

Abstract
Recent advances in reprogramming allow us to turn somatic cells into human induced pluripotent stem cells (hiPSCs). Disease modeling using patient-specific hiPSCs allows the study of the underlying mechanism for pathogenesis, also providing a platform for the development of in vitro drug screening and gene therapy to improve treatment options. The promising potential of hiPSCs for regenerative medicine is also evident from the increasing number of publications (>7000) on iPSCs in recent years. Various cell types from distinct lineages have been successfully used for hiPSC generation, including skin fibroblasts, hematopoietic cells and epidermal keratinocytes. While skin biopsies and blood collection are routinely performed in many labs as a source of somatic cells for the generation of hiPSCs, the collection and subsequent derivation of hair keratinocytes are less commonly used. Hair-derived keratinocytes represent a non-invasive approach to obtain cell samples from patients. Here we outline a simple non-invasive method for the derivation of keratinocytes from plucked hair. We also provide instructions for maintenance of keratinocytes and subsequent reprogramming to generate integration-free hiPSC using episomal vectors.
Keywords: Developmental Biology, Issue 102, Keratinocytes, human induced pluripotent stem cells, hair, integration-free reprogramming, regenerative medicine, episomal vectors
Introduction
The discovery of human induced pluripotent stem cells (hiPSCs) has revolutionized the field of regenerative medicine, providing a feasible method for generation of patient-specific stem cells 1-3. hiPSCs have been successfully generated from various somatic cell types, including fibroblasts 4,5, hematopoietic cells 6,7, renal epithelial cells from urine 8 and keratinocytes 9,10. To date, skin fibroblasts and hematopoietic cells represent the most commonly used cell sources for generating patient-specific iPSCs. Arguably, this is due to the fact that skin biopsies and blood collection are routine medical procedures and large biobanks of patient blood or skin samples have been established in many countries.
In contrast to blood cells and skin fibroblast which require invasive extraction methods, keratinocytes represent an easily accessible cell type for hiPSC generation. Keratinocytes are keratin-rich epithelial cells that form the exterior epidermal barrier of the skin and are also found in nails and hair 11. In particular, keratinocytes can be found on the outer root sheath (ORS) of hair follicles, an external cellular layer that covered the hair shaft together with the inner root sheath (IRS) cells (12, Figure 1). As hair collection is a simple procedure that does not require the assistance of medical personnel, it provides an opportunity for patients to collect and send their own hair samples to laboratories, which would greatly facilitate the collection of patient samples for hiPSC generation. Epidermal keratinocytes also have a higher reprogramming efficiency and faster reprogramming kinetics compared to fibroblasts, adding to the advantages of using keratinocytes as the starting cells for hiPSC generation 9,13. Furthermore, hiPSCs can also be generated using other cell populations within the hair follicle, including the dermal papilla cells located at the base of the hair follicle 14,15.
Previous reports of iPSC generation using hair-derived cells often utilize retroviral or lentiviral-based reprogramming methods 9,14,15. However, these viral methods introduce undesirable genomic integration of foreign transgenes during reprogramming. In comparison, the use of episomal vectors represents a feasible, non-viral reprogramming method to generate integration-free iPSCs 4. We have previously developed a simple, cost-effective and non-viral method to efficiently reprogram keratinocyte into hiPSCs using episomal vectors 13. Here we provide a detailed protocol for the generation of keratinocyte-derived hiPSCs, including the derivation of keratinocytes from plucked hair, expansion and maintenance of the keratinocytes and subsequent reprogramming to generate hiPSCs.
Protocol
The collection of human hair sample from individuals requires ethical approval by the human research ethics committee in the host institutions and should be done in compliance with the institutional guidelines.
1. Isolation of Keratinocytes from Plucked Hair
Thaw Extracellular Matrix (ECM) solution (i.e., Matrigel) on ice O/N.
Using pre-chilled pipette tips, add 200 µl ECM solution to 12 ml of chilled DMEM/F12 medium. Coat a 12-well plate with 1 ml of diluted ECM solution into each well. Incubate the coated plate O/N at 37 °C. Wash the coated plate with pre-warmed DMEM/F12 medium before use.
- Collect primary human hair from the individual’s head by placing fingers close to the root of the hair and plucking the hair in one quick and smooth motion. Check to confirm that each collected hair contains an intact hair follicle and determine the growth phase of the hair under a dissecting microscope (Figure 1).
- Collect 5-10 anagen hairs from each individual to extract keratinocytes. Process plucked hairs as soon as possible to ensure successful derivation of keratinocytes. Note: Figure 1 illustrates the morphology of hairs in different phases of the growth cycle. Anagen hair is in the growth phase, with a visible outer layer of epithelium surrounding the length of the hair, known as the ORS. On the other hand, telogen hair represents hair in the resting phase and does not have a visible ORS, but has a clear ball of cells covering the root of the hair, known as the telogen bulge.
Place the hair samples into a petri dish containing 10 ml of Antibiotic Mix to wash for 5 min at RT.
Using sterilized scissors and forceps, cut off excessive hair shaft, leaving a hair fragment with an intact hair follicle and around 0.5 - 1 cm of hair shaft.
Carefully transfer 1 hair fragment into each well of the ECM-coated plate using forceps.
Using a 1 ml pipette, add approximately 3 drops of Knockout Serum Replacement (KSR) medium (~100 - 200 µl) to the hair samples. Note: Keep the hair fragments in close contact to the plate surface to allow for attachment and subsequent keratinocyte outgrowth. Adding excessive media at this step may causes the hair fragments to float and decreases the chance of successful hair attachment.
Incubate the hair samples in a 37 °C incubator with 5% CO2 and allow the hair follicles to attach O/N. On the following day, gently add another 3 drops of KSR medium (~100 - 200 µl) to keep the hair samples moist.
- Observe the hair samples daily to confirm that the hair follicles have attached successfully. Generally attachment of hair follicles is apparent within 1-3 days. Thick hairs are usually easier to attach compared to fine hairs, as fine hairs have higher tendency to float which hinder the attachment process.
- Once the hair have attached to the coated plate, add 500 µl of KSR medium to the well. Take extra care when adding the medium as hair samples may not have attached firmly. From this point, change media every 2 days. Note: Keratinocyte outgrowths should be visible from the ORS region of the hair fragment after 3-7 days (Figure 2A-B). Allow the keratinocytes to grow up to 14 days before passaging the cells as described in the next section.
2. Maintenance and Passaging of Keratinocytes
Pre-coat a 6-well plate using the Coating Matrix Kit. Add 680 µl of the dilution medium from the kit to each well, followed by 6.8 µl of Coating Matrix solution. Shake the plate to ensure uniform distribution of the Coating Matrix solution on the plate.
Allow the coated plate to incubate for 30 min at RT. Remove the Coating Matrix solution prior to plating of keratinocytes.
Dissociate keratinocytes into a single cell suspension with 500 µl of 0.25% trypsin per well for 2-5 min at 37 °C. Inactivate the trypsin with 500 µl of medium containing serum (e.g., Fetal Bovine Serum (FBS) medium) and collect into a sterile 15 ml tube. Remove the hair fragment using sterilized forceps.
Determine the number of harvested cells using a haemocytometer.
Centrifuge the cells for 3 min at 200 x g. Wash the cells with PBS and repeat centrifugation. Aspirate the PBS and resuspend the cell pellet in fresh Keratinocyte medium.
Plate down 4 × 104 keratinocytes into one well of ECM-coated plate in the presence of Keratinocyte medium. Keep the keratinocytes in a 37 °C incubator with 5% CO2 (Figure 2C). Change media every day and passage cells when the culture is ~80% confluent, which may take approximately 2-7 days depending on the passage number and proliferation rate of the keratinocytes. The derived keratinocytes should be expanded to generate sufficient frozen stocks using standard slow-freezing cryopreservation methods16. Note: The described culture conditions support the expansion of homogenous population of keratinocytes with typical cobblestone morphology (Figure 2C). Occasionally, a few differentiated keratinocytes, with large and flat morphology and have lost replicative potential, may be observed in culture. Characterization of keratinocytes can be performed by immunostaining using antibodies against cytokeratin 14 as previously described 17. Established keratinocyte cell lines should also be tested for mycoplasma after 1-3 passages using commercially available kit.
3. Generation of hiPSCs from Keratinocytes Using Episomal Vectors
Pre-coat a 6-well plate with 0.1% gelatin (2 ml/well). Allow gelatin to coat for at least 20 min at RT.
For reprogramming experiment, use keratinocytes no later than passage 6. Dissociate keratinocytes into a single cell suspension with 1 ml of 0.25% trypsin per well for 2-5 min at 37 °C. Inactivate trypsin with 1 ml of media containing serum (e.g., FBS media) and collect in a 15 ml tube.
Centrifuge cells for 3 min at 200 x g and resuspend cells in Keratinocyte medium. Plate 1 × 105-1.5 × 105 keratinocytes per well in a gelatinized plate O/N.
On the following day (Day 0), check the plate to ensure cells are 50-60% confluent. Note: If keratinocytes are less than ~50% confluent they might not grow well after transfection.
- On Day 0, perform transfection of the episomal reprogramming vectors on the keratinocytes, using a ratio of 8 µl transfection reagent to 2 µg total plasmid DNA.
- Take 0.5 µg for each of the 4 plasmids (pCXLE-eGFP, pCXLE-hOct3/4-shP53F, pCXLE-hSK, pCXLE-hUL) and dilute in 100 µl of Reduced Serum medium (e.g., OPTI-MEM). Note: The plasmid pCXLE-eGFP is used to monitor the transfection efficiencies in keratinocytes and is not required for successful reprogramming.
- Add 8 µl of transfection reagent to the diluted DNA. Mix by flicking the tube. Allow the DNA reaction mix to rest undisturbed at RT for 15 min.
- Replace the Keratinocyte medium with fresh medium.
- Gently add the DNA reaction mix to keratinocyte culture and incubate in a 37 °C incubator with 5% CO2 for 4 hr. Note: It is not recommended to perform prolonged transfection O/N in keratinocytes as they exhibit low survival post-transfection.
- After 4 hr post-transfection, replace the Keratinocyte medium with fresh medium.
On Day 1 (1 day post-transfection), change the keratinocyte medium and check transfection efficiency by estimating the percentage of eGFP positive cells under a fluorescent microscope. Typically > 60-70% transfection efficiency is observed for primary keratinocytes using this protocol.
- On Day 2, repeat transfection as described in step 3.5. Two consecutive transfections would typically achieve > 90% transfection efficiency. Keratinocyte culture usually reaches confluence by the end of the second transfection.
- Prepare a 10 cm dish of mouse embryonic fibroblast (MEF) feeder for each reprogramming reaction. Prepare mitotically inactivated MEF feeders as previously described 18.
- Pre-coat a 10 cm dish with 5 ml of 0.1% gelatin for at least 20 min. Plate 4 × 106 mitotically inactivated MEF per 10 cm dish in FBS medium. Allow the cells to settle and attach O/N in a 37 °C incubator with 5% CO2.
On Day 3, harvest the keratinocytes with 0.25% trypsin as described in step 3.2 and 3.3. Resuspend the keratinocytes in KSR medium and plate 90% of keratinocytes from 1 well of a 6-well plate into a 10 cm MEF feeder dish with KSR medium.
- From Day 4 onwards, change the KSR medium every day.
- Alternatively after Day 18, replace culture medium every second day. Note: Non-reprogrammed keratinocytes will cease to proliferate, while hiPSC colonies with defined boundary that resemble human embryonic stem cells (hESCs) will emerge from Day 14 - 21.
- Isolate hiPSC colonies by manual dissection from Day 21 onwards. Avoid hiPSC colonies that are closely clustered together and only pick hiPSC colonies that are clearly separated from others. Note: hiPSC colonies can be picked earlier, but the colony with defined boundaries might not be obvious at earlier time points given the small colony size. Reprogramming efficiencies may vary between different patient’s keratinocytes, and is also influenced by other factors such as passage number or proliferation rate of keratinocytes.
- For manual dissection of hiPSC colonies, use a 26G needle to cut hiPSC colonies under a dissecting microscope. Cut a hiPSC colony into small pieces around 300 - 600 µm in length. Transfer the pieces from one hiPSC colony into a new MEF feeder plate (2.5 × 104 MEF/cm2) to establish a clonal line of hiPSCs. Initially, culture each hiPSC clonal line in one well of a 12 well plate or organ culture dishes, then expand to 6 well plate format.
- Maintain established clonal lines of hiPSCs in KSR medium on MEF feeders. Once the culture reach 70 - 80% confluency with hiPSC colonies ~1.5 mm in diameter, passage hiPSCs using standard enzymatic passaging methods (Accutase, Dispase or Collagenase IV) according to the manufacturer’s instruction.
Characterize established hiPSC lines to confirm pluripotency and their ability to differentiate into cells representative of the three germ layers in vitro and/or in vivo13,19. Also, routinely test established hiPSC lines for mycoplasma using commercially available kits to ensure they are pathogen-free. Routinely monitor genomic stability of hiPSC lines by karyotyping or array-based copy number variation (CNV) analysis.
Representative Results
The hair goes through 3 different phases of growth cycle: anagen (the growth phase), catagen (the regression phase) and telogen (the rest phase) 20,21. The anagen hair follicle contains multiple layers of epithelium; these layers include the ORS, IRS and the hair shaft (Figure 1). Anagen hair eventually undergoes transition to the catagen phase, which is marked by apoptosis of the ORS and termination of hair shaft differentiation. Finally, catagen hair transition to the telogen phase, where apoptosis ceases and the telogen follicle becomes quiescent with a characteristic telogen bulge 20,21.
Figure 1 illustrates the morphology of a telogen hair and an anagen hair. We typically utilize anagen hair for keratinocyte derivation. Following this protocol, keratinocyte outgrowths can be observed as early as 3 days after hair attachment (Figure 2A) and will continue to proliferate (Figure 2B). In our experience, some anagen hairs may fail to attach or fail to observe keratinocyte outgrowth; thus collect at least 5 - 10 anagen hairs from each individual to ensure successful keratinocyte isolation. Subsequently, the keratinocytes can be passaged onto a new plate and maintained for multiple passages (Figure 2C). It is important to note that there is better growth of keratinocytes on a Coating Matrix with Keratinocyte medium as described in section 2, compared to Matrigel with KSR medium.
Following expansion, the keratinocytes can be reprogrammed to generate hiPSCs as described in section 3. Figure 3A shows a typical keratinocyte-derived hiPSC colony after 32 days of reprogramming. It is common to observe some differentiation at the center of the hiPSC colony. Once manually picked, the derived hiPSCs typically display high nucleus to cytoplasmic ratio and a defined colony boundary (Figure 3B). Established cell lines of hiPSCs can then be characterized for pluripotency as described previously 13,19, such as the expression of pluripotent markers OCT4, NANOG and TRA-160 (Figure 3C-E).
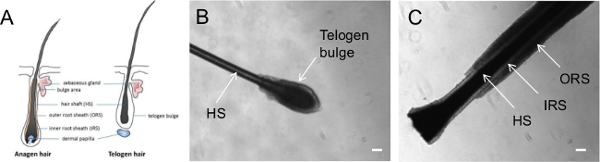 Figure 1. Representative images of plucked hairs at different growth phases. (A) Diagram illustrating hairs in the anagen or telogen phase. Phase contrast images showing a plucked hair in (B) telogen phase and (C) anagen phase. HS = hair shaft; IRS = Inner Root Sheath; ORS = Outer Root Sheath. Scale bar = 100 µm. Please click here to view a larger version of this figure.
Figure 1. Representative images of plucked hairs at different growth phases. (A) Diagram illustrating hairs in the anagen or telogen phase. Phase contrast images showing a plucked hair in (B) telogen phase and (C) anagen phase. HS = hair shaft; IRS = Inner Root Sheath; ORS = Outer Root Sheath. Scale bar = 100 µm. Please click here to view a larger version of this figure.
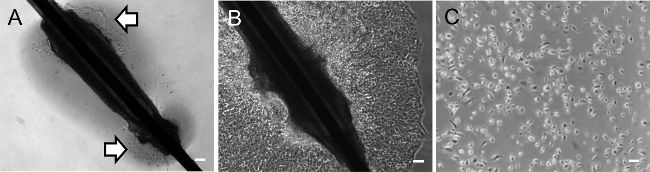 Figure 2. Representative images of hair-derived keratinocytes. Phase contrast images of a plucked hair plated down for (A) 3 days and (B) 10 days. White arrows mark the outgrowth of keratinocyte from a plucked hair. (C) Phase contrast image of Day 3 keratinocyte culture with a typical cobblestone morphology after passaging. Scale bar = 100 µm. Please click here to view a larger version of this figure.
Figure 2. Representative images of hair-derived keratinocytes. Phase contrast images of a plucked hair plated down for (A) 3 days and (B) 10 days. White arrows mark the outgrowth of keratinocyte from a plucked hair. (C) Phase contrast image of Day 3 keratinocyte culture with a typical cobblestone morphology after passaging. Scale bar = 100 µm. Please click here to view a larger version of this figure.
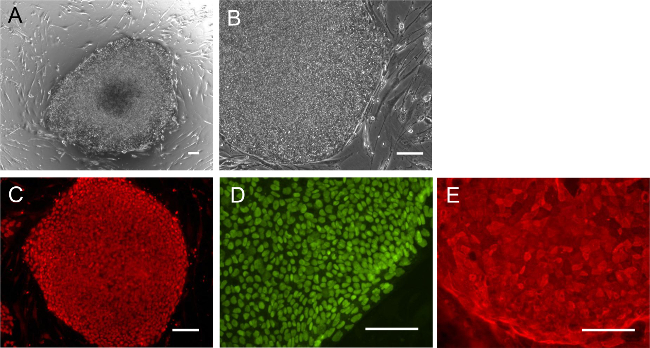 Figure 3. Representative images of keratinocyte-derived hiPSCs. (A) Phase contrast image of a Day 32 reprogramming culture showing a keratinocyte-derived hiPSC colony. (B) Manually selected keratinocyte-derived hiPSCs with morphology similar to hESCs. Immunostaining of hiPSCs with pluripotent markers (C) NANOG and (D) OCT4 and (E) TRA-160 in keratinocyte-derived hiPSCs. Scale bar = 100 µm. Please click here to view a larger version of this figure.
Figure 3. Representative images of keratinocyte-derived hiPSCs. (A) Phase contrast image of a Day 32 reprogramming culture showing a keratinocyte-derived hiPSC colony. (B) Manually selected keratinocyte-derived hiPSCs with morphology similar to hESCs. Immunostaining of hiPSCs with pluripotent markers (C) NANOG and (D) OCT4 and (E) TRA-160 in keratinocyte-derived hiPSCs. Scale bar = 100 µm. Please click here to view a larger version of this figure.
Discussion
Generation of patient-specific hiPSCs offers a unique approach for studying pathogenesis in the diseased cell types in vitro, and also provides a platform for drug screening to identify novel molecules that can rescue the disease phenotypes. This disease modeling approach using hiPSCs has yielded promising results for a variety of diseases, including Long QT syndrome, Huntington’s disease, Parkinson’s disease and amyotrophic lateral sclerosis 22. Several initiatives are already underway to establish large-scale libraries of patient-specific hiPSCs, including consortiums in USA, Europe, Australia, China, South Korea and Japan 23,24.
Here a protocol to establish hiPSCs from hair-derived keratinocytes is described, which has the potential to facilitate and fast-track the establishment of large-scale libraries of disease-specific hiPSCs. This protocol offers two advantages: Firstly, the episomal system utilized in this protocol generates integration-free hiPSCs using a cocktail of six reprogramming factors (OCT4, SOX2, KLF4, L-MYC, LIN28, shRNA for p53) at relatively high efficiency 4. Compared to retroviral or lentiviral-mediated reprogramming methods, the use of episomal vector avoids undesirable genomic integration of foreign transgenes during reprogramming., Therefore, many initiatives favor the use of integration-free reprogramming methods for establishment of large-scale hiPSC libraries, such as episomal vectors, Sendai virus or mRNA, over integrative reprogramming methods 23.
Secondly, hair is easily accessible and can be harvested by the patients themselves without the use of invasive procedures or the attendance of medical staffs. This provides a unique opportunity for patients from different regions to collect and mail in their own hair samples to the laboratory for keratinocyte derivation and subsequent reprogramming. In support of the feasibility of this strategy, our unpublished data indicate that keratinocytes can be successfully isolated from plucked hair that was stored at 4 °C for up to 10 days.
The methodology described here will allow the derivation of keratinocyte from plucked hair. While keratinocytes can be derived from just a single hair, our recommendation is to collect at least 5 anagen hairs for keratinocyte derivation. One limitation of this protocol is that some hairs may not attach well and the keratinocytes may fail to grow. Thicker hair tends to attach better, while fine hair requires reduced amount of culture medium during plating to avoid floating and enhance attachment. Once keratinocytes are derived, it is advisable to expand and freeze down stocks using standard slow-freezing cryopreservation methods 16. Subsequent reprogramming of keratinocytes can be performed following steps described in this protocol. It is important to note that the proliferation rate of the starting cells may affect the reprogramming efficiency, with decreased reprogramming efficiency observed in late passages as cells reach cellular senescence 25-27. In this regard, use keratinocytes no later than passage 6 for reprogramming experiments. In addition, reprogramming efficiencies may vary between different individual’s keratinocytes.
Recent studies suggest widespread genetic mosaicism in multiple types of somatic tissues 28, with ~30% of skin fibroblasts reported to have somatic CNVs in the genome 29. These CNVs may be caused by errors in DNA replication, DNA repair or transposon mobilization. It is also possible that prolonged UV exposure to the skin can cause additional CNVs in epidermal cells including fibroblasts and keratinocytes, but the extent of this is not well understood. As CNVs in keratinocytes will be carried over during the reprogramming process, it is important to perform routine karyotyping or CNV analysis to ensure that the established hiPSCs maintain genomic stability. In summary, here we described procedures for generation of hiPSCs from hair-derived keratinocytes, which could be used for disease modeling and regenerative medicine.
Disclosures
The authors have no conflict of interest.
Acknowledgments
The authors wish to thank Harene Ranjithakumaran and Stacey Jackson for technical support. This work was supported in part by grants from the National Health and Medical Research Council (R.C.B. Wong, A. Pébay), the University of Melbourne (R.C.B. Wong), Retina Australia (R.C.B. Wong, S.S.C. Hung, A. Pébay) and the Ophthalmic Research Institute of Australia (R.C.B. Wong, S.S.C. Hung, A. Pébay); Australian Research Council Future Fellowship (A. Pébay, FT140100047), Cranbourne Foundation Fellowship (R.C.B. Wong); intramural funding from the National Institutes for Health (R.C.B. Wong, S.S.C. Hung) and operational infrastructure support from the Victorian Government.
References
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Park IH, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:U141–U141. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Okita K, et al. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells. 2013;31:458–466. doi: 10.1002/stem.1293. [DOI] [PubMed] [Google Scholar]
- Dowey SN, Huang X, Chou BK, Ye Z, Cheng L. Generation of integration-free human induced pluripotent stem cells from postnatal blood mononuclear cells by plasmid vector expression. Nat Protoc. 2012;7:2013–2021. doi: 10.1038/nprot.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, et al. Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol. 2011;22:1221–1228. doi: 10.1681/ASN.2011010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aasen T, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- Peters A, Zambidis E. Chapter 16. Generation of nonviral integration-free induced pluripotent stem cells from plucked human hair follicles. In: Ye K, Jin S, editors. Human Embryonic and Induced Pluripotent Stem Cells: Lineage-Specific Differentiation Protocols.Springer Protocols Handbooks. Springer; 2012. pp. 203–227. [Google Scholar]
- Fuchs E. Scratching the surface of skin development. Nature. 2007;445:834–842. doi: 10.1038/nature05659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limat A, Noser FK. Serial cultivation of single keratinocytes from the outer root sheath of human scalp hair follicles. J Invest Dermatol. 1986;87:485–488. doi: 10.1111/1523-1747.ep12455548. [DOI] [PubMed] [Google Scholar]
- Piao Y, Hung SS, Lim SY, Wong RC, Ko MS. Efficient generation of integration-free human induced pluripotent stem cells from keratinocytes by simple transfection of episomal vectors. Stem Cells Transl Med. 2014;3:787–791. doi: 10.5966/sctm.2013-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CA, et al. Reprogramming of human hair follicle dermal papilla cells into induced pluripotent stem cells. J Invest Dermatol. 2012;132:1725–1727. doi: 10.1038/jid.2012.12. [DOI] [PubMed] [Google Scholar]
- Muchkaeva IA, et al. Generation of iPS Cells from Human Hair Follice Dermal Papilla Cells. Acta naturae. 2014;6:45–53. [PMC free article] [PubMed] [Google Scholar]
- Naaldijk Y, Friedrich-Stockigt A, Sethe S, Stolzing A. Comparison of different cooling rates for fibroblast and keratinocyte cryopreservation. J Tissue Eng Regen. 2013. [DOI] [PubMed]
- Sporl F, et al. Real-time monitoring of membrane cholesterol reveals new insights into epidermal differentiation. J Invest Dermatol. 2010;130:1268–1278. doi: 10.1038/jid.2009.412. [DOI] [PubMed] [Google Scholar]
- Conner DA, et al. Ausubel FN, et al., editors. Mouse embryo fibroblast (MEF) feeder cell preparation. Current protocols in molecular biology. 2001. p. Unit 23 22. [DOI] [PubMed]
- Pebay A, et al. Essential roles of sphingosine-1-phosphate and platelet-derived growth factor in the maintenance of human embryonic stem cells. Stem Cells. 2005;23:1541–1548. doi: 10.1634/stemcells.2004-0338. [DOI] [PubMed] [Google Scholar]
- Myung P, Ito M. Dissecting the bulge in hair regeneration. J Clin Invest. 2012;122:448–454. doi: 10.1172/JCI57414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso L, Fuchs E. The hair cycle. J Cell Sci. 2006;119:391–393. doi: 10.1242/jcs.02793. [DOI] [PubMed] [Google Scholar]
- Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares FA, Sheldon M, Rao M, Mummery C, Vallier L. International coordination of large-scale human induced pluripotent stem cell initiatives: Wellcome Trust and ISSCR workshops white paper. Stem cell reports. 2014;3:931–939. doi: 10.1016/j.stemcr.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan R, Watt FM. What is the point of large-scale collections of human induced pluripotent stem cells. Nat Biotechnol. 2013;31:875–877. doi: 10.1038/nbt.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utikal J, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:U1145–U1112. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, et al. Proliferation rate of somatic cells affects reprogramming efficiency. J Biol Chem. 2013;288:9767–9778. doi: 10.1074/jbc.M112.403881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, et al. Late passage human fibroblasts induced to pluripotency are capable of directed neuronal differentiation. Cell Transplant. 2011;20:193–203. doi: 10.3727/096368910X514305. [DOI] [PubMed] [Google Scholar]
- Huallachain M, Karczewski KJ, Weissman SM, Urban AE, Snyder MP. Extensive genetic variation in somatic human tissues. Proc Natl Acad Sci U S A. 2012;109:18018–18023. doi: 10.1073/pnas.1213736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abyzov A, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492:438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]