and acidity enhancement within the tyrosine cluster of ketosteroid isomerase, providing further support for quantum delocalization of protons as a mechanism to perturb the tyrosine pKa in the active site.3 The extra stabilization energy mainly comes from the strong interactions between Y57 and Y16, which is further buttressed by Y32 to enforce the right geometry and relative proton affinities. As we modulated the proton affinity balance between the active site residues, KSI-Cl-Y variants showed relatively small but systematic changes in the catalytic rate (kcat) that will be discussed in greater detail elsewhere (Y. Wu and S. G. Boxer, unpublished data). Natarajan et al. recently described the catalytic consequences of substitution with fluorotyrosines at Tyr16 introduced by protein semisynthesis. 25 Negligible changes in KSI’s catalytic efficiency were observed;25 however, it has been suggested that the level of pKa detuning in this case may be too small to significantly perturb the overall energetics of strong H-bonds, leading to the insensitivity.23 The amber suppression strategy in this study allows both larger and multisite energy perturbations to the active site of KSI while minimizing structural changes. Finally, we note that there are interesting parallels between this system and another well-studied example of the effects of delocalization, namely electron hole delocalization, in the special pair (P) cation radical (P+) of photosynthetic reaction centers. In that case, delocalization shifts the P/P+ reduction potential substantially relative to that of a single bacterio(chlorophyll) molecule, making it easier to oxidize, and this is further modulated by local hydrogen bonds.26,27 Thus, in addition to the large diversity of chemical properties among individual amino acids and prosthetic groups, nature also exploits delocalization among multiple copies of amino acids and prosthetic groups to further fine-tune their properties.
Supplementary Material
Acknowledgments
Funding
S.D.F. thanks the Stanford Bio-X interdisciplinary graduate fellowship for support. This work was supported in part by a grant from the National Institutes of Health (Grant GM27738 to S.G.B.).
We are very grateful to Professor Jiangyun Wang from the Institute of Biophysics, Chinese Academy of Sciences, for making available plasmids pEVOL-ClYRS and for advice on the chemical synthesis of 3-chlorotyrosine. We thank Drs. Aina Cohen, Clyde Smith, Pete Dunten (SLAC), Marc Allaire (ALS), and Nicolas Levinson for help with X-ray diffraction data collection and analysis and Stephen Lynch for help with the NMR measurements. We thank Dr. Lu Wang and Professor Tom Markland for useful discussions about this work.
ABBREVIATIONS
- Cl-Y
3-chlorotyrosine where Cl is ortho to the hydroxyl group
- KSI
ketosteroid isomerase from P. putida with additional C-terminal amino acids LVPR
- KSI-Cl-Y
KSI with incorporation of Cl-Y in the active site used to study catalytic properties
- D40N
KSI with the D40N mutation used to study the pKa of the hydrogen bond network
- D40N-Cl-Y
KSI with incorporation of Cl-Y in the background of D40N
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00958.
pKa, 1H NMR, and 13C NMR of [13Cζ]Cl-Y, X-ray crystallography statistics, additional UV–vis and mass spectrometry characterizations of D40N-Cl-Y variants, detailed discussions about the two populations of D40N with supporting pKa and NMR data, and a qualitative illustration of the Morse potential model (PDF)
References
- 1.Fafarman AT, Sigala PA, Schwans JP, Fenn TD, Herschlag D, Boxer SG. Quantitative, directional measurement of electric field heterogeneity in the active site of ketosteroid isomerase. Proc Natl Acad Sci U S A. 2012;109:E299–308. doi: 10.1073/pnas.1111566109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim SW, Cha SS, Cho HS, Kim JS, Ha NC, Cho MJ, Joo S, Kim KK, Choi KY, Oh BH. High-resolution crystal structures of delta5–3-ketosteroid isomerase with and without a reaction intermediate analogue. Biochemistry. 1997;36:14030–14036. doi: 10.1021/bi971546+. [DOI] [PubMed] [Google Scholar]
- 3.Wang L, Fried SD, Boxer SG, Markland TE. Quantum delocalization of protons in the hydrogen-bond network of an enzyme active site. Proc Natl Acad Sci U S A. 2014;111:18454–18459. doi: 10.1073/pnas.1417923111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCubbin JA, Maddess ML, Lautens M. Total synthesis of cryptophycin analogues via a scaffold approach. Org Lett. 2006;8:2993–2996. doi: 10.1021/ol0609356. [DOI] [PubMed] [Google Scholar]
- 5.Kim SW, Kim CY, Benisek WF, Choi KY. Cloning, nucleotide sequence, and overexpression of the gene coding for delta 5–3-ketosteroid isomerase from Pseudomonas putida biotype B. J Bacteriol. 1994;176:6672–6676. doi: 10.1128/jb.176.21.6672-6676.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kraut DA, Sigala PA, Pybus B, Liu CW, Ringe D, Petsko GA, Herschlag D. Testing electrostatic complementarity in enzyme catalysis: hydrogen bonding in the ketosteroid isomerase oxyanion hole. PLoS Biol. 2006;4:e99. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, Jiang L, Li J, Wang L, Yu Y, Zhou Q, Lv X, Gong W, Lu Y, Wang J. Significant Expansion of Fluorescent Protein Sensing Ability through the Genetic Incorporation of Superior Photo-Induced Electron-Transfer Quenchers. J Am Chem Soc. 2014;136:13094–13097. doi: 10.1021/ja505219r. [DOI] [PubMed] [Google Scholar]
- 8.Oltrogge LM, Boxer SG. Short Hydrogen Bonds and Proton Delocalization in Green Fluorescent Protein (GFP) ACS Cent Sci. 2015;1:148–156. doi: 10.1021/acscentsci.5b00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwans JP, Sunden F, Gonzalez A, Tsai Y, Herschlag D. Uncovering the determinants of a highly perturbed tyrosine pKa in the active site of ketosteroid isomerase. Biochemistry. 2013;52:7840–7855. doi: 10.1021/bi401083b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kraut DA, Sigala PA, Fenn TD, Herschlag D. Dissecting the paradoxical effects of hydrogen bond mutations in the ketosteroid isomerase oxyanion hole. Proc Natl Acad Sci U S A. 2010;107:1960–1965. doi: 10.1073/pnas.0911168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leslie AGW. Joint CCP4+ESF-EAMCB Newsletter on Protein Crystallography. Vol. 26. Daresbury Laboratory; Warrington, U.K: 1992. Recent changes to the MOSFLM package for processing film and image plate data. [Google Scholar]
- 12.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 13.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr, Sect D: Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 14.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 15.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 16.DeLano WL. http://www.pymol.org.
- 17.Sigala PA, Fafarman AT, Schwans JP, Fried SD, Fenn TD, Caaveiro JMM, Pybus B, Ringe D, Petsko GA, Boxer SG, Herschlag D. Quantitative dissection of hydrogen bond-mediated proton transfer in the ketosteroid isomerase active site. Proc Natl Acad Sci U S A. 2013;110:E2552–2561. doi: 10.1073/pnas.1302191110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horowitz S, Adhikari U, Dirk LMA, Del Rizzo PA, Mehl RA, Houtz RL, Al-Hashimi HM, Scheiner S, Trievel RC. Manipulating Unconventional CH-Based Hydrogen Bonding in a Methyltransferase via Noncanonical Amino Acid Mutagenesis. ACS Chem Biol. 2014;9:1692–1697. doi: 10.1021/cb5001185. [DOI] [PubMed] [Google Scholar]
- 19.Schwans JP, Kraut DA, Herschlag D. Determining the catalytic role of remote substrate binding interactions in ketosteroid isomerase. Proc Natl Acad Sci U S A. 2009;106:14271–14275. doi: 10.1073/pnas.0901032106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fried SD, Bagchi S, Boxer SG. Extreme electric fields power catalysis in the active site of ketosteroid isomerase. Science. 2014;346:1510–1513. doi: 10.1126/science.1259802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, Mandal R, Sinelnikov I, Xia J, Jia L, Cruz JA, Lim E, Sobsey CA, Shrivastava S, Huang P, Liu P, Fang L, Peng J, Fradette R, Cheng D, Tzur D, Clements M, Lewis A, De Souza A, Zuniga A, Dawe M, Xiong Y, Clive D, Greiner R, Nazyrova A, Shaykhutdinov R, Li L, Vogel HJ, Forsythe I. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37:D603–610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenzie RH. A diabatic state model for donor-hydrogen vibrational frequency shifts in hydrogen bonded complexes. Chem Phys Lett. 2012;535:196–200. [Google Scholar]
- 23.Mckenzie RH. Strong hydrogen bonds can be insensitive to pKa detuning. 2014 ( http://condensedconcepts.blogspot.com/2014/11/strong-hydrogen-bonds-can-be.html)
- 24.Richarz R, Wuthrich K. Carbon-13 NMR chemical shifts of the common amino acid residues measured in aqueous solutions of the linear tetrapeptides H-Gly-Gly-X-L-Ala-OH. Biopolymers. 1978;17:2133–2141. [Google Scholar]
- 25.Natarajan A, Schwans JP, Herschlag D. Using unnatural amino acids to probe the energetics of oxyanion hole hydrogen bonds in the ketosteroid isomerase active site. J Am Chem Soc. 2014;136:7643–7654. doi: 10.1021/ja413174b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivancich A, Artz K, Williams JC, Allen JP, Mattioli TA. Effects of hydrogen bonds on the redox potential and electronic structure of the bacterial primary electron donor. Biochemistry. 1998;37:11812–11820. doi: 10.1021/bi9806908. [DOI] [PubMed] [Google Scholar]
- 27.Mattioli TA, Lin X, Allen JP, Williams JC. Correlation between multiple hydrogen bonding and alteration of the oxidation potential of the bacteriochlorophyll dimer of reaction centers from Rhodobacter sphaeroides. Biochemistry. 1995;34:6142–6152. doi: 10.1021/bi00018a017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.