

Abstract
Stroke research has endured many setbacks in translating neuroprotective therapies into clinical practice. In contrast, the real-world therapy (tPA thrombolysis) rarely produces benefits in mechanical occlusion-based experimental models, which dominate preclinical stroke research. This split between the bench and bedside suggests the need to employ tPA-responsive models in preclinical stroke research. To this end, a simple and tPA-reactive thrombotic stroke model is invented and described here. This model consists of transient occlusion of the unilateral common carotid artery and delivery of 7.5% oxygen through a face mask in adult mice for 30 min, while maintaining the animal rectal temperature at 37.5 ± 0.5 °C. Although reversible ligation of the unilateral carotid artery or hypoxia each suppressed cerebral blood flow only transiently, the combination of both insults caused lasting reperfusion deficits, fibrin and platelet deposition, and large infarct in the middle cerebral artery-supplied territory. Importantly, tail-vein injection of recombinant tPA at 0.5, 1, or 4 hr post-tHI (10 mg/kg) provided time-dependent reduction of the mortality rate and infarct size. This new stroke model is simple and can be standardized across laboratories to compare experimental results. Further, it induces thrombosis without craniectomy or introducing pre-formed emboli. Given these unique merits, the tHI model is a useful addition to the repertoire of preclinical stroke research.
Keywords: Medicine, Issue 102, Tissue plasminogen activator (tPA), Laser speckle contrast imaging, Thrombosis, Virchow’s triad, Edaravone, Reperfusion, No-Reflow
Introduction
Thrombolysis and recanalization is the most effective therapy of acute ischemic stroke in clinical practice1. Yet, the majority of preclinical neuroprotection research was performed in a transient mechanic obstruction model (intraluminal suture middle cerebral artery occlusion) that produces rapid recovery of cerebral blood flow upon removal of the vascular occlusion and shows little to no benefits by tPA thrombolysis. It has been suggested that the dubious choice of stroke models contributed, at least in part, to the difficulty in translating neuroprotective therapy to patients2,3. Hence, there is an increasing call for employing tPA-responsive thromboembolic stroke models in preclinical research, but such models also have technical problems (see Discussion)4-7. Here we describe a new thrombotic stroke model based on unilateral transient hypoxic-ischemic (tHI) insult and its responses to intravenous tPA therapy8.
The tHI stroke model was developed based on the Levine procedure (permanent ligation of the unilateral common carotid artery followed by exposure to transient hypoxia in a chamber) that was invented for experiments with adult rats in 19609. The original Levine procedure faded into obscurity because it only produced variable brain damage, but the same insult caused consistent neuropathology in rodent pups when it was re-introduced by Robert Vannucci and his colleagues as a model of neonatal hypoxic-ischemic encephalopathy (HIE) in 198110. In recent years, some investigators re-adapted the Levine-Vannucci model to adult mice by adjusting the temperature in the hypoxic chamber11. It is plausible that the inconsistent brain lesions in the original Levine procedure may arise from fluctuating body temperatures of adult rodents in the hypoxic chamber. To test this hypothesis, we modified the Levine procedure by administering hypoxic gas through a facemask, while maintaining the rodent core temperature at 37 °C on the surgical table12. As expected, stringent body temperature control greatly increased the reproducibility of HI-induced brain pathology. The HI insult also triggers coagulation, autophagy, and gray- and white-matter injury13. Other investigators have also used the HI model to investigate post-stroke inflammatory responses14.
A unique feature of the HI stroke model is that it closely follows the Virchow’s triad of thrombus formation, including the stasis of blood flow, endothelial injury (e.g. due to HI-induced oxidative stress), and hypercoagulability (HI-induced platelet activation) (Figure 1A)15. As such, the HI model may capture some pathophysiological mechanisms relevant to real-world ischemic stroke. With this idea in mind, we further refined the HI model with reversible ligation of the unilateral common carotid artery (therefore to create a transient HI insult), and tested its responses to tPA thrombolysis with or without Edaravone. Edaravone is a free radical scavenger already approved in Japan to treat ischemic stroke within 24 hr of onset9. Our experiments showed that as brief as 30 min transient HI triggers thrombotic infarction, and that combined tPA-Edaravone treatment confers synergistic benefits8. Here we describe detailed surgical procedures and methodological considerations of the tHI model, which can be used to optimize reperfusion treatments of acute ischemic stroke.
Protocol
This protocol is approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University and follows the National Institutes of Health Guideline for Care and Use of Laboratory Animals.
1. Setup
Prepare the surgical bed on warming pad connected with heat pump at 37 °C for at least 15 min before the surgery. Place a neck roll using the barrel of 3 ml syringe on the surgical bed. Prepare the anesthesia gas with 2% isoflurane in medical air.
Prepare autoclaved forceps, scissors, micro needle holders, hemostat, cotton swabs and sutures. Prepare tissue glue and eye ointment.
Set up the hypoxia system and temperature controllers with heating lamp and rectal probe. Prepare hypoxia gas with 2% isoflurane in 7.5% O2 balanced by 92.5% N2.
One hour before surgery, mice are analgesized by subcutaneous injection of a slow-release Meloxicam (4.0 mg/kg).
2. Transient Cerebral Hypoxia-ischemia (Figure 1B)
Anesthetize 10-13 week old male C57BL/6 mice weighing 22 to 30 g in the anesthesia induction chamber with 3% isoflurane until the animal is unresponsive to foot squeeze, and then remove the hair on right neck using an electronic shaver.
Place mice on the surgical bed connected with 2% isoflurane in medical air at flow rate of 2 L/min. Secure forelimbs stretched out along neck roll at sides using medical tape.
Clean the surgical site for incision with betadine followed by alcohol and then cotton swabs.
Under dissecting microscope, make a 0.5 cm right-cervical incision using a straight forceps and a micro scissors about 0.2 cm lateral from midline skin.
Use a pair of fine serrated forceps to pull apart the fascia and tissue to expose the right common carotid artery (RCCA). Carefully separate the RCCA from the vagal nerve using a pair of fine smooth forceps.
Live knot two precut 5-0 silk suture (releasable) on the RCCA, and then sew up the skin using 4-0 Nylon monofilament suture (Figure 1C).
Apply eye ointment on both eyes to prevent dryness.
- Quickly transfer the mice to hypoxia system and put nose and mouth in face-mask with 2% isoflurane in 7.5% O2 at flow rate of 0.5-1 L/min for 30 min.
- During hypoxia, use temperature controllers with heating lamps to control the rectal temperature at 37.5 ± 0.5 °C. Monitor the respiratory rate at 80-120 breaths/min. The maintenance of the body temperature above 37 °C during the hypoxia is important to create consistent brain infarction. Low respiratory rate usually happens after 20 min hypoxia. Remove the face-mask and allow normal air supply if the respiratory rate drops below to 40. This takes 1-2 min and does not count into the 30 min hypoxia duration.
After hypoxia, transfer mice to a surgical bed and release the two sutures from RCCA. Close the wound using tissue glue, and then return mice to the cage. Exclude the animals if both of two live knots are unexpectedly released after hypoxia.
Monitor the mice for 5-10 min to recover from hypoxia and anesthesia. Place the wetted food in the cage and return it to animal care facility. Note: Animals showing mild to severe circling behavior at 24 hr after tHI are correlated with brain infraction. Most animals with seizure symptoms die before the 24 hr timepoint after tHI.
3. Laser Speckle Contrast Imaging
Note: Although this is not an essential procedure of the tHI model, a two-dimensional laser speckle contrast imaging system16 can be used to characterize the changes of cerebral blood flow (CBF) during or after transient hypoxia-ischemia. To document the alterations of CBF under tHI, record immediately after the step 2.6. Alternatively, to compare CBF recovery after tHI insult, these procedures can be performed following the step 2.10.
Place an anesthetized mouse in the prone position and perform a 1 cm-long midline incision on the scalp with the skull exposed but unopened.
Monitor CBF in both cerebral hemispheres under a blood flow imager according to manufacturer’s protocol and start recording the cerebral blood flow immediately after the CCAO surgery (step 2.6). Continue for 50 min.
Show CBF image with arbitrary units in a 16-color palette and analyze in real-time the selected regions using the MoorFLPI software following the manufacturer’s instructions (Figure 2).
After recording the CBF image, close the scalp with tissue glue and return the animal to the cage.
4. tPA Administration
Inject animals at the tail vein with the solvent or 10 mg/kg recombinant tPA (220-300 μl of 1 mg/m tPA) at 0.5, 1, or 4 hr after tCCAo plus hypoxia (Figure 4).
5. Brain Damage Detection with Several Different Options
Note: To collect brain samples, euthanize the mice at 1, 4 or 24 hr after tHI.
- Perform quantification of infarct volume by in vivo 2,3,5-Triphenyltetrazolium chloride (TTC) method at 24 hrs after tHI insult as previous described.17
- Intraperitoneally inject animals with 1.4 M mannitol solution (~0.1 ml/g body weight) 30 min before transcardial perfusion. Transcardial perfuse mice with PBS followed by 10 ml of 2% TTC.
- Remove the brain of animals with surgical instruments after 10 min and place into 4% paraformaldehyde for fixation overnight and section into 1 mm thickness with a vibratome.
- Snap a series of four sectioned brain slides by digital microscope and quantify the infarct volume as the ratio of the infarcted area (white area in the right side) to the area of the undamaged, contralateral hemisphere using the ImageJ software.
- Alternatively, perform thrombosis formation by immunofluorescence at 1 hr after tHI insult.
- Freeze the fixed brain in O.C.T. compound and section the brains at 12 μm thickness using a cryostat.
- Incubate the brain slide with rabbit anti-fibrinogen antibody (1:100) following by goat anti-rabbit Alexa Fluro 488 dye (1:200) to observe the fluorescence on a fluorescent microscope.
- Alternatively, perform vessel obstruction by tail vein injection of 100 µl 2% Evans blue dye at 4 hr after tHI insult.
- Euthanize the mice and quickly cut head to remove brains into 4% paraformaldehyde after Evans blue injection. Note: It takes 5-10 min for Evans blue circulation with blue color of both fore- and hind limbs.
- Section fixed brains at 100 μm thickness using a sliding microtome and observe the fluorescence using a 680 nm emission filter on a fluorescent microscope.
Representative Results
Two-dimensional laser speckle contrast imaging (LSCI)16 was used to compare the alterations of cerebral blood flow (CBF) by 30 min transient unilateral carotid occlusion (tCCAO), 30 min exposure to hypoxia (7.5% oxygen), and 30 min unilateral carotid ligation under hypoxia (tHI). This experiment revealed that tCCAO under normoxia suppressed the CBF on the carotid ligated hemisphere to ~50% of the baseline value, which quickly recovered to above 85% after release of the carotid occlusion (R in Figure 2A). Exposure to systemic hypoxia alone reduced the CBF to approximately 75% of the baseline value, which transiently rebound to ~130% after returning to the normoxic atmosphere (Figure 2B). In contrast carotid ligation under hypoxia (tHI) quickly reduced CBF in the ipsilateral hemisphere to less than 20% of the baseline value around 10 min, which rarely recovered to above 30% at 20 min after release of the carotid ligation and returning to normoxia. CBF on the contralateral hemisphere (L), however, fluctuated between 20 and 50% during hypoxia, and quickly returned to above 80% after the tHI insult (Figure 2C).
At 24 hr after tCCAo (30 min) or tCCAO plus 30 min hypoxia (tHI), in vivo TTC stain was used to detect infarction17. This analysis showed no obvious injury by the tCCAO insult, but sizable infarction in the middle cerebral artery-supplied territory following tHI insult (Figure 3A). Anti-fibrin(ogen) immunostaining was used to compare the tCCAO- and tHI-injured brains at 1 hour recovery and showed widespread deposition of fibrin(ogen), an indicator of thrombosis, in the tHI-injured, but not tCCAO-challenged mouse brains (Figure 3B, C). Tail-vein injection of Evans blue dye was also used to compare vascular perfusion of tCCAO- and tHI-injured brains at 4 hr recovery. This analysis showed diminished cerebral perfusion and intense extravasation of the Evans blue dye in tHI-injured, but not the tCCAO-challenged mouse brains (Figure 3D, E).
Finally, the outcomes of tHI insult in mice receiving tail-vein injection of vehicle (at 0.5 hr recovery) or recombinant human tPA (Activase, 10 mg/kg, at 0.5, 1, or 4 hours post-tHI) were compared using in vivo TTC stain at 24 hr recovery (Figure 4A). In vehicle-treated mice, the mortality rate at 24 hr post-tHI was 23.8% and only one in 21 tHI-injured mice was beyond the mean and 2 SD (the outlier). The 24 hours mortality rate in mice receiving tPA treatment at 0.5 hr recovery dropped to 8.3%, but this effect was lost when tPA was administrated at 1 or 4 hours after the tHI insult (Figure 4B). Figure 4C plotted the infarct size of all survived mice in the four treatment groups. Of note, both 0.5 and 1 hr tPA-administration significantly reduced infarct size, when compared to vehicle treatment. The 0.5 hr tPA-treatment group also showed a significantly reduced infarct size than the 4 hr tPA-treatment group. Figure 4D showed representative TTC-stain results after each treatment.
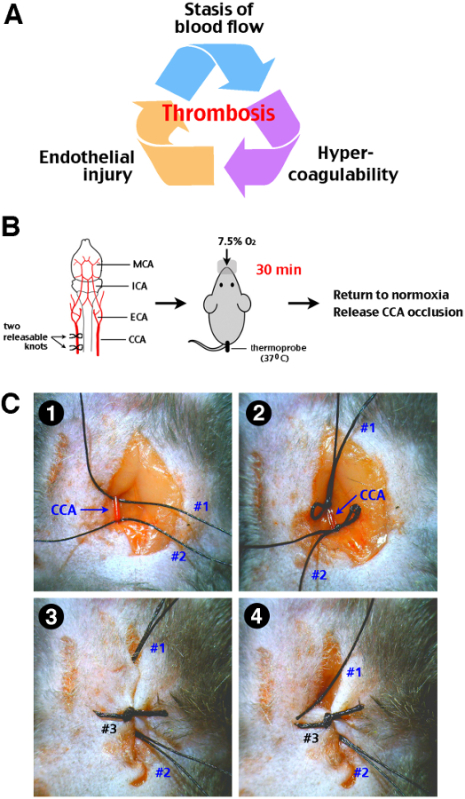 Figure 1:Procedure of transient cerebral hypoxia-ischemia (tHI) insult in adult mice. (A) The Virchow’s triad that propels thrombosis includes stasis of blood flow, endothelial injury, and hypercoagulability of the blood. (B) A schematic diagram of the tHI stroke procedure. Two releasable knots were tied onto the right common carotid artery (CCA), and followed by delivery of 7.5% oxygen via a nose cone for 30 min, while the mouse rectal temperature was maintained at 37-38 °C. After the transient systemic hypoxia, the CCA ligation was released by pulling out one end of the releasable suture knots. MCA, middle cerebral artery; ICA, internal carotid artery; ECA, external carotid artery; CCA, common carotid artery. (C) Surgical procedures for transient right CCA occlusion. 1. Two precut suture (#1 and #2) were placed under an isolated right CAA. 2. Two releasable knots were made. 3. The incision line was closed up by suture #3. Make sure that the ends of suture #1 and #2 were approachable outside the incision line. 4. Carefully pull suture #1 and #2 from outside to release the CCA. When performed gently, this procedure will not cause laceration of the CCA.
Figure 1:Procedure of transient cerebral hypoxia-ischemia (tHI) insult in adult mice. (A) The Virchow’s triad that propels thrombosis includes stasis of blood flow, endothelial injury, and hypercoagulability of the blood. (B) A schematic diagram of the tHI stroke procedure. Two releasable knots were tied onto the right common carotid artery (CCA), and followed by delivery of 7.5% oxygen via a nose cone for 30 min, while the mouse rectal temperature was maintained at 37-38 °C. After the transient systemic hypoxia, the CCA ligation was released by pulling out one end of the releasable suture knots. MCA, middle cerebral artery; ICA, internal carotid artery; ECA, external carotid artery; CCA, common carotid artery. (C) Surgical procedures for transient right CCA occlusion. 1. Two precut suture (#1 and #2) were placed under an isolated right CAA. 2. Two releasable knots were made. 3. The incision line was closed up by suture #3. Make sure that the ends of suture #1 and #2 were approachable outside the incision line. 4. Carefully pull suture #1 and #2 from outside to release the CCA. When performed gently, this procedure will not cause laceration of the CCA.
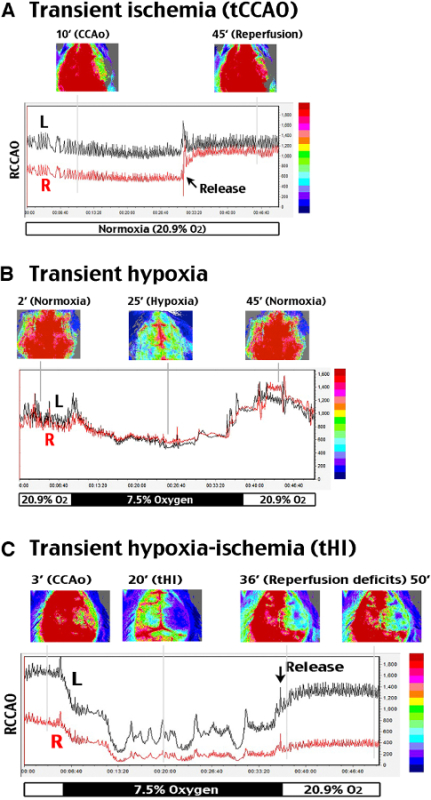 Figure 2:Analysis of cerebral blood flow alterations during and after tHI insult. A two-dimensional laser speckle contrast imaging (LSCI) system was used to evaluate cerebral blood flow (CBF). R (right) indicates the carotid-ligated hemisphere; L (left) is the contralateral hemisphere. (A) tCCAO under normoxia suppressed CBF to ~50% of the baseline value on the carotid-ligated hemisphere (R) for at least 30 min, which recovered to above 85% within 3 min upon release of the carotid ligation. (B) In hypoxia (7.5% oxygen, 30 min) without carotid artery ligation, CBF declined to 76% of the baseline value and transiently rebounded to around 130% after returning to normoxia. (C) In transient carotid ligation under hypoxia (tHI, 30 min), CBF on the carotid-ligated (R) hemisphere quickly dropped to less than 20% of the baseline value, and it rarely recovered to above 30% upon release of the carotid ligation and returning to normoxia. In contrast, CBF on the contralateral (L) hemisphere fluctuated between 20-50% during hypoxia, and quickly returned to >80% of the baseline value after release of carotid ligation and returning to normoxia. Shown are representative CBF tracings for n >4 in each group. The time-points for representative LSCI photographs were marked by grey lines in the representative tracing.
Figure 2:Analysis of cerebral blood flow alterations during and after tHI insult. A two-dimensional laser speckle contrast imaging (LSCI) system was used to evaluate cerebral blood flow (CBF). R (right) indicates the carotid-ligated hemisphere; L (left) is the contralateral hemisphere. (A) tCCAO under normoxia suppressed CBF to ~50% of the baseline value on the carotid-ligated hemisphere (R) for at least 30 min, which recovered to above 85% within 3 min upon release of the carotid ligation. (B) In hypoxia (7.5% oxygen, 30 min) without carotid artery ligation, CBF declined to 76% of the baseline value and transiently rebounded to around 130% after returning to normoxia. (C) In transient carotid ligation under hypoxia (tHI, 30 min), CBF on the carotid-ligated (R) hemisphere quickly dropped to less than 20% of the baseline value, and it rarely recovered to above 30% upon release of the carotid ligation and returning to normoxia. In contrast, CBF on the contralateral (L) hemisphere fluctuated between 20-50% during hypoxia, and quickly returned to >80% of the baseline value after release of carotid ligation and returning to normoxia. Shown are representative CBF tracings for n >4 in each group. The time-points for representative LSCI photographs were marked by grey lines in the representative tracing.
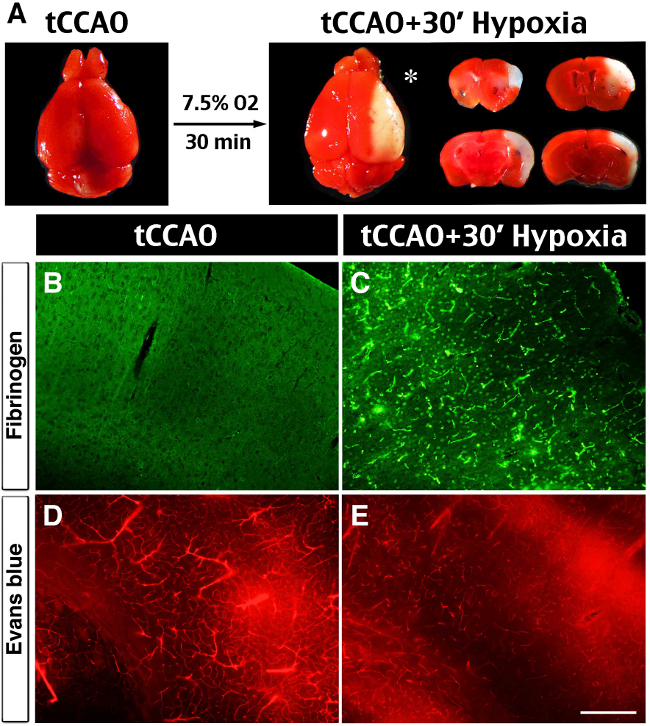 Figure 3:Brain infarct, spontaneous thrombosis and vessel obstruction after the tHI insult. (A) In vivo TTC-stain showed no visible infarction at 24 hr after 30 min transient ligation of the right common carotid artery (tCCAO), but the addition of 30 min hypoxia (7.5% oxygen) to tCCAO produced sizeable infarction in the ipsilateral hemisphere (asterisk), mostly in the middle cerebral artery-supplying area. (B, C) Anti-fibrin(ogen) immunostaining at 1 hr after the tHI insult showed widespread deposits in the ipsilateral hemisphere. In contrast, there was no fibrin(ogen) deposits at 1 hr after the tCCAo (30 min) insult (n >4 for each). (D, E) Cerebral perfusion was evaluated by tail-vein injection of Evans Blue dye at 4 hr after tCCAO (30 min) or tHI (30 min) insult. In post-tCCAO brain, Evans Blue dye filled most of the blood vessels in the ipsilateral hemisphere. In contrast, in post-tHI brain, Evan Blue dye filled fewer blood vessels and was leaked into the parenchyma (n >3). Scale bar: 250 μm.
Figure 3:Brain infarct, spontaneous thrombosis and vessel obstruction after the tHI insult. (A) In vivo TTC-stain showed no visible infarction at 24 hr after 30 min transient ligation of the right common carotid artery (tCCAO), but the addition of 30 min hypoxia (7.5% oxygen) to tCCAO produced sizeable infarction in the ipsilateral hemisphere (asterisk), mostly in the middle cerebral artery-supplying area. (B, C) Anti-fibrin(ogen) immunostaining at 1 hr after the tHI insult showed widespread deposits in the ipsilateral hemisphere. In contrast, there was no fibrin(ogen) deposits at 1 hr after the tCCAo (30 min) insult (n >4 for each). (D, E) Cerebral perfusion was evaluated by tail-vein injection of Evans Blue dye at 4 hr after tCCAO (30 min) or tHI (30 min) insult. In post-tCCAO brain, Evans Blue dye filled most of the blood vessels in the ipsilateral hemisphere. In contrast, in post-tHI brain, Evan Blue dye filled fewer blood vessels and was leaked into the parenchyma (n >3). Scale bar: 250 μm.
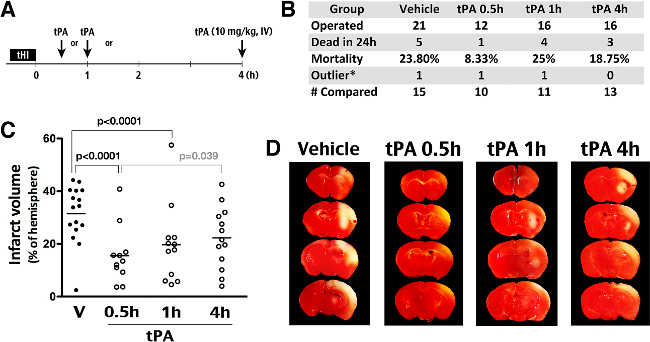 Figure 4:Effects of tPA thrombolysis in the tHI stroke model. (A) Outline of experiments to compare the effects of intravenous tPA administration (10 mg/kg) at 0.5, 1, or 4 hr after the tHI insult. (B) Summary of the number of operated animals, mortality in 24 hr after insults, outliers (the infarct size outside the mean +/- 2 SD), and the number of animals included for comparison of the infarct size. (C) Quantification showed an averaged 32% infarct volume in the vehicle group and significant reduction of infarction in the 0.5 hr (to 16%) and 1 hr (to 20%) groups (Shown are the mean and SEM for each group). The p-values are determined by t-test. (D) Representative TTC-stained brain from animals that were challenged by the tHI insult and received tPA treatment at the indicated time-point after injury. In TTC staining, live tissue showed red color; infarct tissue was pale.
Figure 4:Effects of tPA thrombolysis in the tHI stroke model. (A) Outline of experiments to compare the effects of intravenous tPA administration (10 mg/kg) at 0.5, 1, or 4 hr after the tHI insult. (B) Summary of the number of operated animals, mortality in 24 hr after insults, outliers (the infarct size outside the mean +/- 2 SD), and the number of animals included for comparison of the infarct size. (C) Quantification showed an averaged 32% infarct volume in the vehicle group and significant reduction of infarction in the 0.5 hr (to 16%) and 1 hr (to 20%) groups (Shown are the mean and SEM for each group). The p-values are determined by t-test. (D) Representative TTC-stained brain from animals that were challenged by the tHI insult and received tPA treatment at the indicated time-point after injury. In TTC staining, live tissue showed red color; infarct tissue was pale.
Discussion
Stroke is a major health issue of growing significance for any society with an aging population. Globally, stroke is the second-leading cause of death with an estimated 5.9 million fatal events in 2010, equivalent to 11.1% of all deaths18. Stroke is also the third-leading cause of disability-adjusted life years (DALYs) lost globally in 2010, rising from the fifth position in 199019. These epidemiological data highlight the need of more effective therapies of acute (ischemic) stroke. However, despite intense research in preclinical neuroprotective treatment, tPA-thrombolysis remains the only specific therapy of acute ischemic stroke that is approved by the Food and Drug Administration in the United States, while numerous once promising neuroprotective agents in animal studies failed in clinical trials. The difficulty in translating neuroprotective therapies into patients has many factors, and the current emphasis is on good laboratory practice, meta-analysis of multiple datasets, and international cooperation to improve preclinical stroke research20,21. However, there is a minority opinion suggesting that the translational difficulty is due to a poor choice of mechanical vascular occlusion models (e.g. intraluminal suture MCA occlusion) in the majority of preclinical stroke research to date2,3. Because mechanical vascular occlusion models rarely induce thrombosis and cerebral reperfusion occurs too fast upon the release of mechanical obstruction, these models neither respond to the real-word therapy (tPA fibrinolysis) nor provide a narrow therapeutic window as those in stroke patients. Consequently, the suggested remedy is to emphasize, at least include, thromboembolic stroke models in preclinical stroke research3.
This recommendation, however, has its restraints because current thromboembolic stroke models (exogenous emboli delivery, MCA-injection of thrombin, and photothrombosis) all have certain technical drawbacks4. For the exogenous emboli model, intravascular infusion of emboli results in significant variability in infarct size and location, as well as unpredictable responses to tPA thrombolysis due to differences in clot preparation4,5. Direct injection of thrombin into the MCA branch requires craniectomy, and its utility for optimizing the thrombolytic therapy is yet to be proven4,6. Chemically initiated thromboembolism based on systemic injection of a photosensitive dye (e.g. Rose Bengal or erythrosine B) and irradiation through the exposed skull often produces platelet-only aggregates that do not respond to thrombolysis4,7. Taken together, there is a need of simpler and tPA-responsive thromboembolic models for preclinical stroke research.
The tHI paradigm has four unique advantages as a thromboembolic stroke model. First, the tHI insult wielded endogenous components to form in situ thrombi without the help of exogenous chemicals or pre-formed emboli. Thus, thrombus formation in the tHI model is more relevant to physiological conditions. Second, the tHI model responds favorable to rapid tPA-treatment (at 0.5 and 1 hr post-injury) but not to delayed treatment (at 4 hr). This therapeutic window is similar to those observed in stroke patients. Thus, the tHI model may be utilized for research aimed to improve reperfusion therapy in acute ischemic stroke. Third, the surgical procedures in the tHI model are simple and straightforward, when compared to the intraluminal suture MCA occlusion model. The duration of hypoxia in the tHI model is also controllable. These attributes make the tHI model less susceptible to procedural variations among different laboratories. Finally, the tHI model may shed insights into the mechanisms of incomplete reperfusion despite recanalization of large arteries after thrombolysis, which is a unique challenge in stroke therapy when compared to cardiac ischemia1,22. Hence, the tHI model provides a unique system to study the mechanisms of the cerebral vascular bed-specific dysregulation of hemostasis23.
All experimental brain injury models have limitations, and the tHI model is no exception. Three major technical constraints of the tHI stroke model have been identified in the experiment. First, unlike other stroke models where the insult is limited to the brain, the combination of hypoxia and carotid occlusion leads to peripheral vasodilation and a greater demand for cardiac output12. Thus, when comparing the effects of mouse mutations or neuroprotective agents against the tHI insult, their impacts on the cardiovascular functions shall also be carefully compared. Second, we found that different mouse inbred strains have variable responses to the tHI model, which may be due to uneven patency of the posterior communicating artery24, dissimilar cardiac functions, or a combination of both. Hence, it is recommended that the sex, age, body weight, and mouse strains between two experimental groups be comparable in neuroprotection studies. Finally, the tHI stroke relies on animals to breathe in the hypoxic gas under a lightly anesthetized condition. The effects of anesthesia on stroke outcomes need to be minimized and kept consistent between animals. Nonetheless, as long as researchers are vigilant of these technical details and reduce the variables from animals to animals, the tHI stroke model can be quickly established to yield high consistency of brain infarction.
In summary, tHI is a simple and standardized stroke model that responds favorably to the real-world therapy (tPA thrombolysis) in a clinically relevant temporal window. This new model is a valuable addition to preclinical stroke research, and may help to improve thrombolysis therapy in acute ischemic stroke.
Disclosures
None.
Acknowledgments
This study was supported by the NIH grant NS074559 (to C. K.). We thank all collaborators who contributed to our research articles that the present methodology report is based upon.
References
- Broderick JP, Hacke W. Treatment of acute ischemic stroke: Part I: recanalization strategies. Circulation. 2002;106(12):1563–1569. doi: 10.1161/01.cir.0000030406.47365.26. [DOI] [PubMed] [Google Scholar]
- Hossmann KA. Pathophysiological basis of translational stroke research. Folia Neuropathol. 2009;47(3):213–227. [PubMed] [Google Scholar]
- Hossmann KA. The two pathophysiologies of focal brain ischemia: implications for translational stroke research. J. Cereb. Blood Flow Metab. 2012;32(7):1310–1316. doi: 10.1038/jcbfm.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae IM. Preclinical stroke research--advantages and disadvantages of the most common rodent models of focal ischaemia. Br. J. Pharmacol. 2011;164(4):1062–1078. doi: 10.1111/j.1476-5381.2011.01398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen F, Hilger T, Hoehn M, Hossmann KA. Differences in clot preparation determine outcome of recombinant tissue plasminogen activator treatment in experimental thromboembolic stroke. Stroke. 2003;34(8):2019–2024. doi: 10.1161/01.STR.0000080941.73934.30. [DOI] [PubMed] [Google Scholar]
- Orset C, et al. Mouse model of in situ thromboembolic stroke and reperfusion. Stroke. 2007;38(10):2771–2778. doi: 10.1161/STROKEAHA.107.487520. [DOI] [PubMed] [Google Scholar]
- Watson BD, Dietrich WD, Prado R, Ginsberg MD. Argon laser-induced arterial photothrombosis. Characterization and possible application to therapy of arteriovenous malformations. J. Neurosurgery. 1987;66(5):748–754. doi: 10.3171/jns.1987.66.5.0748. [DOI] [PubMed] [Google Scholar]
- Sun YY, et al. Synergy of combined tPA-edaravone therapy in experimental thrombotic stroke. PLoS One. 2014;9:e98807. doi: 10.1371/journal.pone.0098807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine S. Anoxic-ischemic encephalopathy in rats. Am. J. Pathol. 1960;36:1–17. [PMC free article] [PubMed] [Google Scholar]
- Rice JE3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Annals Neurol. 1981;9(2):131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, et al. Experimental stroke in the female diabetic, db/db, mouse. J. Cereb. Blood Flow Metab. 2001;21(2):52–60. doi: 10.1097/00004647-200101000-00007. [DOI] [PubMed] [Google Scholar]
- Adhami F, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am. J. Pathol. 2006;169(2):566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shereen A, et al. Ex vivo diffusion tensor imaging and neuropathological correlation in a murine model of hypoxia-ischemia-induced thrombotic stroke. J. Cereb. Blood Flow Metab. 2011;31(4):1155–1169. doi: 10.1038/jcbfm.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud JP, Pimentel-Coelho PM, Tremblay Y, Rivest S. The impact of Ly6C low monocytes after cerebral hypoxia-ischemia in adult mice. J. Cereb. Blood Flow Metab. 2014;34(7):e1–e9. doi: 10.1038/jcbfm.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppo GJ. Virchow's triad: the vascular basis of cerebral injury. Rev. Neurol. Dis. 2008;5:12–21. [PMC free article] [PubMed] [Google Scholar]
- Dunn AK. Laser speckle contrast imaging of cerebral blood flow. Annals Biomed. Eng. 2012;40(2):367–377. doi: 10.1007/s10439-011-0469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YY, Yang D, Kuan CY. Mannitol-facilitated perfusion staining with 2,3,5-triphenyltetrazolium chloride (TTC) for detection of experimental cerebral infarction and biochemical analysis. J. Neurosci. Methods. 2012;203(1):122–129. doi: 10.1016/j.jneumeth.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano R, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study. Lancet. 2010;380(9859):2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CJ, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study. Lancet. 2012;380(9859):2197–2223. doi: 10.1016/S0140-6736(12)61689-4. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Macleod MR. Stroke research at a road block: the streets from adversity should be paved with meta-analysis and good laboratory practice. Br. J. Pharm. 2009;157(7):1154–1156. doi: 10.1111/j.1476-5381.2009.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, et al. A concerted appeal for international cooperation in preclinical stroke research. Stroke. 2013;44(6):1754–1760. doi: 10.1161/STROKEAHA.113.000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, et al. Revascularization end points in stroke interventional trials: recanalization versus reperfusion in IMS-I. Stroke. 2005;36(11):2400–2403. doi: 10.1161/01.STR.0000185698.45720.58. [DOI] [PubMed] [Google Scholar]
- Rosenberg RD, Aird WC. Vascular-bed--specific hemostasis and hypercoagulable states. New Eng. J. Med. 1999;340(20):1555–1564. doi: 10.1056/NEJM199905203402007. [DOI] [PubMed] [Google Scholar]
- Majid A, et al. Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke. 2000;31(11):2707–2714. doi: 10.1161/01.str.31.11.2707. [DOI] [PubMed] [Google Scholar]