

Intracellular permeant anions, and not extracellular protons, are the predominant driver of fast gating in the hyperpolarization-activated CLC-2 chloride channel.
Abstract
CLC-2 channels are dimeric double-barreled chloride channels that open in response to hyperpolarization. Hyperpolarization activates protopore gates that independently regulate the permeability of the pore in each subunit and the common gate that affects the permeability through both pores. CLC-2 channels lack classic transmembrane voltage–sensing domains; instead, their protopore gates (residing within the pore and each formed by the side chain of a glutamate residue) open under repulsion by permeant intracellular anions or protonation by extracellular H+. Here, we show that voltage-dependent gating of CLC-2: (a) is facilitated when permeant anions (Cl−, Br−, SCN−, and I−) are present in the cytosolic side; (b) happens with poorly permeant anions fluoride, glutamate, gluconate, and methanesulfonate present in the cytosolic side; (c) depends on pore occupancy by permeant and poorly permeant anions; (d) is strongly facilitated by multi-ion occupancy; (e) is absent under likely protonation conditions (pHe = 5.5 or 6.5) in cells dialyzed with acetate (an impermeant anion); and (f) was the same at intracellular pH 7.3 and 4.2; and (g) is observed in both whole-cell and inside-out patches exposed to increasing [Cl−]i under unlikely protonation conditions (pHe = 10). Thus, based on our results we propose that hyperpolarization activates CLC-2 mainly by driving intracellular anions into the channel pores, and that protonation by extracellular H+ plays a minor role in dislodging the glutamate gate.
INTRODUCTION
CLC-2 is a double-pore homodimeric CLC chloride (Cl−) channel widely expressed in mammalian tissues (Thiemann et al., 1992). CLC-2 regulates neuronal activity by providing background Cl− permeability (Rinke et al., 2010) and is required for colonic electroneutral absorption of NaCl and KCl (Catalán et al., 2012). Malfunctioning CLC-2 channels can result in leukoencephalopathy (Blanz et al., 2007), and their ablation in mice severely degenerates the retina and testes (Bösl et al., 2001; Nehrke et al., 2002). CLC-2 channels open when the voltage (Vm) acts on protopore and common gates that display fast and slow kinetics, respectively. The protopore gate is formed by the negatively charged carboxyl side chain (-CH2-CH2-COO−) of a glutamate (Glu) residue located within the permeation pathway near the outside entry (Dutzler et al., 2002). The atomic structure of the CLC Cl−/H+ exchanger (Dutzler et al., 2002, 2003) suggests that the pore of a CLC Cl− channel is curvilinear with two narrow constrictions (<1 Å) surrounding the protopore gate. The pore radius is ∼2 Å. Although the high energy barrier of this structure precludes Cl− permeation (Miloshevsky and Jordan, 2004), the physical dimensions ensure that the gate interacts both electrostatically and sterically with negatively charged permeant species during the permeation process. Such interactions might regulate gating (Richard and Miller, 1990; Pusch et al., 1995; Chen and Miller, 1996; Rychkov et al., 1996; Sánchez-Rodríguez et al., 2010; Nieto-Delgado et al., 2013). In addition, Vm gating in CLC proteins could be regulated by protonation, either by extracellular or intracellular H+ (Hanke and Miller, 1983; Chen and Chen, 2001; Arreola et al., 2002; Traverso et al., 2006; Niemeyer et al., 2009; Sánchez-Rodríguez et al., 2012). Thus, both Cl− and H+ ions regulate Vm-dependent gating in CLC channels lacking intrinsic Vm sensors. Understanding how these ions contribute to the Vm gating mechanism will shed light onto the CLC channels’ physiology (Miller 2006; Sánchez-Rodríguez et al., 2012; Grieschat and Alekov, 2014).
The opening probability of CLC-2 increases as the membrane potential becomes hyperpolarized (de Santiago et al., 2005). This Vm dependence is not governed by the movement of voltage-sensitive domains; instead, the increased Vm coerces the intracellular Cl− ions into binding sites within the pore. Anions then move through the pore until they reach the protopore gate. We postulate that the gate is then opened by outgoing anions, which undergo an obligate Coulombic repulsive interaction with the gate (Sánchez-Rodríguez et al., 2010, 2012). We predict that pore occupancy by anions provides sufficient electrostatic repulsion for CLC-2 gate opening. This hypothesis agrees with recent observations showing that the glutamate gate is protonated only when Cl− binds to the permeation pathway of the CLC Cl−/H+ exchanger (Picollo et al., 2012). Alternatively, Vm-dependent protonation of the protopore gate by extracellular H+ might weaken the interaction between the gate and its binding site, facilitating opening (Niemeyer et al., 2009). This alternative mechanism requires that the CLC-2 protopore gate is located within the electrical field, as proposed for the CLC Cl−/H+ exchanger (Engh et al., 2007). Moreover, the Vm would need to open the gate at physiological proton concentrations, even when anions are absent from the pore. Therefore, we questioned whether Vm could gate CLC-2 channels: (a) in the presence of poorly permeant anions in the cytosolic side and (b) under low protonation probability conditions.
To answer these questions, we calculated the permeabilities of foreign anions and assessed whether CLC-2 could be activated in the presence of poorly permeant anions in the intracellular side. Among six ions with low permeability ratios (F−, glutamate, gluconate, methanesulfonate, acetate, and sulfate), cystolic-side F−, glutamate, gluconate, and methanesulfonate allowed activation of CLC-2 by hyperpolarization. We also found that CLC-2 could activate under conditions that disfavor protonation.
MATERIALS AND METHODS
Cell culture, transient expression, and electrophysiological recordings
Experiments were conducted on cultured HEK-293 cells transfected with mouse CLC-2 or mouse H538F CLC-2 cDNAs. Whole-cell Cl− currents (ICl(t)) were recorded as described previously (de Santiago et al., 2005; Sánchez-Rodríguez et al., 2010, 2012). ICl(t) was recorded using a control external solution containing (mM): 139 TEA-Cl, 0.5 CaCl2, 20 HEPES, and 100 d-mannitol. The internal solution contained (mM): 140 TEA-Cl, 20 HEPES, and 20 EGTA. The pH of each solution was adjusted to 7.3 with TEA-OH. In these solutions, HEPES was replaced with 20 mM MES or 20 mM CAPS when the pH of external solutions was adjusted to 5.5 (6.5) or 10, respectively. As an alternative internal solution, we also prepared a mixture of 25% SCN− and 75% Cl− containing (mM): 35 TEA-SCN, 105 TEACl, and 20 EGTA. The pH of this solution was adjusted to 10 by adding 30 mM CAPS. External and internal solutions with different foreign anions were prepared by substituting TEACl with the desired TEA-salt. The average tonicities of the external and internal solutions were 387.9 mosm/kg ± 1.9 mosm/kg and 347.3 mosm/kg ± 2.6 mosm/kg, respectively.
Xenopus laevis oocytes were isolated by partial ovariectomy of frogs anaesthetized with 0.1% (wt/vol) tricaine (3-aminobenzoic acid ethyl ester), as described previously (Chang et al., 2005). The surgery protocol complied with the Guide for Care and Use of Laboratory Animals (National Academy of Sciences, 1996). cRNA from rat CLC-2-Δ13-36 (provided by M. Pusch, Istituto di Biofisica, CNR, Genova, Italy) was obtained by in vitro transcription and injected (60–125 ng) into Xenopus oocytes. The cells were used 1–3 d after cRNA injection. Inside-out macropatches were excised by a patch pipette filled with a solution of (mM) 140 TEA-Cl, 40 glycine, and 5 MgCl2 (the MgCl2 was omitted in some excisions). The pH was adjusted to 10 with TEAOH. Because intracellular pH does not affect CLC-2 activation (Sánchez-Rodríguez et al., 2012), the pH of each internal solution was set to 7.3 for enhanced patch stability. The cytosolic side of each excised patch was exposed to internal solutions of different [Cl−] (16–500 mM TEA-Cl) in 20 mM EGTA and 20 mM HEPES. The pH was adjusted to 7.3 with TEAOH.
To record the macroscopic currents from HEK cells and excised macropatches, the cell or patch was maintained at 0 mV, and Vm was changed from 60 or 40 mV to −200 mV in 20-mV steps, and then restored to 60 mV unless otherwise indicated. All of the experiments were performed at ambient temperature (21–23°C). When investigating CLC-2 activation in the presence of internal Glu, acetate, SO42−, or methanesulfonate (MeSO3−), the leak and capacitive currents were subtracted online by a P/8 procedure (Bezanilla and Armstrong, 1977). In this case, the protocol P for generating whole-cell currents consisted of 40-ms hyperpolarizations followed by a 30-ms repolarization to 80 mV. The leak and capacitative currents were generated by a P/8 protocol of opposite polarity. The currents were collected by a pClamp 10 (Molecular Devices) and a sampling card that could record up to 500 kHz. Alternatively, the currents were sampled using a pClamp V8. To avoid electrode polarization in internal solutions lacking Cl−, the patch-clamp electrode inside the holder was embedded in a 3-M KCl/3% agar jacket (Shao and Feldman, 2007). The membrane and reversal potentials were corrected offline by the experimentally measured liquid junction potentials (Neher, 1992).
Analysis
The Vm-dependent activation was determined from normalized conductance Gnorm versus Vm curves (Gnorm(Vm)) as follows: the conductance (G) at each Vm was calculated from the whole-cell current magnitudes (ICl(t)) as G = ICl/(Vm − Vr), where Vr is the reversal potential. The maximum conductance (Gmax) was then estimated by fitting the G versus Vm curves to the Boltzmann equation:
| (1) |
where z is the apparent charge, F is the Faraday constant, R is the gas constant, T is the temperature, and V0.5 is the Vm at which Gnorm = 1/2. The constructed Gnorm(Vm) curves directly reflect the Vm dependence of the apparent open probability (PA).
Instantaneous current–voltage plots were constructed from the magnitudes of the tail currents (Itail) recorded at different Vms. Fitting these plots to linear functions, we obtained the reversal potentials (Vr) under different ionic conditions. In turn, the Vrs were used to calculate the permeability ratio of each anion X relative to Cl− (i.e., PX/PCl). When the external Cl− was replaced by foreign anions, PX/PCl was calculated by Eq. 2, in which ΔVr is the reversal potential shift (ΔVr) induced by the anion X:
| (2) |
Alternatively, when intracellular Cl− was replaced by foreign anions, the permeability ratios were calculated as
| (3) |
The figures and curve fittings were generated by Origin (OriginLab). The experimental data were plotted as the mean ± SEM of the number of independent experiments (n). The dashed black lines in each figure indicate ICl(t) = 0. Where necessary, the significant differences between datasets were evaluated by the paired Student’s t test (with significance defined at the P < 0.05 level).
RESULTS
Intracellular permeant anions facilitate voltage gating
To investigate the contributions of intracellular and extracellular anions to CLC-2 gating, we analyzed the Vm-dependent activation and permeability ratios of several anions. The ICl(t) values of HEK-293 cells expressing WT mouse CLC-2 were measured by conventional whole-cell voltage clamp in external solutions containing Cl−, SCN−, Br−, I−, F−, acetate, glutamate (Glu), gluconate, methanesulfonate, or sulfate (SO42−). The cells were first exposed to an external control solution containing 140 mM Cl− on both sides of the membrane. All of the external Cl− was then replaced by a test anion X−. Alternatively, anions were introduced to the intracellular side at 100% (Cl−, SCN−, I−, and Br−) or 75% (acetate, F−, Glu, gluconate, methanesulfonate, and SO42−). Fig. 1 plots the measured whole-cell ICl(t) (left) and Itail(t) (right) in the presence of external Cl− (Fig. 1 A), external SCN− (Fig. 1 B), and internal Br− (Fig. 1 C). In the presence of external Cl−, the current exhibits the characteristic kinetics of hyperpolarization-activated CLC-2 (de Santiago et al., 2005). The Itail current reverted at −5.4 ± 0.9 mV (n = 28). After replacing all of the external Cl− with SCN−, the onset of ICl(t) and Itail displayed normal kinetics, and ΔVr was −11.4 ± 3.6 mV (n = 5). When all of the intracellular Cl− was replaced by Br−, Vr shifted to 0.5 ± 2.0 mV (n = 12). Subsequently, the ΔVr value after substitution of external Cl− by another anion and the Vr values obtained with different intracellular anions were inserted to Eqs. 2 and 3 to calculate the permeability ratios (PX/PCl). The results are listed in Table 1. The permeability ratios yielded the following selectivity sequence of extracellular anions: SCN− > Cl− > Br− > I− > methanesulfonate = gluconate ≥ acetate = Glu = F− = SO42−. The selectivity sequence of intracellular ions was almost identical (although the permeability ratio of I− changed); namely, SCN− > Br− = Cl− > I− > methanesulfonate = gluconate = acetate = Glu = F− ≥ SO42−. Based on their permeability ratios, the anions were divided into two groups: highly permeant (SCN−, Br−, Cl−, and I−) and poorly permeant (acetate, methanesulfonate, gluconate, Glu, F−, and SO42−). To determine how each anion affects the Vm dependence of the activation, we calculated the normalized conductance Gnorm = G/Gmax (an index of the apparent open probability) at each Vm. Fig. 1 D plots Gnorm(Vm) for the extracellular permeant anions Cl−, SCN−, Br−, and I−. Relative to the Cl− activation curve, the Vm dependences in the presence of SCN− and I− are shifted toward negative voltages, whereas no effects caused by Br− are apparent. Previously, we reported that intracellular Cl− is crucial for gating (Sánchez-Rodríguez et al., 2010). Thus, we constructed the activation curves when highly permeant anions were present on the intracellular side (Fig. 1 E). Intracellular SCN− induced a positive shift in the activation curve relative to the Cl− curve. In contrast, I− shifted the activation curve in the negative direction, whereas Br− again induced no effect. Each curve in D and E of Fig. 1 was fitted to the Boltzmann equation (Eq. 1; black and reddish purple lines in Fig. 1). From these fits, we obtained the Vm values at which the activation is one half of its maximum (V0.5) and the apparent charge times the electrical distance (z × δ). Fig. 1 F plots V0.5 as a function of PX/PCl for permeant anions on the extracellular (closed circles) and intracellular (open circles) sides of the membrane. Whereas the V0.5 values of Cl−, Br−, and I− on the two sides of the membrane do not significantly vary, the V0.5 of SCN− is significantly shifted from approximately −60 mV on the intracellular side to approximately −112 mV on the extracellular side. The V0.5 values of highly permeant intracellular anions are linear functions of their permeability ratios, indicating that highly permeable anions facilitate voltage gating.
Figure 1.

Asymmetric regulation of CLC-2 voltage-dependent activation by permeant anions. (A–C) Examples of whole-cell ICl(t) (left) and Itail(t) (right) recorded in the presence of [Cl−]i = [Cl−]e = 140 mM (A), [Cl−]i = [SCN−]e = 140 mM (B), and [Br−]i = [Cl−]e = 140 mM (C). pHe = pHi 7.3. Cells were held at 0 mV. ICl(t) was recorded between −200 and 40 mV in 20-mV increments and repolarized to 60 mV; Itail(t) was recorded between −60 and 60 mV in 20-mV increments after pulse activation of −120 mV. (D) Gnorm(Vm) curves obtained from cells bathed in solutions containing 140 mM of permeant anions Cl−, SCN−, Br−, or I−. V0.5 and z values yielded by data fits to a Boltzmann equation (continuous lines) are Cl− (squares), −79.9 ± 6.6 mV and −0.94 ± 0.01 (n = 28); SCN− (circles), −111.3 ± 3.8 mV and −1.05 ± 0.06 (n = 5); Br− (upright triangles), −94.3 ± 8.2 mV and −0.98 ± 0.02 (n = 5); I− (inverted triangles), −103 ± 2.3 mV and −0.92 ± 0.01 (n = 5). [Cl−]i = 140 mM, and pHe = pHi 7.3. (E) Gnorm(Vm) curves obtained from cells dialyzed with solutions containing permeant anions Cl−, SCN−, Br−, or I−. Data were fitted as described for B, yielding the following V0.5 and z values: Cl− (squares), −79.9 ± 6.6 mV and −0.94 ± 0.01 (n = 28); SCN− (circles), −59.8 ± 4.1 mV and −0.70 ± 0.03 (n = 6); Br− (upright triangles), −78 ± 2 mV and −0.58 ± 0.05 (n = 12); I− (inverted triangles), −99.4 ± 7.0 mV and −0.68 ± 0.01 (n = 4). [Cl−]e = 140 mM, and pHe = pHi 7.3. (F) V0.5 versus PX/PCl relationship in Vm-dependent activation of CLC-2 in the presence of extracellular and intracellular permeant anions (black and reddish purple symbols, respectively). Continuous line is regression line. Error bars represent mean ± SEM.
Table 1.
Anion selectivity sequence of CLC-2-based on reversal potentials
| Anion | Extracellular | Intracellular | ||||
| ΔVr | Px/PCl | n | Vr | Px/PCl | n | |
| mV | mV | |||||
| Cl− | 0 | 1 | 28 | −5.4 ± 0.9 | 1 | 28 |
| SCN− | −11.4 ± 3.6 | 1.62 ± 0.22 | 5 | 15 ± 0.84 | 1.84 ± 0.06 | 6 |
| Br− | 5.0 ± 0.71 | 0.82 ± 0.02 | 5 | 0.5 ± 2.0 | 1.05 ± 0.08 | 12 |
| I− | 37.6 ± 1.7 | 0.23 ± 0.01 | 5 | −20.5 ± 2.4 | 0.46 ± 0.04 | 4 |
| F− | 36.00 ± 0.63 | 0.00 ± 0.01 | 5 | −34.65 ± 1.97 | 0.01 ± 0.02 | 5 |
| Glu− | 34.53 ± 1.19 | 0.01 ± 0.01 | 5 | −32.80 ± 0.17 | 0.03 ± 0.00 | 4 |
| Methanesulfonate | 31.17 ± 1.11 | 0.06 ± 0.01 | 5 | −31.10 ± 1.16 | 0.06 ± 0.02 | 6 |
| Gluconate | 30.55 ± 0.36 | 0.06 ± 0.01 | 4 | −32.05 ± 1.42 | 0.05 ± 0.02 | 6 |
| Ace− | 33.11 ± 0.92 | 0.03 ± 0.01 | 4 | −32.40 ± 0.77 | 0.04 ± 0.01 | 6 |
| SO42− | 35.89 ± 0.59 | 0.00 ± 0.00 | 3 | −36.06 ± 1.02 | 0.00 ± 0.01 | 6 |
| SCN− > Cl− > Br− > I− > methanesulfonate = gluconate ≥ Ace− = Glu− = F− = SO42− | SCN− > Br− = Cl− > I− > methanesulfonate = gluconate = Ace− = Glu− = F− ≥ SO42− | |||||
Reversal potentials in the presence of poorly permeant anions were determined in solutions containing 25% Cl− plus 75% of targeted poorly permeant anion.
Poorly permeant anions support voltage gating
Previously, we showed that V0.5 decreases with increasing anion permeability (Fig. 1 F). This result agrees with our hypothesis that pore occupancy by intracellular anions is sufficient to activate CLC-2 (Sánchez-Rodríguez et al., 2010). We speculate that under our experimental conditions, hyperpolarizing voltages, which drive the permeant anions into each pore, facilitate channel activation. In this scenario, an anion moving toward the extracellular side pushes the glutamate gate by steric and electrostatic repulsion. What about poorly permeant anions? Although there is limited structural information on how poorly permeant anions (such as F−, Glu, gluconate, methanesulfonate, and acetate) can occupy the pore of CLC channels, x-ray data show that Glu can occupy the anion pathway of the E148A mutant CLC Cl−/H+ exchanger and support valinomycin-induced H+ transfer. Gluconate could also occupy the pore because it can induce H+ transfer in the E148A mutant CLC Cl−/H+ exchanger (Feng et al., 2012). However, in the presence of Glu or gluconate, the WT CLC Cl−/H+ exchanger does not function (Feng et al., 2012). F− can also occupy the anion pathways of both WT and E148A mutant CLC Cl−/H+ exchangers (Lim et al., 2013), but this poorly permeant anion hinders exchanger activity. These data suggest that poorly permeant anions Glu, gluconate, and F− could occupy the pores of CLC proteins including the CLC-2 pore, which is 86% similar to the anion pathway of the CLC Cl−/H+ exchanger (Nieto-Delgado et al., 2013). Thus, we asked, can the mere presence of poorly permeant anions in the pore unlock the channel’s conductive path? To answer our question, we recorded the whole-cell ICl(t) variations in cells dialyzed with solutions (pH 7.3) containing acetate, methanesulfonate, gluconate, Glu, F−, or SO42− (all at 140 mM) in the absence of Cl−. The extracellular solution contained 140 mM Cl− (pH 7.3). Vm was stepped from 40 to −200 mV, and the cells were stimulated for 40 ms at each step. This procedure was followed by repolarization to 80 mV. We reasoned that under these ionic conditions, hyperpolarization would produce no negative current because the anions were poorly permeant; however, if the CLC-2 was opened during the hyperpolarization, repolarization to 80 mV would generate a positive current. Furthermore, if the positive current is generated by influx of external Cl− anions, it should be absent when Cl− is replaced by a poorly permeant anion. Fig. 2 presents the whole-cell currents recorded from six different cells bathed in 140 mM Cl− and dialyzed with solutions containing Cl− (A), Glu (B), methanesulfonate or F− (C), acetate (E), and SO42− (F). As expected, the cells dialyzed with Glu, methanesulfonate, acetate, or SO42− are hyperpolarized and exhibit no negative currents. Surprisingly, large positive tail currents were readily recorded at 80 mV in cells dialyzed with Glu (B) and methanesulfonate or F− (C), but not in cells dialyzed with acetate (E) or SO42− (F). With intracellular F−, we observed more “leak” current than with the other anions. The tail current vanishes when the external Cl− is replaced with Glu (Fig. 2 B, inset), confirming its generation by Cl− influx. Thus, our data indicate that the gate opens during hyperpolarization while the poorly permeant anions Glu, methanesulfonate, or F− reside at the intracellular side. The tail currents observed in cells dialyzed with Glu and hyperpolarized to −160 mV display bi-exponential time courses with time constants of 1.52 ± 0.06 ms and 11.87 ± 0.66 ms (n = 6). Similar time constants were obtained at −180 and −200 mV. CLC-2 was also activated in cells dialyzed with gluconate (not depicted). Fig. 2 D summarizes the Vm-dependent activation in cells dialyzed with Cl−, Glu, methanesulfonate, F−, and gluconate. In the presence of poorly permeant anions, the channel began opening at approximately −60 to −80 mV, whereas in the presence of Cl−, opening started at 0 mV. Furthermore, no saturation was achieved in glutamate, gluconate, or methanesulfonate at the most negative tested voltage (−200 mV). The estimated V0.5 values under different ionic conditions, determined by fitting the activation curves to a Boltzmann function, were (mV) −80 (Cl−), −181 (Glu), −178 (methanesulfonate), −146 (F−), and −189 (gluconate). These values indicate that opening the gate in the presence of poorly permeant anions is more difficult than in the presence of Cl−. If these anions and Cl− travel similar electrical distances from the intracellular side to the glutamate gate, the slopes of their voltage activation curves should be similar. Supporting this assumption, the activation curves are nearly superimposed (Fig. 2 D, inset). From these activation curves, the products of the apparent charges and electrical distances were calculated to be −0.9 (Cl−), −1.4 (Glu), −1.2 (methanesulfonate), −0.96 (F−), and −1.2 (gluconate). We emphasize that these values are rough estimates because the Gnorm versus Vm curves fail to saturate at the most negative Vm (−200 mV) in the presence of glutamate, gluconate, or methanesulfonate. No negative or positive current was recorded in cells dialyzed with acetate or SO42− (Fig. 2, E and F), indicating that CLC-2 was not activated with these ions.
Figure 2.

Voltage-dependent activation of CLC-2 in the presence of intracellular, poorly permeant anions. (A–C and E and F) Whole-cell currents recorded from five cells dialyzed with solutions containing 140 mM Cl− (A), 140 mM Glu (B), 140 mM methanesulfonate or 140 mM F− (C), 140 mM acetate (E), and 140 mM sulfate (F). Cells were bathed in solution containing 140 mM Cl− (pHe = pHi 7.3). B (inset) plots data obtained from the same cell dialyzed with internal solution containing 140 mM Cl− and sequentially bathed in 140 mM Cl− (reddish purple) and 140 mM Glu (black). ICl(t) was recorded between −200 and 40 mV in 20-mV increments, and Itail(t) was recorded at 80 mV. (D) Gnorm(Vm) curves obtained from cells dialyzed with solutions containing poorly permeant anions F−, Glu, gluconate (Gluc), and methanesulfonate (MeSO3−). Continuous lines are fits to Boltzmann equation, yielding the following V0.5 and z values: Cl− (black squares), 79.9 ± 6.6 mV and −0.94 ± 0.01 (n = 28); F− (vermilion triangles), −146.4 ± 3.3 mV and −0.96 ± 0.08 (n = 5); Glu (reddish purple triangles), −180.7 ± 2.3 mV and −1.32 ± 0.02 (n = 6); gluconate (sky blue circles), −189.6 ± 2.8 mV and −1.17 ± 0.05 (n = 8); methanesulfonate (blue triangles), −177.7 ± 3.6 mV and −1.16 ± 0.03 (n = 8). (Inset) Superposition of voltage–activation curves after subtracting corresponding V0.5 values (listed above) under each ionic condition. Error bars represent mean ± SEM.
The observed current in the presence of poorly permeant anions implies that these anions can occupy the CLC-2 pore and open the glutamate gate. Accordingly, we presume that acetate and SO42− cannot open the gate, either because they do not enter the pore or because they bind tightly to a site far from the gate. To partially test this idea, we prepared internal solutions containing chloride (60 mM Cl−) or the following mixtures: 60 mM Cl− plus 80 mM Glu, 60 mM Cl− plus 80 mM acetate, 60 mM Cl− plus 140 mM acetate, or 60 mM Cl− plus 80 mM SO42−. We then recorded the whole-cell currents. As before, the external solution contained 140 mM Cl−. Fig. 3 presents the currents recorded in three cells under different dialysis conditions: 60 mM Cl− (A), 60 mM Cl− plus 80 mM SO42− (B), and 60 mM Cl− plus 80 mM glutamate (C). Remarkably, the ICl(t) kinetics are identical in each cell. Fig. 3 D shows scaled ICl(t) traces from cells dialyzed in Cl− (black), Cl− plus SO2−4 (bluish green), or Cl− plus glutamate (reddish purple). Note that dialysis in Cl− plus glutamate reduces the tail current amplitude. Fig. 3 E summarizes the CLC-2 activation curves obtained in 60 mM Cl− (black), 60 mM Cl− plus 80 mM SO42− (bluish green), and 60 mM Cl− plus 80 mM Glu (reddish purple). In this case, the Vm-dependent activation in the presence of intracellular Cl− plus SO42− is identical to that obtained in the presence of Cl− alone, suggesting that SO42− does not bind in the pore and that Cl− alone opens the gate. In contrast, in the presence of Cl− plus glutamate, the activation is shifted by −13.5 mV relative to the Cl−-only activation curve. This result indicates that Glu also occupies the pore and, together with Cl−, alters the Vm dependence of the channel. Unexpectedly, Cl− plus acetate also altered the CLC-2 activation. Fig. 4 A shows a family of ICl(t) curves obtained from a cell dialyzed with 60 mM Cl− plus 80 mM acetate (pHi 7.3). The green triangles in Fig. 4 D indicate the resulting activation curve. Relative to the activation curve obtained in Cl− alone (continuous black line, from Fig. 3 E), the Cl− plus acetate mixture activation curve is shifted by nearly 60 mV. This result suggests that Cl− and acetate both occupy the pore and that multi-ion occupancy facilitates pore opening. To test the idea that shifting the activation curve requires both Cl− and acetate, we repeated the experiment at pHi 4.2. Under this condition, and considering the pK of acetic acid (4.76), the fraction of free acetate anions decreases to 0.2. Therefore, the activation curve should be almost superimposed on that obtained with Cl− alone. B and C of Fig. 4 plot the ICl(t) traces obtained from cells dialyzed with 60 mM Cl− (pHi 4.2) and 60 mM Cl− plus 80 mM acetate (pHi 4.2), respectively. The currents display noisy behavior with similar kinetics. Indeed, at pHi 4.2, the activation curves are identical in cells dialyzed with Cl− and with Cl− plus acetate (compare reddish purple and blue circles in Fig. 4 D). Surprisingly, at pHi 7.3, the same curves were obtained in 60 mM of intracellular Cl−, indicating that CLC-2 gating is insensitive to pHi (Sánchez-Rodríguez et al., 2010). Collectively, our data show that poorly permeant anions such as Glu, F−, methanesulfonate, and gluconate can open the channel without passing through the pore, whereas acetate and SO2−4 alone cannot interact with the pore.
Figure 3.

Voltage-dependent activation was altered by chloride plus glutamate but not by chloride plus sulfate mixtures. Whole-cell recordings from cells bathed in solution containing [Cl−]e = 140 mM (pHe = pHi 7.3) and dialyzed with 60 mM [Cl−]i (A), 60 mM [Cl−]i plus 80 mM [SO2−4]i (B), and 60 mM [Cl−]i plus 80 mM [Glu]i (C). (D) Comparison of scaled ICl(t) recorded at −140 mV in the presence of Cl− (black), Cl− plus SO2−4 (bluish green), and Cl− plus Glu (reddish purple). (E) Vm dependence of activation obtained from cells dialyzed with 60 mM Cl− (black diamonds), 60 mM Cl− plus 80 mM SO2−4 (bluish green triangles), and 60 mM Cl− plus 80 mM Glu (reddish purple triangles). Continuous lines are Boltzmann fits yielding the following V0.5 and z values: black diamonds, −116.4 ± 2.0 mV and −0.84 ± 0.01 (n = 7); bluish green triangles, −112.5 ± 1.3 mV and −0.91 ± 0.04 (n = 7); reddish purple triangles, −129.9 ± 1.6 mV and −0.78 ± 0.03 (n = 5). Error bars represent mean ± SEM.
Figure 4.

Voltage-dependent activation was altered by chloride plus acetate in a pHi-dependent manner. (A–C) Whole-cell recordings from cells bathed in solution containing [Cl−]e = 140 mM and pHe 7.3 and dialyzed with 60 mM [Cl−]i plus 80 mM [Ace]i with pHi 7.3 (A), 60 mM [Cl−]i with pHi 4.2 (B), and 60 mM [Cl−]i plus 80 mM [Ace]i with pHi 4.2 (C). (D) Vm dependence of activation obtained from cells dialyzed with: bluish green triangles, 60 mM Cl− plus 80 mM acetate, pHi 7.3; blue circles, 60 mM Cl− plus 80 mM acetate, pHi 4.2; reddish purple circles, 60 mM Cl−, pHi 4.2. Continuous black line is corresponding voltage activation curve obtained from cells dialyzed with 60 mM Cl−, pHi 7.3, shown in Fig. 3 E. In all cases, [Cl−]e = 140 mM and pHe 7.3. Continuous lines are Boltzmann fits yielding the following V0.5 and z values: bluish green triangles, −61.8 ± 1.3 mV and −0.86 ± 0.04 (n = 6); blue circles, −112.4 ± 4.1 mV and −0.83 ± 0.05 (n = 5); reddish purple circles, −121.3 ± 1.6 mV and −0.80 ± 0.04 (n = 8). Error bars represent mean ± SEM.
Gate protonation is not crucial for CLC-2 activation
One postulated mechanism of voltage gating in CLC-2 is Vm-dependent protonation of the glutamate gate (Niemeyer et al., 2009). To assess the role of protonation in CLC-2 gating, we performed whole-cell recordings in cells exposed to external solutions at various pH. Here, we ranged the pHe from 6.5 to 10, in which GNorm decreases from its maximum to its minimum, corresponding to the maximum and minimum probability of glutamate gate protonation, respectively (Niemeyer et al., 2009; Sánchez-Rodríguez et al., 2012). Because acetate alone does not open CLC-2 channels, intracellular acetate should induce gating only if the pHe is acidic because the channel is more likely to be protonated. Gating would indicate that the protopore gate opens when protonated, without requiring anions. However, as shown in Fig. 5 A, CLC-2 was not activated by hyperpolarization at pHe 7.3 (black) and 6.5 (bluish green). The internal solution contained 140 mM acetate and its pHi was 6.5. In the presence of intracellular 75% Cl− plus 25% SCN and 140 mM Cl− in the extracellular bath (Fig. 5 B), the CLC-2 was activated under low pronation probability conditions (pHe = pHi 10). Here, the [H+]e of the cellular environment decreases from 10−7.3 M (black) to 10−10 M (sky blue). Although this [H+]e change does not eliminate ICl(t), ICl(t) does decrease to ∼15% of the control value. These results were contrary to our expectations, possibly because Vm-dependent protonation of the glutamate gate in cells dialyzed with acetate was not taking place at pHe 6.5. Therefore, we further decreased the external pH to 5.5. However, at this pH, the activity of WT CLC-2 almost vanishes because residue H538 becomes protonated (Niemeyer et al., 2009). To circumvent this problem, we investigated H538F mutant channels. Fig. 5 C plots the ICl(t) at −120 mV recorded from a cell that was sequentially exposed to pH 7.3 (black) and pH 5.5 (reddish purple). In the H538F mutant, the currents are larger at pHe 5.5 than at pHe 7.3, although the closing rate is slower at pH 5.5. This later result agrees with our previous observation (Sánchez-Rodríguez et al., 2012). The average time constants of the Itail decay are 5.5 ± 0.2 ms and 4.5 ± 0.2 ms at pH 5.5 and 7.3, respectively, indicating stabilized open states at low extracellular pH. As shown in Fig. 5 D, decreasing the external pH increases the magnitude of the apparent open probability by ∼1.7 times. The V0.5 and z values obtained from the activation curves are −138 mV and −0.7, respectively, at pH 7.3 and −98 mV and −0.7, respectively, at pH 5.5. Thus, mutating residue H538 abolished the inhibitory effect at pH 5.5, allowing for almost full channel activation. These data are identical to the findings of Niemeyer et al. (2009). If the mutant channels are open after protonation of the glutamate gate, they must be activated at pHe 5.5, even when poorly permeant anions and Cl− are present in the intracellular and external solutions, respectively. Thus, we recorded the ICl(t) in cells dialyzed with 140 mM acetate. Fig. 5 E plots the results at pHe 7.3 (black) and pHe 5.5 (reddish purple). No currents are evident at either pH, indicating that the H538F mutant cannot be gated in the presence of intracellular acetate even if the glutamate gate is protonated. In contrast, the H538F mutant was activated in Glu-containing internal solution (pHi 7.3) and Cl−-containing (140 mM) external solution at pH 7.3 (Fig. 5 F). Under these conditions, the mutant behaves identically to the WT channel. Collectively, our results demonstrate that conditions favoring protonation of the CLC-2 glutamate gate are insufficient to open the channel but that hyperpolarization can gate the channel under unfavorable protonation conditions.
Figure 5.

Voltage activation of WT and H538F mutant CLC-2 channels does not occur with intracellular acetate and acidic external pH. (A) Representative whole-cell recordings at −120 mV from a single cell expressing WT CLC-2. The cell was dialyzed with internal solution containing 140 mM acetate, pHi 6.5, and bathed in solution containing 140 mM Cl−. The external pH was 7.3 (black) or 6.5 (bluish green). n = 5 samples. (B) Representative traces showing activation of CLC-2 in cells dialyzed with 75% Cl− and 25% SCN−, pHi 10. Cells were hyperpolarized to −120 mV in recording solution with pHe 7.3 (black); pHe was thereafter increased to 10 (sky blue). n = 4 samples. (C) ICl(t) recorded at −120 mV from an HEK-293 cell expressing mutant H538F channels. The cell was sequentially exposed to external solutions of pH 7.3 (black) and pH 5.5 (reddish purple). [Cl−]i = [Cl−]e = 140 mM. Average time constants of tail current decay at pH 5.5 and 7.3 are 4.6 ± 0.2 ms and 3.7 ± 0.3 ms, respectively (n = 5). (D) Vm-dependent activation curves in H538F mutant channels recorded at pH 7.3 (black) and pH 5.5 (reddish purple). Continuous lines are Boltzmann fits yielding the following V0.5 and z values: −138.2 ± 4.0 mV and −0.75 ± 0.02 (pHe 7.3); −98.0 ± 8.0 mV and −0.75 ± 0.02 (pHe 5.5; n = 5). (E) ICl(t) recorded at −120 mV from a cell expressing mutant H538F channels and sequentially exposed to external solutions of pH 7.3 (black) and pH 5.5 (reddish purple). Cell was dialyzed with 140 mM acetate, pHi 7.3, and bathed in solution containing 140 mM Cl− at pHe 7.3 or 5.5 (n = 6). (F) Activation of H538F channels in cells dialyzed with 140 mM [Glu]i and bathed in 140 mM [Cl]e. pHi = pHe 7.3 (n = 3). Error bars represent mean ± SEM.
Gating of CLC-2 requires intracellular Cl−
According to our hypothesis, the relevant intracellular physiological anion Cl− should gate CLC-2. To ensure that increasing [Cl−]i is sufficient to open the channels, we applied solutions containing different [Cl−] to inside-out patches. The extracellular side of each patch was exposed to a solution containing 140 mM Cl− at pH 10.0 (patches exposed to higher pH solutions did not survive). Because the currents in the patches excised from HEK cells were below the recording limit, we switched to Xenopus oocytes expressing a rat CLC-2 clone lacking residues 13–36 (Gründer et al., 1992). To improve patch survival, we set the pHi to 7.3. Previously, we reported that changing the pHi from 4 to 9 exerts no effect on CLC-2 activation (Sánchez-Rodríguez et al., 2012; see also Fig. 4 D). Fig. 6 A plots representative recordings from an excised patch sequentially exposed to intracellular Cl− concentrations of 16.3 mM (black) and 140 mM (bluish green). At low intracellular [Cl−], negligible or no current is apparent between −200 and 40 mV. However, after increasing [Cl−]i to 140 mM, the currents are inwardly rectifying, indicating that intracellular Cl− is indeed required for Vm gating. At negative Vm, the currents exhibit slow activation and tail currents, as observed in whole-cell recordings of HEK cells expressing mouse CLC-2 (Sánchez-Rodríguez et al., 2012). Such responses were absent in patches excised from un-injected oocytes (not depicted). The macroscopic current amplitudes at [Cl−]is ranging from 16.3 to 500 mM (pHe 10) were converted to conductance values, which were then normalized by the conductance measured at [Cl−]i = 500 mM. As shown in Fig. 6 B, the normalized conductance at −170 mV increases with increasing intracellular Cl−, as expected for a ligand binding–dependent phenomenon. We conclude that Vm gating requires internal Cl− and occurs under unfavorable gate protonation conditions.
Figure 6.

Activation of CLC-2 by hyperpolarization in excised patches exposed to increasing [Cl−]i at extracellular pH 10. (A) Macroscopic ICl(t) recorded from an inside-out patch excised from an oocyte expressing rCLC-2-Δ13-36. The intracellular side of the patch was exposed first to 16.3 mM [Cl−]i (black) and then to 140 mM (bluish green). [Cl−]e was 140 mM, and pHe and pHi were set to 10 and 7.3, respectively. (B) [Cl−]i dependence of CLC-2 activation. ICl(t) was measured at −170 mV in solution with pHe 10 and converted to macroscopic conductance (G). At each [Cl−]i, G is normalized by G obtained in 500 mM [Cl−]i (n = 5). Error bars represent mean ± SEM.
DISCUSSION
In this paper, we demonstrated that pore occupancy is responsible for Vm-dependent gating in the CLC-2 channel. To this end, we exposed cells to highly permeant (Cl−, Br−, I−, and SCN−) and poorly permeant anions (acetate, F−, Glu, gluconate, methanesulfonate, and SO42−). Vm gating occurred in cells dialyzed with highly permeant anions and with the poorly permeant anions F−, Glu, gluconate, and methanesulfonate, but was absent in cells dialyzed with acetate or SO42−, even when the external pH was lowered to 5.5, thereby promoting protonation of the glutamate gate. In contrast, Vm-dependent gating occurred at high [Cl−]i and an extracellular pHe of 10, at which gate protonation is unlikely.
Mechanism of gating by pore occupancy
To explain Vm activation by pore occupation, we proposed a model that is focused on the effect of intracellular anions on CLC-2 gating, ignoring the effect of external H+. Protonation is dispensable because it merely stabilizes the open state once the glutamate gate has opened (Sánchez-Rodríguez et al., 2012). Our proposed mechanism, in which pore occupancy by permeant and some nonpermeant anions opens the glutamate gate, is illustrated in Scheme 1. The mechanism proceeds as follows. At positive Vm, the pore remains empty and the glutamate gate (reddish purple) is bound to the pore in the closed conformation C. At negative Vm, the intracellular anions (bluish green circles) are pushed into the pore (CCl conformation). Once inside the pore, the intracellular anions undergo Coulombic interactions with the glutamate gate, initiating gate movement. This mechanism might explain the Vm-dependent gating phenomenon in CLC-2. Under physiological conditions, intracellular Cl− anions will occupy the pore and open the gate. Once the gate is open (OCl conformation), the anions may or may not go through. If the intracellular test anion is allowed to exit through the CLC-2 pore, hyperpolarization followed by positive repolarization will generate negative and positive currents, respectively, provided that Cl− is present in the bath solution. This scenario is consistent with our experiments, in which [Cl−]i increased and activated CLC-2 in inside-out patches. In contrast, if the test anion can occupy the pore from the intracellular side but is forbidden from exiting through the pore, the hyperpolarization produces no current. However, if extracellular Cl− is present in the bath solution, positive repolarization produces a positive current (tail current) only when the gate is opened during hyperpolarization. This situation was experimentally verified in the presence of intracellular F−, Glu, gluconate, and methanesulfonate. Finally, the channel returns to its initial condition when the glutamate gate reverts to the closed conformation under positive Vm. In this mechanism, regardless of their permeability ratios, anions must occupy the pore; otherwise, they cannot interact with the glutamate gate and open the channel. Highly permeable anions (Cl−, Br−, SCN−, and I−) can open the channel because they can enter the pore and pass through the entire permeation pathway. The data showing Vm-dependent activation of CLC-2 in excised patches exposed to increasing intracellular Cl− while the external pH was set to 10 agrees with this idea. However, poorly permeant anions (F−, Glu, gluconate, and methanesulfonate) are clearly prohibited from exiting the pore. Thus, if pore occupancy is the underlying gating mechanism, these anions must occupy the pore to initiate Vm gating. Supporting this hypothesis, our data comparing the voltage-dependent activation in the presence of intracellular Cl− alone, SO42− alone, or intracellular Cl− plus SO42− imply that SO42− cannot occupy the pore because SO42− has low permeability, no Vm gating was observed with this anion alone, and the Vm-dependent gating was identical in cells dialyzed with 60 mM Cl− and 60 mM Cl− plus 80 mM SO42−. Also F− occupies the anion pathway in the CLC Cl−/H+ exchanger (Lim et al., 2013). Furthermore, Glu, and presumably also gluconate, occupies the E148A CLC Cl−/H+ exchanger anion pathway (Feng et al., 2012). Finally, our data indicate that F−, Glu, gluconate, and methanesulfonate sensed the same electrical field. This result is expected if all anions interact with a gate located at a fixed distance. Thus, anions excluded from the pore such as SO42− cannot participate in gating. In contrast, F−, Glu, and gluconate, and possibly methanesulfonate, occupy the CLC-2 pore despite their low permeability and participate in gating. Although permeation is not necessary for gating, pore occupation must be Vm dependent. Such a constraint accounts for the voltage gating induced in the presence of poorly permeant anions.
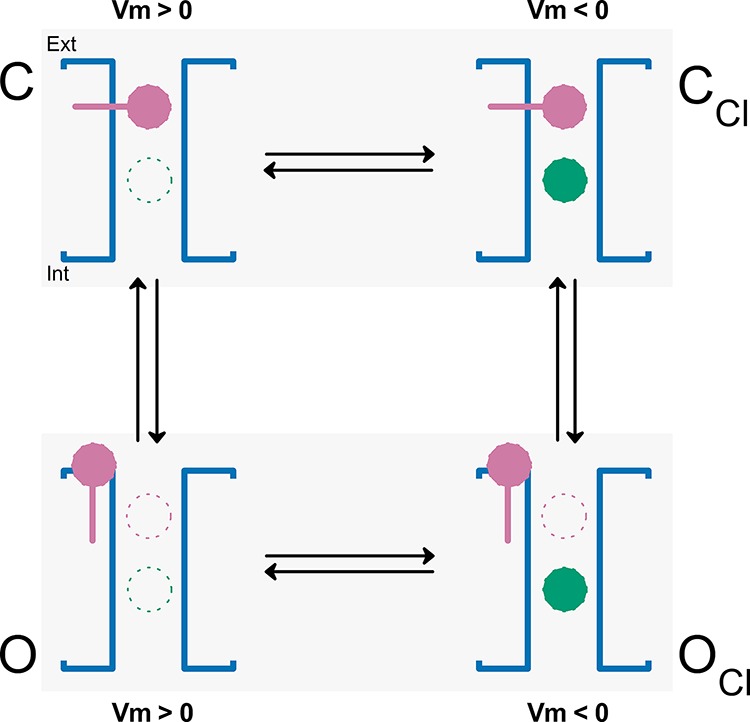 |
(Scheme 1) |
Multi-ion occupancy of the pore is important for voltage gating in CLC-2
Previously, we investigated Cl−/SCN− mixtures in the intracellular solution and reported that multi-ion occupancy is important for Vm-dependent gating in CLC-2 (Sánchez-Rodríguez et al., 2010). In the present experiments, mixed Cl− plus Glu shifted the activation curve to values more negative than those obtained in the presence of Cl− alone. Conversely, in cells dialyzed with acetate plus Cl− at pHi 7.3, the activation curve shifted to the right, as though the channel had become more sensitive to voltage. These two independent datasets support the importance of multi-ion occupancy in CLC-2 gating; both anions must reside in the pore to alter activation. This conclusion was verified by reducing the pHi from 7.3 to 4.2, at which the free acetate is drastically reduced. Under this condition, the changes observed in activation in the presence of Cl− plus acetate at pHi 7.3 were lost. Therefore, at pHi 7.3, the pore is occupied by both acetate and Cl−, whereas at pHi 4.2, it is occupied by Cl− alone. The pore occupation by acetate and Cl− at pHi 7.3 is puzzling, as gating was absent in cells dialyzed with acetate alone at this pHi. We surmise that acetate occupies a site far from the gate and close to the intracellular side but is pushed further into the pore by outflowing Cl− ions. In this manner, multi-ion occupancy could induce changes in activation.
Pore occupancy as a Vm-gating mechanism in other CLC Cl− channels
Our data agree with a previous report in which the binding of two Cl− ions into the permeation pathway of the CLC Cl−/H+ exchanger proved necessary for protonation of the E148 gate (Picollo et al., 2012). That report raised the interesting proposition that pore occupancy could precede protonation in CLC proteins. However, we suspect that pore occupancy cannot be a universal gating mechanism for CLC channels, because CLC-2 fundamentally differs from CLC-0 and CLC-1. For example, intracellular protons are vital for voltage gating in CLC-0 and CLC-1, whereas voltage gating in CLC-2 requires intracellular anions. CLC-0 gating involves the transmembrane movement of protons, and its open probability is modulated by increments in [H+]i (Lísal and Maduke, 2008; Zifarelli et al., 2008). Cl− might allosterically control the protonation in CLC-0 and CLC-1 (Chen and Miller, 1996; Miller, 2006), whereas it is directly responsible for Vm gating of CLC-2. Nevertheless, we must emphasize that gating in both CLC-0 and CLC-1 depends on the external and internal Cl− (Pusch et al., 1995; Rychkov et al., 1996) and can be altered by foreign anions. Extracellular foreign anions cause anomalous mole-fraction behavior of the conductance, permeability, and gating in CLC-0 and CLC-1 (Pusch et al., 1995; Rychkov et al., 1998, 2001). In CLC-1, voltage gating occurs in the presence of both poorly permeant anions such as hydrophobic anions and “impermeant anions” such as glutamate, cyclamate, and methanesulfonate (with permeability ratios of 0.008, 0.007, and 0.008, respectively) (Rychkov et al., 1998, 2001). When present, glutamate, cyclamate, and methanesulfonate shift the activation curve of CLC-1 by 70, 28, and −45 mV, respectively, relative to the activation curve in the presence of Cl− alone (Rychkov et al., 1998). In CLC-0, Vm gating is induced by the permeant anions Br− and NO3−. Replacement of 92.3% of the external Cl− with Glu produces a rightward shift in the reversal potential, accompanied by a rightward shift (approximately −10 mV) in the activation curve and a full closed-to-open transition in the −150 to 100–mV range without changing the slope (Pusch et al., 1995). Based on their effects on CLC-1 gating, Rychkov et al. (1998) categorized anions into three groups: poorly permeant anions that cannot open the channel, anions that open the channel but cannot permeate, and anions that both open and permeate the channel. The present anions tested on CLC-2 can be divided similarly but not identically; the poorly permeant anions affected the CLC channels differently. For example, CLC-1 Vm gating is deactivated by Glu and gluconate, but both anions activate CLC-2; conversely, both CLC-1 and CLC-2 channels are opened by Vm in the presence of methanesulfonate. Although Rychkov et al. (1998) could not determine whether methanesulfonate supports Vm gating in CLC-1, they concluded that permeation and gating are intrinsically linked. If so, then pore occupancy will also be important for CLC-1 Vm gating.
We suspect that pore occupation by permeant species is more important for channel gating than previously thought and deserves further scrutiny. For example, a gating process coupled to ion permeation has been proposed for the KcsA K channel, in which the selectivity filter may also be the gate (VanDongen, 2004). Similar processes have been suggested for CFTR (Wright et al., 2004), GCAC1 in guard cells (Dietrich and Hedrich, 1998), the volume-sensitive Cl− channel (Hernández-Carballo et al., 2010), and the Ca2+-activated Cl− channels TMEM16A and B (Perez-Cornejo et al., 2004; Xiao et al., 2011; Betto et al., 2014). To clarify the roles of permeant species in channel gating, we require additional structural and functional data coupled with computational modeling.
Protonation plays a minor role in dislodging the glutamate gate
We theorized that protonation of the glutamate gate plays a significant role in stabilizing the opened conformation (Sánchez-Rodríguez et al., 2012) but not in triggering channel activation. According to theoretical calculations and experimental results, the estimated pK at which buried Glu residues protonate varies between 2 and 8 (Li et al., 2005). However, the CLC Cl−/H+ exchanger reportedly activates at pKs between 4.6 and 6.2 (Iyer et al., 2002; Picollo et al., 2012; Garcia-Celma et al., 2013). Likewise, the CLC-0 and CLC-2 chloride channels are activated at pKs of ∼6 and 5.9, respectively (Hanke and Miller, 1983; Niemeyer et al., 2009). We anticipate that the pK of the glutamate gate in CLC-2 is below 7 and that its protonation does not initiate gating. Assuming a protonation pK of 6.5 and that the gate is located at 50% (δ = 0.5) within the electrical field, the probability of Vm-dependent protonation can be calculated as:
| (4) |
where [H+]site is [H+] near the site. At −160 mV and [H+]0 of 10−10, 10−7.3, and 10−5.5 M (mimicking our experimental conditions), the protonation probability would be ∼0, 0.5, and 1, respectively. Therefore, if [H+]0 is maintained at 10−10 M, no current is expected under any ionic condition and at any membrane Vm. However, in our experiments conducted at this [H+]0, CLC-2 was opened by hyperpolarizations in excised patches exposed to increasing intracellular Cl−. Conversely, if [H+]0 is 10−7.3, 10−6.5, or 10−5.5 M, the glutamate gate will be protonated and the channel should be opened by hyperpolarization, regardless of the intracellular anion present. However, we observed no positive current in the presence of intracellular acetate or sulfate after strong hyperpolarization. From these estimates, and previous demonstrations that the closing rate of the CLC-2 protopore gate decreases with increasing [H+]e (Sánchez-Rodríguez et al., 2012), we propose that protonation most likely stabilizes the open conformation. In the E268A CLC-5 Cl−/H+ antiporter, transient capacitive currents are independent of both intracellular and extracellular H+ (Zifarelli et al., 2008), indicating that voltage gating occurs through a H+-independent mechanism. However, our data showing a lack of Vm gating in the presence of sulfate could be explained by an alternative mechanism. It has been shown that the glutamate gate in the CLC Cl−/H+ exchanger is unlikely to protonate in the absence of anions (Picollo et al., 2012). Therefore, protonation is prevented when the permeation pathway of CLC-2 is empty, which impedes gating. However, and by the same argument, it is feasible that under physiological conditions, intracellular Cl− is needed first to open the pore and then protonate the gate. Thus, the protonation reaction will stabilize the open conformation.
In conclusion, we propose that voltage gating of the CLC-2 Cl− channel is triggered by pore occupancy, leading to electrostatic interactions between the anions and the pore gate, and culminating in pore opening and ion conduction.
Acknowledgments
The authors thank Drs. Ted Begenisich and Patricia Perez-Cornejo for critical comments to this work.
This work was supported by grants from The National Council for Science and Technology (CONACyT), Mexico (no. 219949) and Fondo de Apoyo a la Investigación–Universidad Autónoma de San Luis Potosí to J. Arreola, and from Ministry of Science and Technology (grant NSC102-2320-B-001-004; Taiwan, ROC) to R.-C. Shieh. J.J. De Jesús-Pérez and A. Castro-Chong are recipients of Graduate Student Fellowships from CONACyT, Mexico (nos. 234820 and 335900, respectively).
The authors declare no competing financial interests.
Author contributions: J.J. De Jesús-Pérez designed research, performed research, and analyzed data. A. Castro-Chong designed research, performed research, and analyzed data. C.Y. Hernández-Carballo performed research. R.-C. Shieh designed research, analyzed data, and wrote the paper. J.A. De Santiago-Castillo analyzed data and modeling. J. Arreola designed research, analyzed data, and wrote the paper.
Merritt C. Maduke served as editor.
References
- Arreola J., Begenisich T., and Melvin J.E.. 2002. Conformation-dependent regulation of inward rectifier chloride channel gating by extracellular protons. J. Physiol. 541:103–112. 10.1113/jphysiol.2002.016485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betto G., Cherian O.L., Pifferi S., Cenedese V., Boccaccio A., and Menini A.. 2014. Interactions between permeation and gating in the TMEM16B/anoctamin2 calcium-activated chloride channel. J. Gen. Physiol. 143:703–718. 10.1085/jgp.201411182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F., and Armstrong C.M.. 1977. Inactivation of the sodium channel. I. Sodium current experiments. J. Gen. Physiol. 70:549–566. 10.1085/jgp.70.5.549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanz J., Schweizer M., Auberson M., Maier H., Muenscher A., Hübner C.A., and Jentsch T.J.. 2007. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J. Neurosci. 27:6581–6589. 10.1523/JNEUROSCI.0338-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bösl M.R., Stein V., Hübner C., Zdebik A.A., Jordt S.E., Mukhopadhyay A.K., Davidoff M.S., Holstein A.F., and Jentsch T.J.. 2001. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl− channel disruption. EMBO J. 20:1289–1299. 10.1093/emboj/20.6.1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalán M.A., Flores C.A., González-Begne M., Zhang Y., Sepúlveda F.V., and Melvin J.E.. 2012. Severe defects in absorptive ion transport in distal colons of mice that lack ClC-2 channels. Gastroenterology. 142:346–354. 10.1053/j.gastro.2011.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.K., Yeh S.H., and Shieh R.C.. 2005. A ring of negative charges in the intracellular vestibule of Kir2.1 channel modulates K+ permeation. Biophys. J. 88:243–254. 10.1529/biophysj.104.052217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.F., and Chen T.Y.. 2001. Different fast-gate regulation by external Cl− and H+ of the muscle-type ClC chloride channels. J. Gen. Physiol. 118:23–32. 10.1085/jgp.118.1.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.Y., and Miller C.. 1996. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 108:237–250. 10.1085/jgp.108.4.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Santiago J.A., Nehrke K., and Arreola J.. 2005. Quantitative analysis of the voltage-dependent gating of mouse parotid ClC-2 chloride channel. J. Gen. Physiol. 126:591–603. 10.1085/jgp.200509310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich P., and Hedrich R.. 1998. Anions permeate and gate GCAC1, a voltage-dependent guard cell anion channel. Plant J. 15:479–487. 10.1046/j.1365-313X.1998.00225.x [DOI] [Google Scholar]
- Dutzler R., Campbell E.B., Cadene M., Chait B.T., and MacKinnon R.. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. 10.1038/415287a [DOI] [PubMed] [Google Scholar]
- Dutzler R., Campbell E.B., and MacKinnon R.. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. 10.1126/science.1082708 [DOI] [PubMed] [Google Scholar]
- Engh A.M., Faraldo-Gómez J.D., and Maduke M.. 2007. The mechanism of fast-gate opening in ClC-0. J. Gen. Physiol. 130:335–349. 10.1085/jgp.200709759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Campbell E.B., and MacKinnon R.. 2012. Molecular mechanism of proton transport in CLC Cl−/H+ exchange transporters. Proc. Natl. Acad. Sci. USA. 109:11699–11704. 10.1073/pnas.1205764109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Celma J., Szydelko A., and Dutzler R.. 2013. Functional characterization of a ClC transporter by solid-supported membrane electrophysiology. J. Gen. Physiol. 141:479–491. 10.1085/jgp.201210927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieschat M., and Alekov A.K.. 2014. Multiple discrete transitions underlie voltage-dependent activation in CLC Cl−/H+ antiporters. Biophys. J. 107:L13–L15. 10.1016/j.bpj.2014.07.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründer S., Thiemann A., Pusch M., and Jentsch T.J.. 1992. Regions involved in the opening of CLC-2 chloride channel by voltage and cell volume. Nature. 360:759–762. 10.1038/360759a0 [DOI] [PubMed] [Google Scholar]
- Hanke W., and Miller C.. 1983. Single chloride channels from Torpedo electroplax. Activation by protons. J. Gen. Physiol. 82:25–45. 10.1085/jgp.82.1.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Carballo C.Y., De Santiago-Castillo J.A., Rosales-Saavedra T., Pérez-Cornejo P., and Arreola J.. 2010. Control of volume-sensitive chloride channel inactivation by the coupled action of intracellular chloride and extracellular protons. Pflugers Arch. 460:633–644. 10.1007/s00424-010-0842-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer R., Iverson T.M., Accardi A., and Miller C.. 2002. A biological role for prokaryotic ClC chloride channels. Nature. 419:715–718. 10.1038/nature01000 [DOI] [PubMed] [Google Scholar]
- Li H., Robertson A.D., and Jensen J.H.. 2005. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 61:704–721. 10.1002/prot.20660 [DOI] [PubMed] [Google Scholar]
- Lim H.H., Stockbridge R.B., and Miller C.. 2013. Fluoride-dependent interruption of the transport cycle of a CLC Cl−/H+ antiporter. Nat. Chem. Biol. 9:721–725. 10.1038/nchembio.1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lísal J., and Maduke M.. 2008. The ClC-0 chloride channel is a ‘broken’ Cl−/H+ antiporter. Nat. Struct. Mol. Biol. 15:805–810. 10.1038/nsmb.1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. 2006. ClC chloride channels viewed through a transporter lens. Nature. 440:484–489. 10.1038/nature04713 [DOI] [PubMed] [Google Scholar]
- Miloshevsky G.V., and Jordan P.C.. 2004. Anion pathway and potential energy profiles along curvilinear bacterial ClC Cl− pores: Electrostatic effects of charged residues. Biophys. J. 86:825–835. 10.1016/S0006-3495(04)74158-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. 1992. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 207:123–131. 10.1016/0076-6879(92)07008-C [DOI] [PubMed] [Google Scholar]
- Nehrke K., Arreola J., Nguyen H.V., Pilato J., Richardson L., Okunade G., Baggs R., Shull G.E., and Melvin J.E.. 2002. Loss of hyperpolarization-activated Cl− current in salivary acinar cells from Clcn2 knockout mice. J. Biol. Chem. 277:23604–23611. 10.1074/jbc.M202900200 [DOI] [PubMed] [Google Scholar]
- Niemeyer M.I., Cid L.P., Yusef Y.R., Briones R., and Sepúlveda F.V.. 2009. Voltage-dependent and -independent titration of specific residues accounts for complex gating of a ClC chloride channel by extracellular protons. J. Physiol. 587:1387–1400. 10.1113/jphysiol.2008.167353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Delgado P.G., Arreola J., and Guirado-López R.A.. 2013. Atomic charges of Cl− ions confined in a model Escherichia coli ClC–Cl−/H+ ion exchanger: a density functional theory study. Mol. Phys. 111:3218–3233. 10.1080/00268976.2013.776709 [DOI] [Google Scholar]
- Perez-Cornejo P., De Santiago J.A., and Arreola J.. 2004. Permeant anions control gating of calcium-dependent chloride channels. J. Membr. Biol. 198:125–133. 10.1007/s00232-004-0659-x [DOI] [PubMed] [Google Scholar]
- Picollo A., Xu Y., Johner N., Bernèche S., and Accardi A.. 2012. Synergistic substrate binding determines the stoichiometry of transport of a prokaryotic H+/Cl− exchanger. Nat. Struct. Mol. Biol. 19:525–531. 10.1038/nsmb.2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M., Ludewig U., Rehfeldt A., and Jentsch T.J.. 1995. Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature. 373:527–531. 10.1038/373527a0 [DOI] [PubMed] [Google Scholar]
- Richard E.A., and Miller C.. 1990. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 247:1208–1210. 10.1126/science.2156338 [DOI] [PubMed] [Google Scholar]
- Rinke I., Artmann J., and Stein V.. 2010. ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J. Neurosci. 30:4776–4786. 10.1523/JNEUROSCI.6299-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov G.Y., Pusch M., Astill D.S., Roberts M.L., Jentsch T.J., and Bretag A.H.. 1996. Concentration and pH dependence of skeletal muscle chloride channel ClC-1. J. Physiol. 497:423–435. 10.1113/jphysiol.1996.sp021778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov G.Y., Pusch M., Roberts M.L., Jentsch T.J., and Bretag A.H.. 1998. Permeation and block of the skeletal muscle chloride channel, ClC-1, by foreign anions. J. Gen. Physiol. 111:653–665. 10.1085/jgp.111.5.653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov G.Y., Pusch M., Roberts M.L., and Bretag A.H.. 2001. Interaction of hydrophobic anions with the rat skeletal muscle chloride channel ClC-1: effects on permeation and gating. J. Physiol. 530:379–393. 10.1111/j.1469-7793.2001.0379k.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Rodríguez J.E., De Santiago-Castillo J.A., and Arreola J.. 2010. Permeant anions contribute to voltage dependence of ClC-2 chloride channel by interacting with the protopore gate. J. Physiol. 588:2545–2556. 10.1113/jphysiol.2010.189175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Rodríguez J.E., De Santiago-Castillo J.A., Contreras-Vite J.A., Nieto-Delgado P.G., Castro-Chong A., and Arreola J.. 2012. Sequential interaction of chloride and proton ions with the fast gate steer the voltage-dependent gating in ClC-2 chloride channels. J. Physiol. 590:4239–4253. 10.1113/jphysiol.2012.232660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X.M., and Feldman J.L.. 2007. Micro-agar salt bridge in patch-clamp electrode holder stabilizes electrode potentials. J. Neurosci. Methods. 159:108–115. 10.1016/j.jneumeth.2006.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiemann A., Gründer S., Pusch M., and Jentsch T.J.. 1992. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 356:57–60. 10.1038/356057a0 [DOI] [PubMed] [Google Scholar]
- Traverso S., Zifarelli G., Aiello R., and Pusch M.. 2006. Proton sensing of CLC-0 mutant E166D. J. Gen. Physiol. 127:51–65. 10.1085/jgp.200509340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDongen A.M. 2004. K channel gating by an affinity-switching selectivity filter. Proc. Natl. Acad. Sci. USA. 101:3248–3252. 10.1073/pnas.0308743101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright A.M., Gong X., Verdon B., Linsdell P., Mehta A., Riordan J.R., Argent B.E., and Gray M.A.. 2004. Novel regulation of cystic fibrosis transmembrane conductance regulator (CFTR) channel gating by external chloride. J. Biol. Chem. 279:41658–41663. 10.1074/jbc.M405517200 [DOI] [PubMed] [Google Scholar]
- Xiao Q., Yu K., Perez-Cornejo P., Cui Y., Arreola J., and Hartzell H.C.. 2011. Voltage- and calcium-dependent gating of TMEM16A/Ano1 chloride channels are physically coupled by the first intracellular loop. Proc. Natl. Acad. Sci. USA. 108:8891–8896. 10.1073/pnas.1102147108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zifarelli G., Murgia A.R., Soliani P., and Pusch M.. 2008. Intracellular proton regulation of ClC-0. J. Gen. Physiol. 132:185–198. 10.1085/jgp.200809999 [DOI] [PMC free article] [PubMed] [Google Scholar]