

Abstract
The red flour beetle, Tribolium castaneum, offers a repertoire of experimental tools for genetic and developmental studies, including a fully annotated genome sequence, transposon-based transgenesis, and effective RNA interference (RNAi). Among these advantages, RNAi-based gene knockdown techniques are at the core of Tribolium research. T. castaneum show a robust systemic RNAi response, making it possible to perform RNAi at any life stage by simply injecting double-stranded RNA (dsRNA) into the beetle’s body cavity.
In this report, we provide an overview of our larval RNAi technique in T. castaneum. The protocol includes (i) isolation of the proper stage of T. castaneum larvae for injection, (ii) preparation for the injection setting, and (iii) dsRNA injection. Larval RNAi is a simple, but powerful technique that provides us with quick access to loss-of-function phenotypes, including multiple gene knockdown phenotypes as well as a series of hypomorphic phenotypes. Since virtually all T. castaneum tissues are susceptible to extracellular dsRNA, the larval RNAi technique allows researchers to study a wide variety of tissues in diverse contexts, including the genetic basis of organismal responses to the outside environment. In addition, the simplicity of this technique stimulates more student involvement in research, making T. castaneum an ideal genetic system for use in a classroom setting.
Keywords: Molecular Biology, Issue 92, RNA interference, RNAi, gene knockdown, red flour beetle, Tribolium castaneum, injection, double-stranded RNA, functional analysis, teaching laboratories
Introduction
The red flour beetle, Tribolium castaneum, is gaining popularity in various fields of biology due in part to the ease of performing RNA interference (RNAi)1-3. RNAi-based gene knockdown techniques allow scientists to perform loss-of-function analyses without utilizing complex genetic methods. T. castaneum show a robust systemic RNAi response, making it possible to perform RNAi at any stage through simple injection of double-stranded RNA (dsRNA) into the beetle’s body cavity4-6. Simultaneous knockdown of multiple genes is also feasible in T. castaneum by injecting two or more different dsRNA molecules at the same time7,8. In addition, a series of hypomorphic phenotypes can be generated by reducing the concentration of injected dsRNA8. These features make RNAi-based reverse genetic techniques attractive alternatives to traditional forward genetics in T. castaneum. Since virtually all T. castaneum tissues are susceptible to extracellular dsRNA molecules9, this technique allows researchers to study a wide variety of tissues in diverse contexts. In addition, although this report focuses on performing RNAi in T. castaneum, many procedures described here are applicable to other insects. Therefore, this protocol is useful for those who wish to perform loss-of-function analyses in their contexts of interest in T. castaneum, as well as for researchers wishing to apply an RNAi-based technique to other insects.
Injecting dsRNA into larvae allows functional analysis at a variety of beetle life stages, including the larval, pupal, and adult stages4,5,10. We have previously reported our overall larval RNAi protocol including molecular biology procedures11. In the current report, we focus on describing the dsRNA injection procedures, which are best explained with visual aides. We provide detailed step-by-step injection procedures as well as good and bad injection examples. This visual protocol complements our previous protocol, and when combined, they provide a more comprehensive view of the larval RNAi procedures in T. castaneum. In addition, we discuss parameters for dsRNA molecules that could affect the success of RNAi, application of RNAi-based assays for physiological research, as well as the applicability of the larval RNAi protocol in a teaching laboratory.
Protocol
1. T. castaneum Stocks and Culturing
Decide on a T. castaneum strain to use for the experiment. NOTE: Several lab-established T. castaneum strains are available. Ga-1 (Georgia-1) is the parental strain of Ga-2 that was used for genome sequencing3. Since Ga-1 shares most DNA polymorphisms (such as single nucleotide polymorphisms, SNPs) with Ga-2, and is healthier than Ga-2 due to less inbreeding, it is ideal for RNAi experiments. pu-11 is another convenient strain to use, as its unique enhanced yellow fluorescent protein (EYFP) expression in the future wing primordia allows us to distinguish the last larval instar from other instars (see Figure S4 of Clark-Hachtel et al. 201312). It is important to document which beetle strain was used in the experiment, as genetic backgrounds appear to significantly affect the RNAi phenotypes13.
Prepare T. castaneum culture flour by adding 5% (by weight) of pre-sieved brewer’s yeast to organic whole-wheat flour (after mixing, store the culture flour at −20 °C). Aliquot the culture flour (at room temperature) into 6 oz plastic Drosophila stock bottles (about 40 g/bottle).
Culture T. castaneum at 30 °C with 70% humidity. Use a humidity-controlled incubator if possible. Add 30-40 adults per culture bottle (male:female ratio 1:1) to start the culture. NOTE: Although mold is rarely an issue at 30 °C, lower temperatures (such as 25 °C) with 70% humidity can cause mold growth in the culture flour. Lower the humidity if this occurs.
Transfer the adults to a new culture bottle every two weeks for subculturing (see step 5.2-5.5 for how to isolate adults from culture flour). NOTE: It takes about three weeks to obtain the last instar larvae. Alternatively, T. castaneum cultures can be kept at a lower temperature (20-25 °C), albeit with a longer culturing period (developmental time at 25 °C is approximately double that at 30 °C).
2. Preparation of Solutions and Instruments for Larval dsRNA Injection
Synthesize dsRNA molecules homologous to the target gene by in vitro transcription. Store at -80 °C. See Philip and Tomoyasu 201111 for detailed molecular biology procedures. Also see discussion for a number of parameters that need to be considered when designing dsRNA molecules.
Make 10 ml of 0.1 M sodium phosphate buffer (pH 7.6 at 25 °C) by mixing 8.5 ml of 1 M Na2HPO4 with 1.5 ml of 1 M NaH2PO4. Check the pH with a pH meter and adjust accordingly.
Make 1 ml of 10x injection buffer by mixing 10 μl of 0.1 M sodium phosphate buffer (pH 7.6 at 25 °C), 100 μl of 0.5 M KCl, 100 μl of green food dye, and 790 μl of double-distilled water (ddH2O). Dilute the 10x injection buffer to make 2x injection buffer (200 μl 10x injection buffer + 800 μl ddH2O). Store both 10x and 2x injection buffers at 4 °C until needed.
- Prepare Sticky Glass Slides.
- Cover an entire glass slide with a form of repositionable glue for larval injections. For adult or pupal injections, make two thin strips of glue along the longer edges of the slide.
- Allow the glue to dry for several days, which ensures that the glue remains tacky enough to adhere beetles to the slide, while also remaining pliant enough to facilitate their removal after injection. NOTE: If the glue stays too sticky, use a finger and tap on the glue, which will reduce the tackiness. Each slide can hold up to 40 or 50 beetles, and can be reused until it fails to securely hold the animals. Alternatively, double-sided tape can also be used, although the tape is sometimes not adhesive enough to hold the larvae.
- Make an Etherization Basket.
- Remove the plunger from a 10 ml disposable syringe, and cut the syringe in half around the 6 ml line.
- Discard the bottom half of the syringe. Briefly heat the cut surface of the top part of the syringe with a gas burner, and quickly push the melted plastic onto a piece of nylon mesh to glue the mesh onto the syringe. Trim away any excess mesh once the plastic hardens.
Prepare the ether bottle. Add 30 ml of ether into a narrow-mouth 100 or 250 ml glass bottle. Add several pieces of tissue paper to enhance the evaporation of ether. NOTE: Ether presents a fire hazard and is also harmful. Always handle ether under a fume hood. Close the lid tightly while not in use. NOTE: Ice can be used as a safer alternative (such as in a classroom setting), although the sedation is less effective.
3. Preparation of Larval Injection Apparatus
Place an X-Y mechanical stage onto a dissecting microscope. NOTE: X-Y mechanical stages are available from major microscope companies. Alternatively, inexpensive microscope stages can also be used for most dissecting microscopes with some modifications (see the Materials table for example).
Place a mechanical needle manipulator near the X-Y mechanical stage.
Assemble an injection syringe. Use a four-way stopcock (with Luer connections) to connect a 30 ml disposable syringe to a glass needle holder, allowing manipulation of the injection syringe without affecting the pressure in the injection needle (see Philip and Tomoyasu 201111 for the detailed injection syringe assembly).
4. Pulling Injection Needles
Pull borosilicate glass needles (B100-50-15, O.D.: 1 mm, I.D.: 0.5 mm, 15 cm length) by a needle puller. NOTE: For Sutter P-87 or P-97, use the setting “Heat = 770, Pull = 45, Vel = 75, Time = 90” or follow Chapter 2 of the Pipette Cookbook: Adherent Cell, C. elegans, Drosophila, & Zebrafish– Recommended Programs. The optimal settings vary depending on the model of the needle puller. The setting for a generic Drosophila injection needle is a good starting point.
Store pulled needles in a plastic case (e.g., a plastic CD case) and secure with removable mounting putty.
5. Isolation and Selection of Larvae
Place a #25 sieve onto a sieve receiver.
Place the contents of the culture bottle (beetles and culture flour) on the sieve, and sift the contents immediately. Do not leave the contents on the sieve without sifting, as larvae will quickly try to burrow through the sieve and get stuck. Aggressively sift the contents to let the flour fall through and leave older larvae (typically 6th and 7th instar larvae), pupae, and adults (along with their cast-off cuticles, exuviae) behind on top of the sieve.
Transfer the materials remaining on the sieve (beetles and exuviae) to the seed pan. Keep tapping the pan to prevent the beetles from escaping.
Remove the exuviae and the flour particles remaining on the surface of the beetles by gently blowing over the seed pan.
Tap the seed pan to move the contents to the bottom of the pan. Then, leave the pan untapped for several seconds. Wait for the adults, which are more mobile than other stages, to come out from the pile. Remove adults by using an art paintbrush (e.g., 1 cm width).
Separate pupae by gently knocking the seed pan while keeping the mouth of the pan slightly downward, as pupae tend to roll faster toward the mouth. Use a paintbrush to remove the pupae, leaving only the larvae on the seed pan.
Place the isolated larvae in a clean Petri dish. Select an appropriate stage of larvae for injection and place them in a separate Petri dish. NOTE: Early last instar larvae (1-2 day after the final larval molt) are often suitable when analyzing the RNAi effect on adult morphologies as well as on metamorphosis. It is, however, sometimes necessary to perform RNAi at the penultimate (or even earlier) stage to evaluate early gene function (e.g., Figures 3E-3H. Also see Clark-Hachtel et al. 201312). Use pupae as a size reference to identify the instar of larvae. The last instar larvae are slightly longer than pupae. Alternatively, pu-11 can be used to identify the last instar larvae as well as to determine the time course of the development of last instar larvae (Figure S4 of Clark-Hachtel et al. 201312).
Keep the selected larvae in a Petri dish until etherization. Place the rest of larvae and other stages of beetles (such as pupae and adults) back into a culture bottle with flour and return it to the incubator.
6. Preparation of Injection Needles
Place a previously pulled glass needle onto a glass microscope slide using either sticky tack or double-sided tape. Using forceps, test the pulled end of the needle while looking through the dissecting microscope to see where the tip bends. Grab the area slightly toward the tip side of where the needle bends with the forceps and twist to break.
Keep good needles and discard others. Consider a good needle as having an opening that is neither too large nor too small (about 0.05 mm diameter. Figures 1A and 1B), and is angled to make penetration of the larval cuticle easier (Figures 1B and 2A-2B). Consider a bad needle as (i) too small, making uptake of dsRNA solution into the needle difficult (step 7) (Figures 1D and 2D), (ii) too large, causing larval injury and possible lethality upon injection (Figures 1C and 2F), (iii) blunt, making penetration of the cuticle difficult and increasing injury.
- Insert the Needle into the Needle Holder.
- First, unscrew the tip of the needle holder and place the back end of the needle into the holder tip. Then, place the rubber gasket under the holder tip (at least 1 cm from the back end of the glass needle).
- Insert the back of the needle into the holder, with the rubber gasket completely inserted into the opening of the needle holder. Screw the tip back onto the holder.
Place the assembled needle holder onto the needle manipulator. Keep the needle holder close to horizontal. Adjust the position of the manipulator, so the tip of the needle is located in the center of the view when looking through the microscope.
7. Frontloading dsRNA Solution into the Needle
Prepare the dsRNA injection solution just prior to injection by adjusting the concentration with ddH2O and mixing the dsRNA solution with equal volumes of 2x injection buffer (step 2.3). Keep the mixed solution on ice. See discussion regarding the dsRNA concentration.
Cut the tip of a 20 μl disposable pipette tip. Pipette 10 μl of the solution into the trimmed pipette tip. Carefully remove the pipette tip from the pipette while keeping the fluid at the tip by providing backpressure. Alternatively, carefully remove the tip and then move the solution to the end of the tip by providing pressure using a finger.
Place the pipette tip with the dsRNA solution on the microscope stage using sticky tack with the opening facing the tip of the needle.
Looking through the microscope, slowly move the loaded tip toward the needle until the tip of the needle is just inside the loaded pipette tip and into the solution.
While looking through the microscope, gently pull back on the plunger of the injection syringe to slowly pull the dsRNA solution into the needle. NOTE: Do not overload the needle. As the end of the dsRNA solution approaches the tip of the needle, slow the rate of pulling and stop loading the solution before the tip of the needle would reach air. If air reaches the tip, the solution will be rapidly drawn into the needle holder, resulting in serious contamination issues. Detailed cleaning procedures of the needle holder are described in Philip and Tomoyasu 201111.
Remove the pressure from the injection syringe and needle holder by opening the stopcock. Then, move the pipette tip away from the tip of the needle. Raise the needle up from the stage to prevent accidentally damaging the needle while placing the larvae on the stage. Remove the emptied pipette tip from the stage.
8. dsRNA Injection
Place the larvae into the etherization basket. Use less than 10 larvae if learning the technique. Use 30-40 larvae in one round once comfortable with the technique.
Place the basket with larvae in the ether bottle and close the lid. NOTE: Ether presents a fire hazard and is also harmful. Perform this step under a fume hood.
Etherize larvae for about 3 min. Check the larvae for movement after 3 min. Place them back in the ether bottle for another 30 sec if they are still moving. Do not etherize for more than 5 min as this may increase lethality. NOTE: The optimal etherization time may vary depending on temperature and humidity.
Transfer the etherized larvae from the basket to the non-stick edge of a glass sticky slide. Using forceps and while looking through the microscope, pick up each larvae one by one and place them onto the slide. Lay them laterally and gently tap down their bodies from head to tail with the forceps, slightly stretching them as you go, to make sure that they are secured onto the slide.
Place the sticky slide with larvae on the microscope stage.
Insert the dsRNA loaded needle gently into the dorsal side of the larvae to avoid damaging the CNS. Also avoid the dorsal midline, as the larval heart is located here. In this and subsequent steps, always use the stage to move the larvae to the needle (instead of using the needle manipulator to move the needle). NOTE: The specific injection site does not matter as RNAi in T. castaneum appears to be systemic. However, it is usually easiest to inject into the dorsal side of the thoracic or abdominal segments.
After inserting the needle into the larvae, pull the needle slightly back to allow room for the dsRNA to enter the larval body cavity.
Push gently on the injection syringe until the larvae turn green (or the color of the injection buffer) and look stretched and full. NOTE: If learning the technique, try to inject as much as possible to determine the amount of solution that can be injected into a larva without significant lethality. It is generally about 0.5-0.7 μl.
Remove the needle from the larvae. While removing the needle, slightly pull back on the injection syringe to avoid dsRNA loss (also be careful not to pull in air).
Inject all larvae on the slide. Make sure to remove larvae that were not successfully injected. Complete injection before larvae wake up (in about 10 min).
Remove the slide with injected larvae from the injection microscope and leave the slide for 5 more min until all larvae recover from the etherization.
With forceps and under a dissecting microscope, gently lift the larvae from head to tail to release them from the sticky slide. Place the newly released larvae in a clean Petri dish. Leave the larvae on the Petri dish for 10-15 min to allow the injection wound to clot.
Place the larvae into a new bottle with clean flour and label appropriately including the strain name, the type of dsRNA injected, concentration of dsRNA, number of larvae injected, and the date.
Culture the injected larvae at 30 °C with 70% humidity. Observe the larvae for RNAi phenotypes regularly. NOTE: The last instar larvae pupate within 7 days. Then, the pupae will eclose into adults after 7 days (if there is no abnormality in timing caused by RNAi).
9. Confirmation of Knockdown and Phenotypic Analyses
Consider assessing efficiency of RNAi knock down by several different molecular biology techniques, such as quantitative reverse transcription-PCR (qRT-PCR) and Western Blotting. See Miller et al. 20127 and Philip and Tomoyasu 201111 for detailed qPCR and Western Blot protocol, respectively.
Analyze RNAi related phenotypes. NOTE: RNAi related phenotypes can be observed at several different stages and under different culture conditions, depending on the genes that were targeted. RNAi can affect the beetle morphology, metamorphosis, physiology, and behavior. How often the presence of phenotype is checked and under what conditions the beetles are reared should be tailored to the specific questions being investigated through RNAi.
Representative Results
One of the key steps for successful injection is to make a good needle. As described in step 6, the tip of the needle needs to be broken prior to injecting T. castaneum. Examples of good and bad needles are shown in Figure 1. A good needle has a sharp and stiff tip, with an approximately 0.05 mm diameter opening (Figure 1B).
Figure 2 presents a successful injection (Figures 2A and 2B) as well as several cases of unsuccessful injections (Figures 2C-2F). With a properly sized injection needle, the needle tip penetrates the larval cuticle with minimal resistance (Figure 2A), and the dsRNA solution (green) flows into the larvae without any leakage (Figure 2B). Do not over-inject, as it will cause overflow of the dsRNA solution even with a properly sized injection needle (Figure 2C). If the tip of the needle is too thin, the needle tip often fails to penetrate the larval cuticle and bends, making the injection difficult (Figure 2D). If this happens, trimming the needle tip sometimes helps. Larger needles are often still usable, albeit with some difficulties in penetrating the larval cuticle (Figure 2E). However, a large amount of the dsRNA solution is easily pushed out from the needle even with slight pressure on the injection syringe, often resulting in an overflow of the dsRNA solution (Figure 2F).
When performing RNAi, it is important to have a positive control to assess that RNAi is working properly, and a negative control to verify that the effect observed is not a non-specific effect of dsRNA or a consequence of injection procedures (such as injury caused by injection, or abnormality from etherization). Injection of dsRNA targeting EYFP can serve as a good positive control when using the pu-11 strain5,7. When EYFP dsRNA is injected in the last larval stage, a significant reduction in EYFP signal in the future wing primordia can be observed as early as one day post injection (Figure 3A). The knockdown of EYFP is complete two days after injection of the EYFP dsRNA (Figure 3B) and this knockdown persists through the pupal stage (Figures 3C and 3D) and throughout the life of the beetle if the dsRNA concentration is high enough (such as 1 μg/μl)7. EYFP dsRNA can also be used as a negative control as it is a non-endogenous gene sequence and should not cause morphological or physiological disruptions (Figures 3A-3D).
We provided images of larval RNAi for vestigial (vg), a critical wing gene12, as another example of how effective this injection-based RNAi technique is in T. castaneum. When dsRNA for vg is injected into penultimate stage larvae (one stage before the last larval stage), a complete loss of the wing structures can be observed in the pupal stage (Figures 3E and 3F). The resulting adult also completely lacks wing structures (Figures 3G and 3H). The RNAi result for vg exemplifies both the robustness and the systemic nature of RNAi in T. castaneum.
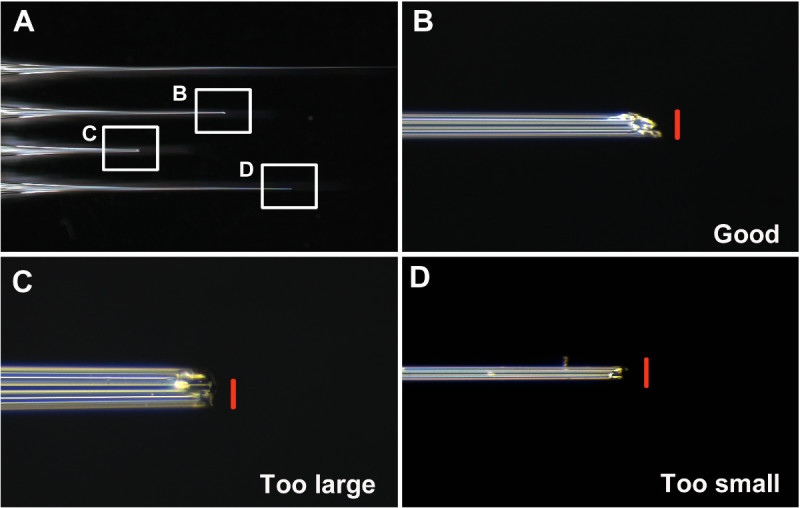 Figure 1: (A) Examples of unbroken, good, and bad needles. (B) The tip of a well-broken good needle. (C) The tip of a broken needle that is too large and blunt. (D) The tip of a broken needle that is too thin. Scale bars (red) are 0.05 mm. Please click here to view a larger version of this figure.
Figure 1: (A) Examples of unbroken, good, and bad needles. (B) The tip of a well-broken good needle. (C) The tip of a broken needle that is too large and blunt. (D) The tip of a broken needle that is too thin. Scale bars (red) are 0.05 mm. Please click here to view a larger version of this figure.
 Figure 2: (A) A properly sized injection needle inserted into the last instar larva. (B) Larva appropriately injected with the green dsRNA solution. (C) Overflow of the dsRNA solution at the injection point caused by over-injection. (D) A thin needle failing to penetrate the larval cuticle. (E) A large injection needle inserted into the last instar larva. (F) Larva injected with a large needle, resulting in overflow and leakage of the dsRNA solution. Arrows indicate the needles. Please click here to view a larger version of this figure.
Figure 2: (A) A properly sized injection needle inserted into the last instar larva. (B) Larva appropriately injected with the green dsRNA solution. (C) Overflow of the dsRNA solution at the injection point caused by over-injection. (D) A thin needle failing to penetrate the larval cuticle. (E) A large injection needle inserted into the last instar larva. (F) Larva injected with a large needle, resulting in overflow and leakage of the dsRNA solution. Arrows indicate the needles. Please click here to view a larger version of this figure.
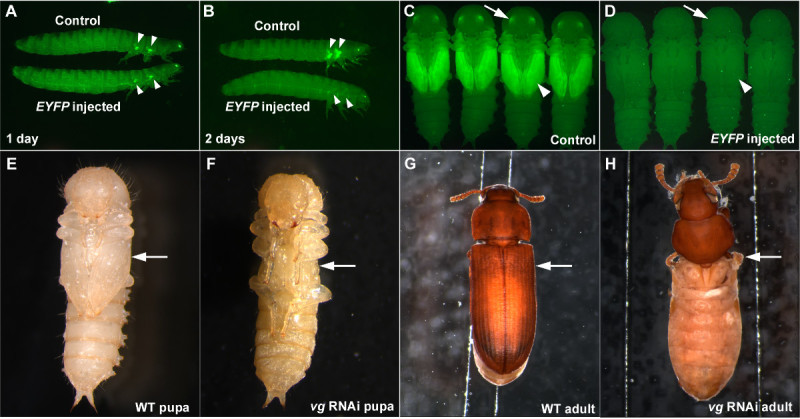 Figure 3:Examples of successful RNAi in T. castaneum. (A-D) Last larval injection of the EYFP dsRNA results in a reduction of EYFP expression. (A) Larvae one day after injection. (B) Larvae two days after injection. (C) Control pupae. (D) Pupae resulting from larval EYFP dsRNA injection. The negative controls are buffer injection (A, B) and dsRed dsRNA injection (C, D). Arrowheads and arrows indicate EYFP expression or lack thereof in the wing primordia and eyes, respectively. (E-H) Penultimate larval RNAi for vg.
(E) Wild-type pupa. (F)
vg RNAi pupa. The lack of wing structures is already visible at the pupal stage (arrow). G) Wild-type adult. (H)
vg RNAi adult. Wing related structures are completely missing (arrow). Please click here to view a larger version of this figure.
Figure 3:Examples of successful RNAi in T. castaneum. (A-D) Last larval injection of the EYFP dsRNA results in a reduction of EYFP expression. (A) Larvae one day after injection. (B) Larvae two days after injection. (C) Control pupae. (D) Pupae resulting from larval EYFP dsRNA injection. The negative controls are buffer injection (A, B) and dsRed dsRNA injection (C, D). Arrowheads and arrows indicate EYFP expression or lack thereof in the wing primordia and eyes, respectively. (E-H) Penultimate larval RNAi for vg.
(E) Wild-type pupa. (F)
vg RNAi pupa. The lack of wing structures is already visible at the pupal stage (arrow). G) Wild-type adult. (H)
vg RNAi adult. Wing related structures are completely missing (arrow). Please click here to view a larger version of this figure.
Discussion
There are a number of important issues that need to be considered to guarantee the success of RNAi, including the length and concentration of the dsRNA molecules, competition among different dsRNA molecules (when attempting multiple knock down), and the possibility of Off-Target Effects (OTE).
dsRNA Length
The length of dsRNA molecules affects the efficiency of the systemic RNAi response, with a longer dsRNA being more efficient to trigger RNAi7,14,15 (though the longer limit of dsRNA is currently unknown). The dsRNA length needs to be longer than 50 bp to induce effective RNAi in T. castaneum7. dsRNA between 150 bp and 500 bp appears to be ideal for RNAi experiments. Although longer dsRNA molecules can also be used, they will have an increased chance of OTE and the gene-cloning step will become increasingly difficult.
dsRNA Concentration
Different degrees of gene knockdown can be achieved depending on the concentration of dsRNA. 1 μg/μl appears to be a reasonable starting concentration, which often produces a near-null phenotype (may vary depending on the gene(s) of interest). RNAi can be performed with a higher concentration (e.g., 7-8 μg/μl) to obtain a stronger RNAi phenotype. RNAi with a serial dilution of dsRNA can sometimes be beneficial to produce a series of hypomorphic phenotypes (Supplemental Data of Tomoyasu et al. 20098, and Borràs-Castells unpublished data).
RNAi Competition
Multiple gene knockdown can be accomplished in T. castaneum by injecting several different dsRNA molecules simultaneously. However, it is also known that having several different dsRNA molecules present within the organism often results in competition between the dsRNAs for access to the RNAi components7,14. It is important to use the same length and the same concentration for all dsRNA when attempting multiple gene knockdown to avoid one dsRNA out-competing the others (although, further adjustments of the dsRNA length and concentration may be required when the expression levels greatly differ among the target genes). We, as well as others, have successfully performed double and triple knockdown (e.g., Tomoyasu et al. 200516, Tomoyasu et al. 200917, and Yang et al. 200918). Although feasible, quadruple RNAi (or more) might be challenging, as it would likely cause significant reduction of RNAi efficiency for all four target genes.
Off-targeting
OTE is an inherent concern for RNAi-based approaches. One way to minimize OTE is to identify regions in the target gene that share similar sequences with other genes and avoid these regions when designing dsRNA. A simple BLAST analysis against the T. castaneum predicted gene set can identify such regions. Several online tools also allow evaluation of potential OTE (e.g., E-RNAi19). Performing RNAi for two non-overlapping regions of the target gene is an easy and efficient way to eliminate the possibility that observed phenotypes are caused by OTE. The possibility of OTE is minimized if RNAi for two non-overlapping regions produce the same phenotypes (unless the two non-overlapping regions share a similar sequence).
Evaluating gene knockdown by means other than phenotypic analyses is often critical to effectively present RNAi-related data. Two major ways to evaluate gene knockdown are qRT-PCR and western blot analysis. qRT-PCR is a convenient way to measure the level of the target mRNA, and has been used in many RNAi-related studies including those in T. castaneum (see Miller et al. 20127 for example). However, caution must be taken, as we have recently seen some cases in which the target mRNA level is up-regulated by RNAi (though the protein product is down-regulated) (Borràs-Castells unpublished data). It is currently unknown if this RNAi induced mRNA up-regulation can be widespread or unique to certain genes. Western blot analysis is another way to confirm gene knockdown. This method is quite reliable as it measures the amount of the final protein product. The requirement of a specific antibody against the protein product of the target gene is a downside to this approach. Utilizing multiple independent measurements in addition to phenotypic analysis will increase the confidence of the phenotypic data obtained by RNAi-based analysis.
Since its conception in T. castaneum, RNAi has primarily been used to study gene function in development and pattern formation. These T. castaneum developmental studies have been highly successful in characterizing evolutionarily conserved and diverged functions of genes (reviewed in Denell 20081 and Klingler 20042). However, RNAi-based studies in T. castaneum are not limited to developmental biology. For example, RNAi can be utilized to study gene function in a wide range of physiological and behavioral responses, including stress tolerance, predation, aggression, mate choice, activity patterns, and defense mechanisms.
One difficulty of applying RNAi to these contexts is the likelihood of pleiotropic effects. Often, genes of interest will have a variety of roles throughout the T. castaneum life cycle, thus making the removal of genes without unintended phenotypic effects difficult. However, the ability to easily perform RNAi at a variety of stages can often be an effective strategy for avoiding these pleiotropic effects. For instance, performing RNAi in adults instead of larvae or pupae might allow us to circumvent unintended lethality caused by gene knockdown during early development. The flexibility of the RNAi response in T. castaneum thus makes this model an attractive choice for adapting RNAi to experiments of gene function in physiological and behavioral responses.
The T. castaneum system is also ideal for use in a teaching laboratory. T. castaneum can be easily cultured on a flour/yeast mixture at room temperature (25 °C) without frequent subculturing, and RNAi techniques in T. castaneum are simple enough to be adapted to a laboratory with young, learning scientists. As RNAi is becoming an essential technique in a variety of biological fields, it is crucial that students are exposed to this technique. The straight-forward nature of the larval RNAi technique in T. castaneum also encourages more students to be involved in research, making T. castaneum a prime candidate for a classroom oriented genetic system.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank the Center for Bioinformatics and Functional Genomics (CBFG) at Miami University for technical support. This work was supported by Miami University start-up grant (YT), and National Science Foundation (YT: IOS 0950964).
References
- Denell R. Establishment of Tribolium as a genetic model system and its early contributions to evo-devo. Genetics. 2008;180:1779–1786. doi: 10.1534/genetics.104.98673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingler M. Tribolium. Curr Biol. 2004;14:R639–R640. doi: 10.1016/j.cub.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Richards S, et al. The genome of the model beetle and pest Tribolium castaneum. Nature. 2008;452:949–955. doi: 10.1038/nature06784. [DOI] [PubMed] [Google Scholar]
- Bucher G, Scholten J, Klingler M. Parental RNAi in Tribolium (Coleoptera) Curr Biol. 2002;12:85–86. doi: 10.1016/s0960-9822(02)00666-8. [DOI] [PubMed] [Google Scholar]
- Tomoyasu Y, Denell RE. Larval RNAi in Tribolium (Coleoptera) for analyzing adult development. Dev Genes Evol. 2004;214:575–578. doi: 10.1007/s00427-004-0434-0. [DOI] [PubMed] [Google Scholar]
- Tomoyasu Y, et al. Exploring systemic RNA interference in insects a genome-wide survey for RNAi genes in Tribolium. Genome biology. 2008;9:R10. doi: 10.1186/gb-2008-9-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SC, Miyata K, Brown SJ, Tomoyasu Y. Dissecting systemic RNA interference in the red flour beetle Tribolium castaneum parameters affecting the efficiency of RNAi. PLoS ONE. 2012;7:e47431. doi: 10.1371/journal.pone.0047431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoyasu Y, Arakane Y, Kramer KJ, Denell RE. Repeated co-options of exoskeleton formation during wing-to-elytron evolution in beetles. Curr Biol. 2009;19:2057–2065. doi: 10.1016/j.cub.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Miller SC, Brown SJ, Tomoyasu Y. Larval RNAi in Drosophila. Dev Genes Evol. 2008;218:505–510. doi: 10.1007/s00427-008-0238-8. [DOI] [PubMed] [Google Scholar]
- Brown SJ, Mahaffey JP, Lorenzen MD, Denell RE, Mahaffey JW. Using RNAi to investigate orthologous homeotic gene function during development of distantly related insects. Evolutio., & development. 1999;1:11–15. doi: 10.1046/j.1525-142x.1999.99013.x. [DOI] [PubMed] [Google Scholar]
- Philip BN, Tomoyasu Y. Gene knockdown analysis by double-stranded RNA injection. Methods Mol Biol. 2011;772:471–497. doi: 10.1007/978-1-61779-228-1_28. [DOI] [PubMed] [Google Scholar]
- Clark-Hachtel CM, Linz DM, Tomoyasu Y. Insights into insect wing origin provided by functional analysis of vestigial in the red flour beetle. Tribolium castaneum, Proceedings of the National Academy of Sciences of the United States of America. 2013;110:16951–16956. doi: 10.1073/pnas.1304332110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzmann P, Schwirz J, Schmitt-Engel C, Bucher G. RNAi phenotypes are influenced by the genetic background of the injected strain. BMC genomics. 2013;14:5. doi: 10.1186/1471-2164-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S, Fleenor J, Xu S, Mello C, Fire A. Functional anatomy of a dsRNA trigger differential requirement for the two trigger strands in RNA interference. Molecular cell. 2000;6:1077–1087. doi: 10.1016/s1097-2765(00)00106-4. [DOI] [PubMed] [Google Scholar]
- Winston WM, Molodowitch C, Hunter CP. Systemic RNAi in C elegans requires the putative transmembrane protein SID-1. Vol. 295. New York, NY: Science; 2002. pp. 2456–2459. [DOI] [PubMed] [Google Scholar]
- Tomoyasu Y, Wheeler SR, Denell RE. Ultrabithorax is required for membranous wing identity in the beetle Tribolium castaneum. Nature. 2005;433:643–647. doi: 10.1038/nature03272. [DOI] [PubMed] [Google Scholar]
- Trauner J, et al. Large-scale insertional mutagenesis of a coleopteran stored grain pest, the red flour beetle Tribolium castaneum, identifies embryonic lethal mutations and enhancer traps. BMC Biol. 2009;7 doi: 10.1186/1741-7007-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, et al. Probing the Drosophila retinal determination gene network in Tribolium (II) The Pax6 genes eyeless and twin of eyeless. Developmental biology. 2009;333:215–227. doi: 10.1016/j.ydbio.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Horn T, Boutros M. E-RNAi a web application for the multi-species design of RNAi reagents--2010 update. Nucleic acids research. 2010;38:332–339. doi: 10.1093/nar/gkq317. [DOI] [PMC free article] [PubMed] [Google Scholar]